

Abstract
The NF-κB family mediates immune and inflammatory responses. In many cancers, NF-κB is constitutively activated and induces the expression of genes that facilitate tumorigenesis. ING4 is a tumor suppressor that is absent or mutated in several cancers. Herein, we demonstrate that in human gliomas, NF-κB is constitutively activated, ING4 expression is negligible, and NF-κB-regulated gene expression is elevated. We demonstrate that an ING4 and NF-κB interaction exists but does not prevent NF-κB activation, nuclear translocation, or DNA binding. Instead, ING4 and NF-κB bind simultaneously at NF-κB-regulated promoters, and this binding correlates with reductions in p65 phosphorylation, p300, and the levels of acetylated histones and H3-Me3K4, while enhancing the levels of HDAC-1 at these promoters. Using a knockdown approach, we correlate reductions in ING4 protein levels with increased basal and inducible NF-κB target gene expression. Collectively, these data suggest that ING4 may specifically regulate the activity of NF-κB molecules that are bound to target gene promoters.
The NF-κB family of transcription factors consists of five structurally similar members that each contains a Rel homology domain and are classified into two groups (13, 14). The first group, p65 (RelA), RelB, and c-Rel, contains a C-terminal transactivation domain (TAD), while the second group, NF-κB1 (p105/p50) and NF-κB2 (p100/p52), lacks a TAD. Through their Rel homology domains, NF-κB members form homo- and heterodimers that bind κB elements. However, only NF-κB dimers that contain p65, RelB, or c-Rel, and thus, a TAD, are able to positively regulate gene expression.
As the prime mediator of inflammation, NF-κB activity must be tightly regulated (13, 14). In unstimulated cells, NF-κB is unable to bind DNA due to interactions with inhibitor of NF-κB (IκB) proteins. In response to cytokines such as tumor necrosis factor alpha (TNF-α) or interleukin-1β (IL-1β) or agents such as phorbol myristate acetate (PMA), IκB kinase (IKK) is activated and phosphorylates the IκB proteins, leading to their ubiquitin-dependent degradation. NF-κB dimers accumulate in the nucleus, bind κB elements, and activate the expression of genes encoding proteins such as matrix metalloproteinase-9 (MMP-9) and cyclooxygenase-2 (COX-2). Although NF-κB activity is normally transient, in many types of human cancers, NF-κB is constitutively activated and may contribute to the processes of tumorigenesis (14). At present, there is little information regarding how NF-κB becomes activated or why NF-κB fails to become inactivated within tumor cells.
Inhibitor of growth 4 (ING4) is a member of the ING tumor suppressor family, which includes ING1, ING2, ING3, and ING5 (4, 9, 12). To date, all ING proteins contain a C-terminal plant homeodomain (PHD), two nuclear localization signals, and two additional motifs termed domain I and II (6, 12). While the functions of domains I and II remain unclear, recent studies of human and yeast cells indicate that the ING proteins use the PHD domains to recognize and bind to histone 3 that is tri-methylated at lysine 4 (H3-Me3K4) (22, 29, 30, 37, 39). Although H3-Me3K4 is typically associated with transcriptionally active DNA, at least one ING protein, ING2, can repress gene expression by binding H3-Me3K4 and recruiting HDAC-1 containing complexes to nearby promoters (37). This suggests that the ING proteins may suppress tumorigenesis by regulating gene expression. Indeed, ING4 can interact with histone acetyltransferases such as p300 and HBO1 and transcription factors, including p53, HIF, and NF-κB (6, 8, 27, 28, 38, 43). However, ING4 expression is reduced in several cancers and is mutated in some cancer cell lines (8, 10, 15). Additionally, multiple alternatively spliced ING4 transcripts were recently identified that encode either cytoplasmic proteins that may not function correctly or proteins that lack some or all of the PHD domain (40). Therefore, in cancers, the loss of ING4 activity by multiple mechanisms may facilitate tumorigenesis. One study demonstrated that levels of ING4 mRNA were inversely correlated with increasing tumor grades of human gliomas (8). This study also demonstrated that ING4 and p65 interact and that ING4 inhibited the ability of NF-κB to activate genes such as IL-8, IL-6, and COX-2, perhaps by inhibiting the ability of p65 to bind DNA (8). These data have yet to be confirmed or extended, and the nature and outcome of an ING4-NF-κB interaction remain to be clarified.
Malignant gliomas are an aggressive, neurologically destructive, and deadly type of tumor of the central nervous system (7, 20). Gliomas are classified according to their proposed cellular origin and clinical presentation (7, 20). Among the gliomas, malignant astrocytomas are the most common and most lethal intracranial tumor. Astrocytomas are typically described as diffusely infiltrative, since glioma cells tend to invade the normal brain, thus rendering these tumors difficult to treat and incurable, despite aggressive and multifocal approaches (7, 20). Therefore, in gliomas, there is a desperate need to focus on the diffusively infiltrative nature and the heterogeneity and development of apoptotic resistance of gliomas, since it is these features that ultimately render gliomas incurable.
To understand the relationship between ING4 and NF-κB in gliomas, we analyzed ING4 expression, NF-κB activation, and levels of select NF-κB-regulated genes in control and glioma tissues. Compared to control samples, glioma tissues demonstrated higher levels of activated NF-κB and NF-κB-regulated genes (MMP-9 and COX-2) and little or no ING4 protein, suggesting that the absence of or reductions in ING4 expression inversely correlate with NF-κB status and gene expression. Using stable glioma cell lines that inducibly express ING4 or glioma cell lines that stably express ING4 shRNA, we demonstrated that ING4 and NF-κB interact and that this interaction does not prevent NF-κB activation or DNA binding ability. Instead, ING4 alters both posttranslational modifications to and protein-protein interactions with p65. Interestingly, ING4 does not appear to equally target all NF-κB molecules. Instead, ING4 may target those NF-κB molecules that are bound to DNA by recognizing two motifs, NF-κB p65 and H3-Me3K4. Once they are present at these promoters, ING4 attenuates NF-κB-mediated activities to reduce the expression of NF-κB-regulated genes. Therefore, in gliomas and other cancers, we propose that the absence of proper ING4 activities may enable already activated NF-κB to perpetually induce the expression of genes that contribute to tumorigenesis.
MATERIALS AND METHODS
Plasmids.
The plasmid carrying human ING4 (pOTB7/ING4) was purchased from Open Biosystems (ATCC Integrated Molecular Analysis of Genomes and their Expression [IMAGE] Consortium number 4309278). The plasmid carrying Flag-ING4 was generated by PCR. The resulting product was analyzed, cloned into the pGem-T plasmid (Promega), and sequenced. The Flag-ING4 (F-ING4) open reading frame (ORF) was subcloned into pcDNA3 or pcDNA4TO to generate pcDNA3/F-ING4 and pcDNA4TO/F-ING4, respectively. To generate the ING4-green fluorescent protein (GFP) expression constructs, PCR was used to remove the stop codon, and the resulting PCR products were sequenced and cloned in frame into pEGFP-N1 to generate pEGFP-N1/ING4-GFP. The plasmids carrying shRNAs specific for either p65 or ING4 were generated by annealing double-stranded oligonucleotides specific for a 19-bp stretch of the p65 ORF or the ING4 ORF into the pBABE-HI-TetO plasmid, which is under the dual control of the tetracycline (Tet) operator and the HI polymerase (Pol) III promoter. Specific sequences are available upon request. The pBABE-HI-TetO plasmid was a generous gift of Xinbin Chen (University of California at Davis, CA). To create Flag-tagged p65, PCR was used to add the Flag epitope in frame to the 5′ end of the p65 ORF. This amplicon was sequenced and subcloned into pcDNA3 to generate pcDNA3/F-p65.
Cell lines.
The U251-MG, U118-MG, and U87-MG glioma cell lines were obtained from ATCC and maintained as previously described (5, 25, 40). U251-MG cells that stably express the Tet repressor (TetR) protein and inducibly express F-ING4 or shRNA specific for p65 (sh-p65), and U118-MG cells that stably express the TetR and that inducibly express shRNA specific for ING4 (shING4) were generated as previously described (26).
Reagents.
PMA was purchased from Calbiochem (Darmstadt, Germany). Recombinant human TNF-α was purchased from R & D Systems (Minneapolis, MN). The secondary peroxidase-conjugated antibodies and enhanced chemiluminescence reagents were purchased from Amersham (Arlington Heights, IL); and the anti-Flag antibodies from Sigma (St. Louis, MO) and the anti-p65 antibodies used in chromatin immunoprecipitation (ChIP) experiments were from Abcam (Cambridge, MA). Normal rabbit and mouse sera, anti-p65, anti-COX-2, anti-p50, anti-CBP, anti-p300, anti-HDAC-1, anti-CDK9, and anti-cyclin T1 antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The anti-RNA Pol II, anti-phosphoserine 2 RNA Pol II, and anti-phosphoserine 5 RNA Pol II antibodies were purchased from Covance (Denver, PA). Protein A/G beads and anti-Ac-H3, anti-Ac-H4, and anti-H3-MeK4 antibodies were purchased from Upstate Cell Signaling Solutions (Charlottesville, VA); anti-ING4 antibodies were from Proteintech (Chicago, IL), Zymed (Carlsbad, CA), and Abcam (Cambridge, MA), and phospho-p65-specific antibodies were from Cell Signaling (Danvers, MA).
Immunohistochemistry.
Formalin-fixed paraffin-embedded tissues were kindly provided by Cheryl Palmer, director of the UAB Brain Tumor Tissue core facility (human IRB approval protocol no. X050415007), and were sectioned at 4- to 6-μm thicknesses and stained as previously described (24) using antibody against ING4 (diluted 1:50; Proteintech, Chicago, IL). As a negative control, adjacent sections were incubated with nonimmune rabbit antiserum. Immune complexes were detected using a tyramide signal amplification protocol according to the manufacturer's instructions (TSA biotin system; Perkin-Elmer Life Sciences Products, Boston, MA). Sections were counterstained with hematoxylin, coverslipped, and imaged.
Immunofluorescence.
U251-MG cells were plated at 3 × 104 cells per well in eight-well chamber slides and grown overnight in complete medium. The next day, the medium was removed and replaced with serum-free medium. Cells were transfected with 0.3 μg of pEGFP-N1/ING4-GFP, using FuGENE 6, and allowed to recover overnight. The next day, cells were stimulated with PMA (50 ng/ml) for 1 h, washed, fixed, permeabilized, and stained as previously described (25).
Simultaneous total RNA and protein purification from human brain tissue.
Resected control and glioma brain tissue samples were obtained from the UAB Brain Tumor Tissue core facility (IRB X050415007), snap-frozen in liquid nitrogen, and stored at −80°C until processed. Tissues were ground with liquid nitrogen in a prechilled mortar and pestle at 4°C. The ground tissue was collected and processed using a Protein and RNA Isolation System kit according to the manufacturer's instructions (Ambion, Austin, TX). RNA was analyzed by reverse transcription-PCR (RT-PCR) as described below.
Total RNA isolation and RT-PCR.
Where indicated, U251-TR/F-ING4 cells were grown in complete medium in the absence or presence of Tet (4 μg/ml) for 24 h, and U251-TR/sh-p65 cells were grown in the absence or presence of Tet for 48 h. Cells were then incubated in serum-free medium in the absence or presence of Tet overnight and then grown in the absence or presence of PMA (50 ng/ml) for various times. Total RNA was isolated as previously described (25). Two micrograms of total RNA were reverse transcribed and analyzed by PCR. Results were analyzed by densitometry and normalized to GAPDH levels and represented as (fold) change over either unstimulated (cell culture) or control tissue (nondiseased, nonmalignant brain tissue).
In vitro binding assay.
Recombinant F-p65 and F-ING4 proteins were generated using a TNT T7 coupled reticulocyte lysate system (Promega, Madison, WI). Reactions occurred in the presence of no template or 1 μg of either pcDNA3/F-p65 or pcDNA3/F-ING4. Equal amounts of reaction mixtures were then mixed and normalized to a final volume of 750 μl with RIPA buffer. Five micrograms of antibodies (rabbit immunoglobulin G [IgG], anti-ING4, or anti-p65) were added and samples rotated overnight at 4°C. Fifty microliters of protein A/G beads were added and samples incubated for 2 h at 4°C with rotation. Samples were washed three times with RIPA buffer, boiled in 2× sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) loading dye and then separated by 10% SDS-PAGE. Proteins were visualized by immunoblotting with a mouse anti-Flag antibody.
Transient transfections and immunoprecipitations.
U251-MG cells were plated at 3 × 106 cells per 10-cm plate. Two micrograms of a plasmid carrying p65 and 2 μg of either an empty pcDNA3 plasmid or the pcDNA3/F-ING4 plasmid were transfected into cells. Cells were grown overnight in complete medium, and then cell lysates were collected as described above. Five hundred micrograms of total protein were immunoprecipitated with 5 μg of anti-p65 monoclonal antibody and analyzed by immunoblotting with anti-Flag antibody as previously described (26). Where indicated, cells were plated at 6 × 107 cells per 15-cm plate in complete medium, in the absence or presence of Tet. At 24 h, cells were incubated in serum-free medium in the absence or presence of Tet for 18 h and then grown in the absence or presence of PMA for 1 h. Cell lysates were prepared as described previously (26). One milligram of total protein was incubated with 5 μg of anti-p65 monoclonal antibody overnight at 4°C. Immunoprecipitates were prepared as described above and analyzed by SDS-PAGE and immunoblotting for ING4 or p300.
Immunoblotting.
Resected brain tissues were prepared as described above. Cultured cells were prepared as previously described (26). Equal amounts of proteins were analyzed by SDS-PAGE and immunoblotting for p65, phosphorylated p65 (p-p65) at serine 536 (S536) and serine 276 (S276), p50, ING4 or the Flag epitope, and actin or GAPDH.
ChIP and re-ChIP assays.
ChIP assays were performed as previously described (19, 25). Immunoprecipitation was performed with 5 μg of the appropriate antibodies, and the immune complexes were absorbed with protein A beads or protein G beads (Upstate Cell Signaling Solutions, Charlottesville, VA) blocked with bovine serum albumin and salmon sperm DNA. For re-ChIP, the chromatin complex was eluted from beads with 10 mM dithiothreitol at 37°C, diluted with RIPA buffer, and then immunoprecipitated with specific antibodies (19). Immunoprecipitated DNA was subjected to semiquantitative PCR and analyzed by gel electrophoresis. Specific primer sequences are available upon request.
RESULTS
NF-κB is constitutively activated in many human gliomas.
NF-κB is constitutively activated in many human cancers including gliomas (42). To confirm these findings, we analyzed the levels of p-p65 (S276) and p-p65 (S536) in eight nonmalignant (control) brain tissue samples and eight samples from glioma patients (glioma). The 16 tissue samples were analyzed by an independent neuropathologist and determined to be nonmalignant (Fig. 1A, lanes 1 to 8), grade III astrocytoma (Fig. 1A, lane 9), or grade IV glioblastoma (Fig. 1A, lanes 10 to 16). Compared to control tissues, which displayed very low levels of p-p65, gliomas displayed elevated levels of p-p65 (S276) and p-p65 (S536). In particular, four glioma samples exhibited pronounced NF-κB phosphorylation (Fig. 1A, lanes 9, 12, 13, and 16). NF-κB activation was also confirmed by measuring the levels of total and phosphorylated IκBα (p-IκBα), and the total levels of p65 and p50. In glioma samples, the levels of total IκBα were reduced, and the levels of p-IκBα were elevated compared to those in control tissues (data not shown). The levels of p50 expression varied slightly among control and glioma samples but were elevated in one glioma sample (Fig. 1A, lane 16). Total p65 levels were comparable among control samples but were considerably higher in five of eight glioma samples (Fig. 1A, lanes 10, 11, 13, 14, and 16). To extend these findings, an additional 8 nonredundant control and 13 glioma tissue samples were evaluated. Using immunoblot analyses, p-p65 (S536) was not observed with any of the control tissues, although p-p65 (S276) was detected in three control tissues (data summarized in Fig. 1D). In contrast, all 13 of the glioma samples demonstrated elevated p-p65 (S276) and p-p65 (S536) levels (data summarized in Fig. 1D), indicating NF-κB activation. Likewise, the levels of total p65 expression were consistent among control tissues but were noticeably elevated in all 13 glioma tissues evaluated (data summarized in Fig. 1D). These results collectively indicate that NF-κB is elevated and constitutively activated in the majority of malignant gliomas.
FIG. 1.

Status of NF-κB phosphorylation, ING4 expression, and NF-κB-regulated gene expression in human gliomas. (A) Thirty micrograms of total protein from fresh snap-frozen brain tissue resected from nonmalignant brain (Control) or patients diagnosed with gliomas (Glioma) was analyzed by immunoblotting for p-p65 (S276), p-p65 (S536), p65, p50, ING4, and GAPDH levels. Control and glioma samples were exposed to film for identical times. (B) The levels of COX-2 and MMP-9 mRNA from control and glioma samples described in the legend to panel A were analyzed by RT-PCR. The sample number corresponds to the lane number assigned to tissues analyzed in panel A. Results presented are representative of two experiments. (C) ING4 immunoreactivity was analyzed in human formalin-fixed, paraffin-embedded control and glioma tissues. Shown are representative photomicrographs of control brain tissue and glioma immunostained for ING4. The first row is stained with hematoxylin and eosin (H & E), the second row is stained with anti-ING4 antibody, and the third row is stained with normal rabbit serum. A total of six control and six glioma tissues were evaluated. (D) Summary of data shown in panels A, B, and C.
NF-κB-regulated genes COX-2 and MMP-9 are elevated in gliomas.
Constitutively activated NF-κB may induce expression of genes that promote tumorigenesis. To evaluate this, when available, the levels of COX-2 and MMP-9 mRNA from the tissue samples shown in Fig. 1A were analyzed. Both of these genes are regulated by NF-κB (3, 11, 35), and each has been implicated in various facets of gliomagenesis (16, 41). mRNA for COX-2 and MMP-9 was detected at low levels in control tissues, and the expression levels of these genes were elevated in several of the glioma tissues analyzed (shown in Fig. 1B and summarized in D). These data suggest that in some gliomas, the levels of COX-2 and MMP-9 expression are elevated and coincide with NF-κB activation.
ING4 expression is low or absent in gliomas.
ING4 expression is low or absent in human gliomas (8). To confirm this finding and to determine whether NF-κB activation correlated with ING4 levels, we analyzed the resected control and glioma tissue shown in Fig. 1A for ING4 protein expression, using immunoblot analyses. ING4 protein was detected in all eight of the control tissues but was detected only in three of the glioma samples analyzed (Fig. 1A, lanes 12, 14, and 16). When it was present, the levels of ING4 protein were reduced in gliomas compared to those in control tissues. To extend these findings, ING4 protein expression was assessed in an additional 12 nonredundant tissue samples using immunohistochemistry experiments. Although ING4 immunoreactivity was detected in the control tissue (n = 6) (Fig. 1C, first column, second row), ING4 immunoreactivity was largely absent or expressed at low levels (less than 25% of all cells) in the glioma samples analyzed (n = 6) (Fig. 1C, second column, second row). These results demonstrate that ING4 protein expression is absent or reduced in glioblastoma tissues. The data from all the control and glioma specimens are summarized in Fig. 1D.
Establishment of an inducible ING4 malignant glioma cell line.
To study the role of ING4, Tet-regulated U251-MG cell lines that inducibly express F-ING4 protein (U251-TR/F-ING4) were generated in human glioma cells. U251-MG cells were chosen as they do not express any detectable ING4 protein or mRNA. In the absence of Tet, neither endogenous nor F-ING4 expression was detected (Fig. 2A, lanes 1 and 3), confirming that U251-MG cells are ING4 null. In the presence of Tet, exogenous F-ING4 expression is induced (Fig. 2A, lanes 2 and 4). In later experiments, PMA was used to activate NF-κB and does not affect F-ING4 expression (Fig. 2, lane 4). These results were also confirmed using antibodies specific for the Flag epitope (data not shown).
FIG. 2.

ING4 reduces COX-2 and MMP-9 expression and binds NF-κB p65. (A) U251-TR/F-ING4 cells were grown in the absence (−) or presence (+) of Tet or PMA for 24 h, and total protein was analyzed by immunoblotting assays with antibodies specific for ING4 and actin. (B) The levels of endogenous ING4 in U87-MG and U118-MG cell lines were compared to exogenous F-ING4 levels in U251-TR/F-ING4 clones 7 and 210 grown in the presence of Tet (+) using immunoblot analyses. Forty micrograms of total protein from each sample was evaluated using two different ING4 antibodies (r = rabbit; g = goat). Clone 210 was chosen for studies outlined herein. (C) U251-TR/F-ING4 cells were grown in the absence or presence of Tet for 24 h and in the absence or presence of PMA for 4 h (COX-2) or 24 h (MMP-9). Total RNA was purified and analyzed by RT-PCR using primers specific for ING4, COX-2, MMP-9, and GAPDH. Densitometry was performed, and the levels of COX-2 and MMP-9 in the absence of Tet and PMA were set at 1, and the (fold) change in induction is shown. Data shown are representative of three experiments. (D) U251-MG (U251) and U251-TR/sh-p65 cells, which inducibly express shRNA specific for p65, were grown in the absence or presence of Tet for 48 h and then in the absence or presence of PMA for 24 h. Forty micrograms of total protein were analyzed by immunoblotting analyses using the antibodies specified. (E) Recombinant F-p65 and F-ING4 proteins were produced by in vitro transcription and translation. Equal volumes of reaction mixtures containing no template (−), F-p65, or F-ING4 were mixed and then immunoprecipitated using normal rabbit serum (IgG), anti-ING4, or anti-p65 antibodies. Immunoprecipitated proteins were analyzed by immunoblotting with anti-Flag antibodies. Arrowheads identify F-ING4 and F-p65 proteins. (F) U251-MG cells were transfected with a plasmid carrying p65 and either an empty plasmid (−) or a plasmid encoding F-ING4 (+). Cells were grown for 24 h, and total protein was collected and immunoprecipitated (IP) with a p65 antibody and analyzed by immunoblotting (IB) with an antibody specific for the Flag epitope. Input samples were analyzed with anti-p65 and Flag antibodies. The location of F-ING4 protein is indicated with an *. (G) U251-MG cells were grown on coverslips and transfected with a plasmid carrying either ING4-GFP or GFP alone. Cells were grown in the absence or presence of PMA for 1 h and then stained for total p65 (p65) or p-p65 (S276). Nuclei were counterstained with 4′,6′-diamidino-2-phenylindole (DAPI) and imaged using indirect immunofluorescent microscopy. The * indicates pictures where p65 and ING4 staining were merged; ‡ indicates pictures where p65, ING4, and DAPI staining were merged.
ING4 levels in U251-TR/F-ING4 cells are comparable to those in gliomas that express ING4 protein.
Next, we determined how the levels of exogenous F-ING4 expression in U251-TR cells compared with endogenous levels of ING4 in other glioma cell lines. Two U251-TR/F-ING4 clones (no. 7 and no. 210) were grown in the absence or presence of Tet for 24 h, and the levels of F-ING4 expression were compared to those of endogenous ING4 protein from U87-MG and U118-MG cells, using two different antibodies. Using a rabbit polyclonal anti-ING4 antibody, we were unable to detect ING4 expression in U87-MG cells, but we determined that the levels of F-ING4 achieved in U251-TR cells are elevated compared to that in U118-MG cells (Fig. 2B, top row). Using a goat polyclonal anti-ING4 antibody, we found that the levels of F-ING4 in U251-TR cells clone no. 210 appeared similar to those in U118-MG cells (Fig. 2B, third row). As such, we chose U251-TR/F-ING4 clone no. 210 for further studies, since this appeared to best approximate physiological ING4 levels.
ING4 inhibits COX-2 and MMP-9 expression.
A previous report has shown that ING4 inhibits the expression of COX-2 (8). To confirm and extend this result, COX-2 and MMP-9 mRNA expression was analyzed by RT-PCR. Although COX-2 and MMP-9 are both activated by NF-κB, they are differentially regulated. COX-2 is optimally expressed at 4 h of PMA stimulation, while we have determined that MMP-9 mRNA is optimally expressed at 24 h (17-19). U251-TR/F-ING4 cells were grown in the absence or presence of Tet for 24 h and in the absence or presence of PMA for 4 h to analyze COX-2 expression or for 24 h to analyze MMP-9 expression. COX-2 and MMP-9 mRNA levels are low in the absence of PMA or in the presence of ING4 alone (Fig. 2C, lanes 1 and 2), but both genes are induced by PMA stimulation (Fig. 2C, lane 3). In the presence of both PMA and ING4, levels of COX-2 and MMP-9 mRNA are diminished (Fig. 2C, compare lanes 3 and 4). ING4 mRNA is expressed upon treatment with Tet (Fig. 2C, lanes 2 and 4). Together, these data indicate that ING4 inhibits PMA-induced COX-2 and MMP-9 mRNA expression.
COX-2 expression is regulated by NF-κB.
To confirm that the induction of COX-2 by PMA is regulated by NF-κB, the levels of COX-2 protein in U251-TR cells, which inducibly express sh-p65, were analyzed. In the absence of PMA or Tet, endogenous p65 mRNA levels are stable and there is minimal expression of COX-2 protein (Fig. 2D, lane 1). When these cells were grown in the presence of Tet for 48 h, the levels of endogenous p65 protein were reduced, and the COX-2 protein level was undetectable (Fig. 2D, lane 2). COX-2 expression was increased by PMA treatment, and these levels were abrogated when endogenous p65 levels were simultaneously reduced (Fig. 2D, compare lanes 3 and 4). For comparison, the levels of the endogenous p65 and COX-2 proteins in the parental U251-MG cells (U251) are also shown (Fig. 2D, lane 5). These results confirm that NF-κB is responsible for PMA-induced COX-2 expression in U251-MG cells.
Interaction between ING4 and NF-κB p65.
Next, we wished to confirm the previous finding that ING4 and p65 interact (8). As such, an in vitro binding assay was performed. In vitro transcription and translation reactions were performed in the absence of any template or in the presence of a plasmid carrying either F-p65 or F-ING4. Equal volumes of these reaction mixtures were then incubated for 4 h and then immunoprecipitated overnight in the presence of normal rabbit serum (IgG), anti-ING4, or anti-p65 antibodies. Samples were then analyzed by immunoblotting using anti-Flag antibodies to allow for the simultaneous detection of both ING4 and p65. As shown in Fig. 2E, normal rabbit serum failed to immunoprecipitate F-p65 or F-ING4 (Fig. 2E, lanes 1 to 4). In contrast, when anti-ING4 antibodies were used, a 29-kDa protein corresponding to F-ING4 was immunoprecipitated (Fig. 2E, lanes 7 and 8). Although F-p65 was not immunoprecipitated by anti-ING4 antibodies in the absence of recombinant F-ING4 (Fig. 2E, lane 6), F-p65 was detected in samples that also contained F-ING4 (Fig. 2E, lane 8). To confirm these results, similar experiments were performed using anti-p65 antibodies. Although F-p65 was immunoprecipitated from samples containing recombinant F-p65 (Fig. 2E, lanes 10 and 12), F-ING4 was detected only in samples containing F-p65 (Fig. 2E, lane 12). These data indicate that ING4 and p65 interact in vitro.
To confirm these results in vivo, parental ING4-null U251-MG cells were transfected with a plasmid carrying p65 and either an empty plasmid or a plasmid carrying F-ING4. Cell lysates were immunoprecipitated using antibodies specific for p65 and were analyzed by immunoblotting with antibody against Flag. A protein band corresponding to the predicted molecular mass of F-ING4 (29 kDa) was immunoprecipitated with p65 in cells transfected with F-ING4 (Fig. 2F, lane 2) but not from cells transfected with an empty vector (Fig. 2F, lane 1).
ING4 does not alter activation or nuclear localization of NF-κB p65.
Through interactions with IκB, NF-κB can accumulate in the cytoplasm. To address whether ING4 similarly affects NF-κB, U251-MG cells were transfected with a plasmid carrying ING4 fused to GFP (ING4-GFP) or with a plasmid expressing GFP alone (GFP). Cells were allowed to recover for 24 h, serum starved, and then stimulated with PMA for 1 h. Expression levels of ING4-GFP or GFP and total p65 and p-p65 (S276) were evaluated by indirect immunofluorescence. In the absence of stimuli, ING4-GFP is detected in the nucleus, while p65 is predominantly cytoplasmic, with moderate levels of nuclear p-p65 (S276) (Fig. 2G, rows 1 and 3). Upon stimulation with PMA, p65 accumulates in the nucleus, and the levels of p-p65 (S276) are elevated (Fig. 2G, rows 2 and 4). These patterns were similar to those experiments in which only GFP was expressed (Fig. 2G, rows 5 to 8), indicating that ING4 does not alter NF-κB activation or translocation to the nucleus.
ING4 alters the transcriptional programs of COX-2 and MMP-9.
The effect of ING4 expression at the endogenous COX-2 and MMP-9 promoters was analyzed using ChIP assays (Fig. 3A). In the absence of stimuli or in the presence of ING4 alone, little p65 is detected at either the COX-2 (Fig. 3A, lanes 1 and 2) or MMP-9 promoter (Fig. 3A, lanes 5 and 6). Upon PMA stimulation, the levels of p65 were increased at both promoters (Fig. 3A, lanes 3 and 7) and do not appear to be affected by ING4 expression (Fig. 3A, lanes 4 and 8). This suggests that ING4 does not prevent NF-κB p65 from binding to the endogenous COX-2 and MMP-9 promoters.
FIG. 3.

ING4 inhibits posttranslational modifications to and interactions with NF-κB at target gene promoters. (A) U251-TR/F-ING4 cells were grown in the absence (−) or presence (+) of Tet for 24 h and then in the absence or presence of PMA for 1 h. ChIP samples were analyzed with antibodies against p65, p-p65 (S536), p50, ING4, p300, HDAC-1, Ac-H3, Ac-H4, RNA Pol II, pS2-RNA Pol II, pS5-RNA Pol II, CDK9, or cyclin T1. ChIP samples were immunoprecipitated with isotype controls (IgG). Prior to immunoprecipitation, equal amounts of sonicated DNA were analyzed by PCR (Input). Results shown are representative of three experiments. (B) U251-TR/F-ING4 cells were grown as described in the legend to panel A and analyzed by immunoblotting using antibodies against ING4 and GAPDH. (C) U251-TR/F-ING4 cells were grown in the absence or presence of Tet for 24 h and then in the absence or presence of TNF-α for 1 h. ChIP samples were analyzed using the antibodies specified.
To determine whether ING4 affected changes in nuclear p-p65 (S276) levels, samples were analyzed using antibodies specific for p-p65 (S276). Unfortunately, in our hands, we were unable to detect p-p65 (S276) at either promoter, using this antibody. It is possible that in vivo, this epitope is masked through protein-protein interactions. However, we were able to analyze the levels of p-p65 (S536), which also correlates with NF-κB activation (44, 45). In the absence of stimuli or in the presence of ING4 alone, no p-p65 (S536) is detected at the COX-2 promoter (Fig. 3A, lanes 1 and 2) or MMP-9 promoter (Fig. 3A, lanes 5 and 6). Upon stimulation with PMA, the levels of p-p65 (S536) are increased at both promoters (Fig. 3A, lanes 3 and 7). However, in the presence of both PMA and ING4, the levels of p-p65 (S536) are reduced (Fig. 3A, lanes 4 and 8). Because ING4 did not reduce the total levels of p65 at either the COX-2 or MMP-9 promoter but rather reduced the levels of p-p65 (S536), these data indicate that ING4 reduces the levels of phosphorylation to p65, while p65 is bound to a promoter.
To determine whether ING4 affected p50, samples were analyzed using anti-p50 antibody (Fig. 3A). In the absence of stimuli, p50 is not detected at the COX-2 promoter (Fig. 3A, lane 1) but is detected at the MMP-9 promoter (Fig. 3A, lane 5). Interestingly, in the presence of ING4 alone, p50 levels were reduced at the MMP-9 promoter (Fig. 3A, lane 6). Furthermore, although p50 is detected at both promoters in the presence of PMA (Fig. 3A, lanes 3 and 7), coexpression of ING4 notably diminished the levels of p50 (Fig. 3A, lanes 4 and 8). These data suggest that ING4 may alter p50's ability to bind DNA.
Next, we determined whether ING4 is present at NF-κB-regulated promoters. Although ING4 is not detected at either the COX-2 or the MMP-9 promoter in the absence of stimuli (Fig. 3A, lanes 1 and 5) or in the presence of PMA alone (Fig. 3A, lanes 3 and 7), moderate levels of ING4 are detected at the MMP-9 promoter upon ING4 expression (Fig. 3A, lane 6). Interestingly, in the presence of both PMA and ING4, elevated levels of ING4 were detected at both the COX-2 and MMP-9 promoters (Fig. 3A, lanes 4 and 8). To confirm that these results were due to properly induced ING4 expression, the ING4 protein was analyzed by immunoblotting, which demonstrated that ING4 was expressed when cells were grown in the presence of Tet (Fig. 3B, lanes 2 and 4). Collectively, these data suggest that ING4 associates with the COX-2 and MMP-9 promoters in vivo.
As described above, we determined that ING4 is present at NF-κB-regulated promoters and can reduce the nuclear levels of p-p65 (data not shown) and p-p65 (S536) bound to DNA (Fig. 3A). Because unphosphorylated p65 interacts with HDAC-1 and p-p65 interacts with CBP/p300 (44, 45), ChIP samples were analyzed using antibodies specific for p300 and HDAC-1. In the absence of PMA (Fig. 3A, lanes 1 and 5) or in the presence of ING4 alone (Fig. 3A, lanes 2 and 6), both promoters are bound by HDAC-1 but not by p300. Upon stimulation with PMA, HDAC-1 levels were reduced and p300 was recruited to these promoters (Fig. 3A, lanes 3 and 7). Interestingly, in the presence of PMA and ING4, the levels of p300 are reduced and HDAC-1 is present at both promoters (Fig. 3A, lanes 4 and 8). These results indicate that ING4 increases the levels of HDAC-1 and reduces the levels of p300 present at NF-κB-regulated promoters. Because ING4 seems to favor HDAC-1 binding over p300 binding, we hypothesized that histone acetylation at these promoters would be altered by ING4. As such, ChIP samples were analyzed using antibodies specific for acetylated histones 3 (Ac-H3) and Ac-H4. In the absence of stimuli, the MMP-9 promoter demonstrates low levels of Ac-H3 and Ac-H4 (Fig. 3A, lane 5), while the COX-2 promoter exhibits moderate levels of Ac-H3 and Ac-H4 (Fig. 3A, lane 1). In response to PMA treatment, the levels of Ac-H3 increase at both promoters (Fig. 3A, lanes 3 and 7), and the levels of Ac-H4 increase at the MMP-9 promoter (Fig. 3A, lane 7). However, the levels of both Ac-H3 and Ac-H4 are substantially reduced in the presence of both PMA and ING4 at both promoters (Fig. 3A, lanes 4 and 8). Therefore, by reducing p-p65 levels, ING4 may promote p65-HDAC-1 interactions and histone deacetylation.
ChIP samples were next analyzed using antibodies for RNA Pol II or RNA Pol II phosphorylated at serine residue 2 (pS2-RNA Pol II) and serine 5 (pS5-RNA Pol II). Although RNA Pol II can loosely associate with DNA, RNA Pol II is activated and transcribing only when phosphorylated at S5 (transcriptional initiation) and S2 (transcriptional elongation) within its C-terminal domain (32). RNA Pol II is present at the COX-2 promoter in the absence of PMA stimulation (Fig. 3A, lane 1) and is present at both promoters in the presence of ING4 or of PMA alone (Fig. 3A, lanes 2, 3, 6, and 7). However, in the presence of both PMA and ING4, both promoters show decreases in RNA Pol II binding (Fig. 3A, lanes 4 and 8). Although pS2-RNA Pol II was not detected at either promoter in the absence of PMA (Fig. 3A, lanes 1 and 4), we did detect moderate levels of pS5-RNA Pol II at both the COX-2 promoter (Fig. 3A, lane 1) and the MMP-9 promoter (Fig. 3A, lane 5). Of note, ING4 expression alone was sufficient to reduce the levels of pS5-RNA Pol II at both promoters (Fig. 3A, lanes 2 and 6). Moreover, while both promoters showed increased levels of pS2-RNA Pol II and pS5-RNA Pol II in response to PMA stimulation (Fig. 3A, lanes 3 and 7), these levels were reduced by the coexpression of ING4 (Fig. 3A, lanes 4 and 8).
RNA Pol II is phosphorylated on S2 and S5 by pTef-b, a cyclin-dependent kinase composed of two subunits, cyclin T1 and CDK9 (31). At NF-κB-regulated promoters, NF-κB interacts with cyclin T1 to induce RNA Pol II activation (1). To assess whether ING4-mediated reductions in RNA Pol II phosphorylation were due to changes in pTef-b binding, we analyzed samples by using antibodies against cyclin T1 and CDK9. CDK9 was detected at the MMP-9 promoter in the absence of PMA (Fig. 3A, lane 5) but was reduced by expression of ING4 alone (Fig. 3A, lane 6). In the presence of PMA, CDK9 was detected at both the COX-2 and the MMP-9 promoters (Fig. 3A, lanes 3 and 7); however, upon coexpression of ING4, the levels of CDK9 at either promoter were reduced (Fig. 3A, lanes 4 and 8). Alone, CDK9 is inactive as a kinase and requires binding of cyclin T1 to stimulate phosphorylation of RNA Pol II (21). In the absence of PMA or in the presence of ING4 alone, cyclin T1 was not associated with the COX-2 (Fig. 3A, lanes 1 and 2) or the MMP-9 promoter (Fig. 3A, lanes 5 and 6). Upon PMA stimulation, cyclin T1 was recruited to both promoters (Fig. 3A, lanes 3 and 7), and this was inhibited by the coexpression of ING4 (Fig. 3A, lanes 4 and 8). Immunoblotting was performed to demonstrate that the effects of ING4 at the COX-2 promoter were not due to reductions in the protein levels (data not shown). These data indicate that ING4 inhibits RNA Pol II phosphorylation by reducing levels of pTef-b at NF-κB promoters. The results of all the ChIP experiments were quantitated by densitometry (see Fig. S1 in the supplemental material).
These experiments were repeated in part using TNF-α, a more physiological stimulus for NF-κB activation. U251-TR/F-ING4 cells were grown in the absence or presence of Tet for 24 h and then in the absence or presence of TNF-α for 1 h. As shown in Fig. 3C, the levels of p65 at the COX-2 promoter are minimal in the absence of Tet (Fig. 3C, lane 1) or TNF-α (Fig. 3C, lane 2). In the presence of TNF-α, the levels of p65 are increased. As described before, the levels of p65 were not altered by coexpression of F-ING4 (Fig. 3C, lanes 3 and 4). For p50, stimulation with TNF-α led to recruitment (Fig. 3C, lane 3), and coexpression of F-ING4 reduced p50 levels (Fig. 3C, lane 4). ChIP samples were also analyzed using antibodies specific for ING4. ING4 was not detected at the COX-2 promoter in the absence of TNF-α or in the presence of either Tet or TNF-α alone (Fig. 3C, lanes 1 to 3). However, in the presence of both TNF-α and Tet, ING4 was detected (Fig. 3C, lane 4). To confirm that the presence of ING4 affected the COX-2 promoter in a fashion similar to that described above, ChIP samples were analyzed using antibodies specific for HDAC-1. In the absence of TNF-α, HDAC-1 is present at the COX-2 promoter (Fig. 3C, lane 1). Interestingly, the levels of HDAC-1 increased upon expression of F-ING4 alone (Fig. 3C, lane 2). However, upon stimulation with TNF-α, the levels of HDAC-1 were diminished (Fig. 3C, lane 3), and in the presence of both TNF-α and ING4, the levels of HDAC-1 were elevated compared to that in TNF-α alone (Fig. 3C, compare lanes 3 and 4). Because these results are consistent with those presented in Fig. 3A, it suggests that ING4 alters events at the COX-2 promoter, regardless of how NF-κB is activated. The results of these ChIP experiments were quantitated by densitometry (see Fig. S2 in the supplemental material).
ING4 does not inhibit the formation of all p65-p300 complexes.
One possible explanation for the findings described above is that an interaction between ING4 and p65 may sterically prevent interactions between p65 and another protein such as p300. As such, we analyzed p65 protein interactions, using coimmunoprecipitation assays. In the absence of PMA or in the presence of ING4 alone, no interaction between p65 and p300 was detected (Fig. 4A, lanes 1 and 2). An interaction between p65 and p300 was detected upon PMA stimulation (Fig. 4A, lane 3); however, this interaction was not grossly altered by the coexpression of ING4 (Fig. 4A, lane 4). Input samples were analyzed by immunoblotting with antibodies against p65 and ING4. These data suggest that ING4 does not grossly disrupt interactions between p65 and p300.
FIG. 4.

ING4 specifically targets promoters bound by NF-κB. (A) U251-TR/F-ING4 cells were grown in the absence (−) or presence (+) of Tet for 24 h and then in the absence or presence of PMA for 1 h. Total protein was collected and immunoprecipitated with antibodies specific for p65. Input and immunoprecipitated proteins were analyzed by immunoblotting using the antibodies specified. (B) U251-TR/F-ING4 cells were grown in the absence or presence of Tet for 24 h and then in the absence or presence of PMA for 1 h. ChIP samples were analyzed using antibodies against p65 (rows 1 to 3) or H3-Me3K4 (row 4) (1° IP). For re-ChIP, immunoprecipitated complexes were extensively washed, eluted, rediluted with immunoprecipitation buffer, and reimmunoprecipitated with antibodies specific for ING4 (rows 1 and 4), p300 (row 2), or HDAC-1 (row 3) (2° IP) and analyzed using COX-2 primers. ChIP samples were prepared as described above and immunoprecipitated with antibodies specific for H3-Me3K4 or isotype controls (IgG). Prior to immunoprecipitation, equal amounts of sonicated DNA were analyzed by PCR (Input). Results shown are representative of two experiments.
ING4 and p65 bind to the COX-2 promoter and promote interactions between p65 and HDAC-1 while inhibiting p65-p300 interactions.
Thus far, we show that ING4 does not prevent p65 from binding to DNA (Fig. 3A, lanes 4 and 8; Fig. 3C, lane 4) and does not disrupt all interactions between p65 and p300 (Fig. 4A, lane 4). However, ING4 does reduce the levels of p-p65 bound to the DNA (Fig. 3A, lanes 4 and 8). Additionally, ING4 reduces binding of p300 in favor of HDAC-1 recruitment (Fig. 3A, lanes 4 and 8). To reconcile these apparent discrepancies, we hypothesized that ING4 may only disrupt p65-p300 interactions when they are bound to the DNA. To assess this possibility, p65-p300-DNA interactions at the COX-2 promoter were analyzed using re-ChIP experiments (Fig. 4B). Initially, samples were immunoprecipitated using p65 antibodies (first round of IP). ChIP complexes were then eluted and reimmunoprecipitated with antibody specific for ING4, p300, or HDAC-1 (second round of IP). No p65-ING4 or p65-p300 complexes were detected at the COX-2 promoter in the absence of PMA stimulation (Fig. 4B, top three panels, lane 1) or upon the ING4 expression alone (Fig. 4B, lane 2). However, upon PMA stimulation, p65 and p300 were detected at the COX-2 promoter (Fig. 4B, lane 3), which was reduced upon ING4 expression (Fig. 4B, lane 4). Conversely, p65 and ING4 were not detected (Fig. 4B, lanes 1 to 3) unless both PMA and ING4 were present (Fig. 4B, lane 4). To assess the potential effects of ING4 on p65 and HDAC-1, samples were analyzed by re-ChIP using p65 and HDAC-1 antibodies (Fig. 4B). In the absence of PMA or in the presence of ING4 alone (Fig. 4B, lanes 1 and 2), p65 and HDAC-1 were detected at the COX-2 promoter. PMA stimulation reduced the levels of p65 and HDAC-1 (Fig. 4B, lane 3), while the coexpression of ING4 appeared to restore and/or enhance p65 and HDAC-1 levels (Fig. 4B, lane 4). Comparable results were obtained for the MMP-9 promoter (data not shown). Together, these data indicate that ING4 enhances the levels of p65 and HDAC-1 and reduces the levels of p65 and p300 at NF-κB-regulated promoters. These results were quantitated by densitometry (see Fig. S3 in the supplemental material).
ING4 binds to the COX-2 promoter, which contains H3-Me3K4 residues.
As described above, our data suggest that ING4 may preferentially bind to promoters that are also bound by p65. Therefore, we hypothesized that ING4 may target these promoters by recognizing p65 and a second motif or protein that is present at the DNA. Recently, studies demonstrated that ING proteins recognize H3-Me3K4 residues using the PHD domain (22, 29, 30, 37, 39). Therefore, ING4 may utilize the PHD to recognize H3-Me3K4 residues (29, 30) and a separate domain to recognize NF-κB p65. To test this, re-ChIP experiments were performed using antibodies specific for H3-Me3K4 (first round of IP) and ING4 (second round of IP). In the absence of stimuli (Fig. 4B, middle panel, lane 1), in the presence of only ING4 (Fig. 4B, lane 2), or upon PMA stimulation (Fig. 4B, lane 3), little ING4 association with H3-Me3K4 was detected. However, in the presence of both PMA and ING4, ING4 and H3-Me3K4 were detected (Fig. 4B, lane 4), suggesting that ING4 is present at promoters that also contain H3-Me3K4. These results were quantitated by densitometry (see Fig. S3 in the supplemental material).
ING4 binding coincides with reductions in the levels of H3-Me3K4 at the COX-2 promoter.
Like histone acetylation, methylation of histones is a posttranslational modification that can regulate gene expression (33, 34). Therefore, the presence of ING4 may alter the H3-Me3K4 levels to attenuate transcription. To determine whether the presence of ING4 affected H3-Me3K4 levels at the COX-2 promoter, ChIP samples were analyzed using antibodies specific for this modification. In the absence of any stimuli, the COX-2 promoter harbors moderate H3-Me3K4 levels (Fig. 4B, bottom panels, lane 1). In the presence of ING4 alone, the levels of H3-Me3K4 at the COX-2 promoter were not affected (Fig. 4B, lane 2), and PMA stimulation did not alter these levels (Fig. 4B, lane 3). However, in the presence of PMA and ING4, the levels of H3-Me3K4 were reduced (Fig. 4B, lane 4). Similar results were obtained for the MMP-9 promoter (data not shown). Therefore, because H3-Me3K4 is an indicator of active gene expression and histone acetylation (2, 36), these data indicate that ING4 may promote demethylation at lysine 4 of histone 3 to attenuate COX-2 and MMP-9 expression. These results were quantitated by densitometry (see Fig. S3 in the supplemental material).
Endogenous ING4 is recruited to the COX-2 promoter.
Thus far, our studies have utilized U251-TR cells that express exogenous ING4. As such, we addressed how reductions in the levels of endogenous ING4 affected an NF-κB-regulated gene such as COX-2. U118-MG cells, which express ING4 (Fig. 2B), were grown in the absence or presence of PMA for 0.5, 1, 2, 4, or 24 h, and samples were analyzed using ChIP assays. As shown in Fig. 5A, the levels of p65 detected at the COX-2 promoter are low (Fig. 5A, lane 1) in the absence of PMA. However, upon stimulation with PMA, the levels of p65 increased at 1 h, peaked at 4 h, and are diminished by 24 h (Fig. 5A, lanes 2 to 6). Additional samples were analyzed using antibodies specific for ING4. In the absence of PMA, there are moderate levels of ING4 present (Fig. 5A, lane 1), which are not altered by PMA stimulation until 2 to 4 h poststimulation, whereupon the levels of ING4 increase (Fig. 5A, lanes 4 and 5). These data demonstrate that endogenous ING4 binds to the promoter of an NF-κB-regulated gene and coincides with events that correlate with reduced gene expression. Interestingly, by 24 h poststimulation, ING4 levels are dramatically reduced (Fig. 5A, lane 6). To determine whether endogenous ING4 correlates with diminished levels of histone acetylation, additional samples were analyzed using antibodies specific for Ac-H3. The COX-2 promoter harbors moderate levels of Ac-H3 in the absence of PMA (Fig. 5A, lane 1), and these levels appear relatively unaffected immediately poststimulation. However, at 2 h poststimulation, the levels of Ac-H3 are diminished (Fig. 5A, lane 4), which coincides with optimal ING4 binding to the COX-2 promoter. We also evaluated the effects of endogenous ING4 on the levels of H3-Me3K4 present at the COX-2 promoter. In the unstimulated state, the levels of H3-Me3K4 are moderate and are immediately reduced upon PMA stimulation (Fig. 5A, compare lanes 1 and 2). These levels remain low until 2 h and appear to recover at 4 to 24 h post-PMA stimulation. The fact that the levels of H3-Me3K4 and ING4 are inversely correlated suggests that while present, ING4 may recruit a demethylating enzyme to reduce the levels of H3-Me3K4 at the COX-2 promoter. The results of these ChIP assays were quantitated by densitometry (see Fig. S4 in the supplemental material).
FIG. 5.

Endogenous ING4 is recruited to the COX-2 promoter. (A) U118-MG cells were grown in the absence (−) or presence (+) of PMA for 0, 0.5, 1, 2, 4, or 24 h. ChIP samples were prepared as described above and immunoprecipitated with antibodies specific for p65, ING4, Ac-H3, H3-Me3K4, or isotype controls (IgG). Prior to immunoprecipitation, equal amounts of sonicated DNA were analyzed by PCR (Input). Results shown are representative of two experiments. (B) Parental U118-MG cells or U118-MG cells that stably express shRNA specific for ING4 (clones 149-3, 149-7, and 498-11) were grown in serum containing medium for 24 h and then 75 μg of total protein was analyzed by immunoblotting using antibodies as described above. (C) Parental U118-MG cells or the 149-3 and 498-11 cell lines were grown in serum containing medium overnight and then serum starved for 24 h. Samples were left untreated (−) or stimulated with PMA for 0, 0.5, 1, 2, or 3 h to study COX-2 mRNA expression or for 3 or 24 h to study MMP-9 mRNA expression. Total RNA was collected, and the levels of COX-2 and MMP-9 mRNA were analyzed using RT-PCR. (D) U118-MG cells or cell lines 149-3 and 498-11 were grown in serum containing medium for 24 h and then serum starved overnight. These cells were left untreated (−) or stimulated with PMA for 1 or 2 h. ChIP samples were analyzed with antibodies against p65, p-p65 (S536), p300, ING4, Ac-H3, or isotype control (IgG). Prior to immunoprecipitation, equal amounts of sonicated DNA were analyzed by PCR (Input).
Establishment of shING4 glioma cell lines.
Stable U118-MG glioma cell lines that constitutively express shRNA molecules specific for ING4 were generated using two different shING4 molecules (no. 149 and 498) designed to target distinct regions of the ING4 ORF. To compare the levels of ING4 protein in parental U118-MG cells with those in the knockdown cell lines, immunoblot analyses were performed. However, to ensure that we could visualize the effects of ING4 shRNA on ING4 protein levels, we analyzed a larger amount of protein (75 μg) than before (Fig. 2B). As shown in Fig. 5B, the use of shING4 molecule 149 reduced ING4 protein levels by approximately 50% (Fig. 5B, lanes 2 and 3), while shING4 molecule 498 reduced the levels of ING4 protein by nearly 100% (Fig. 5B, lane 4). As such, we chose to use both the 149 clone 3 (149-3) line and the 498 clone 11 (498-11) line to determine the effects of reduced ING4 protein expression on NF-κB activity.
Reducing the levels of ING4 protein accelerates COX-2 and MMP-9 expression in a dose-dependent manner.
We hypothesized that reductions in ING4 protein levels would affect the levels of COX-2 and MMP-9 mRNA. To address this, parental U118-MG cells or clones 149-3 and 498-11 were grown in serum-free medium and then in the absence or presence of PMA for 0.5, 1, 2, or 3 h to study COX-2 expression or for 3 or 24 h to study MMP-9 expression. Total RNA was collected, and COX-2 and MMP-9 mRNA levels were evaluated by RT-PCR. In U118-MG cells, low levels of COX-2 mRNA are detected in the absence of any stimulus and are increased by PMA at 3 h (Fig. 5C, first panel, top row). Basal MMP-9 mRNA levels are minimal but increase by 24 h in response to PMA (Fig. 5C, bottom row). When the levels of COX-2 and MMP-9 mRNA were analyzed in the 149-3 cells, which contain approximately one-half the level of ING4 protein, PMA induction of COX-2 and MMP-9 occurred at earlier time points (2 h and 3 h, respectively) (Fig. 5C, middle panel). We also analyzed the levels of COX-2 and MMP-9 mRNA in the 498-11 cells, which express the lowest levels of ING4 protein. As shown in Fig. 5C (panel at right, first and second rows), COX-2 and MMP-9 mRNA levels were increased and induced at earlier time points compared to those in parental U118-MG cells. Thus, these data indicate that ING4 limits NF-κB-mediated expression of these genes.
Reducing the levels of ING4 protein correlates with an increase in positive transcriptional events at the COX-2 promoter in vivo.
Using the knockdown and parental U118-MG cells, we sought to determine whether reducing endogenous ING4 protein levels complemented the data obtained using the overexpression system. As such, ChIP assays were performed with U118-MG, 149-3, or 498-11 cells grown in the absence or presence of PMA for 1 or 2 h. These time points were chosen since changes occur at the COX-2 promoter during this time frame (Fig. 5A, lanes 3 and 4). Initially, we evaluated samples from each cell line by using antibodies for p65. As shown in Fig. 5D, reductions in ING4 protein increased p65 binding to the COX-2 promoter in the absence of any stimuli but did not affect p65 binding at either 1 or 2 h poststimulation (Fig. 5D, top row). In contrast, reductions in ING4 protein did affect the levels of p-p65 (S536) in a dose-dependent manner (Fig. 5D, second row). In particular, the highest levels of p-p65 (S536) were seen at 1 h poststimulation (Fig. 5D, lane 2) in U118-MG cells. In contrast, analyses of the 149-3 (Fig. 5D, lanes 5 and 6) and 498-11 (Fig. 5D, lanes 8 and 9) cells demonstrated elevated and prolonged p-p65 levels. To determine whether these events correlated with changes in the levels of p300, samples were evaluated using antibodies specific for this protein. In the parental U118-MG cells, p300 is present at the COX-2 promoter at 1 h (Fig. 5D, lane 2) but absent at 2 h (Fig. 5D, third row, lane 3). Interestingly, the levels of p300 in both the 149-3 and the 498-11 cells are elevated and prolonged. We next analyzed samples using antibodies specific for ING4 to determine whether the shRNA was effectively reducing the levels of ING4 present at the COX-2 promoter. In the parental U118-MG cells, ING4 binding was increased at 1 h (Fig. 5D, lane 2) and maximal at 2 h poststimulation (Fig. 5D, fourth row, lane 3). In contrast, in the 149-3 cell line, ING4 binding was most obvious at 1 h and was then reduced by 2 h, while no ING4 protein was detected at the COX-2 promoter in the 498-11 cells. Finally, we hypothesized that in the absence of ING4, elevated levels of phosphorylated active NF-κB were bound to p300 while bound to the DNA and thus were able to acetylate nearby histones. Samples were analyzed using antibodies specific for Ac-H3. In the parental cells, Ac-H3 levels were moderate in the untreated and 1-h samples (Fig. 5D, lanes 1 and 2), and these levels were notably reduced by 2 h (Fig. 5D, fifth row, lane 3). However, samples from both the 149-3 (Fig. 5D, lanes 4 to 6) and the 498-11 (Fig. 5D, lane 7 to 9) cells indicate that reductions in ING4 protein correlate with increased and prolonged Ac-H3 at the COX-2 promoter. Thus, these data indicate that ING4 is required to attenuate the transcriptional program of the COX-2 promoter in vivo. The densitometric analyses of these ChIP experiments are presented in Fig. S5 in the supplemental material.
DISCUSSION
NF-κB is a prime mediator of inflammatory and immune responses (13, 14). Normally found in a latent state, NF-κB is activated rapidly by numerous stimuli and induces the expression of genes that operate to minimize insults and eliminate infection. Although tightly regulated at multiple levels, NF-κB is constitutively activated in a number of cancers and is correlated with disease progression (14). At present, how NF-κB becomes activated or why NF-κB remains activated in cancers is unknown. Herein, we present data to show that in gliomas, NF-κB is constitutively activated and coincides with elevated levels of COX-2 and MMP-9, two NF-κB-regulated genes implicated in gliomagenesis (3, 11, 16, 41, 42). We also demonstrate that in gliomas, the ING4 protein expression is largely absent. Also, our analyses of two additional established human glioma cell lines (CH235-MG and CRT-MG) and three primary glioma cultures (0605, 0608, and 0611) revealed that ING4 expression is largely perturbed in gliomas. In particular, we found that the ING4 ORF harbored nonsense mutations in CH235-MG, CRT-MG, and 0611 cells (data not shown) and that both the 0605 and 0608 cells expressed ING4v2 mRNA, which encodes a protein that is mainly cytoplasmic (40). Therefore, because ING4 interacts with p65 and can inhibit NF-κB-induced COX-2 and MMP-9 expression (Fig. 2B), we propose that in gliomas, the absence of ING4 may enable already activated NF-κB to inappropriately induce the expression of genes that enable or promote tumorigenesis.
To ascertain how ING4 modulates NF-κB activity, we generated cell lines that inducibly express F-ING4 in an ING4-null background. Using in vitro binding assays, coimmunoprecipitation studies, immunofluorescence and immunoblot analyses, we show that an NF-κB-ING4 interaction exists but does not prevent NF-κB activation or translocation into the nucleus. As such, ChIP analyses were performed to study the effects of ING4 on the transcriptional programs of COX-2 and MMP-9. Previous studies indicate that both genes are regulated by NF-κB (11, 35) and that each gene is expressed at different times and utilizes different transcriptional programs. For example, the COX-2 promoter contains histones that are moderately acetylated in the unstimulated state, while the MMP-9 promoter has lower levels of acetylated histones (Fig. 3A). In contrast to a previous report (8), our results demonstrated that ING4 expression did not disrupt the ability of p65 to bind DNA, although ING4 did appear to reduce the levels of p50 at both the COX-2 and MMP-9 promoters. However, when p65 and ING4 are coincidentally bound to these promoters, the levels of p-p65 (S536) are reduced. These results led us to hypothesize that reductions in DNA-bound p-p65 levels may disrupt interactions between p65 and p300. Indeed, upon ING4 expression, the levels of p300 were reduced, and the levels of HDAC-1 were enhanced at the COX-2 and MMP-9 promoters. Moreover, these events coincided with reduced levels of histone acetylation, suggesting that ING4 may attenuate NF-κB activation by negatively affecting the local chromatin in order to restrict access of the DNA to transcriptional machinery. In support of this, we observed reductions in the levels of both pTef-b and activated RNA Pol II when ING4 was present, thus indicating that ING4 deactivates the transcriptional program of these genes.
Initially, we hypothesized that ING4 could mediate most of these effects by simply interfering with p65 and its ability to interact with p300. However, the results of coimmunoprecipitation experiments revealed that this interaction was not grossly affected by ING4. Therefore, we speculated that ING4 may discriminately target those p65 molecules that are bound to DNA. Recently, studies showed that ING proteins could recognize and bind H3-Me3K4 (22, 29, 30, 37, 39), thereby providing a potential mechanism by which ING4 could target only NF-κB molecules that are also bound to DNA. In particular, we hypothesized that ING4 specifically targets promoters that are bound by p65 by requiring two motifs, that is, p65 and H3-Me3K4. To address this hypothesis, we performed re-ChIP experiments to assess the effect of ING4 on p65 and either p300 or HDAC-1 while these proteins were bound to a target gene promoter. We determined that in the uninduced state, the COX-2 promoter contains low levels of p65 and HDAC-1. However, upon stimulation with PMA, the levels of p65 and HDAC-1 are diminished and the levels of p65 and p300 are increased. Interestingly, when ING4 is expressed alone, in the absence of additional stimuli, we were unable to detect ING4 at the COX-2 promoter in either the ChIP or re-ChIP experiments. Instead, ING4 was detectable only when the cells were also treated with PMA to activate NF-κB. Additional re-ChIP assays further demonstrated that when ING4 is bound to the DNA, p65 and HDAC-1 are predominately present. This implies that alone, ING4 may be insufficient to bind DNA and/or that ING4 recognizes two motifs at a promoter, that is, NF-κB and H3-Me3K4. This hypothesis would explain why ING4 is able to target NF-κB-regulated promoters. Therefore, we propose that ING4 recognizes and binds two motifs, that is, p65 and H3-MeK4, to restrict its focus to only those NF-κB molecules that are bound to DNA and adjacent to H3-MeK4 molecules. In this fashion, ING4 may recognize and initiate the attenuation of those promoters that are or have been recently activated.
The data described above were generated using cell lines designed to overexpress exogenous F-ING4. As such, we tested these hypotheses with U118-MG glioma cells, which express endogenous ING4. Using ChIP assays over a period of 24 h of PMA stimulation, we determined that the transcriptional program outlined in our overexpression studies was similar to that which occurs in the unmanipulated U118-MG cells. Next, we designed two distinct shRNA molecules specific for ING4 and demonstrated that while each shRNA molecule could reduce the levels of ING4 protein, they did so more or less effectively than one another. This valuable tool allowed us to study the dose-dependent effect of reducing ING4 protein levels on the levels of COX-2 and MMP-9 mRNA and the effects of ING4 on the transcriptional program of COX-2. In the presence of reduced ING4 protein (clone 149-3) or in the absence of ING4 (clone 498-11), COX-2 and MMP-9 mRNA appeared earlier and were elevated and prolonged in expression. Similarly, ChIP studies revealed that reduced ING4 protein levels correlated with changes at the COX-2 promoter that were likely to promote transcription, including sustained levels of p-p65 (s536), p300, and Ac-H3. Therefore, we propose that one function of ING4 is to specifically target NF-κB-regulated promoters to initiate the deactivation process, to prevent prolonged or inappropriate gene expression.
How exactly does ING4 attenuate NF-κB-mediated gene expression? From the ChIP assays, we speculate that in the unstimulated state, a gene regulated by NF-κB may be silent or expressed at low levels due to the presence of HDAC-1 and/or the absence of histone acetylation or phosphorylated RNA Pol II (Fig. 6A). However, in response to stimuli that activate NF-κB, this promoter is induced through binding of NF-κB, p300, and pTef-b (cyclin T1 and CDK9), which induces histone acetylation and RNA Pol II phosphorylation (Fig. 6B). At some time poststimulation, it is possible that ING4 recognizes and directly or indirectly binds to adjacent p65 and H3-Me3K4 molecules, and once bound, ING4 encourages several events, including (i) reductions in the levels of phosphorylated p65 and RNA Pol II, (ii) dismissal of p300, pTef-b, and perhaps p50, (iii) recruitment of HDAC-1, and (iv) reductions in the levels of acetylated and/or methylated histones (Fig. 6C). Using this model, we can envision that in gliomas, where ING4 expression is low or absent, NF-κB molecules that are already activated and bound to DNA may not be inactivated properly (Fig. 6B). Additionally, since H3-Me3K4 may function as the “molecular memory of recent transcriptional activity” (23), the absence of ING4 activity in gliomas may allow high levels of H3-Me3K4 to persist, thus ensuring that these promoters are readily reactivated. Therefore, in gliomas and other cancers where ING4 activity is absent or perturbed, NF-κB may remain inappropriately activated and bound to target gene promoters that are more receptive to transcriptional initiation. As such, we propose that ING4 may be one molecular mechanism by which NF-κB signaling is attenuated at the promoter.
FIG. 6.
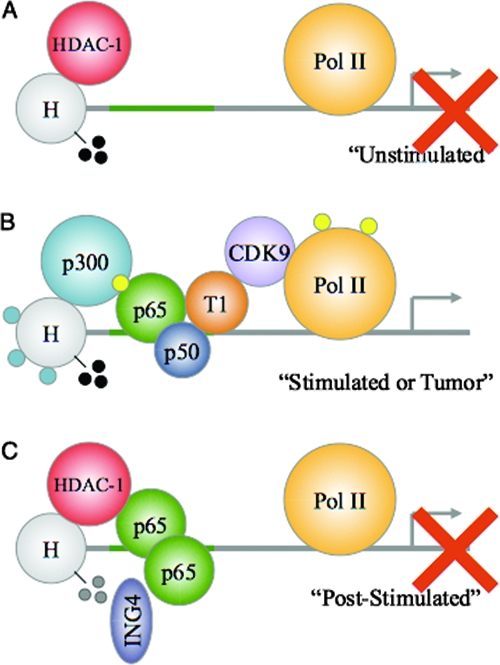
Model of ING4-mediated attenuation of NF-κB activity at target gene promoters. (A) In the unstimulated state, NF-κB-regulated promoters may be bound by HDAC-1 molecules and exhibit low levels of histone acetylation and RNA Pol II (Pol II) phosphorylation. Although these promoters may harbor H3-Me3K4 (black circles), no gene expression is occurring (red X). (B) Upon NF-κB activation (stimulated), an NF-κB molecule containing p-p65 (yellow circles) and p50 binds to the κB element within target gene promoters. HDAC-1 molecules exit the promoter, and p300 molecules interact with p-p65. This results in histone acetylation (blue circles). Additionally, pTef-b interacts with NF-κB to phosphorylate and activate Pol II, thus allowing new gene expression. In tumors, the absence of ING4 may enable aberrant NF-κB signaling. (C) After proper gene expression (poststimulated), ING4 may recognize and simultaneously bind to H3-Me3K4 and p65 molecules that are adjacently bound to DNA. Through these interactions, ING4 reduces p-p65 levels and reduces p65-p300 interactions to favor an increase in p65-HDAC-1 interactions. These interactions also reduce the levels of histone acetylation and the levels of H3-Me3K4 (gray circles), pTef-b, and Pol II phosphorylation to prevent further gene expression (red X). The absence of ING4 in tumors may prevent this necessary attenuation of NF-κB signaling, and thus, these promoters may resemble those described in the legend to panel B (see text for details).
Supplementary Material
Acknowledgments
This work was supported in part by public service grants CA-97247 from the National Cancer Institute (E.N.B. and L.B.N.), NS-54158 from the National Institute of Neurological Disorders and Stroke (E.N.B., S.N., and L.B.N.), IRG-60-001-47 from the American Cancer Society, and CA-13148-31 from the National Cancer Institute (S.N.). The UAB NMDC facility is supported by public service grant P30 NS47466. The CFAR/CCC DNA Sequencing Core is supported by NIH CFAR core grant P30AI27767.
We thank the UAB Neuroscience Molecular Detection Core Facility (NMDC) staff for their assistance with immunohistochemistry experiments. We also thank Cheryl Palmer (University of Alabama at Birmingham) for providing human brain tissue resections, G. Yancey Gillespie and the UAB Brain Tumor Tissue core for providing the glioma and control samples used herein, and Courtney J. Haycraft and Shaun Sparacio for technical assistance. Finally, we recognize the efforts of Maria G. Salazar of the CFAR/CCC DNA Sequencing Core at UAB for expert advice and technical assistance.
Footnotes
Published ahead of print on 8 September 2008.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Barboric, M., R. M. Nissen, S. Kanazawa, N. Jabrane-Ferrat, and B. M. Peterlin. 2001. NF-kappaB binds P-TEFb to stimulate transcriptional elongation by RNA polymerase II. Mol. Cell 8327-337. [DOI] [PubMed] [Google Scholar]
- 2.Bernstein, B. E., M. Kamal, K. Lindblad-Toh, S. Bekiranov, D. K. Bailey, D. J. Huebert, S. McMahon, E. K. Karlsson, E. J. Kulbokas III, T. R. Gingeras, S. L. Schreiber, and E. S. Lander. 2005. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell 120169-181. [DOI] [PubMed] [Google Scholar]
- 3.Buccoliero, A. M., A. Caldarella, C. F. Gheri, A. Taddei, M. Paglierani, M. Pepi, P. Mennonna, and G. L. Taddei. 2006. Inducible cyclooxygenase (COX-2) in glioblastoma: clinical and immunohistochemical (COX-2-VEGF) correlations. Clin. Neuropathol. 2559-66. [PubMed] [Google Scholar]
- 4.Campos, E. I., M. Y. Chin, W. H. Kuo, and G. Li. 2004. Biological functions of the ING family tumor suppressors. Cell. Mol. Life Sci. 612597-2613. [DOI] [PubMed] [Google Scholar]
- 5.Choi, C., O. Kutsch, J. Park, T. Zhou, D. W. Seol, and E. N. Benveniste. 2002. Tumor necrosis factor-related apoptosis-inducing ligand induces caspase-dependent interleukin-8 expression and apoptosis in human astroglioma cells. Mol. Cell. Biol. 22724-736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Doyon, Y., C. Cayrou, M. Ullah, A. J. Landry, V. Cote, W. Selleck, W. S. Lane, S. Tan, X. J. Yang, and J. Cote. 2006. ING tumor suppressor proteins are critical regulators of chromatin acetylation required for genome expression and perpetuation. Mol. Cell 2151-64. [DOI] [PubMed] [Google Scholar]
- 7.Furnari, F. B., T. Fenton, R. M. Bachoo, A. Mukasa, J. M. Stommel, A. Stegh, W. C. Hahn, K. L. Ligon, D. N. Louis, C. Brennan, L. Chin, R. A. DePinho, and W. K. Cavenee. 2007. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 21:2683-2710. [DOI] [PubMed] [Google Scholar]
- 8.Garkavtsev, I., S. V. Kozin, O. Chernova, L. Xu, F. Winkler, E. Brown, G. H. Barnett, and R. K. Jain. 2004. The candidate tumour suppressor protein ING4 regulates brain tumour growth and angiogenesis. Nature 428328-332. [DOI] [PubMed] [Google Scholar]
- 9.Gong, W., K. Suzuki, M. Russell, and K. Riabowol. 2005. Function of the ING family of PHD proteins in cancer. Int. J. Biochem. Cell Biol. 371054-1065. [DOI] [PubMed] [Google Scholar]
- 10.Gunduz, M., H. Nagatsuka, K. Demircan, E. Gunduz, B. Cengiz, M. Ouchida, H. Tsujigiwa, E. Yamachika, K. Fukushima, L. Beder, S. Hirohata, Y. Ninomiya, K. Nishizaki, K. Shimizu, and N. Nagai. 2005. Frequent deletion and down-regulation of ING4, a candidate tumor suppressor gene at 12p13, in head and neck squamous cell carcinomas. Gene 356109-117. [DOI] [PubMed] [Google Scholar]
- 11.He, C. 1996. Molecular mechanism of transcriptional activation of human gelatinase B by proximal promoter. Cancer Lett. 106185-191. [DOI] [PubMed] [Google Scholar]
- 12.He, G. H., C. C. Helbing, M. J. Wagner, C. W. Sensen, and K. Riabowol. 2005. Phylogenetic analysis of the ING family of PHD finger proteins. Mol. Biol. Evol. 22104-116. [DOI] [PubMed] [Google Scholar]
- 13.Hoffmann, A., G. Natoli, and G. Ghosh. 2006. Transcriptional regulation via the NF-κB signaling module. Oncogene 256706-6716. [DOI] [PubMed] [Google Scholar]
- 14.Karin, M., Y. Cao, F. R. Greten, and Z. W. Li. 2002. NF-êB in cancer: from innocent bystander to major culprit. Nat. Rev. Cancer. 2301-310. [DOI] [PubMed] [Google Scholar]
- 15.Kim, S., K. Chin, J. W. Gray, and J. M. Bishop. 2004. A screen for genes that suppress loss of contact inhibition: identification of ING4 as a candidate tumor suppressor gene in human cancer. Proc. Natl. Acad. Sci. USA 10116251-16256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lakka, S. S., S. L. Jasti, C. Gondi, D. Boyd, N. Chandrasekar, D. H. Dinh, W. C. Olivero, M. Gujrati, and J. S. Rao. 2002. Downregulation of MMP-9 in ERK-mutated stable transfectants inhibits glioma invasion in vitro. Oncogene 215601-5608. [DOI] [PubMed] [Google Scholar]
- 17.Ma, Z., M. J. Chang, R. C. Shah, and E. N. Benveniste. 2005. Interferon-gamma-activated STAT-1alpha suppresses MMP-9 gene transcription by sequestration of the coactivators CBP/p300. J. Leukoc. Biol. 78515-523. [DOI] [PubMed] [Google Scholar]
- 18.Ma, Z., H. Qin, and E. N. Benveniste. 2001. Transcriptional suppression of matrix metalloproteinase-9 gene expression by IFN-gamma and IFN-beta: critical role of STAT-1alpha. J. Immunol. 1675150-5159. [DOI] [PubMed] [Google Scholar]
- 19.Ma, Z., R. C. Shah, M. J. Chang, and E. N. Benveniste. 2004. Coordination of cell signaling, chromatin remodeling, histone modifications, and regulator recruitment in human matrix metalloproteinase 9 gene transcription. Mol. Cell. Biol. 245496-5509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maher, E. A., F. B. Furnari, R. M. Bachoo, D. H. Rowitch, D. N. Louis, W. K. Cavenee, and R. A. DePinho. 2001. Malignant glioma: genetics and biology of a grave matter. Genes Dev. 151311-1333. [DOI] [PubMed] [Google Scholar]
- 21.Marshall, R. M., and X. Grana. 2006. Mechanisms controlling CDK9 activity. Front. Biosci. 112598-2613. [DOI] [PubMed] [Google Scholar]
- 22.Martin, D. G., K. Baetz, X. Shi, K. L. Walter, V. E. MacDonald, M. J. Wlodarski, O. Gozani, P. Hieter, and L. Howe. 2006. The Yng1p plant homeodomain finger is a methyl-histone binding module that recognizes lysine 4-methylated histone H3. Mol. Cell. Biol. 267871-7879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ng, H. H., F. Robert, R. A. Young, and K. Struhl. 2003. Targeted recruitment of Set1 histone methylase by elongating Pol II provides a localized mark and memory of recent transcriptional activity. Mol. Cell 11709-719. [DOI] [PubMed] [Google Scholar]
- 24.Nowoslawski, L., B. J. Klocke, and K. A. Roth. 2005. Molecular regulation of acute ethanol-induced neuron apoptosis. J. Neuropathol. Exp. Neurol. 64490-497. [DOI] [PubMed] [Google Scholar]
- 25.Nozell, S., T. Laver, K. Patel, and E. N. Benveniste. 2006. Mechanism of IFN-beta-mediated inhibition of IL-8 gene expression in astroglioma cells. J. Immunol. 177822-830. [DOI] [PubMed] [Google Scholar]
- 26.Nozell, S., Z. Ma, C. Wilson, R. Shah, and E. N. Benveniste. 2004. Class II major histocompatibility complex transactivator (CIITA) inhibits matrix metalloproteinase-9 gene expression. J. Biol. Chem. 27938577-38589. [DOI] [PubMed] [Google Scholar]
- 27.Ozer, A., and R. K. Bruick. 2005. Regulation of HIF by prolyl hydroxylases: recruitment of the candidate tumor suppressor protein ING4. Cell Cycle 41153-1156. [DOI] [PubMed] [Google Scholar]
- 28.Ozer, A., L. C. Wu, and R. K. Bruick. 2005. The candidate tumor suppressor ING4 represses activation of the hypoxia inducible factor (HIF). Proc. Natl. Acad. Sci. USA 1027481-7486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Palacios, A., P. Garcia, D. Padro, E. Lopez-Hernandez, I. Martin, and F. J. Blanco. 2006. Solution structure and NMR characterization of the binding to methylated histone tails of the plant homeodomain finger of the tumour suppressor ING4. FEBS Lett. 5806903-6908. [DOI] [PubMed] [Google Scholar]
- 30.Pena, P. V., F. Davrazou, X. Shi, K. L. Walter, V. V. Verkhusha, O. Gozani, R. Zhao, and T. G. Kutateladze. 2006. Molecular mechanism of histone H3K4me3 recognition by plant homeodomain of ING2. Nature 442100-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peterlin, B. M., and D. H. Price. 2006. Controlling the elongation phase of transcription with P-TEFb. Mol. Cell 23297-305. [DOI] [PubMed] [Google Scholar]
- 32.Phatnani, H. P., and A. L. Greenleaf. 2006. Phosphorylation and functions of the RNA polymerase II CTD. Genes Dev. 202922-2936. [DOI] [PubMed] [Google Scholar]
- 33.Santos-Rosa, H., and C. Caldas. 2005. Chromatin modifier enzymes, the histone code and cancer. Eur. J. Cancer 412381-2402. [DOI] [PubMed] [Google Scholar]
- 34.Santos-Rosa, H., R. Schneider, A. J. Bannister, J. Sherriff, B. E. Bernstein, N. C. Emre, S. L. Schreiber, J. Mellor, and T. Kouzarides. 2002. Active genes are tri-methylated at K4 of histone H3. Nature 419407-411. [DOI] [PubMed] [Google Scholar]
- 35.Schmedtje, J. F., Jr., Y. S. Ji, W. L. Liu, R. N. DuBois, and M. S. Runge. 1997. Hypoxia induces cyclooxygenase-2 via the NF-κB p65 transcription factor in human vascular endothelial cells. J. Biol. Chem. 272601-608. [DOI] [PubMed] [Google Scholar]
- 36.Schneider, R., A. J. Bannister, F. A. Myers, A. W. Thorne, C. Crane-Robinson, and T. Kouzarides. 2004. Histone H3 lysine 4 methylation patterns in higher eukaryotic genes. Nat. Cell Biol. 673-77. [DOI] [PubMed] [Google Scholar]
- 37.Shi, X., T. Hong, K. L. Walter, M. Ewalt, E. Michishita, T. Hung, D. Carney, P. Pena, F. Lan, M. R. Kaadige, N. Lacoste, C. Cayrou, F. Davrazou, A. Saha, B. R. Cairns, D. E. Ayer, T. G. Kutateladze, Y. Shi, J. Cote, K. F. Chua, and O. Gozani. 2006. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature 44296-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shiseki, M., M. Nagashima, R. M. Pedeux, M. Kitahama-Shiseki, K. Miura, S. Okamura, H. Onogi, Y. Higashimoto, E. Appella, J. Yokota, and C. C. Harris. 2003. p29ING4 and p28ING5 bind to p53 and p300, and enhance p53 activity. Cancer Res. 632373-2378. [PubMed] [Google Scholar]
- 39.Taverna, S. D., S. Ilin, R. S. Rogers, J. C. Tanny, H. Lavender, H. Li, L. Baker, J. Boyle, L. P. Blair, B. T. Chait, D. J. Patel, J. D. Aitchison, A. J. Tackett, and C. D. Allis. 2006. Yng1 PHD finger binding to H3 trimethylated at K4 promotes NuA3 HAT activity at K14 of H3 and transcription at a subset of targeted ORFs. Mol. Cell 24785-796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Unoki, M., J. C. Shen, Z. M. Zheng, and C. C. Harris. 2006. Novel splice variants of ING4 and their possible roles in the regulation of cell growth and motility. J. Biol. Chem. 28134677-34686. [DOI] [PubMed] [Google Scholar]
- 41.Van den Steen, P. E., B. Dubois, I. Nelissen, P. M. Rudd, R. A. Dwek, and G. Opdenakker. 2002. Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9). Crit. Rev. Biochem. Mol. Biol. 37375-536. [DOI] [PubMed] [Google Scholar]
- 42.Wang, H., H. Wang, W. Zhang, H. J. Huang, W. S. Liao, and G. N. Fuller. 2004. Analysis of the activation status of Akt, NF-κB, and Stat3 in human diffuse gliomas. Lab. Investig. 84941-951. [DOI] [PubMed] [Google Scholar]
- 43.Zhang, X., K. S. Wang, Z. Q. Wang, L. S. Xu, Q. W. Wang, F. Chen, D. Z. Wei, and Z. G. Han. 2005. Nuclear localization signal of ING4 plays a key role in its binding to p53. Biochem. Biophys. Res. Commun. 3311032-1038. [DOI] [PubMed] [Google Scholar]
- 44.Zhong, H., M. J. May, E. Jimi, and S. Ghosh. 2002. The phosphorylation status of nuclear NF-êB determines its association with CBP/p300 or HDAC-1. Mol. Cell 9625-636. [DOI] [PubMed] [Google Scholar]
- 45.Zhong, H., R. E. Voll, and S. Ghosh. 1998. Phosphorylation of NF-êB p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol. Cell 1661-671. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.