

Abstract
Our norovirus (NoV) surveillance group reported a >4-fold increase in NoV infection in Japan during the winter of 2006-2007 compared to the previous winter. Because the increase was not linked to changes in the surveillance system, we suspected the emergence of new NoV GII/4 epidemic variants. To obtain information on viral changes, we conducted full-length genomic analysis. Stool specimens from 55 acute gastroenteritis patients of various ages were collected at 11 sites in Japan between May 2006 and January 2007. Direct sequencing of long PCR products revealed 37 GII/4 genome sequences. Phylogenetic study of viral genome and partial sequences showed that the two new GII/4 variants in Europe, termed 2006a and 2006b, initially coexisted as minorities in early 2006 in Japan and that 2006b alone had dominated over the resident GII/4 variants during 2006. A combination of phylogenetic and entropy analyses revealed for the first time the unique amino acid substitutions in all eight proteins of the new epidemic strains. These data and computer-assisted structural study of the NoV capsid protein are compatible with a model of antigenic drift with tuning of the structure and functions of multiple proteins for the global outgrowth of new GII/4 variants. The availability of comprehensive information on genome sequences and unique protein changes of the recent global epidemic variants will allow studies of diagnostic assays, molecular epidemiology, molecular biology, and adaptive changes of NoV in nature.
Norovirus (NoV) is a major etiological agent of acute gastroenteritis worldwide and can cause diarrhea in all ages. NoV is relatively stable in water containing chlorine (22), highly infectious in individuals having a functional alpha-1,2 fucosyltransferase (27), and prevalent in natural and living environments (8, 19). It is transmitted through ingestion of contaminated food and water, direct person-to-person contact, and exposure to contaminated airborne vomitus droplets in a semiclosed community (8, 19). NoV commonly causes asymptomatic infection (13, 30, 36), where virus carriers have viral loads similar to those of symptomatic individuals (36). These characteristics allow NoV to spread rapidly and extensively by activities of daily living.
NoV is a nonenveloped virus of the Caliciviridae family. NoV has a single-stranded, positive-sense, polyadenylated RNA genome that encodes three open reading frames (ORFs): ORF1, ORF2, and ORF3 (51). ORF1 encodes a large polyprotein that is cleaved by the viral proteinase into six nonstructural proteins. ORF2 encodes a viral capsid protein (40). ORF3 encodes a VP2 protein that may function as a minor capsid protein for genome packaging (14). As is common in the RNA viruses, NoV in nature is genetically and antigenically highly diverse (16, 20, 21). NoV is tentatively divided into five genogroups (GI to GV) and more than 25 genotypes based on similarities among ORF2 sequences (1, 21). Among them, genogroup II genotype 4 (GII/4) is particularly important in public health, because it is the leading cause of NoV-associated acute gastroenteritis in humans worldwide since the mid-1990s (3, 12, 17, 28, 29, 32, 46).
Notably, the GII/4 epidemic has been augmented periodically during the past ∼15 years. Four NoV pandemics occurred in association with the emergence of new GII/4 variants in the winters of 1995-1996, 2002-2003, 2004-2005, and 2006-2007 (46). In the 2006-2007 epidemic, two variants, 2006a and 2006b, emerged and displaced the resident GII/4 variants in Europe (46). The emergence was associated with an atypical NoV epidemic in Europe (46) and Hong Kong (17). These and other chronological GII/4 variants that caused the global NoV epidemic had amino acid substitutions in the capsid protein (28, 46). Thus, host population immunity may play a role in the evolution of new GII/4 epidemic strains, as seen in the antigenic drift in the influenza virus (28, 46).
In Japan, the GII/4 strains are again the most prevalent since the 2002-2003 epidemic (35-38) and in natural environments (31, 41, 44). As seen in many countries, the rate of NoV infection in Japan increases in the winter. Notably, an atypically high level of NoV activity was noticed in early autumn and was sustained in the winter in 2006-2007 in Japan. The number of outbreaks of NoV infections reported by a nationwide surveillance group increased unusually rapidly in October (n = 72) compared to the previous year (n = 10), reaching peaks in November and December 2006 (n = 428 and 345, respectively), and decreasing during January and February 2007 (n = 164 and 82, respectively) (Infectious Disease Surveillance Center, http://idsc.nih.go.jp/iasr/prompt/graph-ke.html) (43, 52). The total number of infection cases during October and December in 2006 showed an ∼5-fold increase compared to the same season in 2005. We suspected the emergence of new NoV GII/4 variants, because the increase was not linked to changes in the surveillance system. Indeed, studies of the 5′ capsid sequences suggested that a new GII/4 variant strain was circulating in some prefectures in Japan in 2006-2007 (26, 34, 50).
We report here comprehensive information on the near-full-length genomes of the NoV GII/4 variant strains in the atypical Japanese epidemic of 2006-2007. This information was used to examine the molecular mechanisms of the new NoV GII/4 epidemic. Our evolutionary and structural studies support a model of antigenic drift with tuning of multiple viral proteins for the periodic outgrowth of new NoV GII/4 variants.
MATERIALS AND METHODS
Stool specimens.
The Norovirus Surveillance Group of Japan collected stool specimens from 55 NoV GII-positive individuals with acute gastroenteritis. The collection sites were located at 11 different regional public health institutes in Japan (five samples from each institute) (Fig. 1A). The specimens were collected between May and December 2006 (n = 1, 1, 3, 10, 15, and 22 for May, August, September, October, November, and December 2006, respectively). The study also included a single specimen collected in January 2007 and two specimens with unclear collection dates. The specimens were collected from all age groups except ages 10 to 19 years: n = 13, 4, 2, 4, 2, 4, 6, 5, and 4 for ages 0 to 9, 20 to 29, 30 to 39, 40 to 49, 50 to 59, 60 to 69, 70 to 79, 80 to 89, and 99 to 99 years old, respectively. Eleven specimens had no data on age. All stool specimens were stored at −80°C.
FIG. 1.

(A) Geographic locations of 11 sample collection sites in Japan. (B) Schematic illustration of the locations of the primers used in the present study.
Primer design.
NoV GII-specific primers reported previously (21) and newly designed primers were used for the present study. The primer locations are schematically illustrated under the NoV genome structure (Fig. 1B). Each primer was designed to amplify three overlapping fragments of ca. 0.9, 5.2, and 2.5 kb, which cover the N-terminal end of ORF1, most of ORF1, and all of ORF2/3, respectively. cDNAs for 0.9, 5.2, and 2.5-kb regions were synthesized by reverse transcription of viral RNA with antisense primers GII4-r908 (5′-CTC CAA AGA GCT CTG CCA AGA G-3′), GII4r5412 (5′-CCG GCG GTG AAC GCG TTC CCC GC-3′), and Tx30SXN (21) (5′-GAC TAG TTC TAG ATC GCG AGC GGC CGC CCT TTT TTT TTT TTT TTT TTT TTT TTT TTT TT-3′), respectively. The 0.9-kb fragment was amplified by using nested PCR with an outer primer pair, GII4-1F (sense) (5′-GTG AAT GAA GAT GGC GTC TAA CG-3′) and GII4-r908 (antisense), and an inner primer pair, GII4-2F (sense) (5′-GAA TGA AGA TGG CGT CTA ACG ACG-3′) and GII4-r593 (antisense) (5′-GTT GTC AAA GGC TGT GTA GGG-3′). The 5′-half 5.2-kb fragment was amplified by using nested PCR with an outer primer pair, GII4C-22S20 (sense) (5′-CGA CGC TTC CGC TGC CGC TG-3′) and GII4r5412 (antisense), and an inner primer pair, GII4C-64S20 (sense) (5′-CGC AAA ATC TTC AAG TGA CG-3′) and GII4r5295 (antisense) (5′-CGC TCC ATA GTA TTT CAC CTG GAG C-3′). The 3′-half 2.5-kb fragment was amplified by using nested PCR with an outer primer pair, COG-2F (sense) (5′-CAR GAR BCN ATG TTY AGR TGG ATG AG-3′) and Tx30SXN (antisense), and an inner primer pair, G2SKF (sense) (5′-CNT GGG AGG GCG ATC GCA A-3′) and Tx30SXN (antisense) (21). The primers for PCR product sequencing were designed at 26 regions at approximately equal intervals across the NoV genome.
RNA extraction, reverse transcription, and long-range PCR.
A 10% fecal suspension (wt/vol) was prepared with phosphate-buffered saline (PBS; pH 7.2) and centrifuged at 10,000 × g for 10 min. Viral RNA was extracted from the suspension by using a QIAamp viral RNA kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. The RNA extracts were stored at −80°C. Reverse transcription was performed with SuperScript III cDNA synthesis kit (Invitrogen, Carlsbad, CA). Seven microliters of the purified RNA was added to a reaction mixture (final, 20 μl) containing 50 pmol of random hexamar, 25 mM MgCl2 buffer, 10 mM deoxynucleoside triphosphate, 10× RT buffer, 0.1 M dithiothreitol, and 200 U SuperScript III RT. The mixture was incubated at 42°C for 1 h, after which 10 U of RNase H was added at 37°C for 20 min. Long-range PCR was performed with the Expand Long Template PCR System (Roche, Basel, Switzerland). Three microliters of cDNA was added to a reaction mixture (final, 50 μl) containing 350 μM deoxynucleoside triphosphate, 1.75 mM Mg2+, 300 nM PCR primers, and 3.5 U of Expand Long template enzyme. Amplification was performed for 25 to 30 cycles, at 94°C for 10 s, 60°C for 30 s, and 68°C for 5 min per cycle, and a final extension at 68°C for 8 min. Three microliters of the first PCR products was added to the reaction mixture (final 50 μl), and the same PCR condition was used for the second-round amplification.
DNA sequencing.
The PCR products were purified with the QIAquick PCR purification kit and in some cases with a QIAquick gel extraction kit (Qiagen). The purified DNAs were used as a template for direct sequencing. The sequencing reaction was performed in a 96-well microplate using dye-terminator chemistry with BigDye version 3.1 (Applied Biosystems, Foster City, CA) according to the manufacturer's instructions. The reaction products were purified with the Montage SEQ96 sequencing reaction cleanup kit (Millipore, Bedford, MA) and sequenced on an automated ABI 3730 xl DNA analyzer (Applied Biosystems). To obtain full-length genome sequences, fragment sequences of 0.9, 5.2, and 2.5 from the same individual were aligned at an overlapping region by using the Staden Package (http://staden.sourceforge.net).
Phylogenetic analysis.
Nucleotide sequences were aligned with an outgroup by using CLUSTAL W version 1.4 (49). A distance matrix of nucleotide substitutions per site was estimated from the alignment according to Kimura's two-parameter method (23). Neighbor-joining trees (42), maximum-likelihood trees, and UPGMA (unweighted pair-group method with arithmetic averages) trees were generated with 100 bootstrap replicates (10) from the matrix numbers by using MEGA version 3.0 (25). For the phylogenetic analysis of NoV ORF2 complete nucleotide sequences, we included sequences of well-recognized strains identified in the global GII/4 epidemic. They are the <1996 variants (Lordsdale strain [7], GenBank accession no. X86557; Bristol strain [15], accession no. X76716), the 1995-1996 epidemic variants (Grimsby strain, accession no. AJ004864; 95/96-US strain [32], accession no. AF080549; Camberwell strain [5], accession no. U46500), the 2002-2003 epidemic variants (Farmington Hills strain [53], accession no. AY502023; a United Kingdom strain [6], accession no. AY587990), and the 2004-2005 epidemic variants (Hunter strain [3], accession no. DQ078794; a Netherlands strain OB2004-083 [46], accession no. AB303941). We also included the reference sequences of the 2006/2007 epidemic variants, two Netherlands 2006a strains (Terneuzen70/2006/NL and Yerseke38/2006/NL [46], accession nos. EF126964 and EF126963, respectively), two Netherlands 2006b strains (DenHaag89/2006/NL and Nijmegen115/2006/NL [46], accession nos. EF126965 and EF126966, respectively), and a 2006b strain from Kobe, Japan (Kobe034/2006/JP, accession no. AB291542). All other reference sequences used in the present study were obtained from GenBank.
Analysis of amino acid variation.
Amino acid variations at individual positions of viral proteins were calculated according to the method described by Huang et al. (18) on the basis of Shannon's equation (45):
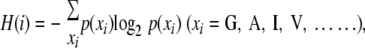 |
where H(i), p(xi), and i indicate the amino acid entropy score of a given position, the probability of occurrence of a given amino acid at the position, and the number of the positions, respectively. An H(i) score of zero indicates absolute conservation, whereas a score of 4.4 indicates complete randomness. The H(i) scores were expressed on the capsid structure constructed by the homology modeling method described below.
Molecular modeling.
The crystal structure of the NoV capsid P domain of the GII/4 VA387 strain at a resolution of 2.00 Å (PDB code: 2OBS (4) was used as the modeling template. P domains of the GII/4 2006b strains have sequence similarities of greater than 90% to that of VA387, which are high enough to construct models with a root mean square distance of ∼1 Å for the main chain between the predicted and actual structures (2). A three-dimensional (3-D) model of the P domain monomer of the earliest 2006b strain in Japan (Aich3/2006/JP) was constructed by using MOE-Align and MOE-Homology in the Molecular Operating Environment (MOE; Chemical Computing Group, Inc., Montreal, Quebec, Canada) as described previously (33, 47). The 3-D structure was thermodynamically optimized by energy minimization using the MOE and an AMBER99 force field (39). A physically unacceptable local structure of the optimized 3-D model was further refined on the basis of Ramachandran plot evaluation by using MOE. The 3-D model of the P-domain dimer was constructed by the superimposition the structures of the P-domain monomer on chains A and B of the NoV capsid oligomer (PDB code 1IHM) (40).
Nucleotide sequence accession numbers.
The DDBJ database accession numbers for the nucleotide sequences reported here are AB447427 to AB447463.
RESULTS
Genome sequencing.
We obtained near-full-length genome sequences for 37 of 55 (67.3%) GII-positive specimens. The genome sequences consist of about 7.5 kb. The initial 22 nucleotides at the 5′ ends of the genomes were from PCR primers. The final 45 nucleotides at the 3′ ends of the genome were excluded from analysis because of the low levels of sequence accuracy. Of the 37 genomes, 35 had no nucleotide insertions or deletions, compared to the reported genome sequence of a 1990s GII/4 strain, Lordsdale (7), whereas 2 of the 37 sequences (Aomori1/2006/JP and Aomori2/2006/JP) had 9 nucleotide deletions in ORF3.
At least one genome sequence was obtained at each of the 11 collection sites (Fig. 1A and Table 1). The 35 genome sequences were collected in May, October, November, and December 2006 and in January 2007 (n = 1, 8, 12, 13, and 1, respectively). The collection months of two specimens were not recorded but were in the study period. The 28 sequences were collected from different age groups (n = 6, 3, 1, 4, 1, 4, 3, 2, and 4 for ages 0 to 9, 20 to 29, 30 to 39, 40 to 49, 50 to 59, 60 to 69, 70 to 79, 80 to 89, and 90 to 99 years old, respectively). The age groups of nine specimens were unavailable.
TABLE 1.
Characteristics of NoV specimens and patients
| Specimen | Geographic origin | Setting | Date | Age (yr) |
|---|---|---|---|---|
| Hokkaido1/2006/JP | Hokkaido | Hospital | 29.Sep.06 | 60 |
| Hokkaido2/2006/JP | Hokkaido | Nursing care center | 10.Nov.06 | 90 |
| Hokkaido3/2006/JP | Hokkaido | Nursing care center | 3.Dec.06 | 0 |
| Hokkaido4/2006/JP | Hokkaido | Nursing care center | 22.Dec.06 | 90 |
| Hokkaido5/2007/JP | Hokkaido | Nursing care center | 3.Jan.07 | 40 |
| Aomori1/2006/JP | Aomori | Kindergarten | 17.Dec.06 | 4 |
| Aomori2/2006/JP | Aomori | Kindergarten | 16.Dec.06 | 4 |
| Aomori4/2006/JP | Aomori | Hotel | 17.Dec.06 | 43 |
| Aomori5/2006/JP | Aomori | Hotel | 17.Dec.06 | 24 |
| Akita1/2006/JP | Akita | Hotel | 13.Nov.06 | Unknown |
| Akita2/2006/JP | Akita | Hospital | 7.Dec.06 | 41 |
| Akita4/2006/JP | Akita | Nursing care center | 18.Dec.06 | Unknown |
| Akita5/2006/JP | Akita | Nursing care center | 19.Dec.06 | 92 |
| Miyagi2/2006/JP | Miyagi | Nursing care center | 14.Dec.06 | 65 |
| Miyagi4/2006/JP | Miyagi | Unknown | Unknown | Unknown |
| Miyagi5/2006/JP | Miyagi | Unknown | Unknown | Unknown |
| Toyama1/2006/JP | Toyama | Hospital | 15.May.06 | 56 |
| Toyama4/2006/JP | Toyama | Nursing care center | 13.Nov.06 | 80 |
| Toyama5/2006/JP | Toyama | Nursing care center | 16.Nov.06 | 89 |
| Aichi3/2006/JP | Aichi | Kindergarten | 19.Oct.06 | 3 |
| Aichi4/2006/JP | Aichi | Nursing care center | 20.Nov.06 | 95 |
| Osaka2/2006/JP | Osaka | Restaurant | 22.Oct.06 | 40 |
| Osaka3/2006/JP | Osaka | Hospital | 1.Nov.06 | 22 |
| Osaka4/2006/JP | Osaka | Nursing care center | 26.Nov.06 | 63 |
| Hiroshima1/2006/JP | Hiroshima | Gymnasium | 3.Oct.06 | 70 |
| Hiroshima2/2006/JP | Hiroshima | Hotel | 14.Oct.06 | 79 |
| Ehime1/2006/JP | Ehime | Nursing care center | 21.Nov.06 | 65 |
| Ehime2/2006/JP | Ehime | Nursing care center | 3.Dec.06 | 78 |
| Ehime5/2006/JP | Ehime | Restaurant | 13.Dec.06 | Unknown |
| Saga1/2006/JP | Saga | Unknown | 18.Nov.06 | 2 |
| Saga4/2006/JP | Saga | Hotel | 22.Oct.06 | Unknown |
| Saga5/2006/JP | Saga | Kindergarten | 10.Nov.06 | 2 |
| Kumamoto1/2006/JP | Kumamoto | Unknown | 6.Oct.06 | Unknown |
| Kumamoto2/2006/JP | Kumamoto | Unknown | 4.Nov.06 | Unknown |
| Kumamoto3/2006/JP | Kumamoto | Unknown | 15.Nov.06 | Unknown |
| Kumamoto4/2006/JP | Kumamoto | Restaurant | 30.Sep.06 | 20 |
| Kumamoto5/2006/JP | Kumamoto | Restaurant | 30.Oct.06 | 34 |
Genetic links of NoV capsid among patients of the 2006-2007 epidemics.
Recently, the complete ORF2 capsid sequences were successfully used to classify chronologically the GII/4 strains during the past ∼15 years in The Netherlands (46). They referred to the two most newly emerged GII/4 epidemic variants in Europe as 2006a and 2006b (46). The genetic relationships of these and the new sequences obtained in the present study were examined by phylogenetic analysis. For the analysis, we included sequences of well-recognized reference strains from the global GII/4 epidemic during the past ∼15 years (see Materials and Methods for the strain names and accession numbers). In addition, we included GII/4 sequences identified in Japan during 1997 and 2006.
A representative neighbor-joining tree shows that the nucleotide sequences of the complete capsid region of the Japanese 2006-2007 strains are divisible into three distinct lineage groups (Fig. 2A, yellow boxes). The first group, cluster I, included 33 of the 37 new sequences (89%) (Fig. 2A, cluster I, yellow boxes, bootstrap value 100/100). The cluster I specimens were from various age groups in all 11 sampling sites between May 2006 and January 2007. The second group, cluster II, included 3 of the 37 (8%) sequences. These were collected in the north of Japan (Aomori1/2006/JP and Aomori2/2006/JP) in December 2006 and in the south of Japan (Saga5/2006/JP) in November 2006 (Fig. 2A, cluster II, yellow boxes, bootstrap value 100/100). The third group, cluster III, included a single sequence collected in central Japan (Osaka2/2006/JP) in October 2006 (Fig. 2A, cluster III, a yellow box, bootstrap value 100/100).
FIG. 2.



Neighbor-joining trees of the nucleotide sequences of the complete ORF2 region (∼1.6 kb) (A) and the 5′-capsid N/S region (228 bases) (B and C) of NoV GII/4. Bootstrap values with 100/100 are indicated at the nodes of the tree. Sequence names are shown in the distinct color boxes on the basis of sampling sites and periods. Yellow boxes, 2006-2007 sequences obtained in the present study; green boxes, 2006/2007 sequences obtained in Europe, China, and Japan in other studies; orange boxes, <2006 sequences in Japan; pink boxes, 2005-2006 sequences in Japan: blue boxes, sequences from the global GII/4 epidemics pre-1996, 1995-1996, 2002-2003, and 2004-2005. Red letters with asterisks indicate sequences of the GII/4 2006a and 2006b epidemic strains in winter 2006-2007 in Europe (46).
Notably, all 33 cluster I sequences from the 2006-2007 Japanese epidemic were grouped with the 2006b sequences from The Netherlands (46) and a 2006 sequence from Japan (Fig. 2A, cluster I, green boxes, bootstrap value 100/100). cluster I was highly unique, and a sequence from China in 2002 (Lanzhou/35666/202/CHN) was only distantly related to it. Three cluster II sequences from the 2006-2007 Japanese epidemic (Aomori1/2006/JP, Aomori2/2006/JP, and Saga5/2006/JP) were grouped with the 2006a sequences from The Netherlands (46) (Fig. 2A, cluster II, bootstrap value 100/100). Cluster II was relatively closely related to a cluster, termed 04/05/AU/NL, that included the 2004-2005 epidemic strains in The Netherlands and Australia. Finally, the single cluster III sequence from the 2006-2007 epidemic (Osaka2/2006/JP) was grouped with sequences of the 2004-2005 epidemic strains in Japan and China (Fig. 2A, cluster III, bootstrap value 100/100). These data suggest that the GII/4 2006b spread dominantly across Japan in 2006.
The 5′-end segment of the VP1 gene, which encodes the capsid N-terminal/shell (N/S) domain (228 bases) (Fig. 1B) (20, 21), has been used to monitor the genotypes of NoV strains in many countries, including Japan. Therefore, we used published sequence information on the N/S region to verify the 2006b dominance in Japan in 2006-2007. A representative neighbor-joining tree shows that 36 of 42 (86%) of the sequences collected during 2006 and 2007 in Japan (26, 34, 50) were grouped with sequences from the cluster I 2006b strains (Fig. 2B, cluster I, green boxes). Only 3 (7%) and 3 (7%) sequences were grouped with sequences from cluster II 2006a and cluster III, respectively (Fig. 2B, clusters II and III, green boxes). Bootstrap values at the branching points of the groups were relatively low, at less than 50/100, probably because the sequences used for the analysis were relatively short. However, the monophyletic relationships among the sequences of clusters I, II, and III were reproducible when the tree was constructed with different algorithms, and the results were consistent with the phylogeny of the complete capsid sequences (Fig. 2A). Together with the sequences obtained in the present study, 69 of the 79 (87%) 5′-capsid sequences obtained across Japan during 2006-2007 were classified as 2006b strains. Only 6 (8%) and 4 (5%) of the 79 sequences were related to the 2006a and 2004-2005 epidemic variant strains, respectively.
Ozawa et al. (36) reported the N/S region sequences obtained across Japan during the winter of 2005-2006. These sequences were used to examine whether or not the 2006a and 2006b strains existed before the atypical Japanese epidemic in 2006-2007. A representative neighbor-joining tree shows that 25 of 36 (69%) of the 2005-2006 sequences (36) were grouped with sequences from the cluster III sequences that emerged in winter 2004-2005 in Japan (Fig. 2C, cluster III, pink boxes). Notably, 2 (6%) and 9 (25%) sequences of the 2005-2006 sequences were grouped with sequences from the cluster I 2006b and cluster II 2006a, respectively (Fig. 2C, clusters I and II, pink boxes). The monophyletic relationships of clusters I, II, and III sequences were reproducible when the tree was constructed with different algorithms. Together, these data suggest that the 2006a and 2006b strains were present in Japan as minorities in winter 2005-2006 and that only the Cluster I 2006b strains dominated over the resident GII/4 and 2006a strains during 2006. There were two conserved amino acid substitutions (S9N and T15A) in the N terminus of the capsid shell domain of the 2006b strains in winter 2006-2007 compared to the 2006b source in Japan in winter 2005-2006.
Genetic links between NoV ORF1, ORF3, and genome among patients in the 2006-2007 epidemics.
Although partial sequences of the 3Dpol region have been reported (46), the molecular phylogenies of full-length ORF1 and ORF3 sequences of the 2006-2007 epidemic variants are unclear. Using phylogenetic analysis, we examined the genetic relationships between the new ORF1 and ORF3 sequences obtained in the present study and those of past epidemic GII/4 variant. For the analysis, we included sequences from Japan, China, Europe, and the United States, which were verified to be GII/4 strains by the genotyping of the ORF2 region.
Representative neighbor-joining trees show that the ORF1, ORF3, and genome sequences of the Japanese 2006-2007 variants again are divisable into three distinct lineage groups. All 33 sequences within cluster I in the ORF2 trees formed a monophyletic group (Fig. 3A, B, and C, cluster I, bootstrap value 100/100). Cluster I included a sequence from China in 2006 in the ORF3 tree (Duan/Beijing/2006/CHN). Cluster I was most closely related to the 02/03 cluster sequences in ORF1 and the genome, whereas in ORF3 it was most closely related to cluster II. The three sequences within cluster II in the ORF2 trees (Aomori1, Aomori2, and Saga5) also formed a unique group in all of the trees (Fig. 3A, B, and C, cluster II, bootstrap value 100/100). Finally, the single cluster III sequence in the ORF2 trees (Osaka2) was again grouped with the 2004-2005 epidemic variants in Japan and China (Fig. 3A, B, and C, cluster III, bootstrap value 100/100). The monophyletic relationships among clusters I, II, and III sequences were reproducible when the tree was constructed with different algorithms.
FIG. 3.



Neighbor-joining trees of the nucleotide sequences of the complete ORF1 (about 5.1 kb) (A), ORF3 (about 0.8 kb) (B), and near-full-length genome (∼7.5 kb) (C) regions of NoV GII/4. Bootstrap values with 100/100 are indicated at the nodes of the tree. Colored boxes indicate the sequences described in Fig. 2.
These trees derived from different genomic regions were consistent for most of the 2006-2007 GII/4 strains investigated. However, the sequences in cluster III exhibited discordant branching orders among ORFs (Fig. 2 and 3). Cluster III was placed within the GII/4 genotype in the ORF2 and ORF3 trees (Fig. 2A and 3B). However, it was placed outside the GII/4 clusters and grouped with the GII sequences; these are referred to as GII/10 and GII/12 in the ORF1 tree (Fig. 3A, bootstrap value 100/100). These data suggest genetic exchanges around the border between ORF1 and ORF2 during the evolutionary histories of the cluster III strains. Potential ancestors for the putative recombination events were not identifiable with currently available genome sequences in the public database. Cluster I 2006b had the longest branch length among all trees examined (Fig. 2 and 3), a finding consistent with its most recent detection.
Amino acid signatures of the 2006-2007 GII/4 epidemic strains.
The amino acid substitutions specific to the cluster I 2006b strains were examined. The deduced amino acid sequences of ORF1, ORF2, and ORF3 of the 33 cluster I 2006b obtained in the present study were aligned with those of the past ∼15 years of epidemic strains described in Fig. 2 and 3. Amino acids specific to clusters I and II were extracted and referred to as amino acid signatures of the 2006b and 2006a epidemic variants, respectively.
Twenty-six amino acid signatures were identified for the cluster I 2006b strains (Fig. 4A). The numbers of the signatures were 6, 1, 2, 1, 1, 3, 7, and 5 in the N-term, NTPase, 3A, VPg, 3Cpro, 3Dpol, VP1 (capsid), and VP2 protein regions, respectively. All seven signatures of the capsid protein were mapped in the protruding P2 domain. Siebenga et al. (46) referred to 48 amino acid positions in the capsid protein as informative sites when at least two epidemic strains had an identical amino acid. Of the 48 positions 7 were perfectly preserved only in the 2006b strains in Japan. The cluster II 2006a strains each had a distinct signature pattern (Fig. 4B). The numbers of 2006a signatures were 5, 4, 1, 6, 3, and 2 in the N-term, NTPase, 3A, 3Dpol, VP1, and VP2 protein regions, respectively. VPg and 3Cpro of 2006a had no signatures.
FIG. 4.


Unique amino acid substitutions of the 2006-2007 GII/4 epidemic strains. The deduced amino acids of NoV GII/4 ORF1s, ORF2s, and ORF3s of the 2006b and 2006a strains were aligned with those of the past chronological epidemic strains. Amino acids are each indicated by one letter code. The positions in the ORFs of the Lordsdale strain are used for the amino acid numbering. Dots indicate amino acids identical to the Lordsdale strain. Amino acid substitutions specific to the 2006b (A) and 2006a (B) strains are shown in boldface italic letters.
Amino acid variation among GII/4 strains.
To examine the levels of variability among the amino acid signature sites, amino acid variation at individual positions of ORF1, ORF2, and ORF3 of the GII/4 strains was examined using the deduced amino acid sequences of ORF1, ORF2, and ORF3 of all strains described in Fig. 3A, Fig. 2A, and Fig. 3B, respectively. Figure 5A shows the distribution of the Shannon entropy scores (18) along the GII/4 ORFs. The data show that some protein regions are particularly variable among the GII/4 strains. These include the N-terminal region of the N-term, 3A, P2 domain of capsid VP1, and the C-terminal region of VP2 (Fig. 5). Twenty-six signatures of the 2006b strains were mostly at variable sites among the GII/4 strains (Fig. 5, asterisks). The signatures were especially concentrated at three regions: the N-terminal region of the N-term, the P2 domain of capsid VP1, and the C-terminal region of VP2 (Fig. 4A and Fig. 5, blue bars).
FIG. 5.

Amino acid variation among the GII/4 epidemic variant proteins. Shannon entropy scores representing variations at individual amino acid positions (18) were calculated using NoV GII/4 ORF1, ORF2, and ORF3 sequences described in Fig. 3A, 2A, and 3B, respectively. The distribution of entropy scores in ORF1, ORF2, and ORF3 is shown along with the amino acid positions. An entropy score of 0 indicates absolute conservation, whereas that of 4.4 indicates complete randomness. Blue bars indicate regions rich in 2006b-specific changes.
When compared to the Lodsdale strain, all cluster I 2006b strains in Japan each had one amino acid insertion at position 395 in the capsid P2 domain, as reported in two Netherlands 2006b strains (46). Two strains in cluster II 2006a (Aomori1/2006/JP and Aomori2/2006/JP) each had three amino acid deletions in the VP2 protein at positions from 164 to 166 of the Lodsdale VP2. The deletions occurred immediately after the 2006a specific amino acid substitution at position 162 (Fig. 4B).
Structural insights into roles of capsid amino acid signatures.
An NoV capsid protein consists of a shell (S) and two protruding (P) domains: P1 and P2 (40). The P domain is exposed on the surface of the capsomer (40). Therefore, the P domain should play critical roles in virus entry into the target and host immune responses, although the precise roles have not been elucidated due to the lack of a tissue culture system to support effective NoV replication. To obtain structural insights into the roles of the amino acid substitutions specific to the 2006b capsid protein, we constructed a 3-D structure model of the VP1 P domain. The X-ray crystal structure of the P domain dimer of VA387 (4) was used for the modeling template, and the structure of the P domain dimer of a 2006b strain, Aichi3/2006/JP, was constructed by homology modeling. The model predicts that the folding of the P2 dimer is identical between the 2006b and the 1995-1996 epidemic strains.
The Shannon entropy scores with the GII/4 capsid P domain in Fig. 5 were expressed on the 3-D structure model (Fig. 6A). Notably, all seven amino acid signatures specific to the capsid domain of the 2006b strains were mapped on the variable P2 domain. Six of the seven signatures—L306, A356, P357, E372, H378, and A412—were positioned on the loops of the P2 domain, whereas the seventh signature, Y352, was positioned on the β-sheet of the P2 domain (Fig. 6A, side view). The side chains of six residues—Y352, A356, P357, E372, H378, and A412—formed accessible outer surfaces of the P2 domain, whereas the side chain of the seventh residue, L306, was positioned toward the P1 domain and was enclosed by neighboring amino acids (Fig. 6A, top view). E372 was located near the β2-β3 loop, which is one of the protruding loops on P2 and is extended by insertion mutation in the GII/3 (24). Thus, the 2006b-specific substitutions in the capsid protein were mostly clustered around regions with which host proteins such as an infection receptor(s) and antibodies can directly interact.
FIG. 6.

Structural model of the VP1 P domain dimer of the NoV GII/4 2006b strain. The model was constructed by homology modeling using the X-ray crystal structure of the P domain dimer of the 1995-1996 epidemic GII/4 strain (4). (A) Shannon entropy scores expressed on the P domain model. (B) Side and top views of the P domain model. Reported functional sites for virus entry into the cells are highlighted. Yellow dot circles, the fucose ring binding sites formed by P-domain dimer (4); cyan chain, an RGD motif (48) on the β2 sheet of the P domain; orange chain indicates an additional RGD-like motif, KGD (46), on the tip of the β4-β5 loop of the P domain. Red sticks indicate side chains of amino acids unique to the 2006b strains.
The side chains of the seven amino acid signatures specific to the capsid domain of the 2006b strains were mapped on the 3-D model along with putative functional sites for virus binding to target cells (Fig. 6B, red sticks). Reported functional sites for virus entry into the cells are highlighted. These include the fucose ring binding sites formed by P domain dimer (4) (yellow dot circles), an RGD motif (48) on the β2 sheet of the P domain (cyan chain), and an additional RGD-like motif, KGD (46), on the tip of the β4-β5 loop of the P domain (orange chain). Mutations in the conserved RGD motif in the P2 domain influence the binding of the viruslike particle of the VA387 strain to the histo-blood group antigens (48). As noted with 2006b European strains (46), an additional RGD-like motif, KGD, was present in the Japanese 2006b P2 domain (Fig. 6, orange residues). The KGD motif also appeared in the <1996 and 2002-2003 GII/4 epidemic strains (46) and not specific to the 2006b strains.
Interestingly, a 2006b-specific amino acid, H378, was positioned near the RGD motif and the newly created KGD motif (Fig. 6, cyan and orange ribbons, respectively). The H378 was also located near the binding site of the fucose ring (4), a part of the putative NoV infection receptor (Fig. 6, yellow dot circles). Consequently, H378, the RGD motif, and the new KGD motif each formed a protein surface rich in charged residues. Y352, A356, P357, and N412 were arranged linearly. These amino acids formed a putative accessible binding cleft on the P2 domain.
Siebenga et al. (46) have also reported the 3-D location of the variable sites using a structural model of the GII/4 capsid protein. They searched the sites where at least two strains had an identical amino acid substitution within a given sequence alignment (informative sites). This method helps one to identify mutations unique to some strains within a variant population. Using an alignment of complete capsid sequences of past epidemic variants, two 2006a strains, and two 2006b strains in Europe, they showed that many informative sites of the GII/4 capsid protein mapped to the surface of the P2 domain. Importantly, all 7 capsid amino acid signatures unique to the 2006b strains identified in the present study were included in the 48 informative sites obtained with the European strains.
DISCUSSION
In this study, we report a total of 37 near-full-length genome sequences of NoV GII/4 strains obtained from 11 sites in Japan during an atypical NoV epidemic that occurred between May 2006 and January 2007. The sequences were used to examine the genetic relationships among the epidemic strains, specific changes in the viral proteins, and the molecular mechanisms underlying the periodic outgrowth of new GII/4 variants in human populations. This is the first report to show that the global epidemic variant strain of NoV GII/4 has conserved amino acid signatures in all eight proteins.
Phylogenetic study of genomic and partial sequences provides basic information on the evolutionary histories of the GII/4 2006b epidemic variant strains (Fig. 2 and 3). The strong monophyly of the 2006b sequences out of past epidemic variants within the GII/4 cluster indicates that the 2006b strains were from an unidentified GII/4 variant source in nature. The branching order of the 2006b sequence cluster suggests that the 2006b source evolved from a common ancestor of the GII/4 02/03 strains, which is consistent with a previous conclusion (46). The constant monophyly of the 2006b sequences across distinct genomic regions within the GII/4 cluster indicates that the 2006b source is not the intergenogroup recombinant.
Notably, the 2006b source was present in Japan in winter 2005-2006 before notification of the atypical epidemic in winter 2006-2007 (Fig. 2C). In the 2005-2006 season, the 2006b source was rather minor compared to the 2004-2005 and 2006a strains (Fig. 2C). However, the minor 2006b strains dominated the resident GII/4 variants in Japan during 2006 (Fig. 2 and 3). Similar replacement of the resident GII/4 variants with the 2006b strains was observed in Europe (46) and Hong Kong (17) during 2006 as well. These data provide additional evidence for the recent global epidemic of the 2006b strains and strongly suggest that the new variant strains had selective advantages over other GII/4 strains that had been circulating before 2006.
Similar replacement of resident GII/4 strains by new GII/4 variant strains has periodically occurred on a global scale during the past ∼15 years and has often been associated with atypical NoV epidemics, as seen in the 2006-2007 epidemic in Japan (3, 12, 17, 28, 29, 32, 46). As Siebenga et al. (46) have discussed, the putative enhancement in the physical stability of virion, viral infectivity, or replication capability in cells alone may not explain the periodic outgrowth of new GII/4 variant strains. Indeed, no studies have indicated that the infectivity of GII/4 or the viral load of GII/4 in a stool specimen is increasing year by year.
Siebenga et al. (46) proposed that host population immunity might possibly act as a selective pressure for the periodic outgrowth phenomenon. They based this hypothesis on their observation that the GII/4 epidemic variants in The Netherlands underwent periodic amino acid substitutions in the capsid proteins. The results of our study are consistent with that hypothesis. The present analysis allowed us to identify the seven conserved amino acid signatures in the capsid of the 2006b strains (Fig. 4A). Remarkably, all seven signatures were arranged on the most protruding P2 domain of the capsid protein. Moreover, six of the seven signatures were positioned on the exposed protruding loops (Fig. 6). Interestingly, the 2006a strain, a recessive new GII/4 strain that appeared in 2006 in Japan, had only two unique signatures in the capsid (Fig. 4). These results provide further evidence that antigenic drift is involved in the outgrowth of new GII/4 strains over circulating GII/4 variants (28, 46).
The 2006b capsid signatures were positioned around the binding sites to putative viral infection receptors (Fig. 6). These amino acids are arranged so that the electrostatic potential on the binding surface and the shape of the ligand-binding cavity can be influenced. The data may imply that some capsid signatures are involved in controlling viral binding properties to infection receptors and thus may provide new bases on which to study the interplay among mutations. It is possible that some antigenic variations on the exposed surface of the capsid P2 domain could result in the functional deterioration or a defect of the capsid protein; therefore, compensatory mutations are required for virus survival. Experiments addressing each of these issues are in progress.
Previous studies focused mostly on substitutions in the capsid protein. Therefore, haplotype and noncapsid genetic changes of the 2006b strains were unclear. In addition to the capsid signatures, the present study discloses 19 changes specific to the 2006b strains (Fig. 4A). The high level of conservation of these 2006b signatures at otherwise variable sites among the GII/4 strains suggest the positive roles of some signatures in the epidemic in humans. Some signatures may be required for the escape from host population immunity, as suggested in the capsid. However, strong immune responses other than those against the capsid have not been reported.
An alternative possible role of conservation may be the maintenance or augmentation of replication fitness of the capsid mutants within a mixed variant population. In this regard, the 2006b signatures outside of the capsid region were particularly prominent in the N-term, and VP2 (Fig. 4A). The N-term is thought to function as a scaffolding protein for assembly of the genome replication complex (9, 11). The VP2 is thought to function as a minor capsid protein for RNA genome packaging (14). Therefore, some amino acid changes in these regions could critically influence intracellular viral replication and might have positive effects on maintaining improved replication fitness of the 2006b strains within a circulating NoV variant population. Further study is necessary to clarify these possibilities.
Another important factor contributing to the NoV diversity is evolutionary bottlenecks. For example, the monomorphic and divergent genotype of the 2006b observed in the present study could have been generated by a bottleneck effect during virus transmission: a few variants might have been transmitted, replicated, and spread to human populations. Therefore, it is possible that some unique genetic changes observed in the 2006b strains were primarily derived from the bottlenecks. On the other hand, given the extremely high levels of NoV infectivity (8, 27), it is likely that competition and natural selection of NoV variants within a mixed virus population occur frequently during virus spread to human populations. Further study is necessary to clarify how bottlenecks and natural selection shape the genetic diversity of NoV global epidemic variants.
Our findings, together with the general observations described in the introduction section, are compatible with a simple scheme of the NoV GII/4 evolution in nature: covariation in the capsid and other proteins confers an advantage in maintaining replication fitness in the face of population immunity, thereby being fixed better in virus that spread in humans during epidemics. When we take into account the ubiquitous existence of NoV in living and natural environments (8, 31, 41, 44), along with high levels of NoV infectivity (8, 27), physical stability (22), and virus production in asymptomatic individuals (36), it appears that humans are likely to be exposed much more frequently to NoVs than had previously been supposed. If this is the case, a human population could maintain certain levels of immunity against resident NoV variants, leading to periodic outgrowth, more severe symptomatic infection, and thus more frequent detection of antigenic variants that restore growth abilities by compensatory mutations.
In summary, we used here viral genome analysis and computational approaches to provide a new insight into the periodic pandemic of the GII/4 strain. A combination of phylogenetic and entropy analyses revealed for the first time the monomorphic and divergent haplotypes of the new epidemic strains. The data and computer-assisted structural study support a model of antigenic drift with tuning of the structure and functions of multiple proteins for the global outgrowth of new GII/4 variants. The availability of information on genome sequences and unique changes of the most recent epidemic variant strains will support studies of diagnostic assays and molecular epidemiology of NoV epidemic variants. Moreover, such information will provide a molecular basis for studying the interplay of viral proteins during the replication and adaptive evolution of NoV in nature.
Acknowledgments
We thank T. Shiino for helpful comments on the manuscript.
This study was supported by grants for Research on Emerging and Re-emerging Infectious Diseases and for Research on Food Safety from the Ministry of Health, Labor, and Welfare of Japan.
Footnotes
Published ahead of print on 4 September 2008.
REFERENCES
- 1.Ando, T., J. S. Noel, and R. L. Fankhauser. 2000. Genetic classification of “Norwalk-like viruses.” J. Infect. Dis. 181(Suppl. 2):S336-S348. [DOI] [PubMed] [Google Scholar]
- 2.Baker, D., and A. Sali. 2001. Protein structure prediction and structural genomics. Science 29493-96. [DOI] [PubMed] [Google Scholar]
- 3.Bull, R. A., E. T. Tu, C. J. McIver, W. D. Rawlinson, and P. A. White. 2006. Emergence of a new norovirus genotype II. 4 variant associated with global outbreaks of gastroenteritis. J. Clin. Microbiol. 44327-333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cao, S., Z. Lou, M. Tan, Y. Chen, Y. Liu, Z. Zhang, X. C. Zhang, X. Jiang, X. Li, and Z. Rao. 2007. Structural basis for the recognition of blood group trisaccharides by norovirus. J. Virol. 815949-5957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cauchi, M. R., J. C. Doultree, J. A. Marshall, and P. J. Wright. 1996. Molecular characterization of Camberwell virus and sequence variation in ORF3 of small round-structured (Norwalk-like) viruses. J. Med. Virol. 4970-76. [DOI] [PubMed] [Google Scholar]
- 6.Dingle, K. E. 2004. Mutation in a Lordsdale norovirus epidemic strain as a potential indicator of transmission routes. J. Clin. Microbiol. 423950-3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dingle, K. E., P. R. Lambden, E. O. Caul, and I. N. Clarke. 1995. Human enteric Caliciviridae: the complete genome sequence and expression of virus-like particles from a genetic group II small round structured virus. J. Gen. Virol. 76(Pt. 9)2349-2355. [DOI] [PubMed] [Google Scholar]
- 8.Estes, M. K., B. V. Prasad, and R. L. Atmar. 2006. Noroviruses everywhere: has something changed? Curr. Opin. Infect. Dis. 19467-474. [DOI] [PubMed] [Google Scholar]
- 9.Ettayebi, K., and M. E. Hardy. 2003. Norwalk virus nonstructural protein p48 forms a complex with the SNARE regulator VAP-A and prevents cell surface expression of vesicular stomatitis virus G protein. J. Virol. 7711790-11797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Felsenstein, J. 1985. Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39783-791. [DOI] [PubMed] [Google Scholar]
- 11.Fernandez-Vega, V., S. V. Sosnovtsev, G. Belliot, A. D. King, T. Mitra, A. Gorbalenya, and K. Y. Green. 2004. Norwalk virus N-terminal nonstructural protein is associated with disassembly of the Golgi complex in transfected cells. J. Virol. 784827-4837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gallimore, C. I., M. Iturriza-Gomara, J. Xerry, J. Adigwe, and J. J. Gray. 2007. Inter-seasonal diversity of norovirus genotypes: emergence and selection of virus variants. Arch. Virol. 1521295-1303. [DOI] [PubMed] [Google Scholar]
- 13.Garcia, C., H. L. DuPont, K. Z. Long, J. I. Santos, and G. Ko. 2006. Asymptomatic norovirus infection in Mexican children. J. Clin. Microbiol. 442997-3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Glass, P. J., L. J. White, J. M. Ball, I. Leparc-Goffart, M. E. Hardy, and M. K. Estes. 2000. Norwalk virus open reading frame 3 encodes a minor structural protein. J. Virol. 746581-6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Green, S. M., K. E. Dingle, P. R. Lambden, E. O. Caul, C. R. Ashley, and I. N. Clarke. 1994. Human enteric Caliciviridae: a new prevalent small round-structured virus group defined by RNA-dependent RNA polymerase and capsid diversity. J. Gen. Virol. 75(Pt. 8)1883-1888. [DOI] [PubMed] [Google Scholar]
- 16.Hansman, G. S., K. Natori, H. Shirato-Horikoshi, S. Ogawa, T. Oka, K. Katayama, T. Tanaka, T. Miyoshi, K. Sakae, S. Kobayashi, M. Shinohara, K. Uchida, N. Sakurai, K. Shinozaki, M. Okada, Y. Seto, K. Kamata, N. Nagata, K. Tanaka, T. Miyamura, and N. Takeda. 2006. Genetic and antigenic diversity among noroviruses. J. Gen. Virol. 87909-919. [DOI] [PubMed] [Google Scholar]
- 17.Ho, E. C., P. K. Cheng, A. W. Lau, A. H. Wong, and W. W. Lim. 2007. Atypical norovirus epidemic in Hong Kong during summer of 2006 caused by a new genogroup II/4 variant. J. Clin. Microbiol. 452205-2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang, C. C., M. Tang, M. Y. Zhang, S. Majeed, E. Montabana, R. L. Stanfield, D. S. Dimitrov, B. Korber, J. Sodroski, I. A. Wilson, R. Wyatt, and P. D. Kwong. 2005. Structure of a V3-containing HIV-1 gp120 core. Science 3101025-1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hutson, A. M., R. L. Atmar, and M. K. Estes. 2004. Norovirus disease: changing epidemiology and host susceptibility factors. Trends Microbiol. 12279-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kageyama, T., M. Shinohara, K. Uchida, S. Fukushi, F. B. Hoshino, S. Kojima, R. Takai, T. Oka, N. Takeda, and K. Katayama. 2004. Coexistence of multiple genotypes, including newly identified genotypes, in outbreaks of gastroenteritis due to Norovirus in Japan. J. Clin. Microbiol. 422988-2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Katayama, K., H. Shirato-Horikoshi, S. Kojima, T. Kageyama, T. Oka, F. Hoshino, S. Fukushi, M. Shinohara, K. Uchida, Y. Suzuki, T. Gojobori, and N. Takeda. 2002. Phylogenetic analysis of the complete genome of 18 Norwalk-like viruses. Virology 299225-239. [DOI] [PubMed] [Google Scholar]
- 22.Keswick, B. H., T. K. Satterwhite, P. C. Johnson, H. L. DuPont, S. L. Secor, J. A. Bitsura, G. W. Gary, and J. C. Hoff. 1985. Inactivation of Norwalk virus in drinking water by chlorine. Appl. Environ. Microbiol. 50261-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kimura, M. 1980. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16111-120. [DOI] [PubMed] [Google Scholar]
- 24.Kumar, S., W. Ochoa, S. Kobayashi, and V. S. Reddy. 2007. Presence of a surface-exposed loop facilitates trypsinization of particles of Sinsiro virus, a genogroup II. 3 norovirus. J. Virol. 811119-1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kumar, S., K. Tamura, and M. Nei. 1993. Molecular evolutionary genetic analysis, version 1.0 Institute of Molecular Evolutionary Genetics, Pennsylvania State University, University Park, PA.
- 26.Kumazaki, M., S. Usuku, and Y. Noguchi. 2007. New variant of norovirus GII/4 strains prevalent in Yokohama City, October 2006-March 2007. Jpn. J. Infect. Dis. 60323-324. [PubMed] [Google Scholar]
- 27.Lindesmith, L., C. Moe, S. Marionneau, N. Ruvoen, X. Jiang, L. Lindblad, P. Stewart, J. LePendu, and R. Baric. 2003. Human susceptibility and resistance to Norwalk virus infection. Nat. Med. 9548-553. [DOI] [PubMed] [Google Scholar]
- 28.Lindesmith, L. C., E. F. Donaldson, A. D. Lobue, J. L. Cannon, D. P. Zheng, J. Vinje, and R. S. Baric. 2008. Mechanisms of GII.4 Norovirus persistence in human populations. PLoS Med. 5e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lopman, B., H. Vennema, E. Kohli, P. Pothier, A. Sanchez, A. Negredo, J. Buesa, E. Schreier, M. Reacher, D. Brown, J. Gray, M. Iturriza, C. Gallimore, B. Bottiger, K. O. Hedlund, M. Torven, C. H. von Bonsdorff, L. Maunula, M. Poljsak-Prijatelj, J. Zimsek, G. Reuter, G. Szucs, B. Melegh, L. Svennson, Y. van Duijnhoven, and M. Koopmans. 2004. Increase in viral gastroenteritis outbreaks in Europe and epidemic spread of new norovirus variant. Lancet 363682-688. [DOI] [PubMed] [Google Scholar]
- 30.Monica, B., S. Ramani, I. Banerjee, B. Primrose, M. Iturriza-Gomara, C. I. Gallimore, D. W. Brown, F. M., P. D. Moses, J. J. Gray, and G. Kang. 2007. Human caliciviruses in symptomatic and asymptomatic infections in children in Vellore, South India. J. Med. Virol. 79544-551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nishida, T., O. Nishio, M. Kato, T. Chuma, H. Kato, H. Iwata, and H. Kimura. 2007. Genotyping and quantitation of noroviruses in oysters from two distinct sea areas in Japan. Microbiol. Immunol. 51177-184. [DOI] [PubMed] [Google Scholar]
- 32.Noel, J. S., R. L. Fankhauser, T. Ando, S. S. Monroe, and R. I. Glass. 1999. Identification of a distinct common strain of “Norwalk-like viruses” having a global distribution. J. Infect. Dis. 1791334-1344. [DOI] [PubMed] [Google Scholar]
- 33.Oka, T., M. Yamamoto, M. Yokoyama, S. Ogawa, G. S. Hansman, K. Katayama, K. Miyashita, H. Takagi, Y. Tohya, H. Sato, and N. Takeda. 2007. Highly conserved configuration of catalytic amino acid residues among calicivirus-encoded proteases. J. Virol. 816798-6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Okada, M., T. Ogawa, H. Yoshizumi, H. Kubonoya, and K. Shinozaki. 2007. Genetic variation of the norovirus GII-4 genotype associated with a large number of outbreaks in Chiba prefecture, Japan. Arch. Virol. 1522249-2252. [DOI] [PubMed] [Google Scholar]
- 35.Okada, M., T. Tanaka, M. Oseto, N. Takeda, and K. Shinozaki. 2006. Genetic analysis of noroviruses associated with fatalities in healthcare facilities. Arch. Virol. 1511635-1641. [DOI] [PubMed] [Google Scholar]
- 36.Ozawa, K., T. Oka, N. Takeda, and G. S. Hansman. 2007. Norovirus infections in symptomatic and asymptomatic food handlers in Japan. J. Clin. Microbiol. 453996-4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Phan, T. G., T. A. Nguyen, T. Kuroiwa, K. Kaneshi, Y. Ueda, S. Nakaya, S. Nishimura, T. Nishimura, A. Yamamoto, S. Okitsu, and H. Ushijima. 2005. Viral diarrhea in Japanese children: results from a one-year epidemiologic study. Clin. Lab. 51183-191. [PubMed] [Google Scholar]
- 38.Phan, T. G., S. Takanashi, K. Kaneshi, Y. Ueda, S. Nakaya, S. Nishimura, K. Sugita, T. Nishimura, A. Yamamoto, F. Yagyu, S. Okitsu, N. Maneekarn, and H. Ushijima. 2006. Detection and genetic characterization of norovirus strains circulating among infants and children with acute gastroenteritis in Japan during 2004-2005. Clin. Lab. 52519-525. [PubMed] [Google Scholar]
- 39.Ponder, J. W., and D. A. Case. 2003. Force fields for protein simulations. Adv. Protein Chem. 6627-85. [DOI] [PubMed] [Google Scholar]
- 40.Prasad, B. V., M. E. Hardy, T. Dokland, J. Bella, M. G. Rossmann, and M. K. Estes. 1999. X-ray crystallographic structure of the Norwalk virus capsid. Science 286287-290. [DOI] [PubMed] [Google Scholar]
- 41.Saitoh, M., H. Kimura, K. Kozawa, O. Nishio, and A. Shoji. 2007. Detection and phylogenetic analysis of norovirus in Corbicula fluminea in a freshwater river in Japan. Microbiol. Immunol. 51815-822. [DOI] [PubMed] [Google Scholar]
- 42.Saitou, N., and M. Nei. 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4406-425. [DOI] [PubMed] [Google Scholar]
- 43.Sakon, N., K. Yamazaki, T. Yoda, T. Tsukamoto, T. Kase, K. Taniguchi, K. Takahashi, and T. Otake. 2007. Norovirus storm in Osaka, Japan, last winter (2006/2007). Jpn. J. Infect. Dis. 60409-410. [PubMed] [Google Scholar]
- 44.Sano, D., Y. Ueki, T. Watanabe, and T. Omura. 2006. Genetic variation in the conservative gene region of Norovirus genogroup II strains in environmental and stool samples. Environ. Sci. Technol. 407423-7427. [DOI] [PubMed] [Google Scholar]
- 45.Shannon, C. E. 1997. The mathematical theory of communication. 1963. MD Comput. 14306-317. [PubMed] [Google Scholar]
- 46.Siebenga, J. J., H. Vennema, B. Renckens, E. de Bruin, B. van der Veer, R. J. Siezen, and M. Koopmans. 2007. Epochal evolution of GGII.4 norovirus capsid proteins from 1995 to 2006. J. Virol. 819932-9941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Song, H., E. E. Nakayama, M. Yokoyama, H. Sato, J. A. Levy, and T. Shioda. 2007. A single amino acid of the human immunodeficiency virus type 2 capsid affects its replication in the presence of cynomolgus monkey and human TRIM5αs. J. Virol. 817280-7285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tan, M., P. Huang, J. Meller, W. Zhong, T. Farkas, and X. Jiang. 2003. Mutations within the P2 domain of norovirus capsid affect binding to human histo-blood group antigens: evidence for a binding pocket. J. Virol. 7712562-12571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 224673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ueki, Y., M. Shoji, C. Sato, Y. Sato, Y. Okimura, N. Saito, H. Otomo, and H. Onodera. 2007. Norovirus GII/4 variants observed in outbreaks of gastroenteritis in Miyagi prefecture between November and December of 2006. Jpn. J. Infect. Dis. 60240-241. [PubMed] [Google Scholar]
- 51.Xi, J. N., D. Y. Graham, K. N. Wang, and M. K. Estes. 1990. Norwalk virus genome cloning and characterization. Science 2501580-1583. [DOI] [PubMed] [Google Scholar]
- 52.Yamagami, T., and S. Hara. 2007. Outbreak of norovirus gastroenteritis involving multiple institutions. Jpn. J. Infect. Dis. 60146-147. [PubMed] [Google Scholar]
- 53.Zheng, D. P., T. Ando, R. L. Fankhauser, R. S. Beard, R. I. Glass, and S. S. Monroe. 2006. Norovirus classification and proposed strain nomenclature. Virology 346312-323. [DOI] [PubMed] [Google Scholar]