

Abstract
The fusogenic human immunodeficiency virus type 1 (HIV-1) gp41 core structure is a stable six-helix bundle formed by its N- and C-terminal heptad repeat sequences. Notably, the negatively charged residue Asp632 located at the pocket-binding motif in the C-terminal heptad repeat interacts with the positively charged residue Lys574 in the pocket formation region of the N-terminal heptad repeat to form a salt bridge. We previously demonstrated that the residue Lys574 plays an essential role in six-helix bundle formation and virus infectivity and is a key determinant of the target for anti-HIV fusion inhibitors. In this study, the functionality of residue Asp632 has been specifically characterized by mutational analysis and biophysical approaches. We show that Asp632 substitutions with positively charged residues (D632K and D632R) or a hydrophobic residue (D632V) could completely abolish Env-mediated viral entry, while a protein with a conserved substitution (D632E) retained its activity. Similar to the Lys574 mutations, nonconserved substitutions of Asp632 also severely impaired the α-helicity, stability, and conformation of six-helix bundles as shown by N36 and C34 peptides as a model system. Furthermore, nonconserved substitutions of Asp632 significantly reduced the potency of C34 to sequestrate six-helix bundle formation and to inhibit HIV-1-mediated cell-cell fusion and infection, suggesting its importance for designing antiviral fusion inhibitors. Taken together, these data suggest that the salt bridge between the N- and C-terminal heptad repeat regions of the fusion-active HIV-1 gp41 core structure is critical for viral entry and inhibition.
Infection with human immunodeficiency virus type 1 (HIV-1) is mediated by its envelope glycoprotein (Env), a type I transmembrane protein which is originally synthesized as the single, glycosylated, polyprotein precursor gp160 and subsequently cleaved by a cellular protease to yield gp120 and gp41 subunits (13, 14, 20, 46, 48). Upon binding of the HIV-1 Env surface subunit gp120 to the cell receptor CD4 and subsequently to a coreceptor (CCR5 or CXCR4), its transmembrane subunit gp41 is released to mediate fusion of viral and cellular membranes (20, 25, 54). Structurally, HIV-1 gp41 consists of extracellular, transmembrane, and cytoplasmic domains (Fig. 1A). Its extracellular domain (ectodomain) contains four major functional regions: a hydrophobic, glycine-rich fusion peptide; an N-terminal heptad repeat (NHR) (also called HR1), a C-terminal heptad repeat (CHR) (also called HR2), and a tryptophan-rich region. In the early 1990s, several peptides derived from the NHR (N peptides) and CHR (C peptides) were found to have potent anti-HIV activity (30, 43, 68, 69). Although their mechanism of action was not known at that time, the unprecedented anti-HIV activity of these peptides opened a new avenue for developing antiviral drugs. A C peptide known as T20 (brand name, Fuzeon) has been successfully developed as a novel class of anti-HIV drugs for clinical use (36, 37, 50).
FIG. 1.
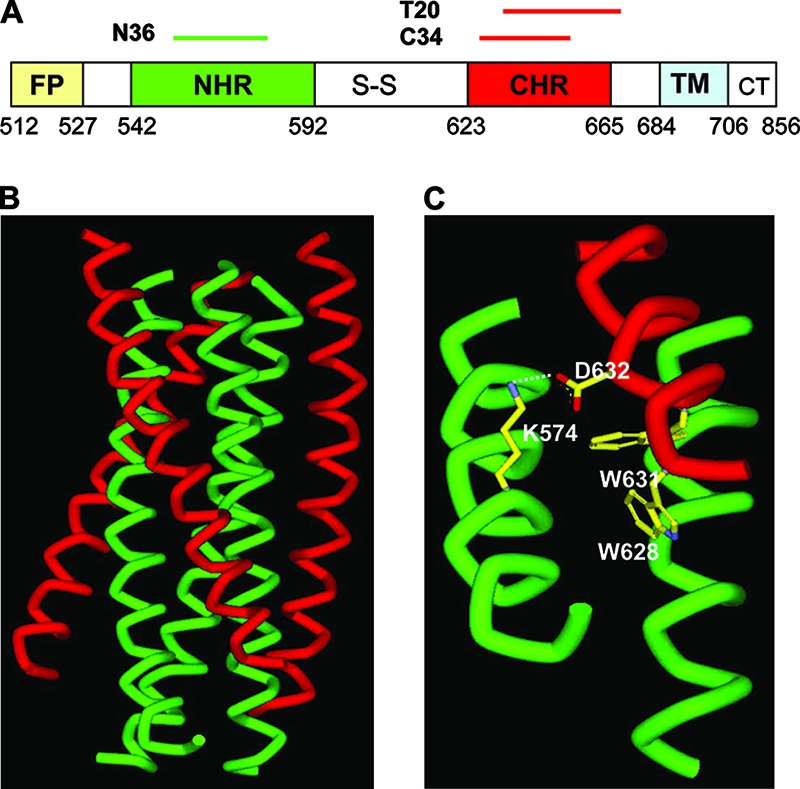
Structure and function of the HIV-1 gp41 core. (A) Schematic view of the gp41 functional regions. FP, fusion peptide; S-S, disulfide bond loop; TM, transmembrane domain; CT, cytoplasmic tail. The residue number for each region corresponds to its position in gp160 of HIV-1HXB2. (B) Crystal structure of the six-helix bundle modeled by the peptides N36 and C34. The N36 helices are green, whereas the C34 helices are red. (C) The salt bridge formed by residues Lys574 in the NHR and Asp632 in the CHR.
The finding of anti-HIV peptides also provided important information to explore the structure of the gp41 molecule. In 1995, Lu et al (42). identified a stable, proteinase-resistant structure comprising two peptides, N51 and C43, derived from a recombinant protein fragment of the gp41 ectodomain by using protein dissection experiments. N51 and C43 associate to form a stable, α-helical trimeric complex of heterodimers, with N51 and C43 helices oriented in an antiparallel fashion (42). Further proteolysis of the N51/C43 complex resulted in the identification of the N36 and C34 peptides (43). Similarly, N36 and C34 form a stable α-helical trimer of NHR-CHR heterodimers, whereas N36 alone is predominantly aggregated and C34 alone remains mostly unfolded (43). X-ray crystallographic studies by three independent groups confirmed that the thermostable subdomain of HIV-1 gp41 folds into a α-helical six-helix bundle, in which three NHR helices form an interior, parallel coiled-coil trimer while three CHR helices pack in an oblique, antiparallel manner into the highly conserved deep hydrophobic grooves on the surface of the N-helical trimer (2, 5, 60, 63). Nuclear magnetic resonance analysis showed that the simian immunodeficiency virus gp41 also formed similar six-helix bundle structure in solution (2). The current model suggests that the six-helix bundle structure formed by NHR and CHR helices represents a core of the fusion-active conformation of HIV-1 envelope, which brings the viral and cellular membranes into close apposition to enable fusion pore formation and virus internalization. Synthetic peptides derived from the NHR and CHR regions of HIV-1 gp41 are thought to target the prehairpin fusion intermediate through interacting with the counterpart regions, thus inhibiting fusion of the virus with the target cell in a dominant-negative manner (4, 6, 19, 53, 69).
The crystal structure of the HIV-1 gp41 core also reveals a highly conserved, hydrophobic cavity on the surface of the NHR helices (2, 5, 60, 63). Considerable evidence suggests that the hydrophobic interaction between the cavity-forming domain in the NHR and the cavity-binding motif in the CHR is highly important for stabilization of the six-helix bundle and viral infectivity (1, 18, 44, 61, 64). Importantly, the deep hydrophobic cavity formed by the internal coiled-coil trimer has been proposed to be an ideal target for designing inhibitors that can disrupt six-helix bundle formation (4, 6, 62). Another important observation from the X-ray structure is that the positively charged residue Lys574 located at the cavity-forming region in the NHR interacts with the negatively charged residue Asp632 at the cavity-binding motif in the CHR to form a salt bridge (Fig. 1C). We have proposed that during the fusion process the formation of the salt bridge between the NHR and CHR of the gp41 core, besides the hydrophobic interaction in the cavity, may play a critical role in viral infection and inhibition thereof (23). We previously demonstrated that the residue Lys574 in the NHR of gp41 is essential for formation of a functional six-helix bundle and virus entry (23). Nonconserved substitutions of Lys574 could completely abolish Env-mediated HIV-1 entry and severely impaired the conformation and stability of the six-helix bundles. The mutant peptides bearing nonconserved substitutions of Lys574 had much less potency to inhibit HIV-1 infection. These data highlight the importance of salt bridge formation in the gp41 core structure (23). In this study, we have focused our efforts on characterizing the functions of residue Asp632 in the gp41 CHR region of HIV-1 by using mutational analysis and biophysical approaches.
MATERIALS AND METHODS
Construction of HIV-1 Env mutants.
The plasmid encoding HIV-1 HXB2 Env was obtained from Kathleen Page and Dan Littman through the NIH AIDS Research and Reference Reagent Program. A panel of HXB2 Env mutants (D632E, D632K, D632R, and D632V) were generated by mutagenesis using the QuikChange XL kit (Stratagene, La Jolla, CA) and verified by DNA sequencing. The primers used for construction of HIV-1 Env mutants were designed and synthesized according to the manufacturer's instructions.
Single-cycle infection assay.
HIV-1 pseudovirus was developed as previously described (22, 32). Briefly, HEK293T cells were cotransfected with a plasmid encoding wild-type or mutant HXB2 Env and a plasmid carrying an Env-defective, luciferase-expressing HIV-1 genome (pNL4-3.luc.RE) by using Fugene 6 reagents (Boehringer-Mannheim, Indianapolis, IN). Supernatants containing HIV-1 pseudovirus were harvested at 48 h posttransfection and clarified by centrifugation at 1,000 × g for 10 min and filtration through a 0.45-μm filter. The virus-containing clarified supernatants were stored at −80°C before use. For single-cycle infection, U87-T4-CXCR4 cells were plated at 104cells/well in 96-well tissue culture plates and grown overnight. The pseudovirus was added to cells and incubated overnight. The culture was refed and incubated for an additional 48 h. Cells were washed with phosphate-buffered saline (PBS) and lysed using the lysis reagent included in a luciferase kit (Promega, Madison, WI). Aliquots of cell lysates were transferred to 96-well Costar flat-bottom luminometer plates (Corning Costar, Corning, NY), followed by addition of luciferase substrate (Promega). Relative light units were determined immediately in an Ultra 384 luminometer (Tecan US).
Synthesis of wild-type and mutant peptides.
Peptides N36 (residues 546 to 581, SGIVQQQNNLLRAIEAQQHLLQLTVWGIKQLQARIL) and C34 (residues 628 to 661, WMEWDREINNYTSLIHSLIEESQNQQEKNEQELL), as well as C34 mutants (D632E, D632K, D632R, and D632V), were synthesized by a standard solid-phase 9-fluorenylmethoxy carbonyl method in the MicroChemistry Laboratory of the New York Blood Center. The peptides were purified to homogeneity (>95% purity) by high-performance liquid chromatography and identified by laser desorption mass spectrometry (PerSeptive Biosystems, Framingham, MA). The concentration of peptides was determined by UV absorbance with theoretically calculated molar extinction coefficients (ɛ) (280 nm) of 5,500 mol/liter−1·cm−1 and 1,490 mol/liter−1·cm−1 based on the number of tryptophan (Trp) residues and tyrosine (Tyr) residues (all the peptides tested contain Trp and/or Tyr), respectively (16).
CD spectroscopy.
A C peptide (C34 or its mutant) was incubated with the N peptide (N36) at 37°C for 30 min (the final concentrations of N peptide and C peptide were 10 μM in 50 mM sodium phosphate and 150 mM NaCl, pH 7.2). The isolated N and C peptides were also tested. Circular dichroism (CD) spectra of these peptides and peptide mixtures were acquired on a Jasco spectropolarimeter (model J-715; Jasco Inc., Japan) at room temperature using a 5.0-nm bandwidth, 0.1-nm resolution, 0.1-cm path length, 4.0-s response time, and 50-nm/min scanning speed. The spectra were corrected by subtraction of a blank corresponding to the solvent. The α-helical content was calculated from the CD signal by dividing the mean residue ellipticity at 222 nm by the value expected for 100% helix formation (−33,000 degrees cm2 dmol−1) (9, 58). Thermal denaturation was monitored at 222 nm by applying a thermal gradient of 2°C/min in the range of 4 to 98°C. The reversibility of the peptide mixtures was measured by performing the reverse melt from 98 to 4°C. The melting curve was smoothened, and the midpoint of the thermal unfolding transition (Tm) values was calculated using Jasco software utilities as described previously (39).
N-PAGE.
Native polyacrylamide gel electrophoresis (N-PAGE) was carried out to determine six-helix bundle formation between the N and C peptides as described previously (41). Briefly, a C peptide (C34 or its mutant) was incubated with N36 at a final concentration of 40 μM at 37°C for 30 min. The mixture was loaded onto a 10- by 1.0-cm precast 18% Tris-glycine gel (Invitrogen, Carlsbad, CA) at 25 μl/per well with an equal volume of Tris-glycine native sample buffer (Invitrogen). Gel electrophoresis was carried out with a constant voltage of 125 V at room temperature for 2 h. The gel was then stained with Coomassie blue and imaged with a FluorChem 8800 imaging system (Alpha Innotech Corp., San Leandro, CA).
Detection of six-helix bundles by ELISA.
A capture enzyme-linked immunosorbent assay (ELISA) as previously described (31) was used to detect the formation of six-helix bundles between the N and C peptides. Briefly, 2 μg/ml immunoglobulin G (IgG) purified from rabbit antisera developed against the N36/C34 complex was applied to wells of a 96-well polystyrene plate (Corning Costar, Acton, MA) in 0.1 M carbonate buffer (pH 9.6) at 4°C overnight. After blocking with 2% nonfat milk, a mixture formed by an N peptide (N36 or its mutant) and C34 at equimolar concentrations (2 μM) was added and incubated at 37°C for 1 h, followed by four washes with PBS containing 0.1% Tween 20. Captured six-helix bundles were detected by sequential addition of NC-1, a mouse monoclonal antibody (MAb) specific for the N36/C34 six-helix bundle that was developed in our laboratory (29); biotin-labeled goat anti-mouse IgG (Sigma, St. Louis, MO); and streptavidin-labeled horseradish peroxidase (SA-HRP) (Zymed, South San Francisco, CA). The reaction was visualized by addition of the substrate 3,3′,5,5′-tetramethylbenzidine, and absorbance at 450 nm was measured with an ELISA plate reader (Tecan US, Research Triangle Park, NC).
Inhibition of six-helix bundle formation by CHR peptides.
The inhibitory activity of CHR peptides (C34 and its mutants) on six-helix bundle formation was measured by a modified ELISA-based method as previously described (24, 24). Briefly, a 96-well polystyrene plate (Costar, Corning Inc., Corning, NY) was coated with six-helix-bundle-specific MAb NC-1 IgG (4 μg/ml in 0.1 M Tris, pH 8.8). The tested peptide at graded concentrations was mixed with C34-biotin (0.25 μM) and incubated with N36 (0.25 μM) at room temperature for 30 min. The mixture was then added to the NC-1-coated plate, followed by incubation at room temperature for 30 min and washing with a washing buffer (PBS containing 0.1% Tween 20) three times. SA-HRP (Invitrogen) and the substrate 3,3′,5,5′-tetramethylbenzidine (Sigma) were then added sequentially. Absorbance at 450 nm was measured using an ELISA reader (Ultra 384; Tecan, Research Triangle Park, NC). The percent inhibition by the peptides and the 50% inhibitory concentrations (IC50s) were calculated as previously described (33).
Cell-cell fusion assay.
A dye transfer assay was used for detection of HIV-1-mediated cell-cell fusion as previously described (32). Briefly, H9/HIV-1IIIB-infected cells were labeled with a fluorescent reagent, calcein-AM (Molecular Probes, Inc., Eugene, OR) and then incubated with MT-2 cells (ratio of 1:5) in 96-well plates at 37°C for 2 h in the presence or absence of tested peptides. The fused and unfused calcein-labeled HIV-1-infected cells were counted under an inverted fluorescence microscope (Zeiss, Germany) with an eyepiece micrometer disc. The percent inhibition of cell-cell fusion and the IC50s were calculated as described before using the GraphPad Prism software (GraphPad Software Inc., San Diego, CA).
Measurement of HIV-1 infectivity.
The inhibitory activity of peptide C34 or its mutants on infection by a laboratory-adapted HIV-1 strain (IIIB) was determined as previously described (32). Briefly, 1 × 104 MT-2 cells were infected with HIV-1 IIIB at 100 50% tissue culture infective doses in 200 μl RPMI 1640 medium containing 10% fetal bovine serum in the presence or absence of the peptides at graded concentrations overnight. The culture supernatants were then removed, and fresh medium was added. On the fourth day postinfection, 100 μl of culture supernatants was collected from each well, mixed with an equal volume of 5% Triton X-100, and assayed for p24 antigen by ELISA. Briefly, the wells of polystyrene plates (Immulon 1B; Dynex Technology, Chantilly, VA) were coated with HIV-specific Ig in 0.85 M carbonate-bicarbonate buffer (pH 9.6) at 4°C overnight, followed by washes with 0.01 M PBS containing 0.05% Tween 20 and blocking with PBS containing 1% dry fat-free milk (Bio-Rad Inc., Hercules, CA). Virus lysates were added to the wells and incubated at 37°C for 1 h. After extensive washes, anti-p24 MAb (183-12H-5C), biotin-labeled anti-mouse IgG1 (Santa Cruz Biotech., Santa Cruz, CA), SA-HRP (Zymed), and 3,3′,5,5′-tetramethylbenzidine (Sigma) were added sequentially. Reactions were terminated by addition of 1 N H2SO4. Absorbance at 450 nm was recorded in an ELISA reader (Ultra 384; Tecan). Recombinant protein p24 (US Biological, Swampscott, MA) was included for establishing a standard dose-response curve.
RESULTS
The salt bridge-forming residue Asp632 in the CHR is essential for Env-mediated HIV-1 entry.
We have recently reported that the salt bridge-forming residue Lys574 in the NHR of HIV-1 gp41 plays an essential role for HIV-1 entry into target cells (23). Here, we used a similar approach to analyze the functionality of residue Asp632, a partner residue in the CHR to form the salt bridge between two heptad repeat regions of the fusogenic gp41 molecule. The negatively charged Asp632 was converted to a negatively charged glutamic acid (Glu) residue, a positively charged lysine (Lys) or arginine (Arg) residue, or a hydrophobic valine (Val) residue by mutagenesis. The point mutations were verified by DNA sequencing, and the expression of HIV-1HXB2 Env glycoprotein was confirmed by radioactive immunoprecipitation assay (data not shown). To determine the effects of these mutations on the Env-mediated HIV-1 entry, a panel of pseudoviruses bearing wild-type or mutant Env glycoprotein were generated and used in a single-cycle complementation assay. As shown in Fig. 2, the conserved mutation of Asp632 (D632E) had no significant effect on viral entry, since the pseudovirus retained infectivity similar to that of the wild-type virus. However, nonconserved substitutions (D632K, D632R, or D632V) completely abolished the infectivity of the pseudoviruses. These results suggest that the salt bridge-forming residue Asp632 in the CHR region of HIV-1 gp41 is also essential for virus entry.
FIG. 2.
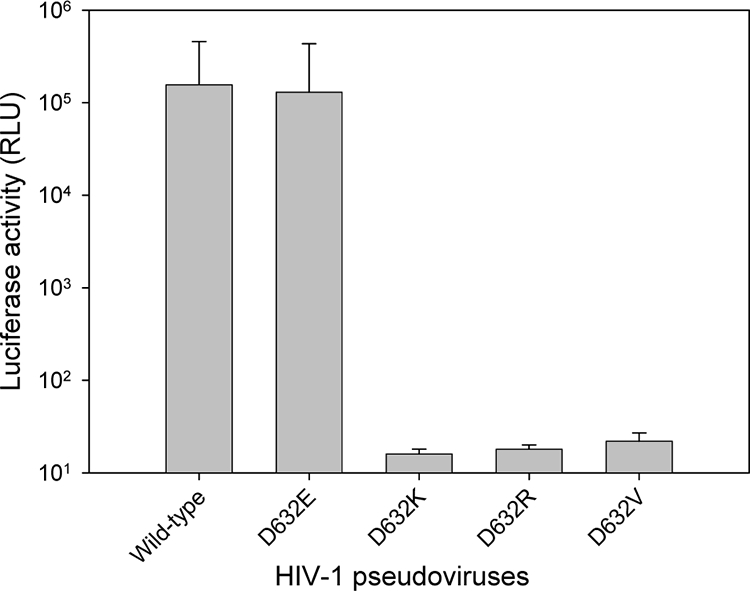
Infectivity of HIV-1 pseudoviruses. Single-cycle infection of the corresponding wild-type or mutant HIV-1HXB2 pseudovirus on U87-T4-CXCR4 cells was measured by a luciferase-based assay. Each sample was tested in triplicate, and data are presented as means ± standard deviations. RLU, relative light units.
Similar to the case for Lys574, substitutions of Asp632 with nonconserved residues severely impair the stability of six-helix bundles.
To investigate the mechanism by which nonconserved substitutions of Asp632 can determine Env-mediated HIV-1 infectivity, we used CD spectroscopy to analyze the interaction of NHR peptide N36 and CHR peptide C34. The counterpart peptides N36 and C34 were included in this study because the thermostable six-helix bundle complex formed by these two peptides has been proposed to be a core structure of the fusion-active gp41 (5). As expected, the equimolar mixture of wild-type C34 and N36 has a typical α-helix conformation, characterized by double minima at 208 and 222 nm (Fig. 3A). The CD spectra of all mutant C34 peptides indicated that they could interact with N36 to form six-helix bundle structures, as shown by the induction of α-helix signals (Fig. 3B to E). However, the α-helicity induced by nonconserved C34 mutations (D632K, D632R, or D632V) significantly decreased whereas the conserved D632E mutation resulted in a slightly increased α-helical signal.
FIG. 3.

The α-helical conformation of the complex formed by N and C peptides analyzed by CD spectroscopy. The final concentration of each peptide in PBS was 10 μM.
The stability of each six-helix bundle structure formed by N and C peptides was determined by thermal denaturation analyses. The signal at 222 nm of the peptide mixture was monitored as the temperature was slowly raised from 4 to 98°C at a scan rate of 2°C/min. The melting curves for the peptide combination are shown in Fig. 4, and their Tm values are shown in Table 1. As a control, the complex of N36 and C34 had a Tm of 64°C (Fig. 4A), consistent with our previous data (24). Similar to the case for residue Lys574, the stability of six-helix bundles was significantly reduced by nonconservative Asp632 substitutions. The D632K, D632R, and D632V mutants had Tm values of 54°C, 55°C, and 58°C, respectively. In contrast, the six-helix bundle bearing the conserved D632E mutation even had a slightly increased Tm value (67°C). These peptide pairs unfolded reproducibly and reversibly as the reverse thermal melts were monitored from 98 to 4°C or heated to 98°C again. These results suggest that substitutions of Asp632 with nonconserved residues severely impair the α-helicity and stability of six-helix bundles formed by NHR and CHR peptides.
FIG. 4.

Thermostability of the α-helical complexes formed by N and C peptides, determined by thermal denaturation analyses. (A to E) The unfolding temperature of each complex was scanned at 222 nm by CD spectroscopy, and their Tm values were calculated. The insets show the first derivative of the curve against temperature, which was used to determine the Tm value. (F) Reversibility of the complexes formed by N and C peptides.
TABLE 1.
Biophysical properties and anti-HIV activity of wild-type and mutant C34
| Peptide | 6-HB formation
|
IC50 (nM)
|
||||
|---|---|---|---|---|---|---|
| [θ]222 | α-Helicity (%) | Tm (oC) | 6-HB | Cell-cell fusion | Infection | |
| C34 | −31,271 | 94.8 | 64 | 125.9 | 5.0 | 1.4 |
| D632E | −32,146 | 97.4 | 67 | 100.0 | 6.6 | 2.2 |
| D632K | −24,926 | 75.5 | 54 | 8,582.2 | 171.2 | 35.5 |
| D632R | −22,776 | 69.0 | 55 | 6,558.4 | 136.0 | 29.3 |
| D632V | −26,259 | 79.6 | 58 | 5,209.6 | 92.6 | 15.2 |
Substitutions of Asp632 with nonconserved residues severely impair the conformation of six-helix bundles.
Our previous studies indicate that the six-helix bundle core structure formed by the peptides N36 and C34 can be visualized by N-PAGE (24). In this study, we used N-PAGE to detect six-helix bundles formed by C34 bearing conservative and nonconservative mutations. Consistent with our previous data, peptide N36, with a net positive charge, showed no band in the native gel, as it migrated up and off the gel, while peptide C34, which carries a net negative charge, showed a specific band (Fig. 5). The conserved D632E mutant gave a band with a migration rate similar to that of the wild-type C34, whereas the peptides with nonconserved mutations (D632K, D632R, and D632V) migrated more slowly. When peptide N36 was mixed with the wild-type and mutant C34 peptides, the specific bands corresponding to the six-helix bundles appeared, but they had different migration rates. Since the migration rates of the six-helix bundles formed by N36 and C34 peptides were dependent on their net charges, molecular sizes, and shapes, it was possible that conservative and nonconservative C34 mutants retained their ability to fold six-helix bundle structures but that the configuration of the six-helix bundles might be changed.
FIG. 5.
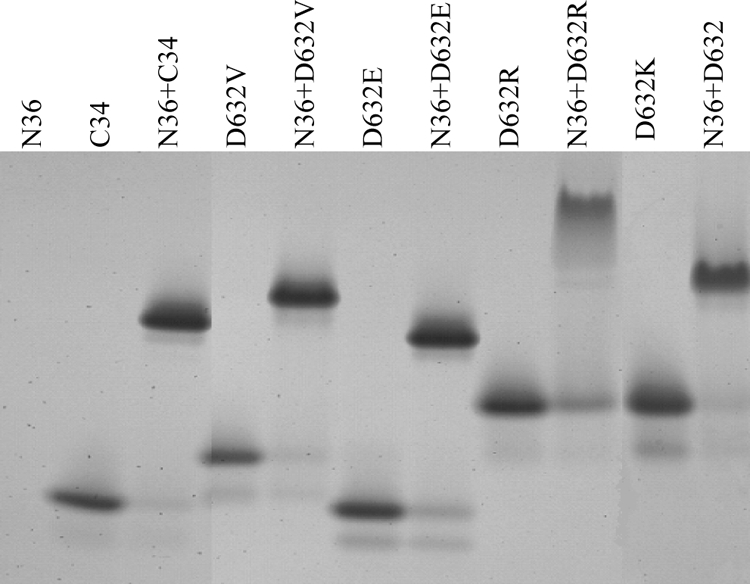
Visualization of six-helix bundles formed between N36 and C34 bearing conserved or nonconserved substitutions by N-PAGE.
We subsequently determined whether the C34 mutants affect the conformation of six-helix bundles with a gp41 core-specific MAb NC-1 (29). Consistently, NC-1 did not react with the isolated peptides N36 and C34 but strongly reacted with the six-helix bundle formed by these two peptides mixed at equal concentrations (Fig. 6). Interestingly, NC-1 recognized the six-helix bundle formed by the D632E mutant similarly to that formed by the wild-type C34, but nonconservative substitutions in the peptide C34 severely damaged its reactivity. These results further suggest that the six-helix bundles might undergo conformational changes after nonconservative substitutions in the CHR peptides and suggest a role of the salt bridge in the formation of a functional conformation of six-helix bundle.
FIG. 6.
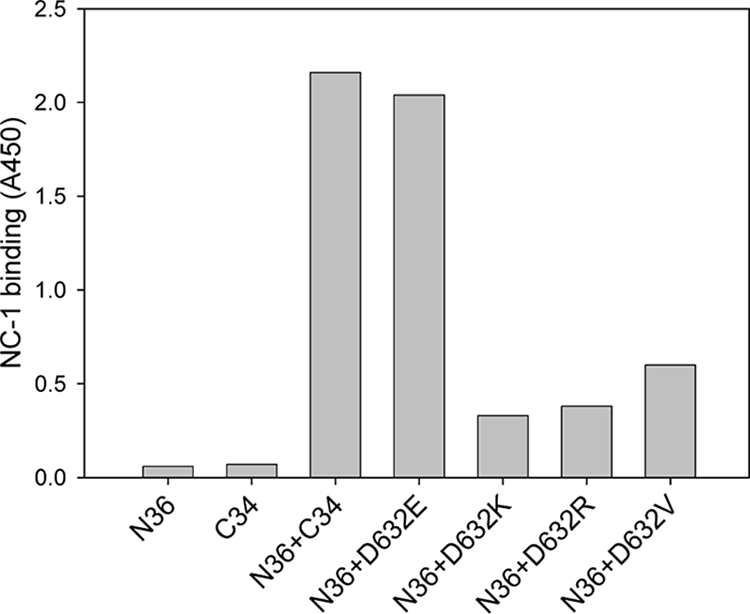
Detection of six-helix bundles formed between N36 and C34 bearing conserved or nonconserved substitutions by ELISA using the gp41 core conformation-specific MAb NC-1.
K574D and D632K double mutations cannot rescue the function of the wild-type salt bridge.
Our data have indicated that single nonconserved substitutions of the salt bridge-forming residues Lys574 and Asp632 can severely impair the conformation and stability of the six-helix bundles and thus inactivate the virus, suggesting the importance of the Lys574-Asp632 salt bridge in the gp41 core structure. It is interesting to know whether Asp574-Lys632 mutations can rescue the function of the wild-type salt bridge. We therefore generated a pseudotyped HIV-1 bearing the K574D and D632K double mutations and used it in a single-cycle complementation assay. From Fig. 7A, we can see that Asp574-Lys632 double mutations cannot rescue the virus infectivity. We also tested K574D and D632K peptides by CD spectroscopy. We found that these two peptides could interact to form a six-helix bundle structure, but this bundle had a significant lower α-helicity (∼60%) than the wild-type or single mutant peptides (Table 1). However, the K574D/D632K bundle had a Tm of 62°C, which is close to that of the wild-type N36/C34 bundle (64°C). These results imply that the Asp574-Lys632 pair may form a salt bridge that stabilizes the six-helix bundle core but that its function for virus infectivity cannot be rescued.
FIG. 7.

Analysis of rescue of salt bridges by Asp574-Lys632 double mutations. (A) Infectivity of HIV-1 pseudovirus bearing K574D and D632K double mutations. Error bars indicate standard deviations. (B) α-Helicity of K574D and D632K peptide complex. (C) Thermostability of K574D and D632K peptide complex.
Nonconserved mutations of Asp632 dramatically impair C34 to inhibit six-helix bundle formation.
The mechanism of action of NHR- or CHR-derived anti-HIV peptides has been considered to inhibit the formation of six-helix bundles in a dominant-negative fashion (6, 62). Considering that the peptide C34 has potent anti-HIV activity, we were interested to know whether mutations affect the ability of C34 to block six-helix bundle formation. For this purpose, an ELISA-based method in which the six-helix bundle-specific MAb NC-1 was used as a capture antibody and peptide C34 was biotinylated was developed (see Materials and Methods). Figure 8A shows that NC-1 can detect the six-helix bundle formed by N36 and biotin-C34 in a dose-dependent manner. We then tested the inhibitory activity of wild-type and mutant C34 on six-helix bundle formation. As shown in Fig. 8B and Table 1, the wild type and the D632E mutant could efficiently block the formation of six-helix bundles; however, this activity for the peptides bearing the D642K, D632R, or D632V mutation dramatically decreased. This result also suggests that the formation of a salt bridge between NHR residue Lys574 and CHR residue Asp632 is critical for the six-helix bundle core structure.
FIG. 8.

Inhibition of six-helix bundle formation by wild-type and mutant C34 peptides. (A) Dose dependence of binding of six-helix bundles formed by N36 and biotin-C34 to MAb NC-1 applied to the plate. Inset, binding to NC-1 of the isolated N36 (250 nM), biotin-C34 (250 nM), and an equimolar mixture of N36 and biotin-C34. Error bars indicate standard deviations. (B) Inhibition of the formation of six-helix bundles by wild-type and mutant C34.
Nonconservative substitutions of Asp632 significantly reduce C34-mediated anti-HIV activity.
Our results described above demonstrated that nonconservative substitutions of Asp632 could dramatically affect the CHR peptides to block six-helix bundle formation, implying that their anti-HIV activity may be compromised. First, we tested their anti-HIV activity by cell-cell fusion assay. From Fig. 9A, we can conclude that the peptides with nonconservative substitutions had much-reduced activity to inhibit HIV-1 mediated cell-cell fusion. The data in Table 1 show that the wild-type C34 and D632E mutant could inhibit the fusion with IC50s of 5.0 and 6.6 nM, respectively. In contrast, the D632K, D632R, and D632V mutants inhibited the fusion with IC50s of only 171.1, 136.0 and 92.6 nM, respectively. Further, the infectivity of HIV-1 IIIB was measured in the presence or absence of the wild-type or mutant peptides by use of a p24-based assay. Consistently, the wild-type C34 and D632E peptides could inhibit HIV-1 infectivity with IC50s of 1.4 and 2.2 nM, respectively (Fig. 9B and Table 1), whereas the peptides with nonconservative substitutions (D642K, D632R, or D632V) possessed much less antiviral activity, with IC50s of 35.5, 29.3, and 15.2 nM, respectively. These results demonstrate that residue Asp632 in the CHR peptides or its ionic interaction with residue Lys574 in the NHR target is highly critical for designing anti-HIV peptides.
FIG. 9.

Anti-HIV activity of wild-type and mutant C34 peptides. (A) Inhibition of HIV-1IIIB-mediated cell-cell fusion. (B) Inhibition of HIV-1IIIB-mediated infection of MT-2 cells. Samples were tested in quadruplicate (A) or triplicate (B). The results are presented as means ± standard deviations.
DISCUSSION
In the present study, we have continued our characterization of the function of the salt bridge formed by the positively charged residue Lys574 in the NHR region and the negatively charged residue Asp632 in the CHR region of the fusion-active HIV-1 gp41 core structure. We show that residue Asp632 is essential for Env-mediated HIV-1 entry. Biophysical characterization demonstrated that this residue determines the conformation and stability of the six-helix bundle structure modeled by NHR peptide N36 and CHR peptide C34. Further, we show that residue Asp632 is also a major determinant of the anti-HIV activity of the peptides. Taking these results together with our previous data on residue Lys574, we propose that the salt bridge between the NHR and CHR of HIV-1 gp41 core is highly critical for virus entry and inhibition.
The structure and function of the HIV-1 gp41 molecule have been extensively studied (10, 15, 25, 35, 51, 55, 56, 66, 67). Considerable evidence suggests that during the fusion process gp41 likely exists in at least three conformations: (i) a metastable prefusogenic state, which is stabilized by interactions with gp120 subunit; (ii) a prehairpin intermediate, formed by insertion of the hydrophobic fusion peptide into the target cell membrane and concurrent folding of the N-terminal trimeric coiled coil; and (iii) the fusogenic trimer-of-hairpins form, in which two α-helical regions, the NHR (adjacent to the N-terminal fusion peptide) and the CHR (near the C-terminal transmembrane segment), associate to form a highly stable six-helix bundle, bringing the viral and cellular membranes into close apposition (6). X-ray crystallographic studies of the gp41 core reveal that the N-terminal homotrimer is packed through the interaction of residues at positions a and d of the characteristic 4-3 heptad repeat sequence [(abcdefg)n] and that its residues at the e and g positions lie on the outside groves to interact with the residues at the a and d positions of the CHR helices (2, 5, 60, 63). A large body of evidence from mutagenesis studies suggested that the helical packing interactions between the central coiled-coil trimer and the C-terminal helix are important determinants of HIV-1 entry and inhibition (3, 7, 8, 21, 27, 28, 44, 45, 47, 52, 57, 58, 61, 64, 65). However, these studies were focused primarily on the predicted critical residues within the NHR helix or the CHR helix, especially the residues at the a, d, e, and g positions of the NHR or the residues at the a and d positions of the CHR. To date, little attention has been paid to the residues responsible for the formation of the salt bridge in the fusogenic gp41 core. We have recently demonstrated that residue Lys574, which is located at the heptad b position of NHR, is essential for HIV-1 entry (23). Here, our mutational analyses indicate that residue Asp632, which is located at the heptad e position of the CHR, is also critical for virus infectivity. Anionic-to-cationic (D632K and D632R) or anionic-to-hydrophobic (D632V) mutations could completely abrogate Env-mediated virus entry, whereas the conserved anionic-to-anionic (D632E) mutation had no significant effect on the infectivity of the pseudoviruses. This result highlights the importance of the salt bridge formed by the NHR and CHR during the viral fusion process.
To explore the mechanism by which single nonconserved substitutions could determine the Env-mediated entry of HIV-1 into target cells, we synthesized a set of wild-type and mutant peptides and used them in biophysical characterization. Similarly, residue Asp632 in peptide C34 was replaced with a positively charged lysine or arginine residue, with the hydrophobic residue valine, or with another negatively charged residue (Glu). CD spectra and thermal denaturation showed that nonconservative substitutions of Asp632 significantly impaired the α-helicity and stability of the six-helix bundle modeled by the peptides N36 and C34, whereas the conservative substitution had no such effects. Although the epitope for MAb NC-1 may be located within the N-helical trimers (12, 56), our data here confirmed further that NC-1 reacts only with the N36/C34 complex and not with the isolated N36 (Fig. 6 and 7). The binding specificity of this conformation-specific antibody suggests that the conformation of the six-helix bundles bearing the nonconservative mutations might undergo an alteration. It is possible that the NHR-CHR salt bridge confers a significant strength to the interhelical interactions and thereby stabilizes the six-helix bundles. The breakage of the salt bridge may disrupt the integrity of the six-helix bundles by changing their conformations or reducing their stability. The resulting destabilization and conformational change of the nonconservative substitution mutants observed here may be related to the fusion-defective phenotypes; this explains how single mutations can completely disrupt the viral infectivity. Further, it can be speculated that the defective phenotype of the mutant Env proteins described above might be a result of the structural perturbations of gp41 caused by nonconservative substitutions.
In an attempt to rescue the function of the Lys574-Asp632 salt bridge, we generated a pseudotyped HIV-1 bearing Asp574-Lys632 double mutations, but this virus had no infectivity (Fig. 7). Biophysical analyses with the mutant peptides (K574D and D632K) suggest that Asp574-Lys632 may form a salt bridge that stabilizes the six-helical bundle, but the α-helicity of this core structure cannot be rescued and thereby this results in virus inactivation. Obviously, our present data cannot rule out the possibility that residues Lys574 and Asp632 have roles other than forming a salt bridge. From the crystal structure of the gp41 core, the interaction of the NHR and CHR helices, especially within the cavity, is largely dependent on hydrophobic contacts but is supplemented by electrostatic interactions (2, 5, 45, 60, 63). The exposed surface of the C-terminal helix is predominantly negative, whereas that of the N-terminal helix is mainly neutral. Therefore, the Asp574-Lys632 double mutations may change the electrostatic potential within the six-helix bundle core and thus abolish its functionality. Further, residue Lys574 is located at the left wall of the cavity and is critical for cavity formation, but nonconserved substitutions of Lys574 may change the configuration of the cavity region. Also, it is possible that nonconserved substitutions of Lys574 or Asp632 can severely damage the α-helicity of the peptide itself (C34 and N36). We speculate that although the possible Asp574-Lys632 salt bridge can strengthen the stability of the six-helix bundle, it cannot maintain the normal conformation of the bundle and rescue its function for virus fusion.
We started to characterize the function of the salt bridge of the gp41 six-helix bundle since our finding of two small-molecule-based anti-HIV fusion inhibitors, NB-2 and NB-64 (32). These two N-substituted pyrrole derivatives were selected from a drug-like chemical library, and they could inhibit infection by both laboratory-adapted and primary HIV-1 strains with distinct genotypes (clades A to G and group O) and phenotypes (R5, X4, and R5X4) at low micromolar levels (32). Computer-aided molecular docking has shown that these compounds fit inside the deep cavity and that their acid groups interact with the positively charged residue Lys574 to form a salt bridge, which may compete off the residue Asp632 and thus block the six-helix bundle formation. Therefore, the salt bridge-forming residue Lys574 may serve as a key determinant of the target of the compounds, and the breakage of the salt bridge formed by Lys574 and Asp632 may be their mechanism of action. Recently, Jacobs et al (26). designed a covalent inhibitor that specifically targets residue Lys574. Using a temperature-arrested state prime wash in vitro assay, they provided evidence for the trapping of a pre-six-helix bundle fusion intermediate conformation by a covalent reaction of Lys574 with a specific anti-HIV peptide composed of a chemical spacer and a reactive moiety positioned strategically to facilitate covalent attachment (26). Therefore, the salt bridge between the NHR and CHR of the gp41 core is critical for the conformation and stability of the six-helix bundle structure and thereby can serve as a potential target for designing anti-HIV peptides and small molecules.
Previous studies have demonstrated that the prototypic peptides derived from the CHR region of HIV-1 gp41, such as C34 and T20, have potent antiviral activity (11, 17, 40). It is generally accepted that peptide C34 has higher activity than T20 in inhibiting HIV-1-mediated cell-cell fusion and infection. Our studies suggest that these two CHR-derived peptides may work by different anti-HIV mechanisms (38, 39). Although they are largely overlapped by their NHR-binding domain, C34 contains the salt bridge residue Asp632 at its N-terminal pocket-binding motif, whereas T20 lacks this residue. It is possible that the salt bridge interaction may occur together with the hydrophobic interactions within the pocket to facilitate the NHR-binding activity of peptide C34 and thereby increase its anti-HIV activity. In line with this hypothesis, C34 with nonconserved mutations (D632K, D632R, and D632V) has much less activity to block the formation of six-helix bundles (Fig. 8) and to inhibit HIV-1-mediated cell-cell fusion and infection (Fig. 9), while C34 with a conserved mutation (D632E) possesses anti-HIV activity similar to that of the wild-type C34, possibly because it maintains its ability to form a salt bridge with residue Lys574. It has been demonstrated that introduction of ion pair interactions or salt bridges into the peptides can increase the α-helicity, stabilize the coiled coils, and thereby improve the pharmacokinetic profiles and anti-HIV activity of the peptides (24, 34, 49, 59). Although the intrahelical salt bridges are important for anti-HIV peptides, our data presented here suggest that formation of such ionic interactions between a fusion inhibitor (peptide or small molecule) and its target sequence (NHR or CHR) should also be considered.
In summary, the significance of our present studies is threefold. First, we demonstrate that residue Asp632 in the CHR of HIV-1 gp41 is critical for six-helix bundle formation and viral entry, which further highlights the importance of the salt bridge of the gp41 core. Second, we propose that the salt bridge of the gp41 core can serve as a molecular target for anti-HIV peptides and small molecules. Third, the formation of a salt bridge with residues within the NHR or CHR is also important for designing novel anti-HIV fusion inhibitors.
Acknowledgments
This work was supported by NIH grant AI 46221, by the 973 program (grant 2006CB504200) from the Chinese Ministry of Science and Technology, and by the Nature Science Foundation of China (grant 30870123).
Footnotes
Published ahead of print on 3 September 2008.
REFERENCES
- 1.Bianchi, E., M. Finotto, P. Ingallinella, R. Hrin, A. V. Carella, X. S. Hou, W. A. Schleif, M. D. Miller, R. Geleziunas, and A. Pessi. 2005. Covalent stabilization of coiled coils of the HIV gp41 N region yields extremely potent and broad inhibitors of viral infection. Proc. Natl. Acad. Sci. USA 10212903-12908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Caffrey, M., M. Cai, J. Kaufman, S. J. Stahl, P. T. Wingfield, D. G. Covell, A. M. Gronenborn, and G. M. Clore. 1998. Three-dimensional solution structure of the 44 kDa ectodomain of SIV gp41. EMBO J. 174572-4584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cao, J., L. Bergeron, E. Helseth, M. Thali, H. Repke, and J. Sodroski. 1993. Effects of amino acid changes in the extracellular domain of the human immunodeficiency virus type 1 gp41 envelope glycoprotein. J. Virol. 672747-2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chan, D. C., C. T. Chutkowski, and P. S. Kim. 1998. Evidence that a prominent cavity in the coiled coil of HIV type 1 gp41 is an attractive drug target. Proc. Natl. Acad. Sci. USA 9515613-15617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chan, D. C., D. Fass, J. M. Berger, and P. S. Kim. 1997. Core structure of gp41 from the HIV envelope glycoprotein. Cell 89263-273. [DOI] [PubMed] [Google Scholar]
- 6.Chan, D. C., and P. S. Kim. 1998. HIV entry and its inhibition. Cell 93681-684. [DOI] [PubMed] [Google Scholar]
- 7.Chen, S. S., C. N. Lee, W. R. Lee, K. McIntosh, and T. H. Lee. 1993. Mutational analysis of the leucine zipper-like motif of the human immunodeficiency virus type 1 envelope transmembrane glycoprotein. J. Virol. 673615-3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen, S. S., S. F. Lee, H. J. Hao, and C. K. Chuang. 1998. Mutations in the leucine zipper-like heptad repeat sequence of human immunodeficiency virus type 1 gp41 dominantly interfere with wild-type virus infectivity. J. Virol. 724765-4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen, Y. H., J. T. Yang, and K. H. Chau. 1974. Determination of the helix and beta form of proteins in aqueous solution by circular dichroism. Biochemistry 133350-3359. [DOI] [PubMed] [Google Scholar]
- 10.Colman, P. M., and M. C. Lawrence. 2003. The structural biology of type I viral membrane fusion. Nat. Rev. Mol. Cell Biol. 4309-319. [DOI] [PubMed] [Google Scholar]
- 11.Debnath, A. K. 2006. Progress in identifying peptides and small-molecule inhibitors targeted to gp41 of HIV-1. Expert Opin. Investig. Drugs 15465-478. [DOI] [PubMed] [Google Scholar]
- 12.Dimitrov, A. S., J. M. Louis, C. A. Bewley, G. M. Clore, and R. Blumenthal. 2005. Conformational changes in HIV-1 gp41 in the course of HIV-1 envelope glycoprotein-mediated fusion and inactivation. Biochemistry 4412471-12479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doms, R. W., P. L. Earl, S. Chakrabarti, and B. Moss. 1990. Human immunodeficiency virus types 1 and 2 and simian immunodeficiency virus Env proteins possess a functionally conserved assembly domain. J. Virol. 643537-3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Earl, P. L., R. W. Doms, and B. Moss. 1990. Oligomeric structure of the human immunodeficiency virus type 1 envelope glycoprotein. Proc. Natl. Acad. Sci. USA 87648-652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eckert, D. M., and P. S. Kim. 2001. Mechanisms of viral membrane fusion and its inhibition. Annu. Rev. Biochem. 70777-810. [DOI] [PubMed] [Google Scholar]
- 16.Edelhoch, H. 1967. Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry 61948-1954. [DOI] [PubMed] [Google Scholar]
- 17.Este, J. A., and A. Telenti. 2007. HIV entry inhibitors. Lancet 37081-88. [DOI] [PubMed] [Google Scholar]
- 18.Follis, K. E., S. J. Larson, M. Lu, and J. H. Nunberg. 2002. Genetic evidence that interhelical packing interactions in the gp41 core are critical for transition of the human immunodeficiency virus type 1 envelope glycoprotein to the fusion-active state. J. Virol. 767356-7362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Furuta, R. A., C. T. Wild, Y. Weng, and C. D. Weiss. 1998. Capture of an early fusion-active conformation of HIV-1 gp41. Nat. Struct. Biol. 5276-279. [DOI] [PubMed] [Google Scholar]
- 20.Gallo, S. A., C. M. Finnegan, M. Viard, Y. Raviv, A. Dimitrov, S. S. Rawat, A. Puri, S. Durell, and R. Blumenthal. 2003. The HIV Env-mediated fusion reaction. Biochim. Biophys. Acta 161436-50. [DOI] [PubMed] [Google Scholar]
- 21.He, Y., J. Cheng, J. Li, Z. Qi, H. Lu, M. Dong, S. Jiang, and Q. Dai. 2008. Identification of a critical motif for the human immunodeficiency virus type 1 (HIV-1) gp41 core structure: implications for designing novel anti-HIV fusion inhibitors. J. Virol. 826349-6358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.He, Y., P. D'Agostino, and A. Pinter. 2003. Analysis of the immunogenic properties of a single-chain polypeptide analogue of the HIV-1 gp120-CD4 complex in transgenic mice that produce human immunoglobulins. Vaccine 214421-4429. [DOI] [PubMed] [Google Scholar]
- 23.He, Y., S. Liu, W. Jing, H. Lu, D. Cai, D. J. Chin, A. K. Debnath, F. Kirchhoff, and S. Jiang. 2007. Conserved residue Lys574 in the cavity of HIV-1 Gp41 coiled-coil domain is critical for six-helix bundle stability and virus entry. J. Biol. Chem. 28225631-25639. [DOI] [PubMed] [Google Scholar]
- 24.He, Y., Y. Xiao, H. Song, Q. Liang, D. Ju, X. Chen, H. Lu, W. Jing, S. Jiang, and L. Zhang. 2008. Design and evaluation of sifuvirtide, a novel HIV-1 fusion inhibitor. J. Biol. Chem. 28311126-11134. [DOI] [PubMed] [Google Scholar]
- 25.Jacobs, A., H. Garg, M. Viard, Y. Raviv, A. Puri, and R. Blumenthal. 2008. HIV-1 envelope glycoprotein-mediated fusion and pathogenesis: implications for therapy and vaccine development. Vaccine 263026-3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jacobs, A., O. Quraishi, X. Huang, N. Bousquet-Gagnon, G. Nault, N. Francella, W. G. Alvord, N. Pham, C. Soucy, M. Robitaille, D. Bridon, and R. Blumenthal. 2007. A covalent inhibitor targeting an intermediate conformation of the fusogenic subunit of the HIV-1 envelope complex. J. Biol. Chem. 28232406-32413. [DOI] [PubMed] [Google Scholar]
- 27.Ji, H., C. Bracken, and M. Lu. 2000. Buried polar interactions and conformational stability in the simian immunodeficiency virus (SIV) gp41 core. Biochemistry 39676-685. [DOI] [PubMed] [Google Scholar]
- 28.Ji, H., W. Shu, F. T. Burling, S. Jiang, and M. Lu. 1999. Inhibition of human immunodeficiency virus type 1 infectivity by the gp41 core: role of a conserved hydrophobic cavity in membrane fusion. J. Virol. 738578-8586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang, S., K. Lin, and M. Lu. 1998. A conformation-specific monoclonal antibody reacting with fusion-active gp41 from the human immunodeficiency virus type 1 envelope glycoprotein. J. Virol. 7210213-10217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiang, S., K. Lin, N. Strick, and A. R. Neurath. 1993. HIV-1 inhibition by a peptide. Nature 365113. [DOI] [PubMed] [Google Scholar]
- 31.Jiang, S., K. Lin, L. Zhang, and A. K. Debnath. 1999. A screening assay for antiviral compounds targeted to the HIV-1 gp41 core structure using a conformation-specific monoclonal antibody. J. Virol. Methods 8085-96. [DOI] [PubMed] [Google Scholar]
- 32.Jiang, S., H. Lu, S. Liu, Q. Zhao, Y. He, and A. K. Debnath. 2004. N-substituted pyrrole derivatives as novel human immunodeficiency virus type 1 entry inhibitors that interfere with the gp41 six-helix bundle formation and block virus fusion. Antimicrob. Agents Chemother. 484349-4359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang, S. B., K. Lin, and A. R. Neurath. 1991. Enhancement of human immunodeficiency virus type 1 infection by antisera to peptides from the envelope glycoproteins gp120/gp41. J. Exp. Med. 1741557-1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kammerer, R. A., V. A. Jaravine, S. Frank, T. Schulthess, R. Landwehr, A. Lustig, C. Garcia-Echeverria, A. T. Alexandrescu, J. Engel, and M. O. Steinmetz. 2001. An intrahelical salt bridge within the trigger site stabilizes the GCN4 leucine zipper. J. Biol. Chem. 27613685-13688. [DOI] [PubMed] [Google Scholar]
- 35.Korazim, O., K. Sackett, and Y. Shai. 2006. Functional and structural characterization of HIV-1 gp41 ectodomain regions in phospholipid membranes suggests that the fusion-active conformation is extended. J. Mol. Biol. 3641103-1117. [DOI] [PubMed] [Google Scholar]
- 36.Lalezari, J. P., J. J. Eron, M. Carlson, C. Cohen, E. DeJesus, R. C. Arduino, J. E. Gallant, P. Volberding, R. L. Murphy, F. Valentine, E. L. Nelson, P. R. Sista, A. Dusek, and J. M. Kilby. 2003. A phase II clinical study of the long-term safety and antiviral activity of enfuvirtide-based antiretroviral therapy. AIDS 17691-698. [DOI] [PubMed] [Google Scholar]
- 37.Lalezari, J. P., K. Henry, M. O'Hearn, J. S. Montaner, P. J. Piliero, B. Trottier, S. Walmsley, C. Cohen, D. R. Kuritzkes, J. J. Eron, Jr., J. Chung, R. DeMasi, L. Donatacci, C. Drobnes, J. Delehanty, and M. Salgo. 2003. Enfuvirtide, an HIV-1 fusion inhibitor, for drug-resistant HIV infection in North and South America. N. Engl. J. Med. 3482175-2185. [DOI] [PubMed] [Google Scholar]
- 38.Liu, S., W. Jing, B. Cheung, H. Lu, J. Sun, X. Yan, J. Niu, J. Farmar, S. Wu, and S. Jiang. 2007. HIV gp41 C-terminal heptad repeat contains multifunctional domains. Relation to mechanisms of action of anti-HIV peptides. J. Biol. Chem. 2829612-9620. [DOI] [PubMed] [Google Scholar]
- 39.Liu, S., H. Lu, J. Niu, Y. Xu, S. Wu, and S. Jiang. 2005. Different from the HIV fusion inhibitor C34, the anti-HIV drug Fuzeon (T-20) inhibits HIV-1 entry by targeting multiple sites in gp41 and gp120. J. Biol. Chem. 28011259-11273. [DOI] [PubMed] [Google Scholar]
- 40.Liu, S., S. Wu, and S. Jiang. 2007. HIV entry inhibitors targeting gp41: from polypeptides to small-molecule compounds. Curr. Pharm. Des. 13143-162. [DOI] [PubMed] [Google Scholar]
- 41.Liu, S., Q. Zhao, and S. Jiang. 2003. Determination of the HIV-1 gp41 fusogenic core conformation modeled by synthetic peptides: applicable for identification of HIV-1 fusion inhibitors. Peptides 241303-1313. [DOI] [PubMed] [Google Scholar]
- 42.Lu, M., S. C. Blacklow, and P. S. Kim. 1995. A trimeric structural domain of the HIV-1 transmembrane glycoprotein. Nat. Struct. Biol. 21075-1082. [DOI] [PubMed] [Google Scholar]
- 43.Lu, M., and P. S. Kim. 1997. A trimeric structural subdomain of the HIV-1 transmembrane glycoprotein. J. Biomol. Struct. Dyn. 15465-471. [DOI] [PubMed] [Google Scholar]
- 44.Lu, M., M. O. Stoller, S. Wang, J. Liu, M. B. Fagan, and J. H. Nunberg. 2001. Structural and functional analysis of interhelical interactions in the human immunodeficiency virus type 1 gp41 envelope glycoprotein by alanine-scanning mutagenesis. J. Virol. 7511146-11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Malashkevich, V. N., D. C. Chan, C. T. Chutkowski, and P. S. Kim. 1998. Crystal structure of the simian immunodeficiency virus (SIV) gp41 core: conserved helical interactions underlie the broad inhibitory activity of gp41 peptides. Proc. Natl. Acad. Sci. USA 959134-9139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McKeating, J. A., A. McKnight, and J. P. Moore. 1991. Differential loss of envelope glycoprotein gp120 from virions of human immunodeficiency virus type 1 isolates: effects on infectivity and neutralization. J. Virol. 65852-860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mo, H., A. K. Konstantinidis, K. D. Stewart, T. Dekhtyar, T. Ng, K. Swift, E. D. Matayoshi, W. Kati, W. Kohlbrenner, and A. Molla. 2004. Conserved residues in the coiled-coil pocket of human immunodeficiency virus type 1 gp41 are essential for viral replication and interhelical interaction. Virology 329319-327. [DOI] [PubMed] [Google Scholar]
- 48.Moore, J. P., J. A. McKeating, R. A. Weiss, and Q. J. Sattentau. 1990. Dissociation of gp120 from HIV-1 virions induced by soluble CD4. Science 2501139-1142. [DOI] [PubMed] [Google Scholar]
- 49.Otaka, A., M. Nakamura, D. Nameki, E. Kodama, S. Uchiyama, S. Nakamura, H. Nakano, H. Tamamura, Y. Kobayashi, M. Matsuoka, and N. Fujii. 2002. Remodeling of gp41-C34 peptide leads to highly effective inhibitors of the fusion of HIV-1 with target cells. Angew. Chem. Int. 412937-2940. [DOI] [PubMed] [Google Scholar]
- 50.Palella, F. J., Jr., K. M. Delaney, A. C. Moorman, M. O. Loveless, J. Fuhrer, G. A. Satten, D. J. Aschman, S. D. Holmberg, et al. 1998. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. N. Engl. J. Med. 338853-860. [DOI] [PubMed] [Google Scholar]
- 51.Quintana, F. J., D. Gerber, S. C. Kent, I. R. Cohen, and Y. Shai. 2005. HIV-1 fusion peptide targets the TCR and inhibits antigen-specific T cell activation. J. Clin. Investig. 1152149-2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rimsky, L. T., D. C. Shugars, and T. J. Matthews. 1998. Determinants of human immunodeficiency virus type 1 resistance to gp41-derived inhibitory peptides. J. Virol. 72986-993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Root, M. J., M. S. Kay, and P. S. Kim. 2001. Protein design of an HIV-1 entry inhibitor. Science 291884-888. [DOI] [PubMed] [Google Scholar]
- 54.Roux, K. H., and K. A. Taylor. 2007. AIDS virus envelope spike structure. Curr. Opin. Struct. Biol. 17244-252. [DOI] [PubMed] [Google Scholar]
- 55.Sackett, K., and Y. Shai. 2003. How structure correlates to function for membrane associated HIV-1 gp41 constructs corresponding to the N-terminal half of the ectodomain. J. Mol. Biol. 33347-58. [DOI] [PubMed] [Google Scholar]
- 56.Sackett, K., Y. Wexler-Cohen, and Y. Shai. 2006. Characterization of the HIV N-terminal fusion peptide-containing region in context of key gp41 fusion conformations. J. Biol. Chem. 28121755-21762. [DOI] [PubMed] [Google Scholar]
- 57.Shu, W., H. Ji, and M. Lu. 2000. Interactions between HIV-1 gp41 core and detergents and their implications for membrane fusion. J. Biol. Chem. 2751839-1845. [DOI] [PubMed] [Google Scholar]
- 58.Shu, W., J. Liu, H. Ji, L. Radigen, S. Jiang, and M. Lu. 2000. Helical interactions in the HIV-1 gp41 core reveal structural basis for the inhibitory activity of gp41 peptides. Biochemistry 391634-1642. [DOI] [PubMed] [Google Scholar]
- 59.Spek, E. J., A. H. Bui, M. Lu, and N. R. Kallenbach. 1998. Surface salt bridges stabilize the GCN4 leucine zipper. Protein Sci. 72431-2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tan, K., J. Liu, J. Wang, S. Shen, and M. Lu. 1997. Atomic structure of a thermostable subdomain of HIV-1 gp41. Proc. Natl. Acad. Sci. USA 9412303-12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang, S., J. York, W. Shu, M. O. Stoller, J. H. Nunberg, and M. Lu. 2002. Interhelical interactions in the gp41 core: implications for activation of HIV-1 membrane fusion. Biochemistry 417283-7292. [DOI] [PubMed] [Google Scholar]
- 62.Weiss, C. D. 2003. HIV-1 gp41: mediator of fusion and target for inhibition. AIDS Rev. 5214-221. [PubMed] [Google Scholar]
- 63.Weissenhorn, W., A. Dessen, S. C. Harrison, J. J. Skehel, and D. C. Wiley. 1997. Atomic structure of the ectodomain from HIV-1 gp41. Nature 387426-430. [DOI] [PubMed] [Google Scholar]
- 64.Weng, Y., and C. D. Weiss. 1998. Mutational analysis of residues in the coiled-coil domain of human immunodeficiency virus type 1 transmembrane protein gp41. J. Virol. 729676-9682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Weng, Y., Z. Yang, and C. D. Weiss. 2000. Structure-function studies of the self-assembly domain of the human immunodeficiency virus type 1 transmembrane protein gp41. J. Virol. 745368-5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wexler-Cohen, Y., B. T. Johnson, A. Puri, R. Blumenthal, and Y. Shai. 2006. Structurally altered peptides reveal an important role for N-terminal heptad repeat binding and stability in the inhibitory action of HIV-1 peptide DP178. J. Biol. Chem. 2819005-9010. [DOI] [PubMed] [Google Scholar]
- 67.Wexler-Cohen, Y., and Y. Shai. 2007. Demonstrating the C-terminal boundary of the HIV 1 fusion conformation in a dynamic ongoing fusion process and implication for fusion inhibition. FASEB J. 213677-3684. [DOI] [PubMed] [Google Scholar]
- 68.Wild, C., T. Greenwell, and T. Matthews. 1993. A synthetic peptide from HIV-1 gp41 is a potent inhibitor of virus-mediated cell-cell fusion. AIDS Res. Hum. Retroviruses 91051-1053. [DOI] [PubMed] [Google Scholar]
- 69.Wild, C., T. Oas, C. McDanal, D. Bolognesi, and T. Matthews. 1992. A synthetic peptide inhibitor of human immunodeficiency virus replication: correlation between solution structure and viral inhibition. Proc. Natl. Acad. Sci. USA 8910537-10541. [DOI] [PMC free article] [PubMed] [Google Scholar]