

Abstract
Hepatitis C virus (HCV) infection is frequently associated with the development of hepatocellular carcinoma (HCC), which is one of the male-dominant diseases. Androgen signaling in liver may be related to carcinogenesis. In this study, we investigated whether HCV proteins cross talk with the androgen receptor (AR) signaling pathway for promotion of carcinogenesis. We have demonstrated that HCV core protein alone or in context with other HCV proteins enhances AR-mediated transcriptional activity and further augments in the presence of androgen. Subsequent study suggested that HCV core protein activates STAT3, which in turn enhances AR-mediated transcription. This activity was blocked by a pharmacological inhibitor of the Jak/Stat signaling pathway, AG490. Vascular endothelial growth factor (VEGF) is a target gene of AR in liver and plays an important role in angiogenesis. Therefore, we examined whether HCV infection modulates VEGF expression in hepatocytes. Our results demonstrated that HCV enhances VEGF expression and facilitates tube formation in human coronary microvascular endothelial cells in the presence of AR. Together, our results suggest that HCV core protein acts as a positive regulator in AR signaling, providing further insight into oncogenic potential in the development of HCC in HCV-infected individuals.
Hepatitis C virus (HCV) infection affects nearly 4 million people and is the most common cause of cirrhosis and hepatocellular carcinoma (HCC) in the United States (23). The current approved therapy for treatment for HCV is pegylated interferon in combination with ribavirin (43). Although several advances have shown promise for improving the management of HCV infection, it nevertheless remains as a major health problem (2, 7, 60). Regardless of the etiology, chronic liver diseases progress at unequal rates in the two sexes, with the major sequelae, such as cirrhosis and HCC, being more common in men than in women (16, 54). The significance of androgen receptor (AR) expression in HCC and the surrounding liver has been studied extensively (18, 31, 33). With this background, we decided to investigate the relationship between AR and HCC after HCV infection, knowing that human liver expresses both estrogen receptors and ARs.
AR is a ligand-dependent transcription factor that belongs to the nuclear receptor superfamily (14, 17). The transcriptional activation function of AR is not only essential for normal sexual development in men but is also implicated in the progression of cancer (9). AR has a modular structure containing the N terminus harboring transcriptional activation domain(s), a central DNA-binding domain, and a C-terminal ligand-binding domain. Binding of androgen to the ligand-binding domain induces conformational change in AR and leads to the shuttling of receptor from the cytoplasm to the nucleus, where it forms a homodimer and is recruited to the AR element (ARE). ARE is present in the regulatory elements on the target genes such as vascular endothelial growth factor (VEGF) and transforming growth factor β1 and regulates the growth and proliferation of hepatocytes (6, 51).
Members of the signal transducer and activator of transcription (STAT) family of transcription factors have been implicated in transformation, tumor cell survival, invasion, and metastasis (15). Activation of STAT3, in particular, has been detected in diverse malignancies, including liver (32, 52). STAT3 is also constitutively activated in immune cells in the tumor microenvironment (53). Janus-activated kinase 1 activates STAT3 by phosphorylating at Ser-727 and at Tyr-705 (8, 28, 42, 45). The cooperation of both serine and tyrosine phosphorylation is necessary for full activation of STAT3 (3, 11). Activation of the AR N-terminal domain by interleukin-6 (IL-6) via STAT3 signal pathways has been reported (48). STAT3 is known to enhance the transactivation of AR in the presence of ligand (12).
We have shown that HCV core protein induces STAT3 signaling pathway (5). HCV NS5A also activates STAT3 by inducing phosphorylation at Tyr-705 (41, 59). In the present study, we have shown that HCV core protein alone or infection with cell culture-grown HCV enhances AR activity in the presence of androgen. HCV core increased STAT3 phosphorylation both at Ser-727 and at Tyr-705, which in turn activates AR. We have also observed that HCV infection enhances VEGF mRNA expression and induces in vitro angiogenesis in the presence of androgen.
MATERIALS AND METHODS
Plasmids and reagents.
The plasmid expressing human AR protein (pSG5-AR) (26) and the AR-response element-directed luciferase reporter plasmid (pARE3-Luc) were kindly provided by Chawnshang Chang (University of Rochester, NY). HCV protein expression vectors HCV core, NS5A, and FL (amino acids 1 to 2962) generated from genotype 1a (clone H77) were described previously (40, 41). 5α-Androgen-17β-ol-3-one (DHT; Sigma-Aldrich, St. Louis, MO) and kinase inhibitors AG490 and LY294002 (Calbiochem, San Diego, CA) were purchased. The reagents were dissolved in dimethyl sulfoxide or ethanol and were added to cell culture medium at appropriate dilutions.
Cell lines.
Human hepatoma cell lines (Huh-7 and HepG2), immortalized human hepatocytes (IHH), and prostate cancer cells (PC-3) were used as described previously (21). Cells were maintained in Dulbecco modified Eagle medium (DMEM; Cambrex, Walkersville, MD) containing 10% fetal bovine serum, 200 U of penicillin G/ml, and 200 μg of streptomycin/ml at 37°C in a 5% CO2 incubator. Human coronary microvascular endothelial cells (HCMECs) were grown in special medium from Lonza.
Generation of cell culture-grown HCV.
HCV genotype 1a (clone H77) or genotype 2a (clone JFH1) was grown as previously described (21, 55). For experiments with HCV-infected cells, a 0.02 focus-forming unit was used.
Luciferase assay.
Huh-7 or IHH cells grown in six-well dishes were transfected with 2 μg of plasmid DNA by using Lipofectamine (Invitrogen, Carlsbad, CA). Cells were incubated in DMEM containing 0.5% fetal bovine serum after transfection, and then treated for 24 h with 10 nM DHT or its control solvent in DMEM on the second day. At 48 h posttransfection, cells were lysed with reporter lysis buffer (Promega, Madison, WI), and the luciferase activity was determined by using a luminometer (Optocomp II; MGM Instruments, Hamden, CT) as described previously (29).
RNA extraction and real-time reverse transcription-PCR (RT-PCR).
Total RNA was isolated from cells by using the RNeasy minikit (Qiagen, Valencia, CA). cDNA synthesis was performed using random hexamers. PCR amplification was performed on cDNA templates using primers specific for AR (sense primer [5′-AAGGCTATGAATGTCAGCCCA-3′] and antisense primer [5′-CATTGAGGCTAGAGAGCAAGGC-3′]) (20) and for VEGF (sense primer [5′-CGAAACCATGAACTTTCTGC-3′] and antisense primer [5′-CCTCAGTGGGCACACACTCC-3′]). The primers for GAPDH (glyceraldehyde-3-phosphate dehydrogenase) were chosen as described previously (21). For RNA quantitation, real-time PCR was performed by using a QTaq one-step qRT-PCR Sybr kit (Clontech, Mountain View, CA) with ABI Prism 7500 (Applied Biosystems, Foster City, CA). The GAPDH housekeeping gene was used for normalization, and data were analyzed by the comparative threshold cycle method. Relative quantification of gene expression using the 2−ΔΔCT method correlated with the absolute gene quantification obtained by standard curve (44). Each real-time PCR assay was performed in triplicate.
Immunoblot analysis.
Cells were lysed using sodium dodecyl sulfate sample buffer. Proteins were subjected to electrophoresis on 10% polyacrylamide gel and transferred onto a nitrocellulose membrane. The membrane was probed with an antibody for STAT3 (Santa Cruz Biotechnology, Santa Cruz, CA) or for phospho Y705-STAT3 or phospho S727STAT3 (Cell Signaling Technology, Danvers, MA). Proteins were detected by using an enhanced chemiluminescent ECL Western blot substrate (Pierce, Rockford, IL). Protein bands were scanned by image analyzer (Amersham Molecular Dynamics, Sunnyvale, CA) to quantify using ImageQuant software.
Tube formation assay.
Plastic cell culture wells were coated with Matrigel (BD Biosciences, San Jose, CA) on ice. After coating, plates were kept at 37°C for 30 min. HCMECs were mixed with conditioned media from mock-infected, AR-transfected, and/or HCV-infected Huh-7 cells and seeded on top Matrigel. After 18 h of incubation, at least three images were acquired from each well per condition by using a phase-contrast microscope with ×10 objective lens. The number and length of tubes were analyzed by using Scion image.
RESULTS
HCV enhances AR-responsive gene expression in the presence of androgen.
The AR is expressed in normal liver, but its expression and activation is increased in tumor tissue and in the surrounding liver tissue of patients with HCC (27, 33). We initially examined AR mRNA expression level by real-time RT-PCR in three different hepatic cell lines. Huh-7 cells displayed less AR expression than IHH and HepG2 cells by 9.2- and 11.5-fold, respectively (Fig. 1A). Our results are in agreement with the recent reports that differential AR expression occurs in hepatocytes (51, 61). Since Huh-7 and IHH cell lines support HCV growth (21, 55), we chose to use these two cell lines for subsequent studies.
FIG. 1.
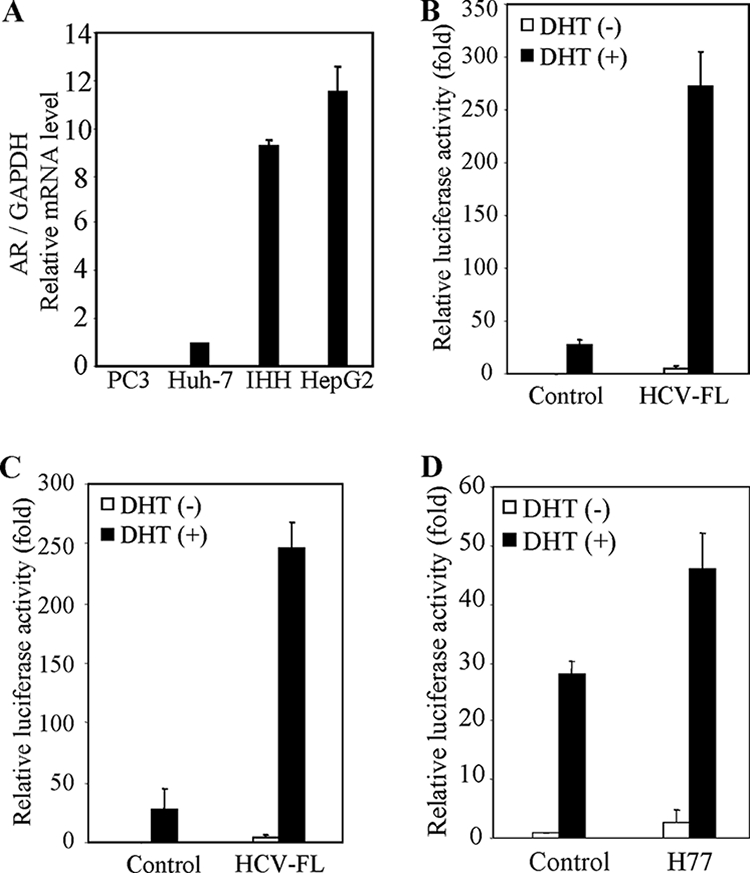
HCV infection enhances AR activation in the presence of DHT. (A) AR mRNA expression varies in different hepatocyte cell lines. RNAs from Huh-7, IHH, and HepG2 cells were subjected to real-time RT-PCR for AR and GAPDH gene expression. The PC-3 (AR-negative) cell line was used as a negative control. The results are presented as relative expression levels of AR normalized with GAPDH housekeeping gene as an internal control. The expression level of AR in Huh-7 cells was arbitrarily chosen as 1 for comparison. (B) HCV protein expression enhances androgen-dependent AR-responsive gene expression. IHH were cotransfected with pARE-luciferase reporter construct (pARE3-luc) and HCV-FL expression plasmid DNA or empty vector (negative control) in the presence or absence of DHT. Cell lysates were prepared after 48 h of transfection, and the luciferase activity was measured. The relative luciferase activity of the negative control in the absence of DHT was arbitrarily set as 1 for comparison. Luciferase activities are presented as an average from three independent experiments (mean ± the standard deviation [SD]). (C) Huh-7 cells were similarly used for transfection with HCV-FL. Experiments were performed as described in panel B for measurement of luciferase activity. (D) IHH were infected with cell culture-grown HCV (clone H77) for 24 h, followed by transfection with pARE3-luc. Experiments were performed as described in panel B for measurement of the luciferase activity. The lowest level of significance was P < 0.001.
To examine the effects of HCV proteins on the AR signaling pathway, we investigated AR-mediated transcriptional activation. The reporter gene pARE3-Luc, containing a luciferase gene linked to androgen response elements, was transfected into Huh-7 or IHH, together with pSG5-AR and HCV-FL. The cells were then treated with 10 nM DHT or left untreated. Transfection with HCV-FL enhanced a modest level of ARE-luciferase activity in both Huh-7 cells and IHH compared to the vector-transfected control (Fig. 1B and C). Interestingly, cotransfection of pSG5-AR and HCV-FL significantly increased reporter activity in the presence of DHT. We also examined whether HCV infection enhances AR activation. In the presence of androgen, the transfection of AR into HCV-infected IHH increased reporter activity compared to mock-infected cells (Fig. 1D). Similar results were also observed in Huh-7 cells (data not shown). Thus, our results suggested that HCV proteins increase AR activity.
HCV core protein is involved in AR activation.
We next examined the function of a specific HCV protein in AR activation. HCV core and NS5A proteins have been implicated in promotion of cell growth regulation (22, 24, 30, 38, 39). To examine the role of core and NS5A on AR activation, we analyzed ARE-luciferase activity in cotransfection experiments using Huh-7 cells in the presence of androgen. Cotransfection of pSG5-AR and CMV-core increased reporter activity to ∼24-fold compared to cells transfected with pSG5-AR alone (Fig. 2). However, introduction of HCV NS5A did not increase ARE reporter activity. These results suggested that HCV core protein is responsible for HCV-induced AR transcriptional activation.
FIG. 2.
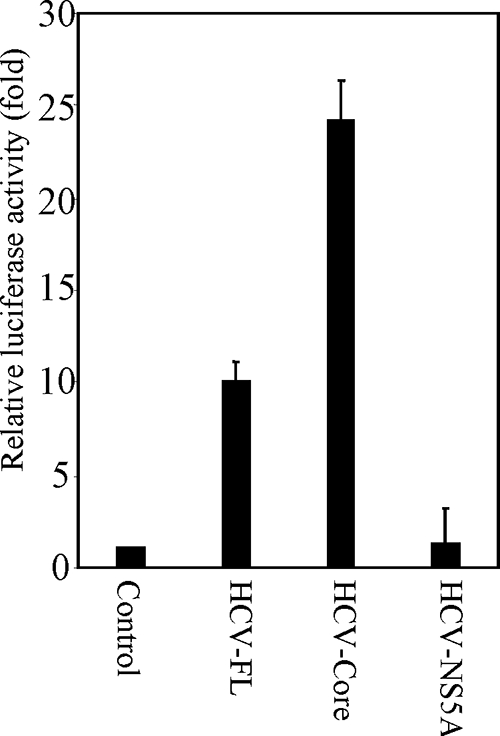
HCV core protein enhances AR activation. Huh-7 cells were transfected with pARE3-luc reporter plasmid DNA, pSG5-AR, and HCV-FL, HCV-core, HCV-NS5A, or vector alone. Cells were treated with 10 nM DHT 24 h posttransfection, and the luciferase activity was measured. The luciferase activity is presented as an average from three independent experiments (mean ± the SD). The lowest level of significance was P < 0.001.
HCV induced AR activation associates with JAK/STAT signaling pathway.
We examined a direct association between HCV core and AR by coimmunoprecipitation to delineate the mechanism underlying HCV core-mediated AR activation. Cell lysates were prepared from Huh-7 cells cotransfected with Flag-tagged core and pSG5-AR expression constructs and immunoprecipitated with Flag antibody. The blot was probed with an antibody to AR. We did not observe coprecipitation of AR and HCV core under our experimental conditions. Therefore, we postulate that HCV core protein may activate specific signaling pathway(s) that affects AR and its transcriptional activity in a ligand-independent manner. AR is activated by the phosphatidylinositol 3-kinase (PI3K)/AKT and JAK/STAT3 signaling pathway (48). We used inhibitors for PI3K/AKT (LY294002) and JAK/STAT3 (AG490) to test the involvement of a downstream kinase for the resulting AR activation. IHH or Huh-7 cells were cotransfected with HCV-FL and AR constructs and treated with specific inhibitors in the presence of DHT. The reporter activity was significantly reduced in the presence of AG490, but not with LY294002 (Fig. 3A). The enhancement HCV core-mediated ARE gene expression was also inhibited by AG490 (Fig. 3B). Huh-7 cells transiently transfected with HCV core plasmid DNA (1.8 μg) demonstrated the expression of HCV core protein by Western blot analysis (Fig. 3C). Therefore, the inhibitory effect of AG490 indicated that JAK and downstream STAT3 activation might be involved in HCV-induced AR transcriptional activation.
FIG. 3.
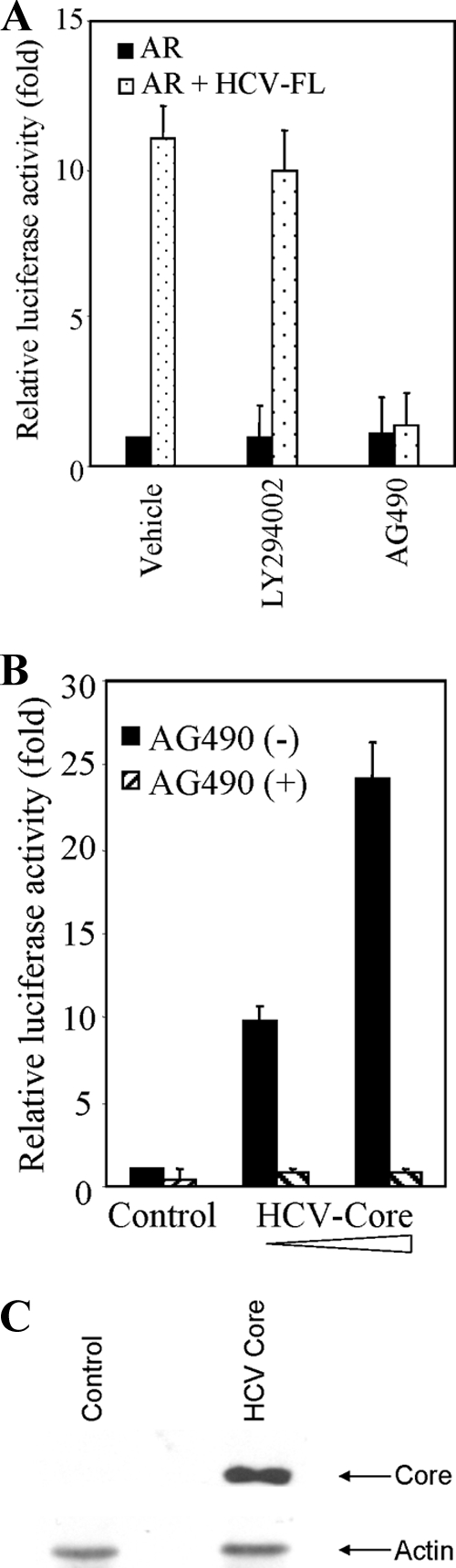
Involvement of STAT3 in HCV-induced AR activation. (A) Huh-7 cells were cotransfected with pSG5-AR and HCV-FL or vector alone. Cells are treated with 10 nM DHT and 20 nM LY294002 or 50 μM AG490. Luciferase activity was measured and is presented as an average from three independent experiments (mean ± the SD). (B) Huh-7 cells were cotransfected with pSG5-AR and HCV-core (0.6 and 1.8 μg) or vector alone (control). Transfected cells were treated with 10 nM DHT in presence or absence of 50 μM AG490, and the luciferase activity was measured. The lowest level of significance was P < 0.001. (C) Huh-7 cells were transfected with vector control or HCV-core plasmid DNA (1.8 μg), and cell lysates were analyzed after 48 h of transfection for core protein expression by Western blot analysis using a rabbit anti-core antibody. The blot was reprobed with antibody to actin as an internal control.
STAT3 augments HCV core induced AR activation.
We investigated phosphorylation status of STAT3 to understand how STAT3 signaling affects AR gene transactivation in the presence of HCV protein(s). Huh-7 or IHH were cotransfected with AR and HCV-FL, HCV-core, HCV-NS5A, or vector alone. Our results demonstrated a significant activation of S727STAT3 in the presence of HCV-FL or HCV core (Fig. 4A). On the other hand, activation of Y705STAT3 was observed from both core and NS5A-expressing cells. HCV-infected IHH also displayed phosphorylation of S727STAT3 (Fig. 4B). Therefore, phosphorylation of S727 in STAT3 appeared to be important for enhancement of AR activity. HCV core expression was observed from virus-infected cell lysates. To examine AR mRNA upregulation during HCV infection, we performed semiquantitative RT-PCR analysis from the RNA of mock- or HCV-infected IHH. The results showed that the endogenous AR mRNA is increased compared to mock-infected IHH (Fig. 4C), suggesting that HCV infection affects AR activation through STAT3 signaling.
FIG. 4.
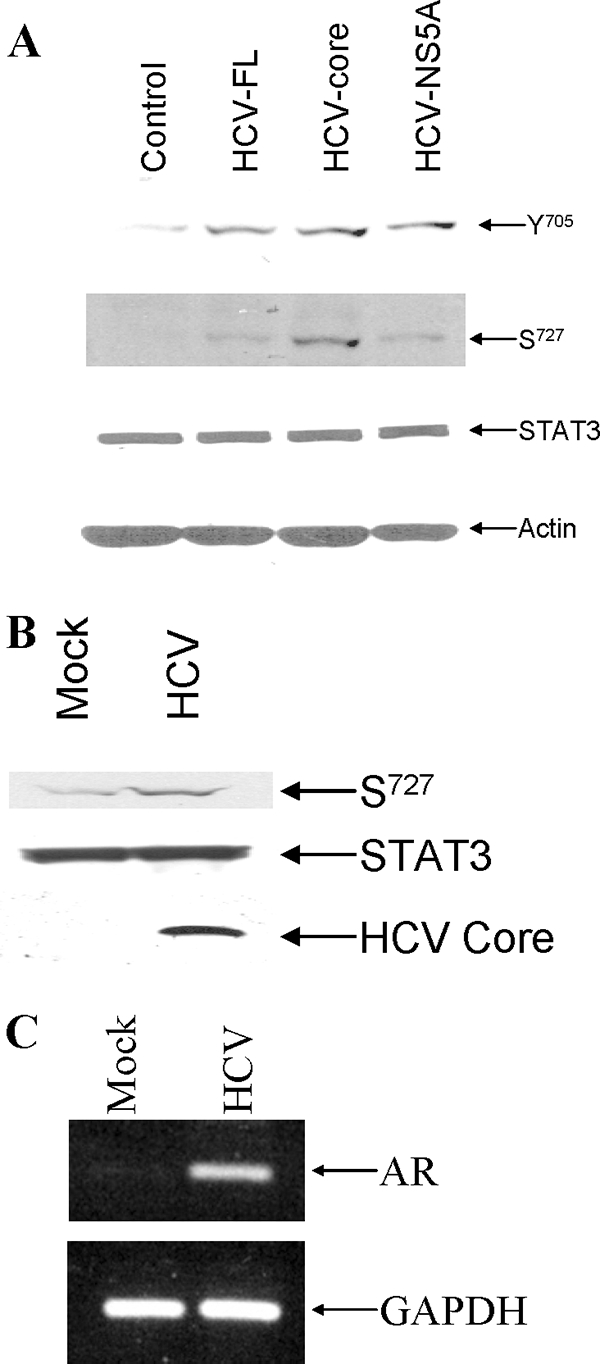
HCV augments the phosphorylation status of STAT3. (A) Huh-7 cells were transfected with HCV-FL, HCV-core, HCV-NS5A, or vector control and treated with 10 nM DHT. The phosphorylation status of STAT3 and total STAT3 were detected by Western blot analysis using anti-phospho-STAT3 (pSer-727 or pTyr-705) or anti-STAT3 antibody. (B) Mock- or HCV-infected IHH were treated with 10 nM DHT. The results of Western blot analysis of S727STAT3 and total STAT3 are shown. The expression of HCV core protein is shown from HCV-infected cell lysates by Western blot analysis using a rabbit anti-core antibody. (C) IHH were infected with HCV or left uninfected (mock). Total RNA was isolated after 72 h of infection, and RT-PCR was performed for AR mRNA expression.
HCV enhances VEGF mRNA expression.
We examined the functional role of HCV-mediated AR activation. VEGF is one of the AR-responsive genes in the liver (6). Recently, Nasimuzzaman et al. (35) have shown that HCV induces expression and secretion of VEGF. To determine whether HCV infection in the presence of DHT and AR induces VEGF transcription, total cellular RNA was extracted from mock- and HCV-infected Huh-7 cells. The level of VEGF mRNA was quantified by real-time RT-PCR. The results showed an increase in the VEGF mRNA level in HCV-infected cells in the presence of AR (Fig. 5A). Thus, our results indicated that HCV infection promotes VEGF mRNA synthesis in the presence of AR.
FIG. 5.

AR expression enhances the HCV-mediated VEGF mRNA level and the formation of capillarylike structure in HCMECs. (A) Real-time PCR for analysis of VEGF induction. Relative expression of VEGF gene in HCV-infected Huh-7 cells (▪) and mock-infected cells (□). The results presented are based on the relative expression level of housekeeping GAPDH gene. The lowest level of significance was P < 0.0001. (B) HCMECs (5 × 104 cells/well) were incubated with different CMs for 18 h, and tube formation between cells was observed and captured by digital camera. CM from control Huh-7 cells (a), cells transfected with AR (b), cells infected with HCV (c), or cells infected with HCV and transfected with AR (d) was used. (C) Quantification of the total tube length. The tube length of HCMECs incubated with CM from Huh-7 cells was set as 1, and other experimental conditions described in panel B were compared. The lowest level of significance was P < 0.005. (D) Tube numbers were counted. The results are presented as the mean ± the SD from at least three independent experiments.
AR expression enhances HCV-mediated capillarylike structure in HCMECs.
VEGF acts as a paracrine growth factor of endothelial cells. The tube formation using endothelial cells is a widely used method to demonstrate angiogenesis in vitro. Therefore, we performed in vitro angiogenesis assay to determine whether enhanced VEGF expression increases tube formation in endothelial cells. For this, conditioned medium (CM) from Huh-7 cells with or without HCV and AR was added to HCMECs grown on Matrigel. Tube formation of HCMECs increased when CM from AR-transfected cells or HCV-infected cells were used (Fig. 5B). This effect was pronounced from CM from HCV-infected cells expressing AR. The length and number of capillarylike tube structures were scored (Fig. 5C and D), suggesting that AR expression together with HCV proteins enhances angiogenesis.
DISCUSSION
The inflammatory immune responses of the host to HCV antigens induce hepatocyte damage, which is followed by the regeneration of hepatocytes and the development of fibrosis, important features in the pathogenesis of HCC. Men have a higher prevalence of HCC than women, varying between 2:1 and 4:1 (56). Etiological factors such as HBV and HCV play a role in this process, and genetic and hormonal factors may also be important (10, 34, 36).
Androgen is a ligand and major activator of AR signaling (1, 27, 46). Androgen binds to AR, inducing a conformational change, and then AR forms a homodimer and is phosphorylated at several sites. These active ARs stabilize the ligand-receptor homodimer, making the ligand-receptor complex for translocation to the nucleus. The AR-ligand complex then initiates gene transcription by binding to specific AREs in the promoter regions of target genes. After DNA binding, RNA polymerase machinery is recruited to the initiation site and the transcription of AR-regulated genes begins (13).
There are other well-known AR activation pathways, including STAT3 (48) and PI3K/AKT (57) in an androgen-independent manner. We and others have shown that HCV can activate various cytoplasmic signaling pathways, including JAK/STAT (5, 35, 41) and PI3K/AKT (4, 58). Because HCV replicates in the cytoplasm, irrespective of androgen treatment, it may activate AR by affecting preexisting cytosolic cellular signaling pathways. We have shown that HCV activates STAT3 phosphorylating S727 and enhances AR activation. The presence of DHT further increases AR activity. IL-6, an inflammatory cytokine, induces upregulation of AR expression through the activation of STAT3 in prostate cancer cell line LNCaP (25).
Translocation of STAT3 into the nucleus needs both phosphorylated Y705STAT3 and phosphorylated S727STAT3. HCV core can increase both the phosphorylated forms of STAT3 (Fig. 6). However, the effect of NS5A on phosphorylated S727STAT3 is weaker. This may be a reason why NS5A did not activate androgen response gene expression in spite of containing several proline-rich sequences consistent with Src homology 3-binding sites found in cellular signaling molecules (47).
FIG. 6.

Proposed model for HCV-induced AR activation. Inflammatory cytokine IL-6 activates STAT3 signaling. HCV also enhances the phosphorylation of Tyr-705 and Ser-727 of STAT3 through the core protein and enhances AR expression. AG490, a JAK-1 protein inhibitor, inhibits the JAK/STAT signaling pathway, suggesting an association of STAT3 as a downstream modulator.
The early stage of hepatocarcinogenesis is not well understood pathologically and clinically. Even early HCC lesions are more often overt HCC that already have an arterial blood supply, rather than extremely well differentiated and supplied by the portal vein as generally believed (19). It is well known that vascular (portal) invasion portends a poor prognosis (37). The status of angiogenesis in HCC correlates with disease progression and prognosis. VEGF is a primary androgen-responsive gene in the liver, which is the subset regulated directly by AR-occupied AREs (6). VEGF expression in HCC tissues was thought to be related to the histological grade (49). VEGF plays a central role in mediating angiogenesis in HCC (50). We have shown that AR enhances VEGF mRNA expression in HCV-infected Huh-7 cells more efficiently than in mock-infected cells. Moreover, androgen and HCV strongly induce angiogenesis. We have demonstrated that STAT3 is phosphorylated at S727 in the presence of HCV core, which subsequently activates AR. VEGF is modulated by AR in neoplastic growth. Therefore, we hypothesize that HCV-mediated activation of AR may increase VEGF function, resulting in the development of angiogenesis in liver. In conclusion, we have identified HCV core as an activator of AR, which may have implications for HCC as a male-dominant disease.
Acknowledgments
We thank Chawnshang Chang for providing the AR plasmid DNAs, Arvind Patel for anti-core antibody, and Len Grosso for helping us to measure the genome copy number of HCV.
This study was supported by research grants AI045144 and AI 065535 (R.B.R.) from the National Institutes of Health.
Footnotes
Published ahead of print on 3 September 2008.
REFERENCES
- 1.Alimirah, F., J. Chen, Z. Basrawala, H. Xin, and D. Choubey. 2006. DU-145 and PC-3 human prostate cancer cell lines express androgen receptor: implications for the androgen receptor functions and regulation. FEBS Lett. 5802294-2300. [DOI] [PubMed] [Google Scholar]
- 2.Andersson, K., and R. T. Chung. 2006. Hepatitis C virus in the HIV-infected patient. Clin. Liver Dis. 10303-320. [DOI] [PubMed] [Google Scholar]
- 3.Aziz, M. H., H. T. Manoharan, J. M. Sand, and A. K. Verma. 2007. Protein kinase Cɛ interacts with Stat3 and regulates its activation that is essential for the development of skin cancer. Mol. Carcinog. 46646-653. [DOI] [PubMed] [Google Scholar]
- 4.Banerjee, S., K. Saito, M. Ait-Goughoulte, K. Meyer, R. B. Ray, and R. Ray. 2008. Hepatitis C virus core protein upregulates serine phosphorylation of insulin receptor substrate-1 and impairs the downstream Akt/protein kinase B signaling pathway for insulin resistance. J. Virol. 822606-2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Basu, A., K. Meyer, K. K. Lai, K. Saito, A. M. Di Bisceglie, L. E. Grosso, R. B. Ray, and R. Ray. 2006. Microarray analyses and molecular profiling of Stat3 signaling pathway induced by hepatitis C virus core protein in human hepatocytes. Virology 349347-358. [DOI] [PubMed] [Google Scholar]
- 6.Bolton, E. C., A. Y. So, C. Chaivorapol, C. M. Haqq, H. Li, and K. R. Yamamoto. 2007. Cell- and gene-specific regulation of primary target genes by the androgen receptor. Genes Dev. 212005-2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bortolotti, F., G. Verucchi, C. Camma, G. Cabibbo, L. Zancan, G. Indolfi, R. Giacchino, M. Marcellini, M. G. Marazzi, C. Barbera, G. Maggiore, P. Vajro, S. Bartolacci, F. Balli, A. Maccabruni, M. Guido and Italian Observatory for HCV Infection and Hepatitis C in Children. 2008. Long-term course of chronic hepatitis C in children: from viral clearance to end-stage liver disease. Gastroenterology 1341900-1907. [DOI] [PubMed] [Google Scholar]
- 8.Carballo, M., M. Conde, R. El Bekay, J. Martin-Nieto, M. J. Camacho, J. Monteseirin, J. Conde, F. J. Bedoya, and F. Sobrino. 1999. Oxidative stress triggers STAT3 tyrosine phosphorylation and nuclear translocation in human lymphocytes. J. Biol. Chem. 27417580-17586. [DOI] [PubMed] [Google Scholar]
- 9.Chen, C. D., D. S. Welsbie, C. Tran, S. H. Baek, R. Chen, R. Vessella, M. G. Rosenfeld, and C. L. Sawyers. 2004. Molecular determinants of resistance to antiandrogen therapy. Nat. Med. 1033-39. [DOI] [PubMed] [Google Scholar]
- 10.Chiu, C. M., S. H. Yeh, P. J. Chen, T. J. Kuo, C. J. Chang, P. J. Chen, W. J. Yang, and D. S. Chen. 2007. Hepatitis B virus X (HBx) protein enhances androgen receptor-responsive gene expression depending on androgen level. Proc. Natl. Acad. Sci. USA 1042571-2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Decker, T., and P. Kovarik. 2000. Serine phosphorylation of STATs. Oncogene 192628-2637. [DOI] [PubMed] [Google Scholar]
- 12.De Miguel, F., S. O. Lee, S. A. Onate, and A. C. Gao. 2003. Stat3 enhances transactivation of steroid hormone receptors. Nucleic Recept. 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Edwards, J., and J. M. S. Bartlett. 2005. The androgen receptor and signal-transduction pathways in hormone-refractory prostate cancer. Part 1: modifications to the androgen receptor. BJU Int. 951320-1326. [DOI] [PubMed] [Google Scholar]
- 14.Gelmann, E. P. 2002. Molecular biology of the androgen receptor. J. Clin. Oncol. 203001-3015. [DOI] [PubMed] [Google Scholar]
- 15.Germain, D., and D. A. Frank. 2007. Targeting the cytoplasmic and nuclear functions of signal transducers and activators of transcription 3 for cancer therapy. Clin. Cancer Res. 135665-5669. [DOI] [PubMed] [Google Scholar]
- 16.Giannitrapani, L., M. Soresi, E. La Spada, M. Cervello, N. D'Alessandro, and G. Montalto. 2006. Sex hormones and risk of liver tumor. Ann. N. Y. Acad. Sci. 1089228-236. [DOI] [PubMed] [Google Scholar]
- 17.Heinlein, C. A. 2004. Androgen receptor in prostate cancer. Endocrinol. Rev. 25276-308. [DOI] [PubMed] [Google Scholar]
- 18.Iqbal, M. J., M. L. Wilkinson, P. J. Johnson, and R. Williams. 1983. Sex steroid receptor proteins in foetal, adult, and malignant human liver tissue. Br. J. Cancer 48791-796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iwamoto, S., H. Sanefuji, and K. Okuda. 2003. Computed tomography angiographic findings in hepatocellular carcinoma less than 2 cm detected during follow-up in 29 patients. J. Gastroenterol. Hepatol. 181076-1080. [DOI] [PubMed] [Google Scholar]
- 20.Jie, X., C. Lang, Q. Jian, L. Chaoqun, Y. Dehua, S. Yi, J. Yanping, X. Luokun, Z. Qiuping, W. Hui, G. Feili, J. Boquan, J. Youxin, and T. Jinquan. 2007. Androgen activates PEG10 to promote carcinogenesis in hepatic cancer cells. Oncogene 265741-5751. [DOI] [PubMed] [Google Scholar]
- 21.Kanda, T., A. Basu, R. Steele, T. Wakita, J. S. Ryerse, R. Ray, and R. B. Ray. 2005. Generation of infectious hepatitis C virus in immortalized human hepatocytes. J. Virol. 804633-4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koike, K. 2007. Hepatitis C virus contributes to hepatocarcinogenesis by modulating metabolic and intracellular signaling pathways. J. Gastroenterol. Hepatol. 22(Suppl. 1)S108-S111. [DOI] [PubMed] [Google Scholar]
- 23.Lee, W. M., J. L. Dienstag, K. L. Lindsay, A. S. Lok, H. L. Bonkovsky, M. L. Shiffman, G. T. Everson, A. M. Di Bisceglie, T. R. Morgan, M. G. Ghany, C. Morishima, E. C. Wright, J. E. Everhart, et al. 2004. Evaluation of the HALT-C Trial: pegylated interferon as maintenance therapy for chronic hepatitis C in previous interferon nonresponders. Control Clin. Trials 25472-492. [DOI] [PubMed] [Google Scholar]
- 24.Levremo, M. 2006. Viral hepatitis and liver cancer: the case of hepatitis C. Oncogene 253834-3847. [DOI] [PubMed] [Google Scholar]
- 25.Lin, D.-L., M. C. Whitny, Z. Yao, and E. T. Kelly. 2001. Interleukin-6 induces androgen responsiveness in prostate cancer cells through up-regulation of androgen receptor expression. Clin. Cancer Res. 71773-1781. [PubMed] [Google Scholar]
- 26.Litvinov, I. V., C. Chang, and J. T. Isaacs. 2004. Molecular characterization of the commonly used human androgen receptor expression vector, pSG-AR. Prostate 58319-324. [DOI] [PubMed] [Google Scholar]
- 27.Ma, W.-L., C.-L. Hsu, M.-H. Wu, C.-T. Wu, C.-C. Wu, J.-J. Lai, Y.-S. Jou, C.-W. Chen, S. Yeh, and C. Chang. 2008. Androgen receptor is a new potential therapeutic target for the treatment of hepatocellular carcinoma. Gastroenterology 135947-955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Madamanchi, N. R., S. Li, C. Patterson, and M. S. Runge. 2001. Reactive oxygen species regulate heat-shock protein 70 via the JAK/STAT pathway. Arterioscler. Thromb. Vasc. Biol. 21321-326. [DOI] [PubMed] [Google Scholar]
- 29.Majumder, M., A. K. Ghosh, R. Steele, R. Ray, and R. B. Ray. 2001. Hepatitis C virus NS5A physically associates with p53 and regulates p21/waf1 gene expression in a p53-dependent manner. J. Virol. 751401-1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Majumder, M., R. Steele, A. K. Ghosh, X. Y. Zhou, L. Thonburg, R. Ray, N. J. Phillips, and R. B. Ray. 2003. Expression of hepatitis C virus nonstructural 5A protein in the liver of transgenic mice. FEBS Lett. 555528-532. [DOI] [PubMed] [Google Scholar]
- 31.Maruyama, S., N. Nagasue, D. K. Dhar, A. Yamanoi, O. N. Ei-Assal, K. Satoh, and K. Okita. 2001. Preventive effect of FK143, a 5α-reductase inhibitor, on chemical hepatocarcinogenesis in rats. Clin. Cancer Res. 72096-2104. [PubMed] [Google Scholar]
- 32.Merchant, J. L. 2008. What lurks beneath: IL-11, via Stat3, promotes inflammation-associated gastric tumorigenesis. J. Clin. Investig. 1181628-1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nagasue, N., A. Itoh, H. Yukaya, and Y. Ogawa. 1985. Androgen receptors in hepatocellular carcinoma and surrounding parenchyma. Gastroenterology 89643-647. [DOI] [PubMed] [Google Scholar]
- 34.Nagasue, N., Y. C. Chang, T. Hayashi, G. Galizia, H. Kohno, T. Nakamura, and H. Yukaya. 1989. Androgen receptor in hepatocellular carcinoma as a prognostic factor after hepatic resection. Ann. Surg. 209424-427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nasimuzzaman, M., G. Waris, D. Mikolon, D. G. Stupack, and A. Siddiqui. 2007. Hepatitis C virus stabilizes hypoxia-inducible factor 1alpha and stimulates the synthesis of vascular endothelial growth factor. J. Virol. 8110249-10257. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 36.Naugler, W. E., T. Sakurai, S. Kim, S. Maeda, K. Kim, A. M. Elsharkawy, and M. Karin. 2007. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science 317121-124. [DOI] [PubMed] [Google Scholar]
- 37.Okuda, K. 2002. Natural history of hepatocellular carcinoma including fibrolamellar and hepato-cholangiocarcinoma variants. J. Gastroenterol. Hepatol. 17401-405. [DOI] [PubMed] [Google Scholar]
- 38.Ray, R. B., L. M. Lagging, K. Meyer, and R. Ray. 1996. Hepatitis C virus core protein cooperates with ras and transforms primary rat embryo fibroblasts to tumorigenic phenotype. J. Virol. 704438-4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ray, R. B., and R. Ray. 2001. Hepatitis C virus core protein: intriguing properties and functional relevance. FEMS Microbiol. Lett. 202149-156. [DOI] [PubMed] [Google Scholar]
- 40.Saito, K., M. Ait-Goughoulte, S. M. Truscott, K. Meyer, A. Blazevic, G. Abate, R. B. Ray, D. F. Hoft, and R. Ray. 2008. Hepatitis C virus inhibits cell surface expression of HLA-DR, prevents dendritic cell maturation and induces interleukin-10 production. J. Virol. 823320-3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sarcar, B., A. K. Ghosh, R. Steele, R. Ray, and R. B. Ray. 2004. Hepatitis C virus NS5A mediated STAT3 activation requires cooperation of Jak1 kinase. Virology 32251-60. [DOI] [PubMed] [Google Scholar]
- 42.Schaefer, L. K., S. Wang, and T. S. Schaefer. 1999. c-Src activates the DNA binding and transcriptional activity of Stat3 molecules: serine 727 is not required for transcriptional activation under certain circumstances. Biochem. Biophys. Res. Commun. 266481-487. [DOI] [PubMed] [Google Scholar]
- 43.Schiff, E. R. 2007. Emerging strategies for pegylated interferon combination therapy. Nat. Clin. Pract. Gastroenterol. Hepatol. 4(Suppl. 1)S17-S21. [DOI] [PubMed] [Google Scholar]
- 44.Schmittagen, T. D., B. A. Zakrajsek, A. G. Mills, V. Gorn, M. J. Singer, and M. W. Reed. 2000. Quantitative reverse transcription-polymerase chain reaction to study mRNA decay: comparison of endpoint and real-time methods. Anal. Biochem. 285194-204. [DOI] [PubMed] [Google Scholar]
- 45.Shen, Y., K. Schlessinger, X. Zhu, E. Meffre, F. Quimby, D. E. Levy, and J. E. Darnell, Jr. 2004. Essential role of STAT3 in potential survival and growth revealed by mice lacking STAT3 serine 727 phosphorylation. Mol. Cell. Biol. 24407-419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sinha-Hikim, I., W. E. Taylor, N. F. Genzalez-Cadavid, W. Zheng, and S. Bhasin. 2004. Androgen receptor in human skeletal muscle and cultured muscle satellite cells: up-regulation by androgen treatment. J. Clin. Endocrinol. Metab. 895245-5255. [DOI] [PubMed] [Google Scholar]
- 47.Tan, S.-L., H. Nakao, Y. He, S. Vijaysri, P. Neddermann, B. L. Jacobs, B. J. Mayer, and M. G. Katze. 1999. NS5A, a nonstructural protein of hepatitis C virus, binds growth factor receptor-bound protein 2 adaptor protein in a Src homology 3 domain/ligand-dependent manner and perturbs mitogenic signaling. Proc. Natl. Acad. Sci. USA 965533-5538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ueda, T., N. Bruchovsky, and M. D. Sadar. 2002. Activation of the androgen receptor N-terminal domain by interleukin-6 via MAPK and STAT3 signal transduction pathways. J. Biol. Chem. 2777076-7085. [DOI] [PubMed] [Google Scholar]
- 49.Yamaguchi, R., H. Yano, A. Iemura, S. Ogasawara, M. Haramaki, and M. Kojiro. 1998. Expression of endothelial growth factor in human hepatocellular carcinoma. Hepatology 2868-77. [DOI] [PubMed] [Google Scholar]
- 50.Yang, Z. F., and R. T. Poon. 2008. Vascular changes in hepatocellular carcinoma. Anat. Rec. 291721-734. [DOI] [PubMed] [Google Scholar]
- 51.Yoon, G., J. Y. Kim, Y. K. Choi, Y. S. Won, and I. K. Lim. 2006. Direct activation of TGF-β1 transcription by androgen and androgen receptor complex in Huh7 human hepatoma cells and its tumor in nude mice. J. Cell. Biochem. 97393-411. [DOI] [PubMed] [Google Scholar]
- 52.Yoshikawa, H., K. Matsubara, G. S. Qian, P. Jackson, J. D. Groopman, J. E. Manning, C. C. Harris, and J. G. Herman. 2001. SOCS-1, a negative regulator of the JAK/STAT pathway, is silenced by methylation in human hepatocellular carcinoma and shows growth-suppression activity. Nat. Genet. 2829-35. [DOI] [PubMed] [Google Scholar]
- 53.Yu, H., M. Kortylewski, and D. Pardoll. 2007. Crosstalk between cancer and immune cells: role of STAT3 in the tumor microenviroment. Nat. Rev. Immunol. 741-51. [DOI] [PubMed] [Google Scholar]
- 54.Yu, M. W., and C. J. Chen. 1994. Hepatitis B and C viruses in the development of hepatocellular carcinoma. Crit. Rev. Oncol. Hematol. 1771-91. [DOI] [PubMed] [Google Scholar]
- 55.Wakita, T., T. Pietschmann, T. Kato, T. Date, M. Miyamoto, Z. Zhao, K. Murthy, A. Habermann, H. G. Krausslich, M. Mizokami, R. Bartenschlager, and T. J. Liang. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11791-796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wands, J. R. 2007. Hepatocellular carcinoma and sex. N. Engl. J. Med. 3571974-1976. [DOI] [PubMed] [Google Scholar]
- 57.Wang, Y., J. I. Kreisberg, and P. M. Ghosh. 2007. Cross-talk between the androgen receptor and the phosphatidylinositol 3-kinase/Akt pathway in prostate cancer. Curr. Cancer Drug Targets 7591-604. [DOI] [PubMed] [Google Scholar]
- 58.Waris, G., D. J. Felmlee, F. Negro, and A. Siddiqui. 2007. Hepatitis C virus induces proteolytic cleavage of sterol regulatory element binding proteins and stimulates their phosphorylation via oxidative stress. J. Virol. 818122-8130. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 59.Waris, G., J. Turkson, T. Hassanein, and A. Siddiqui. 2005. Hepatitis C virus (HCV) constitutively activates STAT-3 via oxidative stress: role of STAT-3 in HCV replication. J. Virol. 791569-1580. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 60.Williams, R. 2006. Global challenges in liver disease. Hepatology 44521-526. [DOI] [PubMed] [Google Scholar]
- 61.Zheng, Y., W. L. Chen, W. L. Ma, C. Chang, and J. H. Ou. 2007. Enhancement of gene transactivation activity of androgen receptor by hepatitis B virus X protein. Virology 363454-461. [DOI] [PMC free article] [PubMed] [Google Scholar]