

Abstract
We previously reported that cells harboring the hepatitis C virus (HCV) RNA replicon as well as those expressing HCV NS3/4A exhibited increased sensitivity to suboptimal doses of apoptotic stimuli to undergo mitochondrion-mediated apoptosis (Y. Nomura-Takigawa, et al., J. Gen. Virol. 87:1935-1945, 2006). Little is known, however, about whether or not HCV infection induces apoptosis of the virus-infected cells. In this study, by using the chimeric J6/JFH1 strain of HCV genotype 2a, we demonstrated that HCV infection induced cell death in Huh7.5 cells. The cell death was associated with activation of caspase 3, nuclear translocation of activated caspase 3, and cleavage of DNA repair enzyme poly(ADP-ribose) polymerase, which is known to be an important substrate for activated caspase 3. These results suggest that HCV-induced cell death is, in fact, apoptosis. Moreover, HCV infection activated Bax, a proapoptotic member of the Bcl-2 family, as revealed by its conformational change and its increased accumulation on mitochondrial membranes. Concomitantly, HCV infection induced disruption of mitochondrial transmembrane potential, followed by mitochondrial swelling and release of cytochrome c from mitochondria. HCV infection also caused oxidative stress via increased production of mitochondrial superoxide. On the other hand, HCV infection did not mediate increased expression of glucose-regulated protein 78 (GRP78) or GRP94, which are known as endoplasmic reticulum (ER) stress-induced proteins; this result suggests that ER stress is not primarily involved in HCV-induced apoptosis in our experimental system. Taken together, our present results suggest that HCV infection induces apoptosis of the host cell through a Bax-triggered, mitochondrion-mediated, caspase 3-dependent pathway(s).
Hepatitis C virus (HCV) often establishes persistent infection to cause chronic hepatitis, liver cirrhosis, and hepatocellular carcinoma, which is a significant health problem around the world (56). Although the exact mechanisms of HCV pathogenesis, such as viral persistence, liver cell injury, and carcinogenesis, are not fully understood yet, an accumulating body of evidence suggests that apoptosis of hepatocytes is significantly involved in the pathogenesis of HCV (1, 2, 9). It is widely accepted that apoptosis of virus-infected cells is an important strategy of the host to protect itself against viral infections. Apoptotic cell death can be mediated either by the host immune responses through the function of virus-specific cytotoxic T lymphocytes and/or by viral proteins themselves that trigger an apoptotic pathway(s) of the host cell.
Apoptotic pathways can be classified into two groups: the mitochondrial death (intrinsic) pathway and the extrinsic cell death pathway initiated by the tumor necrosis factor (TNF) family members (31, 63). Mitochondrion-mediated apoptosis is initiated by a variety of apoptosis-inducing signals that cause the imbalance of the major apoptosis regulator, the proteins of the Bcl-2 family, such as Bcl-2, Bax, and Bid. For example, the proapoptotic protein Bax accumulates on mitochondria after being activated and triggers an increase in the permeability of the outer mitochondrial membrane. Consequently, the mitochondria release cytochrome c and other key molecules that facilitate apoptosome formation to activate caspase 9. This, in turn, activates downstream death programs, such as caspase 3 and poly(ADP-ribose) polymerase (PARP). The mitochondria also release apoptosis-inducing factor and endonuclease G to facilitate caspase-independent apoptosis. On the other hand, the extrinsic cell death pathway involves the activation of caspase 8 through binding to the adaptor protein Fas-associated protein with death domain (FADD), which in turn activates caspase 3 to facilitate cell death.
There have been many studies regarding the HCV protein(s) that is directly involved in apoptosis, identifying the protein as either proapoptotic or antiapoptotic, and some data are inconsistent. For example, core (5, 13, 36, 73), E1 (15, 16), E2 (12), NS3 (48), NS4A (43), and NS5A and NS5B (57) have been reported to induce apoptosis. On the other hand, there are reports showing that core (40, 49, 51), E2 (35), NS2 (21), NS3 (58), and NS5A (33, 67) function as antiapoptotic proteins. However, whether the virus as a whole is proapoptotic or antiapoptotic needs to be studied in the context of virus replication, which is believed to be much more dynamic than mere expression of a viral protein(s).
We previously reported that replication of an HCV RNA replicon rendered the host cell prone to undergoing mitochondrion-mediated apoptosis upon suboptimal doses of apoptosis-inducing stimuli (43). Recently, an efficient virus infection system using a particular clone of HCV genotype 2a and a highly permissive human hepatocellular carcinoma-derived cell line has been developed (37, 38, 66, 71). In this study, by using the virus infection system, we examined the possible effect of HCV infection on the fate of the host cell. We report here that HCV infection induces apoptosis via the mitochondrion-mediated pathway, as demonstrated by the increased accumulation of the proapoptotic protein Bax on the mitochondria, decreased mitochondrial transmembrane potential, and mitochondrial swelling, which result in the release of cytochrome c from the mitochondria and the activation of caspase 3.
MATERIALS AND METHODS
Cells.
The Huh7.5 cell line (6), a highly HCV-susceptible subclone of Huh7 cells, was a kind gift from C. M. Rice, Center for the Study of Hepatitis C, The Rockefeller University. The cells were propagated in Dulbecco's modified Eagle medium supplemented with 10% heat-inactivated fetal bovine serum and 0.1 mM nonessential amino acids.
Virus.
The virus stock used in this study was prepared as described below. The pFL-J6/JFH1 plasmid, encoding the entire viral genome of a chimeric strain of HCV genotype 2a, J6/JFH1 (37), was kindly provided by C. M. Rice. The plasmid was linearized by XbaI digestion and in vitro transcribed by using T7 RiboMAX (Promega, Madison, WI) to generate the full-length viral genomic RNA. The in vitro-transcribed RNA (10 μg) was transfected into Huh7.5 cells by means of electroporation (975 μF, 270 V) using Gene Pulser (Bio-Rad, Hercules, CA). The cells were then cultured in complete medium, and the supernatant was propagated as an original virus (J6/JFH1-passage 1 [J6/JFH1-P1]). Since the infectious titer of the original virus was not high enough for infection of all the cells in the culture at once, an adapted strain of the virus was obtained by passaging the virus-infected cells 47 times. The adapted virus (J6/JFH1-P47), which is a pool of adapted mutants, possesses 10 amino acid mutations (K78E, T396A, T416A, N534H, A712V, Y852H, W879R, F2281L, M2876L, and T2925A) and a single nucleotide mutation in the 5′-untranslated region (U146A) and produces a much higher titer of infectivity in Huh7.5 cell cultures than the original J6/JFH1-P1 (our unpublished data). Virus infection was performed at a multiplicity of infection of 2.0. Culture supernatants of uninfected cells served as a control (mock preparation).
Virus infectivity was measured by indirect immunofluorescence analysis, as described below, and expressed as cell-infecting units/ml.
Cell viability/proliferation assay.
Huh7.5 cells were seeded in 96-well plates at a density of 1.0 × 104 cells/well and cultured overnight. The cells were then infected with the virus or the mock preparation, and, at different time points, cell viability/proliferation was determined by the WST-1 assay (Roche, Mannheim, Germany), as described previously (43).
Detection of apoptosis.
The degree of apoptosis was measured by using a Cell Death Detection ELISAPlus kit (Roche), which is based on the determination of cytoplasmic histone-associated DNA fragments, according to the manufacturer's protocol. In brief, cells cultured in a 96-well plate were centrifuged at 200 × g for 10 min at 4°C to remove the supernatant. After the cells were lysed with lysis buffer, the plate was centrifuged at 200 × g for 10 min to separate the cytoplasmic and nuclear fractions. Twenty microliters of supernatant was placed in each well of a streptavidin-coated 96-well plate. Subsequently, a mixture of biotin-labeled anti-histone antibody and peroxidase-labeled anti-DNA antibody was added and wells were incubated for 2 h at room temperature. After wells were washed three times to remove the unbound components, peroxidase activities were determined photometrically with 2,2′-azino-diethyl-benzthiazolin sulfonate as a substrate and measured by using a microplate reader (Bio-Rad).
Caspase enzymatic activities.
Activities of caspase 3, 8, and 9 were measured by using Caspase-Glo 3/7, 8, and 9 assays (Promega), respectively, according to the manufacturer's instructions. In brief, a proluminescence caspase 3/7, 8, or 9 substrate, which consists of aminoluciferin (substrate for luciferase) and the tetrapeptide sequence DEVD, LETD, or LEHD (cleavage site for caspase 3/7, 8, or 9, respectively), was added to cultured cells in each well of a 96-well plate, and the plate was incubated for 30 min at room temperature. In the presence of caspase 3/7, 8, or 9, aminoluciferin was liberated from the proluminescence substance and utilized as a substrate for the luciferase reaction. The resultant luminescence in relative light units was measured by using a Luminescencer-JNR AB-2100 (Atto, Tokyo, Japan).
Cell fractionation.
Cells were fractionated by using a mitochondrial isolation kit (Pierce, Rockford, IL), according to the manufacturer's instructions. Briefly, 2 × 107 cells were harvested and suspended in reagent A containing a protease inhibitor cocktail (Roche). The cell suspension was mixed with buffer B, vortexed for 5 min, and then mixed with reagent C. The nuclei and unbroken cells were removed by centrifugation at 700 × g for 10 min at 4°C, and the supernatant was used as cell lysate. The cell lysate was further centrifuged at 3,000 × g for 15 min at 4°C. The pellet obtained, which was considered the mitochondrial fraction, was washed once with reagent C and dissolved in a lysis buffer containing 10 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1% NP-40, and a protease inhibitor cocktail. The remaining supernatant was further centrifuged at 100,000 × g for 30 min at 4°C, and the resultant supernatant was collected as a cytosolic fraction.
To verify successful mitochondrial fractionation, the cytosolic and mitochondrial fractions were analyzed by immunoblotting, as described below, using antibody against Tim23, a mitochondrion-specific protein.
Analysis of the mitochondrial transmembrane potential.
The mitochondrial transmembrane potential was measured by flow cytometry using the cationic lipophilic green fluorochrome rhodamine 123 (Rho123; Sigma, St. Louis, MO), as described previously (43). Briefly, cells (7 × 105) were harvested, washed twice with phosphate-buffered saline (PBS), and incubated with Rho123 (0.5 μg/ml) at 37°C for 25 min. The cells were then washed twice with PBS, and Rho123 intensity was analyzed by a flow cytometer (Becton Dickinson, San Jose, CA). A total of 10,000 events were collected per sample. Mean fluorescence intensities were measured by calculating the geometric mean for each histogram peak.
Detection of morphological changes of the mitochondria.
Mitochondrial morphology was analyzed by two different methods. (i) For fluorescence microscopy, Huh7.5 cells seeded on glass coverslips in a 24-well plate were incubated for 30 min at 37°C with 100 nM MitoTracker (Molecular Probes, Eugene, OR). After being washed twice with PBS, the cells were fixed with 3.7% paraformaldehyde and observed under a confocal laser scanning microscope (Carl Zeiss, Oberkochen, Germany). When needed, the fixed cells were subjected to indirect immunofluorescence to confirm HCV infection, as described below. (ii) Electron microscopy was performed as described previously (23, 43). In brief, cells were fixed with 4% paraformaldehyde and 0.2% glutaraldehyde for 30 min at room temperature. After being washed with PBS, the cells were collected, dehydrated in a series of 70%, 80%, and 90% ethanol, embedded in LR White resin (London Resin, Berkshire, United Kingdom), and kept at −20°C for 2 days to facilitate resin polymerization. After ultrathin sectioning, samples were etched in 3% H2O2 for 5 min at room temperature and washed with PBS. Sections were stained with uranyl acetate and lead citrate and observed under a transmission electron microscope (JEM 1299EX; JOEL, Tokyo, Japan).
Detection of mitochondrial superoxide.
Cells seeded on glass coverslips in a 24-well plate were incubated with 5 μM MitoSOX Red (Molecular Probes) at 37°C for 10 min. After being washed with warm Hanks' balanced salt solution with calcium and magnesium (Invitrogen, Carlsbad, CA), the cells were fixed with 3.7% paraformaldehyde and observed under a confocal laser scanning microscope (Carl Zeiss). When needed, the fixed cells were subjected to indirect immunofluorescence to confirm HCV infection, as described below.
Indirect immunofluorescence.
Cells seeded on glass coverslips in a 24-well plate at a density of 6 × 104 cells/well were infected with HCV or left uninfected. At different time points after virus infection, the cells were fixed with 3.7% paraformaldehyde in PBS for 15 min at room temperature and permeabilized in 0.1% Triton X-100 in PBS for 15 min at room temperature. After being washed with PBS twice, cells were consecutively stained with primary and secondary antibodies. Primary antibodies used were anti-active caspase 3 rabbit polyclonal antibody (Promega) and an HCV-infected patient's serum. Secondary antibodies used were Cy3-conjugated donkey anti-rabbit immunoglobulin G (IgG; Chemicon, Temecula, CA), Alexa Fluor 594-conjugated goat anti-human IgG (Molecular Probes), and fluorescein isothiocyanate (FITC)-conjugated goat anti-human IgG (MBL, Nagoya, Japan). The cells were washed with PBS, counterstained with Hoechst 33342 solution (Molecular Probes) at room temperature for 10 min, mounted on glass slides, and observed under a confocal laser scanning microscope (Carl Zeiss). The specificity of this immunostaining was confirmed by using mouse monoclonal antibody against HCV core protein (C7-50; Abcam, Tokyo, Japan).
To analyze the possible localization of the activated Bax on mitochondrial membranes, cells were incubated with MitoTracker and subjected to immunofluorescence analysis using rabbit polyclonal antibody against activated Bax (NT antibody; Upstate, Lake Placid, NY). This antibody is directed toward N-terminal residues 1 to 21 of Bax in an N-terminal conformation-dependent manner and specifically recognizes the active form of Bax, in which this segment is exposed in response to apoptotic stimuli (64).
Immunoblotting.
Cells were lysed in a buffer containing 10 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1% NP-40, and a protease inhibitor cocktail (Roche). After two freeze-thaw cycles, cell debris was removed by centrifugation. Protein quantification was carried out using a bicinchoninic acid protein assay kit (Pierce). Equal amounts of soluble proteins (4 to 20 μg) were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto a polyvinylidene difluoride membrane (Millipore, Bedford, MA), which was then incubated with the respective primary antibody. The primary antibodies used were mouse monoclonal antibodies against cytochrome c (A-8; Santa Cruz Biotechnology, Santa Cruz, CA), HCV NS3 (Chemicon), Tim23, Bax and Bcl-2 (BD Biosciences Pharmingen, San Diego, CA); rabbit polyclonal antibodies against Bak (Upstate), caspase 3, and PARP (Cell Signaling Technology, Danvers, MA); and goat polyclonal antibodies against glucose-regulated protein 78 (GRP78) and GRP94 (Santa Cruz Biotechnology). Horseradish peroxidase-conjugated goat anti-mouse IgG (MBL), goat anti-rabbit IgG (Bio-Rad), and donkey anti-goat IgG (Santa Cruz Biotechnology) were used as secondary antibodies. In some experiments, a commercial kit that facilitates the antigen-antibody reaction (Can Get Signal; Toyobo, Osaka, Japan) was used to obtain stronger signals. The respective protein bands were visualized by means of an enhanced chemiluminescence (GE Healthcare, Buckinghamshire, United Kingdom), and the intensity of each band was quantified by using NIH Image J. Protein loading was normalized by probing with goat antibody against actin (Santa Cruz Biotechnology) as a primary antibody.
Statistical analysis.
The two-tailed Student t test was applied to evaluate the statistical significance of differences measured from the data sets. A P value of <0.05 was considered statistically significant.
RESULTS
HCV infection induces caspase 3-dependent apoptosis in Huh7.5 cells.
We first examined virus growth in Huh7.5 cells. HCV grew efficiently in the culture, and virus titers in the supernatant reached a plateau level at 2 days postinfection (Fig. 1A). Immunofluorescence analysis revealed that >95% of the cells were infected with HCV on the same day (Fig. 1B). To examine the possible impact of HCV infection on the cells, we measured the cell viability/proliferation at 0, 2, 4, and 6 days postinfection. As shown in Fig. 1C, the proliferation of HCV-infected cells was significantly slower than that of the mock-infected control. Similar results were obtained when the parental Huh7 cells were used for HCV infection (data not shown). The observed delay in cell proliferation was associated with an increase in cell death, seen as cell rounding and floating in the culture (data not shown) and in cellular DNA fragmentation (Fig. 1D). As DNA fragmentation is a hallmark of apoptosis, our data suggest that HCV infection induces apoptosis in Huh7.5 cells.
FIG. 1.

HCV infection induces apoptosis in Huh7.5 cells. (A) Virus infectivity in the culture supernatants of HCV-infected cells. (B) Detection of HCV antigens in the cells. Huh7.5 cells mock inoculated or inoculated with HCV were subjected to indirect immunofluorescence analysis to detect HCV antigens (red staining) using an HCV-infected patient's serum and Alexa Fluor 594-conjugated goat anti-human IgG at 2, 4, and 6 days postinfection (dpi). Nuclei were counterstained with Hoechst 33342 (blue staining). Scale bar, 50 μm. (C) Cell viability/proliferation was measured for HCV-infected cultures and the mock-inoculated controls. Proliferation of the control cells at day 0 postinfection was arbitrarily expressed as 1.0. Data represent means ± standard deviations (SD) of three independent experiments. *, P < 0.01, compared with the control. (D) DNA fragmentation was measured as an index of apoptotic cell death for HCV-infected cultures and the mock-inoculated controls. DNA fragmentation of the control cells at 4 days postinfection was arbitrarily expressed as 1.0. Data represent means ± SD of three independent experiments. *, P < 0.01, compared with the control.
The J6/JFH1-P47 strain of HCV used in this study possesses adaptive mutations compared to the original strain (J6/JFH1-P1). Therefore, we compared the impacts of the two strains on cell viability/proliferation and DNA fragmentation. While both strains caused inhibition of cell proliferation and an increase in DNA fragmentation, J6/JFH1-P47 appeared to exert a stronger cytopathic effect than J6/JFH-P1 (data not shown).
To further verify that HCV infection induces apoptotic cell death, we analyzed caspase 3 activities in HCV-infected Huh7.5 cells and the mock-infected control. As shown in Fig. 2A, caspase 3 activities in HCV-infected cells increased to levels that were 2.2, 6.0, and 12 times higher than that in the control cells at 2, 4, and 6 days postinfection, respectively. We also examined HCV-induced caspase 3 activation by immunoblot analysis. Activation of caspase 3 requires proteolytic processing of its inactive proenzyme into the active 17-kDa and 12-kDa subunit proteins. The anti-caspase 3 antibody used in this analysis recognizes 35-kDa procaspase 3 and the 17-kDa subunit protein. At 6 days postinfection, activated caspase 3 was detected in HCV-infected cells but not in the mock-infected control (Fig. 2B, second row from the top). Analysis of the death substrate PARP, which is a key substrate for active caspase 3 (61), also demonstrated that the uncleaved PARP (116 kDa) was proteolytically cleaved to generate the 89-kDa fragment in HCV-infected cells but not in the mock-infected control (Fig. 2B, third row). Cleavage of PARP facilitates cellular disassembly and serves as a marker of cells undergoing apoptosis (44).
FIG. 2.

HCV infection activates caspase 3 in Huh7.5 cells. (A) Caspase 3 activities in cells infected with HCV and mock-infected controls. The caspase 3 activity of the control cells at day 0 postinfection was arbitrarily expressed as 1.0. *, P < 0.05; †, P < 0.01 (compared with the control). Data represent means ± standard deviations (SD) of three independent experiments. (B) Immunoblot analysis to detect the activated form of caspase 3 (∼17 kDa) and cleavage product of PARP (∼85 kDa) in HCV-infected cells and the mock-infected control at 2, 4, and 6 days postinfection (dpi). Huh7.5 cells treated with actinomycin D (ActD; 50 ng/ml) for 30 h served as a positive control. Amounts of actin were measured as an internal control to verify an equal amount of sample loading. (C) Huh7.5 cells infected with HCV or mock infected were subjected to indirect immunofluorescence analysis at 6 dpi. Cells treated with ActD (50 ng/ml) for 30 h served as a positive control. After fixation and permeabilization, the cells were incubated with anti-active caspase 3 rabbit polyclonal antibody followed by Cy3-labeled donkey anti-rabbit IgG (top) and with an HCV-infected patient's serum followed by FITC-labeled goat anti-human IgG (middle). The cells were then stained with Hoechst 33342 for the nuclei (bottom). Scale bar, 20 μm. (D) Quantification of active caspase 3-expressing cells. The percentages of cells expressing active caspase 3 were determined for HCV-infected cultures and mock-infected controls. Data represent means ± SD of three independent experiments. *, P < 0.05, compared with the control. (E) Nuclear translocation of active caspase 3 in HCV-infected cells. Subcellular localization of active caspase 3 in HCV-infected cells was examined by indirect immunofluorescence analysis at 6 days postinfection as described in the legend for panel C. Scale bar, 5 μm.
In order to further confirm these observations, indirect immunofluorescence staining was performed by using an anti-active caspase 3 antibody that specifically recognizes the newly exposed C terminus of the 17-kDa fragment of caspase 3 but not the inactive precursor form. As shown in Fig. 2C, the activated form of caspase 3 was clearly observed in HCV-infected cells but not in the mock-infected control at 6 days postinfection. The activation of caspase 3 was observed also at 4 days postinfection (data not shown). We found that caspase 3 activation was detectable in 12% and 21% of HCV antigen-positive cells at 4 and 6 days postinfection, respectively, whereas it was detectable only minimally in mock-infected cells at the same time points (Fig. 2D). These results strongly suggest that HCV-induced cell death is caused by caspase 3-dependent apoptosis. We also observed nuclear translocation of active caspase 3 in HCV-infected cells (Fig. 2E). This result is consistent with previous reports (28, 70) that activated caspase 3 is located not only in the cytoplasm but also in the nuclei of apoptotic cells. Concomitantly, nuclear condensation and shrinkage were clearly observed in the caspase 3-activated cells. As the activation and nuclear translocation of caspase 3 occur before the appearance of the nuclear change, not all caspase 3-activated cells exhibited the typical nuclear changes. Taken together, these results indicate that HCV-induced apoptosis is associated with activation and nuclear translocation of caspase 3.
HCV infection induces the activation of the proapoptotic protein Bax.
The proteins of the Bcl-2 family are known to directly regulate mitochondrial membrane permeability and induction of apoptosis (63). Therefore, we examined the expression levels of proapoptotic proteins, such as Bax and Bak, and antiapoptotic protein Bcl-2 in HCV-infected Huh7.5 cells and the mock-infected control. The result showed that expression levels of Bak or Bcl-2 did not differ significantly between HCV-infected cells and the control. Interestingly, however, Bax accumulated on the mitochondria in HCV-infected cells to a larger extent than in the mock-infected control (Fig. 3A), with the average amount of mitochondrion-associated Bax in HCV-infected cells being 2.7 times larger than that in the control cells at 6 days postinfection (Fig. 3B).
FIG. 3.

HCV infection induces Bax activation in Huh7.5 cells. (A) Accumulation of Bax on the mitochondria in HCV-infected Huh7.5 cells. Cytosolic and mitochondrial fractions as well as whole-cell lysates were prepared from HCV-infected cells and the mock-infected control at 6 days postinfection and analyzed by immunoblotting using antibodies against Bax, Bak, Bcl-2, NS3, Tim23, and actin. Amounts of Tim23, a mitochondrion-specific protein, were measured to verify equal amounts of mitochondrial fractions. Amounts of actin were measured to verify equal amounts of whole-cell lysates and cytosolic fractions. (B) The intensities of the bands of mitochondrion-associated Bax in HCV-infected cells and the mock-infected control were quantified. The intensity of the mock-infected control was arbitrarily expressed as 1.0. Data represent means ± standard deviations (SD) of three independent experiments. *, P < 0.01, compared with the control. (C) Conformational change of Bax in HCV-infected cells. Huh7.5 cells infected with HCV and the mock-infected control were subjected to indirect immunofluorescence analysis at 6 days postinfection. After incubation with MitoTracker (middle row), the cells were incubated with an antibody specific for the N terminus of Bax (NT antibody), followed by Alexa Fluor 488-labeled goat anti-rabbit IgG (top row). Merged images are shown on the bottom. Scale bar, 10 μm. (D) Quantification of activated Bax-positive cells. The percentages of cells expressing activated Bax were determined for HCV-infected cultures and the mock-infected control. Data represent means ± SD of three independent experiments. *, P < 0.01, compared with the control.
In response to apoptotic stimuli, Bax undergoes a conformational change to expose its N and C termini, which facilitates translocation of the protein to the mitochondrial outer membrane (32). Thus, the conformational change of Bax represents a key step for its activation and subsequent apoptosis. We therefore investigated the possible conformational change of Bax in HCV-infected cells by using a conformation-specific NT antibody that specifically recognizes the Bax protein with an exposed N terminus. As shown in Fig. 3C, Bax staining with the conformation-specific NT antibody was readily detectable in HCV-infected cells at 6 days postinfection whereas there was no detectable staining with the same antibody in the mock-infected control. Moreover, the activated Bax was shown to be colocalized with MitoTracker, a marker for mitochondria, in HCV-infected cells. The conformational change of Bax was observed in 10% and 15% of HCV-infected cells at 4 and 6 days postinfection, respectively (Fig. 3D). This result was consistent with what was observed for caspase 3 activation in HCV-infected cells (Fig. 2D). Taken together, these results suggest that HCV infection triggers conformational change and mitochondrial accumulation of Bax, which lead to the activation of the mitochondrial apoptotic pathway.
HCV infection induces the disruption of the mitochondrial transmembrane potential, release of cytochrome c from mitochondria, and activation of caspase 9.
The accumulation of Bax on the mitochondria is known to decrease the mitochondrial transmembrane potential and increase its permeability, which result in the release of cytochrome c and other key molecules from the mitochondria to the cytoplasm to activate caspase 9. Therefore, we examined the possible effect of HCV infection on mitochondrial transmembrane potential in Huh7.5 cells. Disruption of the mitochondrial transmembrane potential was indicated by decreased Rho123 retention and, hence, decreased fluorescence. As shown in Fig. 4, HCV-infected cells showed ∼50% and ∼70% reductions in Rho123 fluorescence intensity compared with the mock-infected control at 4 and 6 days postinfection, respectively.
FIG. 4.

HCV infection induces disruption of the mitochondrial transmembrane potential in Huh7.5 cells. (A) Huh7.5 cells infected with HCV and the mock-infected control were stained with Rho123 and subjected to flow cytometric analysis to measure the mitochondrial transmembrane potential at 4 and 6 days postinfection (dpi). The red and black lines represent Rho123 staining of HCV-infected cells and the mock-infected control, respectively. The green profiles represent staining of the cells with PBS alone. (B) Mean fluorescence intensities of HCV-infected cells and the mock-infected control at 4 and 6 dpi. Data represent means ± standard deviations (SD) of three independent experiments. *, P < 0.05; †, P < 0.01 (compared with the control).
Recent studies have indicated that loss of mitochondrial membrane potential leads to mitochondrial swelling, which is often associated with cell injury (27, 50). Also, we and other investigators have reported that HCV NS4A (43), core (53), and p7 (22) target mitochondria. We therefore analyzed the effect of HCV infection on mitochondrial morphology. Confocal fluorescence microscopic analysis using MitoTracker revealed that mitochondria began to undergo morphological changes at 4 days postinfection and that approximately 40% of HCV-infected cells exhibited mitochondrial swelling and/or aggregation compared with the mock-infected control at 6 days postinfection (Fig. 5A and B). It should also be noted that mitochondrial swelling and/or aggregation was seen in a region different from the “membranous web,” where the HCV replication complexes accumulate to show stronger expression of HCV antigens. This observation implies the possibility that an indirect effect(s) of HCV infection, in addition to a direct effect of an HCV protein, as observed for NS3/4A (43), is involved in mitochondrial swelling and/or aggregation.
FIG. 5.

HCV infection induces mitochondrial morphology changes in Huh7.5 cells. (A) Fluorescence microscopy analysis. Mitochondrial morphologies of HCV-infected cells and the mock-infected control at 6 days postinfection were examined by confocal microscopy. The cells were directly incubated with MitoTracker (upper row) and then stained for HCV antigens by using an HCV-infected patient's serum, followed by FITC-labeled goat anti-human IgG (bottom row). Scale bar, 5 μm. (B) Quantification of swollen mitochondrion-positive cells. The percentages of cells exhibiting swollen and/or aggregated mitochondria were determined for HCV-infected cultures and the mock-infected control. Data represent means ± standard deviations of three independent experiments. *, P < 0.01, compared with the control. (C) Electron microscopic analysis. Mitochondrial morphologies of HCV-infected cells and the mock-infected control at 6 days postinfection were examined by electron microscopy. Arrows indicate mitochondria. Scale bar, 1 μm.
Electron microscopic analysis also demonstrated swelling and structural alterations of mitochondria in HCV-infected cells, whereas mitochondria remained intact in the mock-infected control (Fig. 5C). This result suggests a detrimental effect of HCV infection on the volume homeostasis and morphology of mitochondria and is consistent with previous observations that liver tissues from HCV-infected patients showed morphological changes in mitochondria (3).
Mitochondrial swelling and the morphological change of mitochondrial cristae are associated with cytochrome c release (27, 54). We then examined the effect of HCV infection on cytochrome c release in Huh7.5 cells. The result clearly demonstrated cytochrome c release from the mitochondria to the cytoplasm in HCV-infected cells but not in the mock-infected control (Fig. 6A). The release of cytochrome c from mitochondria is known to induce activation of caspase 9 (31). We then analyzed caspase 9 activities in the cells. As shown in Fig. 6B, caspase 9 activities in HCV-infected cells increased to levels that were ca. five times higher than that in the control cells at 4 and 6 days postinfection.
FIG. 6.

HCV infection induces cytochrome c release and caspase 9 activation in Huh7.5 cells. (A) Cytochrome c release. Mitochondrial and cytosolic fractions were prepared from HCV-infected cells and the mock-infected control at 6 days postinfection and analyzed by immunoblotting using antibodies against cytochrome c, Tim23, NS3, and actin. Can Get Signal (Toyobo, Osaka, Japan) was used to obtain stronger signals for cytochrome c. Amounts of Tim23 and actin were measured to verify equal amounts of mitochondrial and cytosolic fractions, respectively. Also, Tim23 was used to show successful separation of mitochondria. (B) Caspase 9 activation. Caspase 9 activities in cells infected with HCV and mock-infected controls were measured at 0, 2, 4, and 6 days postinfection. The caspase 9 activity of the control cells at day 0 postinfection was arbitrarily expressed as 1.0. Data represent means ± standard deviations (SD) of three independent experiments. *, P < 0.05, compared with the control. (C) HCV infection induces a marginal degree of caspase 8 activation. Caspase 8 activities in cells infected with HCV and mock-infected controls were measured at 0, 2, 4, and 6 days postinfection. The caspase 8 activity of the control cells at day 0 postinfection was arbitrarily expressed as 1.0. Data represent means ± SD of three independent experiments. *, P < 0.05, compared with the control.
HCV infection induces a marginal degree of caspase 8 activation.
In addition to the mitochondrial death (intrinsic) pathway described above, the extrinsic cell death pathway, which is initiated by the TNF family members and mediated by activated caspase 8 (31, 62), is also the focus of attention in the study of apoptosis. Therefore, we examined caspase 8 activities in HCV-infected cells and the mock-infected control. As shown in Fig. 6C, caspase 8 activities in HCV-infected cells increased to a level that was ca. two times higher than that in the control cells at 4 and 6 days postinfection. This increase was much smaller than that observed for caspase 9 activation (Fig. 6B).
HCV infection induces increased production of mitochondrial reactive oxygen species (ROS).
The production of ROS, such as superoxide, by mitochondria is the major cause of cellular oxidative stress (8), and a possible link between ROS production and Bax activation has been reported (18, 42). Therefore, we next examined the mitochondrial ROS production in HCV- and mock-infected cells by using MitoSOX, a fluorescent probe specific for superoxide that selectively accumulates in the mitochondrial compartment. As shown in Fig. 7A and B, approximately 25% of HCV-infected cells displayed a much higher signal than did the mock-infected control. This result suggests that oxidative stress is induced by HCV infection.
FIG. 7.

HCV infection induces increased production of mitochondrial superoxide in Huh7.5 cells. (A) Mitochondrial superoxide production in HCV-infected cells and the mock-infected control was examined at 6 days postinfection. Cells were directly incubated with MitoSOX (upper row) and then stained for HCV antigens by using an HCV-infected patient's serum, followed by FITC-labeled goat anti-human IgG (bottom row). Scale bar, 10 μm. (B) Quantification of MitoSOX-stained cells. The percentages of cells stained with MitoSOX were determined for HCV-infected cultures and the mock-infected control. Data represent means ± standard deviations of three independent experiments. *, P < 0.05, compared with the control.
HCV infection does not induce ER stress.
It is well known that HCV nonstructural proteins form the replication complex on the endoplasmic reticulum (ER) membrane (4, 19, 39, 46). It was recently reported that HCV infection (55) as well as the transfection of the full-length HCV replicon (17) and the expression of the entire HCV polyprotein (14) induced an ER stress response. Therefore, we tested whether HCV infection in our system induces ER stress. We adopted increased expression of GRP78 and GRP94 as indicators of ER stress (34) and, as a positive control, used tunicamycin to induce ER stress (20, 25). As had been expected, the expression levels of GRP78 and GRP94 were markedly increased in Huh7.5 cells when cells were treated with tunicamycin for 48 h (Fig. 8, right). On the other hand, HCV infection did not alter expression levels of GRP78 or GRP94 at 2, 4, or 6 days postinfection compared with those for the mock-infected control (Fig. 8). This result suggests that ER stress, if there is any, is marginal and does not play an important role in HCV-induced apoptosis in Huh7.5 cells.
FIG. 8.
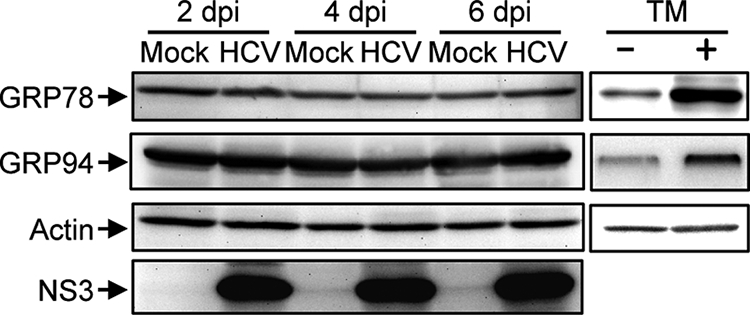
HCV infection does not induce ER stress in Huh7.5 cells. Huh7.5 cells infected with HCV and mock-infected controls were harvested at 2, 4, and 6 days postinfection (dpi), and the whole-cell lysates were subjected to immunoblot analysis using antibodies against GRP78, GRP94, NS3, and actin. Amounts of actin were measured to verify equal amounts of sample loading. Huh7.5 cells treated with tunicamycin (TM; 5 μg/ml) for 48 h served as a positive control.
DISCUSSION
The mitochondrion is an important organelle for cell survival and death and plays a crucial role in regulating apoptosis. An increasing body of evidence suggests that apoptosis occurs in the livers of HCV-infected patients (1, 2, 9) and that HCV-associated apoptosis involves, at least partly, a mitochondrion-mediated pathway (2). In those clinical settings, however, it is not clear whether apoptosis is mediated by host immune responses through the activity of cytotoxic T lymphocytes or whether it is mediated directly by HCV replication and/or protein expression itself. In experimental settings, ectopic expression of HCV core (13, 36), E2 (12), and NS4A (43) has been shown to induce mitochondrion-mediated apoptosis in cultured cells. However, these observations need to be verified in the context of virus replication. The recent development of an efficient HCV infection system in cell culture (37, 66, 71) has allowed us to investigate whether HCV replication directly causes apoptosis. In the present study, we have demonstrated that HCV infection induces Bax-triggered, mitochondrion-mediated, caspase 3-dependent apoptosis, as evidenced by increased accumulation of Bax on mitochondria and its conformational change (Fig. 3), decreased mitochondrial transmembrane potential (Fig. 4), and mitochondrial swelling (Fig. 5), which lead to the release of cytochrome c from the mitochondria (Fig. 6A) and subsequent activation of caspase 9 and caspase 3 (Fig. 6B and 2, respectively).
We also observed increased production of mitochondrial superoxide in HCV-infected cells (Fig. 7). This result is consistent with previous observations that expression of the entire HCV polyprotein (47) or HCV replication (60) enhanced production of ROS, including superoxide, through deregulation of mitochondrial calcium homeostasis. ROS, which are produced through the mitochondrial respiratory chain (8), were reported to trigger conformational change, dimerization, and mitochondrial translocation of Bax (18, 42). It is likely, therefore, that activation of Bax in HCV-infected cells is mediated, at least partly, through increased production of ROS in the mitochondria. Kim et al. (29) reported that ROS is a potent activator of c-Jun N-terminal protein kinase, which can phosphorylate Bax, leading to its activation and mitochondrial translocation. In this connection, HCV core protein has been shown to play a role in generating mitochondrial ROS (30). It was also reported that HCV core protein bound to the 14-3-3ɛ protein to dissociate Bax from the Bax/14-3-3ɛ complex, thereby promoting the Bax translocation to the mitochondria (36).
In addition to the caspase 9 activation that is mediated through the mitochondrial death (intrinsic) pathway, caspase 8 activation was seen in HCV-infected cells, though to a lesser extent (Fig. 6B and C). Caspase 8 is a key component of the extrinsic death pathway initiated by the TNF family members (31, 62). This pathway involves death receptors, such as Fas, TNF receptor, and TNF-related apoptosis-inducing ligand (TRAIL) receptors, which transduce signals to induce apoptosis upon binding to their respective ligands (52). In HCV-infected patients, the Fas-mediated signal pathway is involved in apoptosis of virus-infected hepatocytes (24). It was also reported that HCV (JFH1 strain) infection induced apoptosis through a TRAIL-mediated pathway in LH86 cells (72). On the other hand, a caspase 9-mediated activation of caspase 8, which is considered a cross talk between the intrinsic and the extrinsic death pathways, in certain cell systems was also reported (10, 11, 65). Whether the observed caspase 8 activation in HCV-infected cells was mediated through the extrinsic death pathway initiated by a cytokine(s) produced in the culture or whether it was mediated through the cross talk between the intrinsic and the extrinsic death pathways awaits further investigation. In this connection, activated caspase 8 is known to cleave the proapoptotic protein Bid to generate the Bid cleavage product truncated Bid (tBid), which facilitates the activation of Bax (63, 68). Under our experimental conditions, however, tBid was barely detected in HCV-infected cells even at 6 days postinfection (data not shown). It is thus likely that caspase 8 activation is marginal and is not the primary cause of Bax activation in our experimental system.
HCV protein expression and HCV RNA replication take place primarily in the ER or an ER-like membranous structure (39, 46). Like other members of the family Flaviviridae, such as dengue virus (69), Japanese encephalitis virus (69), West Nile virus (41), and bovine viral diarrhea virus (26), HCV has been reported to induce ER stress in the host cells (5, 14, 17, 55, 60). ER stress is triggered by perturbations in normal ER function, such as the accumulation of unfolded or misfolded proteins in the lumen. On the other hand, in response to ER stress, the unfolded protein response (UPR) is activated to alleviate the ER stress by stimulating protein folding and degradation in the ER as well as by inhibiting protein synthesis (7). The UPR of the host cell is disadvantageous for progeny virus production and may therefore be considered an antiviral host cell response. It was reported that, to counteract the disadvantageous UPR so as to maintain viral protein synthesis, HCV RNA replication suppressed the IRE1-XBP1 pathway, which is responsible for protein degradation upon UPR (59). Also, HCV E2 was shown to inhibit the double-stranded RNA-activated protein kinase-like ER-resident kinase (PERK), which attenuates protein synthesis during ER stress by phosphorylating the alpha subunit of eukaryotic translation initiation factor 2 (45). It is reasonable, therefore, to assume that HCV-infected cells may not necessarily exhibit typical responses to ER stress. In fact, our results revealed that HCV infection in Huh7.5 cells did not enhance expression of GRP78 and GRP94, which are ER stress-induced chaperone proteins (Fig. 8). Our result thus implies the possibility that ER stress is not crucially involved in HCV-induced apoptosis in Huh7.5 cells. Taking advantage of this phenomenon, we could demonstrate that an ER stress-independent, mitochondrion-mediated pathway plays an important role in HCV-induced apoptosis. In this connection, Korenaga et al. (30) reported that HCV core protein increased ROS production in isolated mitochondria, independently of ER stress, by selectively inhibiting electron transport complex I activity.
In this study, we observed that increased ROS production, Bax activation, and caspase 3 activation were detectable in approximately 15% to 25% of HCV antigen-positive Huh7.5cells at 6 days postinfection (Fig. 7B, 3D, and 2D, respectively). On the other hand, >90% of the cells in the cultures were confirmed positive for HCV antigens (Fig. 1B). These results imply the possibility that HCV establishes persistent infection in Huh7.5 cells, with a minor fraction of virus-infected cells beginning to undergo apoptosis after a prolonged period of time. Alternatively, it is possible that Huh7.5 cells, though being derived from a cell line (6), are a mixture of two sublineages, with one sublineage being apoptosis prone and the other apoptosis resistant. To test the latter possibility, further cloning of Huh7.5 cells is now under way in our laboratory.
In conclusion, our present results collectively suggest that HCV infection induces apoptosis through a Bax-triggered, mitochondrion-mediated, caspase 3-dependent pathway.
Acknowledgments
We are grateful to C. M. Rice (Center for the Study of Hepatitis C, The Rockefeller University) for providing pFL-J6/JFH1 and Huh7.5 cells.
This work was supported in part by grants-in-aid for scientific research from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) and the Ministry of Health, Labor and Welfare, Japan.
This study was carried out as part of the Program of Founding Research Centers for Emerging and Reemerging Infectious Diseases, MEXT, Japan. This study was also part of the 21st Century Center of Excellence Program at Kobe University Graduate School of Medicine.
Footnotes
Published ahead of print on 3 September 2008.
REFERENCES
- 1.Bantel, H., A. Lügering, C. Poremba, N. Lügering, J. Held, W. Domschke, and K. Schulze-Osthoff. 2001. Caspase activation correlates with the degree of inflammatory liver injury in chronic hepatitis C virus infection. Hepatology 34758-767. [DOI] [PubMed] [Google Scholar]
- 2.Bantel, H., and K. Schulze-Osthoff. 2003. Apoptosis in hepatitis C virus infection. Cell Death Differ. 10S48-S58. [DOI] [PubMed] [Google Scholar]
- 3.Barbaro, G., G. Di Lorenzo, A. Asti, M. Ribersani, G. Belloni, B. Grisorio, G. Filice, and G. Barbarini. 1999. Hepatocellular mitochondrial alterations in patients with chronic hepatitis C: ultrastructural and biochemical findings. Am. J. Gastroenterol. 942198-2205. [DOI] [PubMed] [Google Scholar]
- 4.Bartenschlager, R., M. Frese, and T. Pietschmann. 2004. Novel insights into hepatitis C virus replication and persistence. Adv. Virus Res. 63171-180. [DOI] [PubMed] [Google Scholar]
- 5.Benali-Furet, N. L., M. Chami, L. Houel, F. De Giorgi, F. Vernejoul, D. Lagorce, L. Buscail, R. Bartenschlager, F. Ichas, R. Rizzuto, and P. Paterlini-Bréchot. 2005. Hepatitis C virus core triggers apoptosis in liver cells by inducing ER stress and ER calcium depletion. Oncogene 244921-4933. [DOI] [PubMed] [Google Scholar]
- 6.Blight, K. J., J. A. McKeating, and C. M. Rice. 2002. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. 7613001-13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boyce, M., and J. Yuan. 2006. Cellular response to endoplasmic reticulum stress: a matter of life or death. Cell Death Differ. 13363-373. [DOI] [PubMed] [Google Scholar]
- 8.Brookes, P. S. 2005. Mitochondrial H+ leak and ROS generation: an odd couple. Free Radic. Biol. Med. 3812-23. [DOI] [PubMed] [Google Scholar]
- 9.Calabrese, F., P. Pontisso, E. Pettenazzo, L. Benvegnù, A. Vario, L. Chemello, A. Alberti, and M. Valente. 2000. Liver cell apoptosis in chronic hepatitis C correlates with histological but not biochemical activity or serum HCV-RNA levels. Hepatology 311153-1159. [DOI] [PubMed] [Google Scholar]
- 10.Camacho-Leal, P., and C. P. Stanners. 2008. The human carcinoembryonic antigen (CEA) GPI anchor mediates anoikis inhibition by inactivation of the intrinsic death pathway. Oncogene 271545-1553. [DOI] [PubMed] [Google Scholar]
- 11.Chae, Y. J., H. S. Kim, H. Rhim, B. E. Kim, S. W. Jeong, and I. K. Kim. 2001. Activation of caspase-8 in 3-deazaadenosine-induced apoptosis of U-937 cells occurs downstream of caspase-3 and caspase-9 without Fas receptor-ligand interaction. Exp. Mol. Med. 4284-292. [DOI] [PubMed] [Google Scholar]
- 12.Chiou, H. L., Y. S. Hsieh, M. R. Hsieh, and T. Y. Chen. 2006. HCV E2 may induce apoptosis of Huh-7 cells via a mitochondrial-related caspase pathway. Biochem. Biophys. Res. Commun. 345453-458. [DOI] [PubMed] [Google Scholar]
- 13.Chou, A. H., H. F. Tsai, Y. Y. Wu, C. Y. Hu, L. H. Hwang, P. I. Hsu, and P. N. Hsu. 2005. Hepatitis C virus core protein modulates TRAIL-mediated apoptosis by enhancing Bid cleavage and activation of mitochondria apoptosis signaling pathway. J. Immunol. 1742160-2166. [DOI] [PubMed] [Google Scholar]
- 14.Christen, V., S. Treves, F. H. Duong, and M. H. Heim. 2007. Activation of endoplasmic reticulum stress response by hepatitis viruses up-regulates protein phosphatase 2A. Hepatology 46558-565. [DOI] [PubMed] [Google Scholar]
- 15.Ciccaglione, A. R., C. Marcantonio, A. Costantino, M. Equestre, and M. Rapicetta. 2003. Expression of HCV E1 protein in baculovirus-infected cells: effects on cell viability and apoptosis induction. Intervirology 46121-126. [DOI] [PubMed] [Google Scholar]
- 16.Ciccaglione, A. R., C. Marcantonio, E. Tritarelli, M. Equestre, F. Magurano, A. Costantino, L. Nicoletti, and M. Rapicetta. 2004. The transmembrane domain of hepatitis C virus E1 glycoprotein induces cell death. Virus Res. 1041-9. [DOI] [PubMed] [Google Scholar]
- 17.Ciccaglione, A. R., C. Marcantonio, E. Tritarelli, M. Equestre, F. Vendittelli, A. Costantino, A. Geraci, and M. Rapicetta. 2007. Activation of the ER stress gene gadd153 by hepatitis C virus sensitizes cells to oxidant injury. Virus Res. 126128-138. [DOI] [PubMed] [Google Scholar]
- 18.D'Alessio, M., M. De Nicola, S. Coppola, G. Gualandi, L. Pugliese, C. Cerella, S. Cristofanon, P. Civitareale, M. R. Ciriolo, A. Bergamaschi, A. Magrini, and L. Ghibelli. 2005. Oxidative Bax dimerization promotes its translocation to mitochondria independently of apoptosis. FASEB J. 191504-1506. [DOI] [PubMed] [Google Scholar]
- 19.Egger, D., B. Wölk, R. Gosert, L. Bianchi, H. E. Blum, D. Moradpour, and K. Bienz. 2002. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J. Virol. 765974-5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elbein, A. D. 1987. Inhibitors of the biosynthesis and processing of N-linked oligosaccharide chains. Annu. Rev. Biochem. 56497-534. [DOI] [PubMed] [Google Scholar]
- 21.Erdtmann, L., N. Franck, H. Lerat, J. Le Seyec, D. Gilot, I. Cannie, P. Gripon, U. Hibner, and C. Guguen-Guillouzo. 2003. The hepatitis C virus NS2 protein is an inhibitor of CIDE-B-induced apoptosis. J. Biol. Chem. 27818256-18264. [DOI] [PubMed] [Google Scholar]
- 22.Griffin, S., D. Clarke, C. McCormick, D. Rowlands, and M. Harris. 2005. Signal peptide cleavage and internal targeting signals direct the hepatitis C virus p7 protein to distinct intracellular membranes. J. Virol. 7915525-15536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hidajat, R., M. Nagano-Fujii, L. Deng, M. Tanaka, Y. Takigawa, S. Kitazawa, and H. Hotta. 2005. Hepatitis C virus NS3 protein interacts with ELKS-δ and ELKS-α, members of a novel protein family involved in intracellular transport and secretory pathways. J. Gen. Virol. 862197-2208. [DOI] [PubMed] [Google Scholar]
- 24.Jarmay, K., G. Karacsony, Z. Ozsvar, I. Nagy, J. Lonovics, and Z. Schaff. 2002. Assessment of histological feature in chronic hepatitis C. Hepatogastroenterology 49239-243. [PubMed] [Google Scholar]
- 25.Jiang, C. C., L. H. Chen, S. Gillespie, K. A. Kiejda, N. Mhaidat, Y. F. Wang, R. Thorne, X. D. Zhang, and P. Hersey. 2007. Tunicamycin sensitizes human melanoma cells to tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by up-regulation of TRAIL-R2 via the unfolded protein response. Cancer Res. 675880-5888. [DOI] [PubMed] [Google Scholar]
- 26.Jordan, R., L. Wang, T. M. Graczyk, T. M. Block, and P. R. Romano. 2002. Replication of a cytopathic strain of bovine viral diarrhea virus activates PERK and induces endoplasmic reticulum stress-mediated apoptosis of MDBK cells. J. Virol. 769588-9599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaasik, A., D. Safiulina, A. Zharkovsky, and V. Veksler. 2007. Regulation of mitochondrial matrix volume. Am. J. Physiol. Cell Physiol. 292C157-C163. [DOI] [PubMed] [Google Scholar]
- 28.Kamada, S., U. Kikkawa, Y. Tsujimoto, and T. Hunter. 2005. Nuclear translocation of caspase-3 is dependent on its proteolytic activation and recognition of a substrate-like protein(s). J. Biol. Chem. 280857-860. [DOI] [PubMed] [Google Scholar]
- 29.Kim, B. J., S. W. Ryu, and B. J. Song. 2006. JNK- and p38 kinase-mediated phosphorylation of Bax leads to its activation and mitochondrial translocation and to apoptosis of human hepatoma HepG2 cells. J. Biol. Chem. 28121256-21265. [DOI] [PubMed] [Google Scholar]
- 30.Korenaga, M., T. Wang, Y. Li, L. A. Showalter, T. Chan, J. Sun, and S. A. Weinman. 2005. Hepatitis C virus core protein inhibits mitochondrial electron transport and increases reactive oxygen species (ROS) production. J. Biol. Chem. 28037481-37488. [DOI] [PubMed] [Google Scholar]
- 31.Kumar, S. 2007. Caspase function in programmed cell death. Cell Death Differ. 1432-43. [DOI] [PubMed] [Google Scholar]
- 32.Lalier, L., P. F. Cartron, P. Juin, S. Nedelkina, S. Manon, B. Bechinger, and F. M. Vallette. 2007. Bax activation and mitochondrial insertion during apoptosis. Apoptosis 12887-896. [DOI] [PubMed] [Google Scholar]
- 33.Lan, K. H., M. L. Sheu, S. J. Hwang, S. H. Yen, S. Y. Chen, J. C. Wu, Y. J. Wang, N. Kato, M. Omata, F. Y. Chang, and S. D. Lee. 2002. HCV NS5A interacts with p53 and inhibits p53-mediated apoptosis. Oncogene 214801-4811. [DOI] [PubMed] [Google Scholar]
- 34.Lee, A. S. 2001. The glucose-regulated proteins: stress induction and clinical applications. Trends Biochem. Sci. 26504-510. [DOI] [PubMed] [Google Scholar]
- 35.Lee, S. H., Y. K. Kim, C. S. Kim, S. K. Seol, J. Kim, S. Cho, Y. L. Song, R. Bartenschlager, and S. K. Jang. 2005. E2 of hepatitis C virus inhibits apoptosis. J. Immunol. 1758226-8235. [DOI] [PubMed] [Google Scholar]
- 36.Lee, S. K., S. O. Park, C. O. Joe, and Y. S. Kim. 2007. Interaction of HCV core protein with 14-3-3ɛ protein releases Bax to activate apoptosis. Biochem. Biophys. Res. Commun. 352756-762. [DOI] [PubMed] [Google Scholar]
- 37.Lindenbach, B. D., M. J. Evans, A. J. Syder, B. Wölk, T. L. Tellinghuisen, C. C. Liu, T. Maruyama, R. O. Hynes, D. R. Burton, J. A. McKeating, and C. M. Rice. 2005. Complete replication of hepatitis C virus in cell culture. Science 309623-626. [DOI] [PubMed] [Google Scholar]
- 38.Lindenbach, B. D., P. Meuleman, A. Ploss, T. Vanwolleghem, A. J. Syder, J. A. McKeating, R. E. Lanford, S. M. Feinstone, M. E. Major, G. Leroux-Roels, and C. M. Rice. 2006. Cell culture-grown hepatitis C virus is infectious in vivo and can be recultured in vitro. Proc. Natl. Acad. Sci. USA 1033805-3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lindenbach, B. D., and C. M. Rice. 2005. Unravelling hepatitis C virus replication from genome to function. Nature 436933-938. [DOI] [PubMed] [Google Scholar]
- 40.Marusawa, H., M. Hijikata, T. Chiba, and K. Shimotohno. 1999. Hepatitis C virus core protein inhibits Fas- and tumor necrosis factor alpha-mediated apoptosis via NF-κB activation. J. Virol. 734713-4720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Medigeshi, G. R., A. M. Lancaster, A. J. Hirsch, T. Briese, W. I. Lipkin, V. Defilippis, K. Früh, P. W. Mason, J. Nikolich-Zugich, and J. A. Nelson. 2007. West Nile virus infection activates the unfolded protein response, leading to CHOP induction and apoptosis. J. Virol. 8110849-10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nie, C., C. Tian, L. Zhao, P. X. Petit, M. Mehrpour, and Q. Chen. 2008. Cysteine 62 of Bax is critical for its conformational activation and its proapoptotic activity in response to H2O2-induced apoptosis. J. Biol. Chem. 28315359-15369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nomura-Takigawa, Y., M. Nagano-Fujii, L. Deng, S. Kitazawa, S. Ishido, K. Sada, and H. Hotta. 2006. Non-structural protein 4A of Hepatitis C virus accumulates on mitochondria and renders the cells prone to undergoing mitochondria-mediated apoptosis. J. Gen. Virol. 871935-1945. [DOI] [PubMed] [Google Scholar]
- 44.Oliver, F. J., G. de la Rubia, V. Rolli, M. C. Ruiz-Ruiz, G. de Murcia, and J. M. Murcia. 1998. Importance of poly(ADP-ribose) polymerase and its cleavage in apoptosis. J. Biol. Chem. 27333533-33539. [DOI] [PubMed] [Google Scholar]
- 45.Pavio, N., P. R. Romano, T. M. Graczyk, S. M. Feinstone, and D. R. Taylor. 2003. Protein synthesis and endoplasmic reticulum stress can be modulated by the hepatitis C virus envelope protein E2 through the eukaryotic initiation factor 2α kinase PERK. J. Virol. 773578-3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pawlotsky, J. M., S. Chevaliez, and J. G. McHutchison. 2007. The hepatitis C virus life cycle as a target for new antiviral therapies. Gastroenterology. 1321979-1998. [DOI] [PubMed] [Google Scholar]
- 47.Piccoli, C., R. Scrima, G. Quarato, A. D'Aprile, M. Ripoli, L. Lecce, D. Boffoli, D. Moradpour, and N. Capitanio. 2007. Hepatitis C virus protein expression causes calcium-mediated mitochondrial bioenergetic dysfunction and nitro-oxidative stress. Hepatology 4658-65. [DOI] [PubMed] [Google Scholar]
- 48.Prikhod'ko, E. A., G. G. Prikhod'ko, R. M. Siegel, P. Thompson, M. E. Major, and J. I. Cohen. 2004. The NS3 protein of hepatitis C virus induces caspase-8-mediated apoptosis independent of its protease or helicase activities. Virology 32953-67. [DOI] [PubMed] [Google Scholar]
- 49.Ray, R. B., K. Meyer, R. Steele, A. Shrivastava, B. B. Aggarwal, and R. Ray. 1998. Inhibition of tumor necrosis factor (TNF-α)-mediated apoptosis by hepatitis C virus core protein. J. Biol. Chem. 2732256-2259. [DOI] [PubMed] [Google Scholar]
- 50.Safiulina, D., V. Veksler, A. Zharkovsky, and A. Kaasik. 2006. Loss of mitochondrial membrane potential is associated with increase in mitochondrial volume: physiological role in neurones. J. Cell. Physiol. 206347-353. [DOI] [PubMed] [Google Scholar]
- 51.Saito, K., K. Meyer, R. Warner, A. Basu, R. B. Ray, and R. Ray. 2006. Hepatitis C virus core protein inhibits tumor necrosis factor alpha-mediated apoptosis by a protective effect involving cellular FLICE inhibitory protein. J. Virol. 804372-4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schulze-Osthoff, K., D. Ferrari, M. Los, S. Wesselborg, and M. E. Peter. 1998. Apoptosis signaling by death receptors. Eur. J. Biochem. 254439-459. [DOI] [PubMed] [Google Scholar]
- 53.Schwer, B., S. Ren, T. Pietschmann, J. Kartenbeck, K. Kaehlcke, R. Bartenschlager, T. S. Yen, and M. Ott. 2004. Targeting of hepatitis C virus core protein to mitochondria through a novel C-terminal localization motif. J. Virol. 787958-7968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Scorrano, L., M. Ashiya, K. Buttle, S. Weiler, S. A. Oakes, C. A. Mannella, and S. J. Korsmeyer. 2002. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev. Cell 255-67. [DOI] [PubMed] [Google Scholar]
- 55.Sekine-Osajima, Y., N. Sakamoto, K. Mishima, M. Nakagawa, Y. Itsui, M. Tasaka, Y. Nishimura-Sakurai, C. H. Chen, T. Kanai, K. Tsuchiya, T. Wakita, N. Enomoto, and M. Watanabe. 2008. Development of plaque assays for hepatitis C virus-JFH1 strain and isolation of mutants with enhanced cytopathogenicity and replication capacity. Virology 37171-85. [DOI] [PubMed] [Google Scholar]
- 56.Shepard, C. W., L. Finelli, and M. J. Alter. 2005. Global epidemiology of hepatitis C virus infection. Lancet Infect. Dis. 5558-567. [DOI] [PubMed] [Google Scholar]
- 57.Siavoshian, S., J. D. Abraham, C. Thumann, M. P. Kieny, and C. Schuster. 2005. Hepatitis C virus core, NS3, NS5A, NS5B proteins induce apoptosis in mature dendritic cells. J. Med. Virol. 75402-411. [DOI] [PubMed] [Google Scholar]
- 58.Tanaka, M., M. Nagano-Fujii, L. Deng, S. Ishido, K. Sada, and H. Hotta. 2006. Single-point mutations of hepatitis C virus that impair p53 interaction and anti-apoptotic activity of NS3. Biochem. Biophys. Res. Commun. 340792-799. [DOI] [PubMed] [Google Scholar]
- 59.Tardif, K. D., K. Mori, R. J. Kaufman, and A. Siddiqui. 2004. Hepatitis C virus suppresses the IRE1-XBP1 pathway of the unfolded protein response. J. Biol. Chem. 27917158-17164. [DOI] [PubMed] [Google Scholar]
- 60.Tardif, K. D., G. Waris, and A. Siddiqui. 2005. Hepatitis C virus, ER stress, and oxidative stress. Trends Microbiol. 13159-163. [DOI] [PubMed] [Google Scholar]
- 61.Tewari, M., L. T. Quan, K. O'Rourke, S. Desnoyers, Z. Zeng, D. R. Beidler, G. G. Poirier, G. S. Salvesen, and V. M. Dixit. 1995. Yama/CPP32 beta, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly (ADP-ribose) polymerase. Cell 81801-809. [DOI] [PubMed] [Google Scholar]
- 62.Thorburn, A. 2004. Death receptor-induced cell killing. Cell. Signal. 16139-144. [DOI] [PubMed] [Google Scholar]
- 63.Tsujimoto, Y. 2003. Cell death regulation by the Bcl-2 protein family in the mitochondria. J. Cell. Physiol. 195158-167. [DOI] [PubMed] [Google Scholar]
- 64.Upton, J. P., A. J. Valentijn, L. Zhang, and A. P. Gilmore. 2007. The N-terminal conformation of Bax regulates cell commitment to apoptosis. Cell Death Differ. 14932-942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Viswanath, V., Y. Wu, R. Boonplueang, S. Chen, F. F. Stevenson, F. Yantiri, L. Yang, M. F. Beal, and J. K. Andersen. 2001. Caspase-9 activation results in downstream caspase-8 activation and bid cleavage in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinson's disease. J. Neurosci. 219519-9528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wakita, T., T. Pietschmann, T. Kato, T. Date, M. Miyamoto, Z. Zhao, K. Murthy, A. Habermann, H. G. Kräusslich, M. Mizokami, R. Bartenschlager, and T. J. Liang. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11791-796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang, J., W. Tong, X. Zhang, L. Chen, Z. Yi, T. Pan, Y. Hu, L. Xiang, and Z. Yuan. 2006. Hepatitis C virus non-structural protein NS5A interacts with FKBP38 and inhibits apoptosis in Huh7 hepatoma cells. FEBS Lett. 5804392-4400. [DOI] [PubMed] [Google Scholar]
- 68.Wei, M. C., W. X. Zong, E. H. Cheng, T. Lindsten, V. Panoutsakopoulou, A. J. Ross, K. A. Roth, G. R. MacGregor, C. B. Thompson, and S. J. Korsmeyer. 2001. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292727-730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yu, C. Y., Y. W. Hsu, C. L. Liao, and Y. L. Lin. 2006. Flavivirus infection activates the XBP1 pathway of the unfolded protein response to cope with endoplasmic reticulum stress. J. Virol. 8011868-11880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhivotovsky, B., A. Samali, A. Gahm, and S. Orrenius. 1999. Caspases: their intracellular localization and translocation during apoptosis. Cell Death Differ. 6644-651. [DOI] [PubMed] [Google Scholar]
- 71.Zhong, J., P. Gastaminza, G. Cheng, S. Kapadia, T. Kato, D. R. Burton, S. F. Wieland, S. L. Uprichard, T. Wakita, and F. V. Chisari. 2005. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. USA 1029294-9299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhu, H., H. Dong, E. Eksioglu, A. Hemming, M. Cao, J. M. Crawford, D. R. Nelson, and C. Liu. 2007. Hepatitis C virus triggers apoptosis of a newly developed hepatoma cell line through antiviral defense system. Gastroenterology 1331649-1659. [DOI] [PubMed] [Google Scholar]
- 73.Zhu, N., A. Khoshnan, R. Schneider, M. Matsumoto, G. Dennert, C. Ware, and M. M. C. Lai. 1998. Hepatitis C virus core protein binds to the cytoplasmic domain of tumor necrosis factor (TNF) receptor 1 and enhances TNF-induced apoptosis. J. Virol. 723691-3697. [DOI] [PMC free article] [PubMed] [Google Scholar]