

Abstract
Three types of editosomes, each with an identical core containing six related KREPA proteins, catalyze the U insertion and deletion RNA editing of mitochondrial mRNAs in trypanosomes. Repression of expression of one of these, KREPA3 (also known as TbMP42), shows that it is essential for growth and in vivo editing in both procyclic (PF) and bloodstream (BF) life cycle stages of Trypanosoma brucei. RNA interference knockdown results in editosome disruption and altered in vitro editing in PFs, while repression by regulatable double knockout results in almost complete loss of editosomes in BFs. Mutational analysis shows that the KREPA3 zinc fingers and OB-fold domain are each essential for growth and in vivo editing. Nevertheless, KREPA3 with mutated zinc fingers incorporates into editosomes that catalyze in vitro editing and thus is not essential for editosome integrity, although stability is affected. In contrast, the OB-fold domain is essential for editosome integrity. Overall, KREPA3, especially its OB-fold, functions in editosome integrity, and its zinc fingers are essential for editing in vivo but not for the central catalytic steps. KREPA3 may function in editosome organization and/or RNA positioning.
RNA editing in trypanosomes creates mature mitochondrial (mt) mRNAs by the insertion and deletion of uridylates (U's) as specified by the sequences of small guide RNAs (gRNAs) (6, 52). It employs several multiprotein complexes, one of which is the editosome that sediments at ∼20S in glycerol gradients and contains the enzymes that catalyze the key catalytic steps of pre-mRNA cleavage, U insertion or deletion, and RNA ligation (32, 50, 53). The pre-mRNA becomes partially duplexed with gRNA 3′ to the editing site (ES), after which editing occurs by cycles of enzymatic steps. The pre-mRNA is cleaved at the ES by the KREN1 endonuclease that is specific for deletion sites or by KREN2 or KREN3, which are specific for different insertion sites. One or more U's are added to the 5′ mRNA fragment by the KRET2 3′ terminal uridylyl transferase or U's are removed by the KREX1 or KREX2 3′ exonuclease, with the number of added or removed U's specified by the gRNA sequence. The processed 5′ fragment is then religated with the 3′ fragment by the KREL1 or KREL2 RNA ligase (3, 10-12, 15, 16, 27, 45, 48, 56). Multiple cycles of editing extend the amount of pre-mRNA/gRNA duplex, generally in the 5′ direction, until they are completely complementary. The editing of most mRNAs requires multiple gRNAs with those specifying editing of the more 3′ regions creating a pre-mRNA sequence that is complementary to the 5′ “anchor” sequence of the subsequent gRNA thus providing for their orderly use. Other complexes are involved in editing, including the MRP1/MRP2, RBP16, TbRGG1, and RET1 complexes, which appear to preprocess and traffic the mRNAs and gRNAs and regulate editing (2, 33, 34, 42, 57). Other proteins that are not associated with these complexes and have not yet been identified may function as accessory factors.
There are three types of editosomes with distinct editing site cleavage specificities. Each of them has a distinct endonuclease, KREN1, KREN2 or KREN3, and one or two other unique proteins, as well as an identical “core” of proteins (Fig. 1) (11, 38). The identical “core” contains six related A-type kinetoplastid RNA editing proteins (KREPA1 to KREPA6), each of which has a C-terminal OB-fold domain, while the three largest (KREPA1, -A2, and -A3) also have two C2H2 zinc finger (ZF) motifs (39, 62). KREPA1 and KREPA2 each form different heterotrimeric subcomplexes in vivo and in vitro that catalyze the postcleavage steps of insertion and deletion editing, respectively (14, 15, 19, 28, 37, 47, 53). KREPA1 associates with KRET2 and KREL2 resulting in an insertion subcomplex that adds U's and ligates the products as specified by the gRNA. Similarly, KREPA2 associates with KREX2 and KREL1 in a deletion subcomplex that catalyzes U removal and ligation as specified by gRNA. The binary interactions of these A proteins with their specific enzymes enhance the catalytic activity of the latter. KREPA proteins appear to play roles in both the architecture and the function of the editosomes. Loss of expression of KREPA1, KREPA2, KREPA4, and KREPA6 proteins results in partial or complete loss of editosomes (19, 29, 37, 46, 54). Brecht et al. (8) reported that recombinant KREPA3 binds double-stranded RNA (dsRNA) and dsDNA and has endo-exo-RNase activity that localizes to the OB-fold domain and that knockdown had little effect on 20S editosome integrity. However, Law et al. (30) found that KREPA3 was important for editosome stability and endonuclease activity but not U addition, U removal, or RNA ligase activities. These findings have not been resolved with each other or the identification of three editing endonucleases and two U-specific exonucleases (10, 11, 27, 45, 56) but may reflect experimental differences, as well as roles for KREPA3 that affect editosome structure and its enzymatic activities.
FIG. 1.
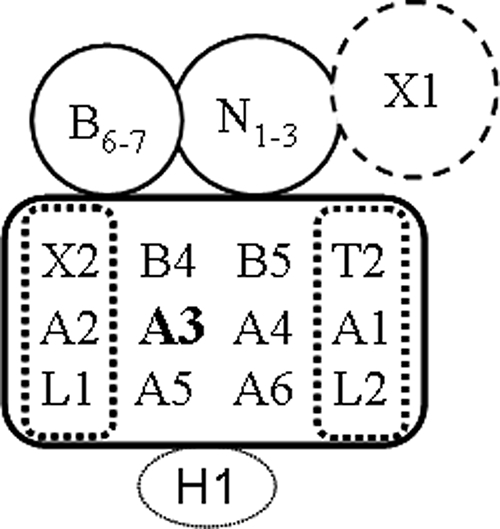
Schematic representation of 20S editosomes. Each of the three editosomes consists of a common core of proteins (rectangle) with an insertion subcomplex consisting of KREPA1, KRET2, and KREL2 and a deletion subcomplex of KREPA2, KREX2, and KREL1. KREPA3 is in the core of all editosomes (bold). Each editosome has a different endonuclease, KREN1, KREN2, or KREN3 (N1-3) and a unique KREPB protein (B6-7). Only KREN1 editosomes contain KREX1 (X1). KREN1 deletion endonuclease editosomes contain KREPB8 and KREX1, KREN2 insertion endonuclease editosomes contain KREPB7, and KREN3 insertion endonuclease editosomes contain KREPB6. The KREH1 helicase is found in biochemically but not TAP tag-purified editosomes.
C2H2-ZF proteins display diversity in ZF number, position in the protein, amino acid spacing, flanking sequence, and in their ligands which include dsDNA, dsRNA, DNA-RNA hybrids, single-stranded RNA, and protein (22). The binding modes differ among the ligands (7, 31, 61). Binding of C2H2 ZFs to DNA can be sequence specific and binding to RNA shows enormous diversity and can be sequence specific or not, whereas ZFs typically function to form homo- or heterodimers between proteins. The two ZFs of KREPA1, KREPA2, and KREPA3 are widely separated by one to several hundred amino acids, which is atypical for most C2H2 ZFs. Except for the more C-terminal ZF of KREPA1 (that has additional amino acids between the second cysteine and the third histidine), their ZFs share the (F/T)XCX2CX3FX5ΨX2HX4H pattern that is found in many other ZF proteins that have roles in DNA, RNA, and protein binding. The ZFs of Trypanosoma brucei KREPA1, KREPA2, and KREPA3, as well as the KREPA2 ortholog of Leishmania tarentolae, LC-4, have been proposed to act in protein-protein interaction or substrate RNA recognition and binding (25).
Different OB-fold domains have little sequence similarity but share a conserved topological architecture consisting of five β-strands forming two antiparallel β-sheets that support a common binding face for all its ligands, which includes nucleic acids, oligosaccharides, and a few proteins (1, 35, 55). The loops connecting the β-strands vary widely in length and conformation and accommodate the various modes of binding of different ligands. The C-terminal regions of KREPA1, KREPA2, KREPA3, and KREPA6 are predicted to have OB-fold domains based on their structural organization and key types of amino acids, but the OB-fold of KREPA4 is more similar to an S1-motif (46). The OB-fold domains of KREPA1 and KREPA2 have been proposed to play roles in substrate RNA recognition and binding and the coordination of the TUTase and ligase, or exoUase and ligase enzymatic steps in editing, respectively (47). Both KREPA4 and KREPA6 display RNA-binding activity with a preference for the gRNA oligo(U) tail and were hypothesized to use their OB-fold domain to coordinate the forward progression of RNA editing steps (46, 54). The OB-fold of KREPA3 was proposed to have both RNA recognition and ribonucleolytic activities (8, 36).
We show here that KREPA3 is essential for RNA editing and the survival of both PF and BF parasites. We find that KREPA3 is important for editosome integrity since knockdowns in PFs generate smaller complexes with the U addition, U removal, and ligation steps of editing that are characteristic of the heterotrimeric insertion and deletion editosome subcomplexes. We also show that KREPA3 bridges these subcomplexes within the editosome core. The deletion and insertion endonucleolytic activities are reduced but are only associated with the ∼20S editosomes. We also show that transcriptional inactivation of KREPA3 in BFs, which appears more complete than RNA interference (RNAi) knockdown, results in essentially complete loss of editosomes and the in vitro catalytic activities. Exclusive expression of KREPA3 proteins with mutations in one or both ZFs or deletion of the OB-fold resulted in ablation of RNA editing in vivo and cell death. Tagged KREPA3 with one or both ZFs mutated incorporate into ∼20S editosomes, and these catalyze all activities in vitro. In contrast, tagged KREPA3 with the OB-fold deleted does not incorporate into editosomes nor rescue ∼20S editosomes upon loss of wild-type (WT) KREPA3. Thus, the ZFs and OB-fold domain are indispensable for KREPA3 function in vivo. However, KREPA3 is not responsible for the central catalytic steps of editing, although it may affect these steps indirectly through editosome structure. Hence, KREPA3 appears to function in RNA binding and editosome organization, which may coordinate the steps of editing and/or affect the processivity of editing.
MATERIALS AND METHODS
Plasmid constructs.
The plasmid expressing tetracycline (tet)-inducible RNAi for KREPA3 was constructed according to the report of Brecht et al. (8). A 701-bp fragment from the 3′ end of KREPA3 was amplified by using the primers 5′- ATACTCGAGCTGCTGCTAATTCTGC-3′ and 5′-ATAAAGCTTTCACACCTTCAACACTG-3′, digested with XhoI and HindIII, and inserted into similarly digested pZJM (59) to create pZJM-A3. The restriction sites are underlined. The plasmids used for generating the regulatable knockout (RKO) cell line, BF KREPA3-RKO, were based on a previous strategy (48, 60) with some modifications. Distal 5′ and 3′ KREPA3 open reading frame (ORF) flanking sequences located outside the flanking sequences used for pLEW90-A3-KO2 (below) were amplified from T. brucei 427 genomic DNA by using the primers 5′-ATAGCGGCCGCGCACACGTGTGGACATTATC-3′ and 5′-ATAACGCGTCTCGAGAACTTATGGGGTAGAAGACG-3′ and the primers 5′-ATATCTAGAATTTAAATACCAAGCGAGAAAGGGAAGA-3′ and 5′- ATAAGGCCTGCGGCCGCTAATGCTGTTATGGGCGCTG-3′, respectively. The PCR products were digested with NotI and MluI and with XbaI and StuI, respectively, and inserted into similarly digested pLEW13 to generate the construct pLEW13-A3-KO1 for replacement of the first KREPA3 allele. The 5′ and 3′ flanking sequences adjacent to the KREPA3 ORF were amplified from genomic DNA by using the primers 5′-ATAGCGGCCGCTTGGCGGAAGAAAGAGAACG-3′ and 5′-ATAACGCGTCTCGAGGGACGAGACATGAGGTGATG-3′ and the primers 5′-ATATCTAGAATTTAAATTGGTGCCTCAATATGATGGT-3′ and 5′-ATAAGGCCTGCGGCCGCTTGGGACCCATCGCTATTTG-3′, respectively. The PCR products were digested with NotI and MluI and with XbaI and StuI, respectively, and inserted into similarly digested pLEW13 to generate the construct pLEW13-A3-KO2. The XhoI-EcoRV fragment from pLEW13-A3-KO2 was then replaced with the XhoI-StuI fragment from pLEW90 to generate pLEW90-A3-KO2 for replacement of the second KREPA3 allele. The full-length KREPA3 ORF was amplified from genomic DNA by using the primers 5′-CCTCGAGCCACCATGAAGCGTGTTACTTCAC-3′ and 5′- CGGATCCTTATCACACCTTCAACACTGACC-3′ and cloned into pLEW79 after digestion with XhoI and BamHI to generate pLEW79-KREPA3.
To generate KREPA3 mutants with a tandem affinity purification (TAP) tag at the C terminus, full-length KREPA3 and KREPA3 with a deletion of the C-terminal OB-fold domain were amplified by using the primers 5′-CCTCGAGCCACCATGAAGCGTGTTACTTCAC-3′ and 5′- CGGATCCCACCTTCAACACTGACC-3′ and the primers 5′-CCTCGAGCCACCATGAAGCGTGTTACTTCAC-3′ and 5′-CGGATCCCCAATGTGCAGCCACTTC-3′, respectively. After digestion with XhoI and BamHI, the products were cloned into pLEW79-MHTAP (23, 40) to create pLEW79-A3-TAP and pLEW79-A3ΔOB-TAP. Mutagenesis of the ZF cysteines to alanines was performed by using a QuikChange II XL site-directed mutagenesis kit (Stratagene, CA). Mutation of the first ZF (C53A/C56A) was performed with the oligonucleotides 5′-CACTCCCTCCTTTCAGGCCGGCGAAGCCGGTAAGGCTTTTCGTC-3′ and 5′-GACGAAAAGCCTTACCGGCTTCGCCGGCCTGAAAGGAGGGAGTG-3′, mutation of the second ZF (C183A/C186A) was performed with the oligonucleotides 5′-GGACAAAAAAACATTTGTCGCGACGATTGCTCAGAAAACATTTCGG-3′ and 5′-CCGAAATGTTTTCTGAGCAATCGTCGCGACAAATGTTTTTTTGTCC-3′, and the resulting plasmids named pLEW79-A3ZFm1-TAP and pLEW79-A3ZFm2-TAP. To generate pLEW79-A3ZFm1&2-TAP, a HindIII and BamHI fragment containing the ZFm2 was excised from pLEW79-A3ZFm2-TAP and inserted into pLEW79-A3ZFm1-TAP digested with the same enzymes. All of the mutations were confirmed by sequencing. To generate KREPA3-RKO cell lines with constitutively expressed TAP-tagged WT or mutant KREPA3 alleles, the TAP-tagged KREPA3 WT and mutant genes were amplified from the plasmids described above by using the primers 5′-CCTCGAGCCACCATGAAGCGTGTTACTTCAC-3′ and 5′-ATTCATGATCAGGTTGACTTCCCCGCGGAATTC-3′, digested with XhoI and BclI, and inserted individually into pHD1344tub (10), which targets integration of constitutively expressed ectopic alleles into β-tubulin locus.
Transfections and repression of KREPA3 expression.
The T. brucei PF 29-13 cell line (60) was transfected with 10 μg of NotI-linearized pZJM-A3, and stable transfectants were screened as described previously (59). The resulting cell line was named PF KREPA3-RNAi and maintained in SDM-79 medium plus 10% fetal bovine serum with 15 μg of G418/ml, 25 μg of hygromycin/ml, and 2.5 μg of phleomycin/ml. dsRNA was induced by adding 1 μg of tet/ml. Growth of the cells was monitored in the absence or in the presence of tet and counted daily with a Coulter counter. The cells were maintained between 1.0 × 106 and 2.0 × 107 cells/ml.
The BF KREPA3-RKO cell line was generated as previously described (48, 60) and maintained in HMI-9 medium plus 10% fetal bovine serum with multiple selection markers (below). Briefly, wild-type BF 427 cells were electroporated with NotI-linearized plasmids in a stepwise order; first with pLEW13-A3-KO1 and selection with 2.5 μg of G418/ml to replace one of the KREPA3 alleles, then with pLEW79-KREPA3 and selection with 2.5 μg of phleomycin/ml to insert the tet-regulatable ectopic KREPA3 allele into the ribosomal DNA spacer region, and finally with pLEW90-A3-KO2 and selection with 5 μg of hygromycin/ml in addition to 1 μg of tet/ml to induce ectopic KREPA3 expression and replace the second KREPA3 endogenous allele. Correct integration of each plasmid was confirmed by PCR. The expression of ectopic KREPA3 was repressed by washing and culturing the cells in the medium minus tet. Growth of the cells was monitored in the presence or absence of tet and diluted daily to 1.0 × 105 to 2.0 × 105 cells/ml.
The pHD1344tub plasmids with TAP-tagged WT or mutant KREPA3 genes for constitutive expression from the tubulin locus were linearized with NotI and transfected into KREPA3-RKO cells independently. The resistant clones were selected with 0.1 μg of puromycin/ml and designated RKO-KREPA3 WT-TAP, ZFm1-TAP, ZFm2-TAP, ZFm1&2-TAP, and ΔOB-TAP. Expression of the tagged genes was determined by Western blotting.
RNA isolation and real-time RT-PCR analysis.
Total RNA was harvested from the cell lines using the TRIzol LS reagent (Gibco-BRL) according to the manufacturer's instructions. Real-time reverse transcription-PCR (RT-PCR) was performed as described previously (9). Briefly, 10 μg of RNA was treated with DNase I by using a DNA-free kit (Ambion). Then, 4.5 μg of the treated RNA was reverse transcribed by using random hexamers and TaqMan RT reagents (Applied Biosystems) to generate the cDNA templates for real-time PCR. At the same time, a control reaction without reverse transcriptase was carried out to rule out contamination by genomic DNA. The cDNA reactions were diluted 1:7 in water as the template for most PCRs and further diluted 1:50 for the PCR of high abundance of internal control 18S rRNA or β-tubulin. Each PCR included 2.5 μl of cDNA, 10 μl of forward and reverse primers (each at 0.75 μM), and 12.5 μl of Sybr green PCR master mix (Applied Biosystems) mixed in a well of a 96-well plate (Applied Biosystems), and each kind of mRNA was analyzed in triplicate. The sequences of all of the used primers had been described (9). The amplification conditions were 50°C for 2 min and 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min, using an ABI Prism 7000 thermocycler. Relative changes of target amplicons were determined by comparing the level in KREPA3-repressed cells to that in cells in which KREPA3 was expressed after normalization to 18S rRNA or β-tubulin by using the Pfaffl method (43), with PCR efficiencies calculated by linear regression using LinregPCR (44).
Crude mitochondria preparation and glycerol gradient sedimentation.
Crude mitochondria were prepared from roughly 2 × 1010 PF KREPA3-RNAi cells or 3 × 109 BF KREPA3-RKO or RKO KREPA3 WT-TAP or ZFm2-TAP cells in the absence or presence of 1 μg of tet/ml essentially as described previously (18). The mitochondria from PFs were lysed with 1.5 ml of mt lysis buffer (20 mM HEPES [pH 7.9], 10 mM magnesium acetate, 100 mM KCl, 1 mM EDTA) containing protease inhibitors and centrifuged twice at 13,000 rpm for 10 min at 4°C. The cleared lysates were loaded onto 11-ml 10 to 30% glycerol gradients and centrifuged at 38,000 rpm for 12 h at 4°C by using an SW40 rotor (Beckman). Twenty-four 500-μl fractions were collected from top to bottom. The crude mitochondria from BFs were lysed with 600 μl of mt lysis buffer, and the cleared lysates were loaded onto 4.5-ml glycerol gradients and centrifuged at 44,000 rpm for 9 h at 4°C by using an SW55 rotor (Beckman). Twenty-four fractions were collected from top to bottom. All of the fractions were flash frozen in liquid nitrogen and stored at −80°C for Western blotting and in vitro enzymatic assays.
In vitro RNA editing assays.
The standard in vitro RNA editing activities assay has been described previously. Briefly, 5′CL18 and 3′CL13pp with gPCA6-2A RNAs were used for a precleaved insertion editing assay specifying the insertion of two U's, and U5 5′CL and U5 3′CL with gA6[14]PC-del RNAs were used for precleaved deletion editing specifying removal of four U's (20, 21). 5′CL18 and U5 5′CL were 5′ labeled with [γ-32P]ATP by T4 kinase. A6short/TAG.1 pre-mRNA and D34 gRNA were RNA substrates for deletion site cleavage assays, and A6-eES1 pre-mRNA and gA6[14] gRNA were RNA substrates for insertion site cleavage assays (10, 13, 24, 49). A6short/TAG.1 and A6-eES1 mRNAs were 3′ labeled with 32P-labeled pCp with T4 RNA ligase. Portions (10 μl) of individual or pooled gradient fractions or TEV eluates after purification on immunoglobulin G (IgG) beads were incubated with the different substrate RNAs for enzymatic reactions. The reaction products were resolved on 7 M urea-polyacrylamide gels and visualized on Storm PhosphorImager screens (Molecular Dynamics).
RNA ligase adenylation assays were performed as follows. mt gradient fractions (5 μl) were incubated with 5 μCi of [α-32P]ATP in a 20-μl reaction system (12.5 mM KCl, 12.5 mM HEPES [pH 7.9], 5 mM magnesium acetate, 0.25 mM dithiothreitol) at 28°C for 5 min and then stopped by the addition of 10 μl of 3× sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) loading dye. The proteins were resolved by SDS-10% PAGE, and the radiolabeled proteins were detected by using a PhosphorImager.
Coimmunoprecipitation and purification of TAP-tagged proteins.
The monoclonal antibody (MAb) specific for KREPA2 (39) was first incubated with goat anti-mouse IgG-conjugated magnetic beads (Dynal) for 2 h at 4°C with rotation. The crude mt lysates from uninduced or induced PF KREPA3-RNAi cells were lysed with IPP200 containing a protease inhibitor cocktail with 1% Triton X-100 (Roche). The cleared lysate was incubated with the KREPA2 MAb coated beads for 2 h at 4°C with rotation. The beads were washed with IPP200 three times, and 1× SDS loading buffer was added and boiled for 5 min. The starting lysate, the supernatant after immunoprecipitation, and the precipitates were resolved by SDS-10% PAGE, and a Western blot was performed with the specific MAbs against KREPA1, KREPA2, KREPA3, and KREL1 (39).
BF KREPA3-RKO or tagged RKO-KREPA3 WT or mutant cells (2 × 108) grown in the presence or absence of tet for 3 days were harvested and lysed with IPP150 containing a cocktail of protease inhibitors (Roche) plus 1% Triton X-100. The whole-cell lysates were incubated with IgG-conjugated agarose beads for 2 h and then cleaved by TEV protease for 2 h at 16°C. The starting whole-cell lysates and the TEV eluates were resolved by SDS-10% PAGE and assayed by Western blotting with MAbs against KREPA1, KREPA2, KREPA3, and KREL1 and polyclonal antibodies against KREPA6 (47) or PGK (41), the level of PGK being a control. Portions (10 μl) of the same TEV eluates were also used for in vitro RNA editing assay.
RESULTS
KREPA3 is essential for growth and in vivo RNA editing.
The effect of KREPA3 repression on cell growth was examined in both the PF and the BF stages of T. brucei. Expression of KREPA3 was repressed in PFs by generating an RNAi cell line in 29-13 PFs (PF KREPA3-RNAi) that expresses dsRNA to a portion of KREPA3 mRNA under the control of tet-inducible promoter as previously described (8). Expression of KREPA3 was blocked in BFs by generating a regulatable knockout cell line (BF KREPA3-RKO) by disrupting both endogenous alleles by homologous recombination and inserting a tet-regulatable ectopic KREPA3 allele (KREPA3 Reg) into the ribosomal DNA locus as described in Materials and Methods. Cell growth was inhibited dramatically upon KREPA3 repression by tet-induced RNAi in PFs (Fig. 2A) and completely blocked in BFs by the withdrawal of tet (Fig. 2B), while the nonrepressed PF and BF control cells grew at the normal rate. KREPA3 protein was not detected in PFs or BFs after 6 or 4 days of repression, respectively, (insets, Fig. 2A and B) as determined by Western analysis with a KREPA3 MAb. Some motile PF RNAi cells were seen by microscope after 12 days of RNAi induction with tet, but only cell debris was seen 6 days after repression in the BF RKO cells after tet withdrawal. The motile PF cells but not BF cells may indicate less complete KREPA3 repression with RNAi compared to the conditional knockout, as has been commonly observed, or it may reflect the slower growth rate of PFs, other differences between life cycles stages, or some combination thereof. Real-time PCR analysis of total RNA from the cells showed that the mRNA levels for the KREPA1, KREPA2, KREPA5, and KREPA6 editosome proteins, which are related to KREPA3, were unaffected by KREPA3 repression by RNAi induction or RKO (data not shown), indicating that the repression was specific to KREPA3. Overall, the dramatic repression of growth of both PFs and BFs by repression of KREPA3 expression indicates that this protein is essential in both life cycle stages of T. brucei.
FIG. 2.

KREPA3 repression is essential for growth and in vivo RNA editing in BFs and PFs. Growth of PF KREPA3-RNAi (A) and BF KREPA3-RKO (B) cells in which KREPA3 was expressed or repressed. KREPA3 expression was repressed by the addition of tet in PFs to induce RNAi expression and the withdrawal of tet from BFs to repress KREPA3 transcription. The insets show Western analysis of lysates from PF cells at day 6 and BF cells at day 4 using a KREPA3 MAb. The positive controls are pooled 20S fractions of a glycerol gradient purified mt lysate from WT PF mitochondria in panel A or a BF cell lysate in panel B. (C and D) The relative abundance of pre-edited, edited, and never-edited ND4 and COI mRNAs in cells in which KREPA3 was repressed compared to cells in which it was expressed in PF KREPA3-RNAi cells at day 6 (C) and BF KREPA3-RKO cells at day 4 (D). The relative change in each target amplicon was determined by real-time RT-PCR of total RNA and normalized to β-tubulin and 18S rRNA. Note the log scale and that 1 represents no difference, >1 is an increase, and <1 is a decrease.
The effect of KREPA3 repression on RNA editing in vivo was assessed in both PF and BF stages by real-time PCR. Total RNA was isolated from nonrepressed PF KREPA3-RNAi or BF KREPA3-RKO cells, as well as from these cells after 6 or 4 days of repression of KREPA3 expression, respectively, when growth inhibition was manifested. The levels of all edited and pre-edited mRNAs except ND9, as well as the never-edited ND4 and COI mRNAs, were determined and normalized to the β-tubulin mRNA and 18S rRNA internal controls. The levels of ND4 and COI mRNAs in the repressed cells relative to those in the nonrepressed cells were unaffected, and the levels of pre-edited mRNAs were unchanged or increased to various amounts (Fig. 2C and D). In contrast, the relative levels of edited mRNAs were reduced dramatically in both PF and BF cells after KREPA3 repression. All but 2 of the 11 edited mRNAs tested were reduced 90% or more (Fig. 2C and D). The level of edited CYb mRNA was below detection in BFs (data not shown). Most of the edited mRNAs were reduced more in BFs than PFs, as especially evident for COII mRNA, perhaps due to a lower efficiency of KREPA3 repression by RNAi compared to the regulatable double knockout. The lack of a concomitant increase in the levels of some pre-mRNAs upon a dramatic reduction in the edited RNA may reflect the inherent differential turnover rates of these RNAs. Thus, KREPA3 is essential for RNA editing in vivo in both PFs and BFs. The extensive reduction of edited mRNA upon KREPA3 knockdown suggests that it might be structurally necessary for editosome integrity and/or play other critical roles in the RNA editing process in vivo.
KREPA3 repression in PFs affects editosome stability and in vitro editing.
The effect of KREPA3 repression in PFs on editosome stability and integrity was examined by coimmunoprecipitation and sedimentation in glycerol gradients. Lysates from the PF KREPA3-RNAi cells in which KREPA3 was expressed or repressed were incubated with a KREPA2 MAb bound to IgG-coated magnetic beads. The supernatants and immunoprecipitates were examined by Western analysis using a mixture of MAbs specific for KREPA1, KREPA2, KREL1, and KREPA3 (Fig. 3A). Although repression of KREPA3 was inhibited significantly after 6 days of RNAi induction, the other editosome proteins were evident in the total lysate (input). The loading control is not shown. KREPA2 and KREL1 were both immunoprecipitated from lysates whether or not KREPA3 expression was repressed, but most KREPA1 remained in the supernatant when KREPA3 was repressed. This is consistent with previous findings that KREL1 interacts with KREPA2 and occurs in a deletion subcomplex, along with KREX2, while KREPA1, KREL2, and KRET2 occur in an insertion subcomplex (47). Glycerol gradient sedimentation of cleared crude mt lysates from these cells revealed a shift in the peak of the editosome components from ∼20S to a smaller S region upon KREPA3 repression, as seen using the cocktail of the four MAbs or by auto-adenylation of KREL1 and KREL2 (Fig. 3B). KREPA1 and adenylatable KREL1 and KREL2 shifted to smaller S region of the gradient upon KREPA3 repression than did the KREPA2 and KREL1 proteins seen by Western analysis (Fig. 3B). Hence, the subcomplex containing KREPA1 and KREL2 may have fewer components than that with KREPA2 and KREL1. The peak of KREL1 protein in the greater S value region of the gradient is seen by Western blotting but not autoadenylation, implying that it is adenylated and hence primed to ligate RNA. Some editosomes remain in the ∼20 to 40S region following KREPA3 repression despite the extensive loss of KREPA3. This may reflect a less-than-complete elimination of KREPA3 since a faint signal for KREPA3 can be detected in this region with longer exposures and/or the presence of editosomes that lack this protein. Overall, the data indicate that KREPA3 is required for editosome integrity and critical to bridge the interaction between insertion and deletion subcomplexes, since most insertion subcomplexes failed to associate with the deletion subcomplexes.
FIG. 3.

Disruption of editosomes and effect on in vitro RNA editing activities upon KREPA3 repression by RNAi in PFs. (A) Western analysis of crude mt lysates from PF KREPA3-RNAi cells in which KREPA3 was expressed or repressed for 6 days before (input) and after immunoprecipitation with a KREPA2 MAb. The Western used a mixture of MAbs for the editosome components indicated on the right. Note the presence of only KREPA2 and KREL1 in the precipitate. The 20S positive control was described in Fig. 2A. (B) Western and adenylation analyses of glycerol gradient fractions of crude mt lysates from cells as in panel A. The Western blot analyses used a mixture of MAbs as indicated, and the adenylation assays reveal both KREL1 and KREL2. These fractions were used for in vitro precleaved insertion (C) and precleaved deletion (D) assays and for endonuclease assays specific for A6 insertion (E) and deletion (F) sites. The RNA substrates are described in Materials and Methods. The radiolabeled input RNAs (I), input RNA with two U's added (+2U), or four U's removed (−4U), ligated products of unprocessed 5′ and 3′ input RNAs (Lig) and edited (Ed) products are indicated. The position of the specific, gRNA-directed cleavage products are indicated by an arrow. gRNA was omitted from control reactions (g-) and RNase T1-digested substrate RNA (T1) was used as a marker. Positive control reactions (+) were performed using a fraction of ∼20S editosome that contains peak editing activity, which was omitted from negative control reactions (−).
KREPA3 repression also resulted in a shift of the in vitro precleaved U-insertion/ligation and U-deletion/ligation activities to a smaller S value in the gradients, from fraction 15-23 to fraction 11-17 (Fig. 3C and D), corresponding to the shift of the editosome proteins. KREPA3 repression did not result in an obvious reduction of U-insertion and U-deletion activities. However, the TUTase and exoUase activities were somewhat affected since ligated products with one U added (Fig. 3C) and three U's removed (Fig. 3D) were enhanced upon KREPA3 repression, possibly due to alterations of the editosome integrity. In contrast to the precleaved editing activities, the insertion and deletion site endonuclease activities remained at ∼20 to 40S and were reduced in amount upon KREPA3 repression compared to the control cells (Fig. 3E and F). The amount of remaining endonuclease activities correlates with the residual level of editosomes rather than the KREPA3 level which was determined by phosphorimager quantitation (data not shown). Thus, the reduction of endonuclease activities appears to be due to editosome disruption rather than loss of KREPA3. Overall, these data indicate that repression of KREPA3 expression affected the in vitro editing activities but that this effect is mainly attributable to the altered editosome structure. This differs from the conclusions of Brecht et al. (8) as discussed below. The ∼20S editosomes retained in PF cells upon KREPA3 repression are competent for the in vitro RNA editing activities but are insufficient to support cell growth and in vivo editing.
Editosome loss upon KREPA3 repression in BFs.
Repression of KREPA3 expression in BF KREPA3-RKO cells resulted in a dramatic loss of editosomes and in vitro editing activities (Fig. 4) in contrast to the partial effect observed in PF KREPA3-RNAi cells. Western analysis of crude mt lysates with the antibody cocktail for the four editosome proteins showed that these proteins were lost from BF KREPA3-RKO cells after 4 days of repression of KREPA3 expression (Fig. 4A). MAb66 was used to detect the 3-methylcrotonoyl-coenzyme A carboxylase β protein (A. Panigrahi, unpublished data), which has no relationship to RNA editing, and was used as a loading control. A very weak signal was discernible in the KREPA2 region of the blot upon KREPA3 repression in BFs, but KREPA1, KREL1, and KREPA3 were undetectable (Fig. 4A, left panel). This is in contrast to the essentially unchanged levels of KREPA1, KREPA2, and KREL1 proteins upon KREPA3 repression by RNAi in PFs (Fig. 4A, right panel). It is likely that the difference in the amount of editosome proteins remaining after KREPA3 repression is due to the different methods used to repress KREPA3 expression or the differences in the editosomes between these life cycle stages. Glycerol gradient sedimentation analysis of cleared crude mt lysates from the BF KREPA3-RKO cells in which KREPA3 was expressed or repressed for 2 or 4 days revealed a dramatic loss of editosomes. All tested editosome components (KREPA1, KREPA2, KREL1, and KREPA6) substantially decreased in the ∼20 to 40S region after 2 days of KREPA3 repression with small amounts of these components seen in the upper fractions (Fig. 4B, middle panel). No editosome proteins were detected in the ∼20S to 40S region after 4 days of KREPA3 repression and only small amounts of KREPA1 and KREPA2 were found in the top gradient fractions (Fig. 4B, lower panel). Not surprisingly, the precleaved insertion (TUTase and ligase), precleaved deletion (ExoUase and ligase), and the insertion and deletion site endonuclease activities in pooled ∼20 to 40S fractions were dramatically reduced after 2 days and undetectable after 4 days of KREPA3 repression, a finding concordant with the loss of editosomes (Fig. 4C, D, E, and F). Therefore, KREPA3 is structurally necessary for editosome stability and integrity in BFs since loss of KREPA3 results in complete loss of editosomes, its components, and the in vitro editing activities.
FIG. 4.

KREPA3 repression in BFs by conditional knockout results in loss of the editosome and in vitro RNA editing activities. (A) Different effect of KREPA3 repression on the editosome in BFs compared to PFs. Western analysis of crude mt lysates from BF KREPA3-RKO (left panel) and PF KREPA3-RNAi (right panel) cells in which KREPA3 was expressed or repressed for 4 or 6 days, respectively, using a cocktail of MAbs as indicated. The 20S positive control was described in Fig. 2A, and MAb66 was used as a loading control. (B) Effect of KREPA3 repression in BF KREPA3-RKO cells on editosome sedimentation. Western analysis of glycerol gradient fractions of crude mt lysates from BF KREPA3-RKO cells in which KREPA3 was expressed or repressed for 2 or 4 days using a cocktail of MAbs as indicated. The 20S positive control was as in Fig. 2B and MAb66 was used as a control. The peak editosome fractions 19 to 23 (∼20 to 40S region) were pooled and used for in vitro precleaved insertion (C) and precleaved deletion (D) assays and endonuclease assays specific for A6 insertion (E) and deletion (F) sites. Assay designations are as in Fig. 3.
ZF mutation or OB-fold deletion in KREPA3 is lethal and blocks editing in vivo.
The KREPA3 ZF motifs and OB-fold domain were found to be critical for KREPA3 function in vivo. The effects of mutating one or both ZFs or deleting the OB-fold domain on cell growth were examined by generating transgenic BF cells that can exclusively express mutant KREPA3 proteins. The mutations replaced both cysteines with alanines in one or both C2H2 ZF motifs or deleted the OB-fold domain as diagrammed in Fig. 5A. WT and mutant KREPA3 alleles were inserted into the β-tubulin locus of BF KREPA3-RKO cells, where they are constitutively expressed. These alleles were TAP-tagged to discriminate them from the tet-regulated WT KREPA3 allele, KREPA3 Reg, from the rRNA locus and to allow for purification of the proteins. The resulting cells were designated RKO-KREPA3WT-TAP, RKO-KREPA3ZFm1-TAP, RKO-KREPA3ZFm2-TAP, RKO-KREPA3ZFm1&2-TAP, and RKO-KREPA3ΔOB-TAP. All of the cells grew at the same rate in the presence of tet, i.e., when KREPA3 Reg was expressed. Withdrawal of tet resulted in the cessation of cell growth after 4 days for all of the KREPA3 mutants (Fig. 5B). No live cells, but only cell debris, could be observed at day 6, a finding similar to the consequence of repression of KREPA3 expression in BF KREPA3-RKO cells. Growth of RKO-KREPA3 WT-TAP cells was inhibited to some extent upon KREPA3 Reg repression, and cells continued a slower growth rate without ultimate death (Fig. 5B). This is presumably due to the effect of the TAP tag since the expression of KREPA3 Reg without any tag supported cell growth at a normal rate (Fig. 2B). Western analysis with the KREPA3 MAb revealed that the withdrawal of tet essentially eliminated expression of the KREPA3 Reg allele after 3 days (Fig. 5C). It also revealed that the level of the TAP-tagged KREPA3ZFm1 and ZFm1&2 proteins was lower than the levels of the WT and ZFm2 proteins in similar numbers of cells. The same result was observed using the PAP reagent that is specific for the protein A component of the TAP tag (data not shown), which indicated that the ZF mutation had no effect on recognition of KREPA3 by the MAb. Therefore, both the ZFs and the OB-fold domain are necessary for KREPA3 function in vivo, since mutations of these motifs are lethal, similar to the effect of KREPA3 repression in BF KREPA3-RKO cells.
FIG. 5.

Mutation of the ZFs or deletion of the OB-fold in KREPA3 results in cell death and loss of in vivo RNA editing. (A) Schematic representation of WT and mutant TAP-tagged KREPA3. The two cysteines of one or both ZFs were mutated to alanines resulting in ZFm1-TAP (C53A/C56A), ZFm2-TAP (C183A/C186A), and ZFm1&2-TAP (C53A/C56A/C183A/C186A) and truncation of KREPA3 before the OB-fold was named ΔOB-TAP. (B) Cell growth in the presence of constitutively expressed WT or mutant TAP-tagged KREPA3 when KREPA3 Reg was expressed (E) or repressed (R). Constitutively expressed WT or mutant TAP-tagged KREPA3 alleles were integrated into the β-tubulin locus of BF KREPA3-RKO cells, and the expression of KREPA3 Reg was controlled by adding or withdrawing tet. (C) Western analysis of KREPA3 Reg and TAP-tagged KREPA3 allele expression using a KREPA3 MAb. KREPA3 Reg expression was efficiently repressed upon withdrawal of tet for 3 days. (D) The relative abundance of pre-edited, edited, and never-edited mRNAs in cells exclusively expressing WT or mutant TAP-tagged KREPA3 alleles in which KREPA3 Reg was repressed for 3 days compared to cells in which it was expressed was detected by real-time RT-PCR. The relative change in the amplicons for pre-edited and edited A6, COIII, and COII and never-edited ND4 were normalized to 18S rRNA. Note the log scale and that 1 represents no difference, >1 is an increase, and <1 is a decrease.
Exclusive expression of each ZF mutation or the OB-fold deletion (ΔOB) of KREPA3 abolished editing in vivo to an extent similar to the loss of KREPA3 expression, as revealed by real-time PCR. Total RNA was isolated from the parental BF KREPA3-RKO cells and the BF KREPA3-RKO cells expressing TAP-tagged KREPA3 WT, ZFm2 or ΔOB mutant alleles with KREPA3 Reg expression or repression for 3 days. The abundance of pre-edited and edited A6, COIII, and COII and never-edited ND4 mRNAs was assayed by real-time PCR, and the relative mRNAs levels were normalized to 18S rRNA as an internal control (Fig. 5D). The relative mRNA abundance was also normalized to β-tubulin with the same results (data not shown). Repression of KREPA3 Reg expression which results in exclusive expression of TAP-tagged KREPA3ZFm2 or KREPA3ΔOB essentially abolished in vivo editing. The relative abundance of edited mRNA decreased to the same level as that of the parental BF KREPA3-RKO cells in the absence of KREPA3 Reg expression, i.e., by ca. 95 to 99%, while the pre-edited mRNA levels were unchanged or slightly increased (Fig. 5D). Exclusive expression of the TAP-tagged WT KREPA3 protein, which somewhat inhibited growth, resulted in a 30 to 60% inhibition of in vivo editing (Fig. 5D). In all of these cases, the relative abundance of never-edited ND4 mRNA was essentially unchanged. Thus, these data indicate that the ZF motifs and OB-fold domain are necessary for KREPA3 function and for RNA editing in vivo.
The C-terminal TAP tag and ZF mutations affect KREPA3 editosome association.
The effect of mutation of the second ZF on the association of KREPA3 with the editosome was first estimated by editosome sedimentation. Crude mt lysates from RKO-KREPA3 WT-TAP or ZFm2-TAP cells in the presence or absence of KREPA3 Reg expression were subjected to glycerol gradient sedimentation. Western analysis was performed on the gradient fractions using a panel of editosome MAbs with similarly treated lysates of KREPA3-RKO cells in the absence of KREPA3 Reg expression serving as a control. A portion of the KREPA3 WT-TAP and ZFm2-TAP proteins were found to incorporate into ∼20-40S complexes, along with the KREPA1 and KREL1 editosome components when KREPA3 Reg was repressed, in contrast to the virtual lack of all editosome proteins in this region in the control cells (Fig. 6A). Thus, both the tagged WT and ZFm2 proteins were able to restore the presence of ∼20S editosomes. Most of the tagged WT KREPA3 sedimented in the ∼5 to 10S region when KREPA3 Reg was expressed, although some of it shifted to larger S values when the expression of the untagged KREPA3 was eliminated, i.e., when KREPA3 Reg repressed for 3 days. The levels of KREPA1 and KREL1 were much lower in the cells exclusively expressing the WT-TAP protein than in cells also expressing the untagged KREPA3 Reg (Fig. 6A). Thus, the C-terminal tag hampered the ability of the protein to efficiently remain incorporated in ∼20S editosomes, and untagged KREPA3 appears to compete with the tagged version for incorporation into 20S editosomes. This is consistent with the partial inhibition of cell growth and in vivo editing in RKO WT-TAP cells in the absence of KREPA3 Reg expression. Parallel studies revealed that the tagged KREPA3 ZFm2 protein was primarily located in a smaller S value region of the gradient than WT-TAP when KREPA3 Reg was expressed and did not shift to a larger S region after KREPA3 Reg expression was repressed (Fig. 6A). Also, KREL1 shifted to a smaller S value region of the gradient upon repression of KREPA3 Reg expression, which was not the case with WT tagged KREPA3 (Fig. 6A). Considering that expression of ZFm2-TAP completely blocked growth and in vivo editing, this result suggests that the ZFm2 protein is involved in protein and/or RNA interaction in a fashion that affects editosome integrity, as well as in vivo function, which is different from the WT version.
FIG. 6.

Effect of KREPA3 ZF disruption on editosome sedimentation. (A) Western analysis of glycerol gradient fractions of crude mt lysates of BF RKO-KREPA3 WT-TAP and ZFm2-TAP cells in which KREPA3 Reg was expressed (E) or repressed (R) for 3 days. Western analysis was performed with antibodies against KREPA1, KREL1, and KREPA3. Similarly prepared lysates from the BF KREPA3-RKO parental cells with KREPA3 expression repressed for 3 days serves as the control. Compared to the control, exclusive expression of either KREPA3 WT-TAP or ZFm2-TAP rescued the presence of the ∼20 to 40S editosomes but to various amounts. Also, the KREPA3 WT-TAP and ZFm2-TAP proteins displayed different distributions on the glycerol gradients. (B) Western analysis and editing assays of the pooled ∼5 to 10S and ∼20 to 40S gradient fractions from WT-TAP with KREPA3 Reg repression in panel A. Western analysis was performed with the MAb mixture against KREPA1, KREPA3, and KREL1. The editing assays and designations are described in Fig. 3. The substantial amount of KREPA3-WT TAP protein in the pooled ∼5 to 10S fractions does not catalyze any of the editing activities.
The effect of the tag and/or mutations in one or both ZFs on the association of KREPA3 with other editosome components in the presence or absence of untagged WT protein was explored further by purification of the tagged complexes. The tagged complexes were immunoprecipitated with IgG beads from KREPA3-RKO and its derivative cells expressing TAP-tagged KREPA3 WT or ZF mutant proteins with KREPA3 Reg expressed or repressed for 3 days. The complexes were eluted by TEV protease cleavage, and the eluates and starting whole-cell lysates were analyzed by Western analysis with the panel of editosome MAbs. The studies clearly showed that the tagged WT protein did not associate with most editosomes in cells expressing KREPA3 Reg but did when KREPA3 Reg expression was repressed, although it was expressed at similar levels as shown by Western analysis of whole-cell lysates (Fig. 7A). This is consistent with the sedimentation results in Fig. 6A. Compared to the WT-TAP cells with KREPA3 Reg repressed, similar amounts of the tested editosome components were coprecipitated with tagged KREPA3 proteins with one or both ZFs mutated, even in the cells expressing KREPA3 Reg (Fig. 6A), which indicated that ZF disruption had little effect on KREPA3-mediated protein interactions. These results also suggested that the association of editosome with WT versus the ZF mutants might be different in protein and/or RNA. Extra bands were observed when the TEV eluates were probed with the KREPA3 MAb, which might be protease degradation products and untagged KREA3 Reg (labeled with an asterisk; this only appeared with KREPA3 Reg expression, and the size is similar to KREPA3). In addition, purification of the tagged complexes from RKO WT-TAP and ZFm2-TAP cells using the TAP procedure and identification of the proteins by liquid chromatography-tandem mass spectrometry (LC-MS/MS) showed that most of the editosome proteins were copurified with KREPA3 ZFm2-TAP similar to what was observed with WT-TAP (data not shown). Thus, these data revealed that both ZFs of KREPA3 are not required for most protein interactions in the editosome, suggesting that the ZFs might function in RNA binding or RNA processing. However, we cannot exclude the possibility that the ZFs might be involved in other unknown protein interactions or the conformation of the entire editosome.
FIG. 7.

Effect of KREPA3 ZF disruption on interaction with the editosome and editing activities in vitro. (A) Western analysis of cell lysates and the complexes copurified with TAP-tagged WT or ZF mutant KREPA3 proteins in the presence or absence of KREPA3 Reg. Lysates of equal numbers of BF KREPA3-RKO, RKO-KREPA3 WT-TAP, ZFm1-TAP, ZFm2-TAP, and ZFm1&2 cells with KREPA3 Reg expressed (E) or repressed (R) for 3 days were purified on IgG beads and eluted by TEV cleavage. The TEV eluates were probed with the antibodies against KREPA1, KREPA2, KREPA3, KREL1, and KREPA6, and the cell lysates were probed with antibodies against KREPA3, KREPA6, and PGK, the latter serving as a control. The 20S positive control was as in Fig. 2B. Roughly similar amounts of editosome proteins were purified with the TAP-tagged KREPA3 ZF mutants whether KREPA3 was expressed or not. In contrast, significant amounts of editosome proteins were purified with the WT-TAP protein only when KREPA3 Reg was repressed. The TEV eluates were used for in vitro precleaved insertion (B) and deletion (C) assays and insertion (D) and deletion (E) endonuclease cleavage assays. The activity assays and designations are as in Fig. 3. The tagged KREPA3 ZF mutant complexes contain all of the in vitro editing activities.
Analysis of glycerol gradient fractions containing primarily TAP-tagged KREPA3 alone (Fig. 6A and B) showed that KREPA3 does not have any editing enzymatic activities. The ∼5 to 10S and the ∼20 to 40S gradient fractions from the RKO-KREPA3 WT-TAP cells with KREPA3 Reg expression repressed (Fig. 6A) were pooled and subjected to Western analysis and in vitro editing assays (Fig. 6B). Western analysis showed that the tagged protein was the most abundant protein detected in the ∼5 to 10S fractions, whereas the ∼20 to 40S fractions also contained KREPA1 and KREL1. Virtually none of the specific editing enzymatic activities were detected in the pooled ∼5 to 10S fractions containing the WT KREPA3-TAP protein. The U-removal products in the ∼5 to 10S fractions are probably due to the nonspecific cleavage by other nucleases since they were also detected in the ∼5 to 10S fractions from KREPA3-RKO cells in the absence of KREPA3 Reg expression (data not shown). As expected, the pooled ∼20 to 40S fractions exhibited all of the in vitro editing activities. Hence, KREPA3 does not appear to catalyze the specific enzymatic steps of editing.
The effects of ZF mutation on in vitro precleaved U-insertion and U-deletion editing (Fig. 7B and C) and on insertion or deletion site endonuclease cleavage (Fig. 7D and E) were assayed using the TEV eluates as shown in Fig. 7A. All of the purified editing complexes had enzymatic activities consistent with the level of editosome proteins copurified with TAP-tagged KREPA3 WT or ZF mutants. In addition, mutation of the KREPA3 ZFs had no direct effect on these activities. Stronger editing activities were observed in the WT-TAP containing complexes when KREPA3 Reg was repressed than when it was expressed, which is due to more incorporation of WT-TAP protein into editosomes upon KREPA3 repression (Fig. 6A and 7A). In contrast, the TEV eluates from the cells with the ZF mutants in the presence of KREPA3 Reg expression had a slightly decreased level of U-insertion activity upon KREPA3 Reg repression, which corresponded to the reduction of KREPA1 protein (Fig. 7A) representing an insertion subcomplex. Overall, the editosomes rescued by the tagged KREPA3 ZF mutants had all the editing activities similar to those rescued by tagged KREPA3 WT protein, indicating that disruption of the ZFs had no effect on the enzymatic activities of editing in vitro. Thus, the KREPA3 ZFs are not critical for the in vitro enzymatic activities or protein interactions in the editosome but are essential for the survival of cells and editing in vivo.
KREPA3 OB-fold deletion abolishes interaction with the editosome.
The effect of deletion of the OB-fold of KREPA3 on the editosome was examined. Tagged KREPA3ΔOB protein was purified from lysates of BF RKO-KREPA3ΔOB-TAP cells in which KREPA3 Reg was expressed or repressed for 3 days using IgG beads, and the protein was eluted by cleavage with TEV protease. Western analysis was performed on the whole-cell lysates (Fig. 8A) and the TEV eluates (Fig. 8B) using the panel of editosome MAbs. KREPA1 and KREPA6 were not detected in whole-cell lysates of KREPA3ΔOB-TAP cells following KREPA3 Reg repression or in TEV eluates whether or not KREPA3 Reg was expressed (Fig. 8A and B). Also, no editosome components were found in the purified KREPA3ΔOB-TAP TEV eluates by LC-MS/MS (data not shown). In addition, the crude mt lysate from the cells in the presence of KREPA3 Reg expression was sedimented on glycerol gradients, and the fractions were subjected to Western analysis using the MAb cocktail (Fig. 8C). KREPA3ΔOB-TAP sedimented at smaller S values than the editosome, indicating that it was not associated with the editosome but was not found at the top of the gradient. This suggested that KREPA3ΔOB-TAP may aggregate or interact with other proteins. Thus, exclusive expression of KREPA3ΔOB not only is incapable of rescuing cell death but also incapable of recovering editosomes following repression of KREPA3 Reg expression. This suggests that the OB-fold, which may function in nucleotide binding, is critical for protein interactions in the editosome.
FIG. 8.

The KREPA3 OB-fold domain is necessary for editosome integrity. Western analysis of purified tagged KREPA3ΔOB protein from BF RKO-KREPA3ΔOB-TAP cells with KREPA3 Reg expressed (E) or repressed (R) for 3 days using IgG beads and eluted by TEV cleavage. (A and B) Whole-cell lysates (A) and TEV eluates (B) were probed with the panel of editing antibodies. KREPA1 and KREPA6 were undetectable in the total cell lysates upon repression of KREPA3 Reg expression. PGK was used as a loading control. After purification, no editosome components were detected in the TEV eluates. (C) Western analysis of gradient fractions of crude mt lysates from. BF RKO-KREPA3ΔOB-TAP cells in the presence of KREPA3 Reg expression. Western analysis was performed with the panel of editing MAbs. KREPA3ΔOB-TAP did not cosediment with the editosome.
DISCUSSION
We show here that KREPA3 is essential for RNA editing and editosome integrity (Fig. 3A and B) but not for the U-addition, U-removal, or RNA ligase catalytic steps of editing in T. brucei (Fig. 3C to F). RNAi-mediated knockdown of KREPA3 expression inhibited growth of PFs, as was observed previously (8, 30), and resulted in a dramatic reduction in the abundance of edited, but not pre-edited mRNAs. KREPA3 knockdown resulted in partial disruption of 20S editosomes into insertion and deletion subcomplexes which have in vitro U-insertion/ligation or U-deletion/ligation activities and which can be separately immunoprecipitated. Editosome endonuclease activities were reduced and only detected in the 20S editosomes that remained (Fig. 3E and F). tet-dependent transcriptional knockdown of a single remaining KREPA3 gene in BFs resulted in almost complete loss of editosomes, loss of edited, but not unedited, mRNAs, and consequently loss of in vitro editing activities. Exclusive expression of TAP-tagged KREPA3 mutated in one or both ZFs or with the OB-fold deleted was lethal, i.e., did not rescue cell growth upon inactivation of WT KREPA3 expression. Exclusive expression of TAP-tagged WT KREPA3 protein rescued cell growth, although at an inhibited level. RNA editing was somewhat inhibited in the tagged WT cells but was dramatically inhibited in cells exclusively expressing mutated or truncated KREPA3 proteins. Tagged KREPA3 proteins with one or both ZFs mutated were incorporated into 20S editosomes that had in vitro U-insertion/ligation, U-deletion/ligation, and insertion and deletion endonuclease activities. However, KREPA3 ZF mutations resulted in editosome destabilization since subcomplexes accumulated upon mutation of ZF2. KREPA3 that lacks the OB-fold was not incorporated into editosomes. Thus, the ZFs are essential in vivo and important for editosome stability, while the OB-fold is essential in vivo and critical for editosome integrity.
The results presented here and elsewhere indicate that while KREPA3 might affect their activity it does not directly catalyze the 3′ TUTase, 3′ exoUase, or ligase steps in editing since subcomplexes devoid of KREPA3 catalyze these activities (30). It is also unlikely that KREPA3 has editing endonuclease activity itself, although reports indicate that recombinant KREPA3 has nuclease activity (8, 36). KREN1, KREN2, and KREN3 are endonucleases as shown by mutation of residues in their RNase III domains which block editing in vivo and eliminate endonucleolytic cleavage by editosomes in vitro (10, 11, 56). Also, high concentrations of a KREN1-CBP fusion protein isolated from insect cell expression showed in vitro specific U-deletion endonuclease activity without the presence of KREPA3 (26). In addition, endonuclease activities that remain following knockdown of KREPA3 expression are only detected in ∼20 to 40S complexes, and their activity levels are proportional to editosome rather than KREPA3 levels (determined by phosphorimager quantitation normalized to KREPA1 and KREPA2 [data not shown]). Similarly, endonuclease activity has only been detected in 20 to 40S editosomes in studies that knock down expression of other editosome components and shift editosomes to lower S values (19, 29, 48). In contrast, mutations of KREPA3 Zn fingers affect editosome structure and/or stability as discussed below, and the remaining editosomes retain the endonuclease and other editing catalytic activities. Nevertheless, KREPA3 might affect, or even be essential for, the editing endonuclease activities. The +1 U insertion and −3 U deletion products are increased upon KREPA3 repression (Fig. 3C and D), implying that KREPA3 might enhance and/or coordinate the activities of the editing enzymes, similar to KREPA1 and KREPA2 (15, 47). These results, along with the reconstitution of endonuclease activity upon supplementation of KREPA3-depleted extracts with recombinant KREPA3 reported by Law et al. (30), suggests that KREPA3 enhances or is needed for endonuclease activity either indirectly through its effect on editosome structure or by direct interaction with the KREN endonucleases. The addition of recombinant KREPA3 might have restored the endonuclease editing activities to KREPA3-depleted extracts by assembling the residual subcomplexes into ∼20S editing complexes.
KREPA3 is essential for editosome structure and stability, since editosomes are disrupted by KREPA3 RNAi knockdown in PFs and lost in BFs upon transcription inactivation. This difference likely reflects a greater reduction of KREPA3 mRNA and protein levels in the latter. The quantitative PCR studies reveal a somewhat greater knockdown of KREPA3 in the BFs than PFs, and KREPA3 protein is faintly detectable in PF but not BF Western blots (Fig. 3B versus 4B and Fig. 4A). In addition, the reduction in edited mRNA was generally greater in the BF than PF knockdowns. Edited cyB mRNA was undetectable in BFs, and edited COII mRNA was reduced much more in BFs than in PFs. This probably reflects the developmental regulation of editing in which there is very little of these two edited mRNAs in BFs (5, 17). Although studies to date have revealed no sedimentation or composition differences between BF and PF editosomes, composition or other editosome differences between the life cycle stages cannot be excluded. Both ZFs are important for ∼20S editosome stability. KREPA3 with mutated ZFs gets incorporated into editosomes, but the abundance of these complexes is diminished and subcomplexes are generated. Exclusive expression of KREPA3 without its OB-fold results in the virtual absence of editosomes. This indicates that proteins that are not incorporated into editosomes are degraded.
The dramatic loss of editosomes upon KREPA3 knockdown reported here mirrors similar results of knockdown of KREPA4, KREPA6, KREPB4, or KREPB5 (4, 29, 46, 54, 58) and suggests that there is a network of interactions among these proteins that is required for editosome stability and perhaps assembly. The loss of some or all of the editosome proteins upon knockdown of one does not appear to be due to changes in mRNA levels for the proteins (58; this study) but may be due to instability or degradation of the editosome proteins when not incorporated into editosomes. As reported elsewhere, yeast two-hybrid analyses showed direct interaction between KREPA3 and KREPA2, KREPA6, and KREPB5 and also showed direct interaction between KREPA6 and KREPA1, KREPA2, KREPA3, and KREPA4 (A. Schnaufer, unpublished data). In addition, we could only detect KREPA6 copurifying with ∼5S TAP-tagged KREPA3 subcomplexes using LC-MS/MS analysis, indicating a high-affinity interaction between KREPA3 and KREPA6 (data not shown). Previous findings showed that KRET2 and KREL2 copurify with tagged KREPA1 and KREX2 and KREL1 copurify with tagged KREPA2 as insertion and deletion editosome subcomplexes, respectively (47). KREPA1 and KREPA2, like KREPA3, each has two ZFs and a C-terminal OB-fold and are critical for the stability of the insertion and deletion subcomplexes, respectively (14, 19, 37). Knockdown of KREPA1 or KREPA2 does not result in dramatic loss of editosomes but rather in a shift to lower S values consistent with loss of the insertion or deletion subcomplexes. Mutation of the ZFs of the KREPA2 (TbMP63) ortholog in Leishmania (LC-4) resulted in disruption of the editosome (25), implying a complex stabilizing role similar to that of the ZFs of KREPA3. The partial KREPA3 knockdown in PFs resulted in KREPA1 insertion subcomplexes with a smaller S value than KREPA2 deletion subcomplexes (Fig. 3B). Correspondingly, KREPA1 RNAi knockdown results in larger (37) subcomplexes, while KREPA2 RNAi knockdowns results in smaller (∼5S) subcomplexes (19). These data together suggest that the deletion subcomplex is more stably associated with more proteins than is the insertion subcomplex, possibly with proteins such as KREPB4 and KREPB5 and/or those in the endonuclease subcomplex.
The KREPA3, KREPA4, KREPA6, and possibly KREPA5 proteins appear to form a structural core of the editosome that bridges the insertion and deletion subcomplexes via association with KREPA1 and KREPA2 (Fig. 1) (29, 46, 47, 54). This core also appears to link to the endonuclease subcomplex via KREPA3 and KREPB5 interaction. The editosome disruption that results from expression of KREPA3 with a C-terminal tag illustrates that KREPA3 might be located in the inner part of the editosome and that the tag may interfere with the normal editosome structure (Fig. 6A). The shift of tagged WT KREPA3 to higher S regions of the gradient upon repression of untagged KREPA3 indicates that it can integrate into editosomes but that it is at a competitive disadvantage with the untagged KREPA3. The TAP tag inhibited cell growth (Fig. 5B), but its replacement with a short c-myc tag resulted in cells that grew at a normal rate (data not shown), suggesting that the effect of the tag was on structure. Despite these effects, tagged KREPA3 with or without mutated ZFs can integrate into editosomes, while elimination of its OB-fold not only abolished this integration but also its interaction with KREPA1, KREPA2, and KREPA6 (Fig. 8A and B). Indeed, no editosome proteins were found by LC-MS/MS to be associated with purified TAP-tagged KREPA3ΔOB (data not shown). Thus, the OB-fold is critical for KREPA3's interaction with other editosome proteins. As will be reported elsewhere, KREPA3, KREPA4, KREPA6, and perhaps the less-studied KREPA5, form a network of interactions that involve the OB-folds (A. Schnaufer, unpublished data). Hence, as illustrated by the effect of expression of KREPA3 from which the OB-fold is deleted, the OB-folds of these proteins appear to function in protein-protein interaction. However, this does not preclude the possibility that the OB-fold domain of KREPA3 and the other KREPA proteins might also affect substrate recognition, binding, and perhaps translocation, as was suggested for KREPA1 (TbMP81) and KREPA2 (TbMP63) (47, 62).
Editosomes with mutated KREPA3 ZFs, while less stable, are capable of catalyzing the endonuclease, 3′ TUTase, 3′ exoUase, and RNA ligase steps of editing in vitro. Overexpression of the TAP-tagged ZF mutants of KREPA1, KREPA2, and KREPA3 in PFs also did not exhibit a dominant-negative effect (data not shown). Nevertheless, the KREPA3 ZF mutations block editing in vivo and are lethal when exclusively expressed. Since KREPA3 binds RNA (8), a likely possibility is that the ZFs function in RNA recognition and binding. C2H2 ZFs in general can nonspecifically bind the dsRNA helix backbone and can specifically recognize individual bases that bulge out of a structurally rigid element (51). Hence, the KREPA3 ZFs may bind the gRNA/pre-mRNA duplex in a sequence-nonspecific manner or via some conserved secondary structure. They may also function in editing processivity. The roles of the two ZFs may differ since mutation of the first ZF had a greater effect on the amount of the tagged protein and editosome stability, although mutation of either one inhibited growth. They may function along with the two ZFs in KREPA1 and KREPA2 and the total of eight U1-like Zn fingers in the three KREN and five KREPB proteins to shuttle the substrate RNA among editosome catalytic sites to accomplish editing of the multiple sites, including both insertion and deletion sites.
Acknowledgments
This study was supported by NIH grant AI014012 to K.D.S. Research was conducted using equipment made possible by the Economic Development Administration-U.S. Department of Commerce and the M. J. Murdock Charitable Trust.
We thank J. Carnes for help with real-time PCR, A. Schnaufer for advice on cell line construction, and M. Parsons for MAb66. We also thank the members of Stuart laboratory for extensive and helpful discussion.
Footnotes
Published ahead of print on 15 September 2008.
REFERENCES
- 1.Agrawal, V., and K. V. Kishan. 2003. OB-fold: growing bigger with functional consistency. Curr. Protein Pept. Sci. 4195-206. [DOI] [PubMed] [Google Scholar]
- 2.Aphasizhev, R., I. Aphasizheva, R. E. Nelson, and L. Simpson. 2003. A 100-kD complex of two RNA-binding proteins from mitochondria of Leishmania tarentolae catalyzes RNA annealing and interacts with several RNA editing components. RNA 962-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aphasizhev, R., I. Aphasizheva, and L. Simpson. 2003. A tale of two TUTases. Proc. Natl. Acad. Sci. USA 10010617-10622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Babbarwal, V. K., M. Fleck, N. L. Ernst, A. Schnaufer, and K. D. Stuart. 2007. An essential role of KREPB4 in RNA editing and structural integrity of the editosome in Trypanosoma brucei. RNA 13737-744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Besteiro, S., M. P. Barrett, L. Riviere, and F. Bringaud. 2005. Energy generation in insect stages of Trypanosoma brucei: metabolism in flux. Trends Parasitol. 21185-191. [DOI] [PubMed] [Google Scholar]
- 6.Blum, B., N. Bakalara, and L. Simpson. 1990. A model for RNA editing in kinetoplastid mitochondria: “guide” RNA molecules transcribed from maxicircle DNA provide the edited information. Cell 60189-198. [DOI] [PubMed] [Google Scholar]
- 7.Brayer, K. J., and D. J. Segal. 2008. Keep your fingers off my DNA: protein-protein interactions mediated by C2H2 zinc finger domains. Cell Biochem. Biophys. 50111-131. [DOI] [PubMed] [Google Scholar]
- 8.Brecht, M., M. Niemann, E. Schlüter, U. F. Müller, K. Stuart, and H. U. Göringer. 2005. TbMP42, a protein component of the RNA editing complex in African trypanosomes has endo-exoribonuclease activity. Mol. Cell 17621-630. [DOI] [PubMed] [Google Scholar]
- 9.Carnes, J., and K. Stuart. 2007. Working together: the RNA editing machinery in Trypanosoma brucei, p. 143-164. In H. U. Göringer (ed.), RNA editing, vol. 20. Springer Verlag, Heidelberg, Germany. [Google Scholar]
- 10.Carnes, J., J. R. Trotter, N. L. Ernst, A. G. Steinberg, and K. Stuart. 2005. An essential RNase III insertion editing endonuclease in Trypanosoma brucei. Proc. Natl. Acad. Sci. USA 10216614-16619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carnes, J., J. R. Trotter, A. Peltan, M. Fleck, and K. Stuart. 2008. RNA editing in Trypanosoma brucei requires three different editosomes. Mol. Cell. Biol. 28122-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cruz-Reyes, J., and B. Sollner-Webb. 1996. Trypanosome U-deletional RNA editing involves guide RNA-directed endonuclease cleavage, terminal U exonuclease, and RNA ligase activities. Proc. Natl. Acad. Sci. USA 938901-8906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cruz-Reyes, J., A. Zhelonkina, L. Rusché, and B. Sollner-Webb. 2001. Trypanosome RNA editing: simple guide RNA features enhance U deletion 100-fold. Mol. Cell. Biol. 21884-892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drozdz, M., S. S. Palazzo, R. Salavati, J. O'Rear, C. Clayton, and K. Stuart. 2002. TbMP81 is required for RNA editing in Trypanosoma brucei. EMBO J. 211791-1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ernst, N. L., B. Panicucci, R. P. Igo, Jr., A. K. Panigrahi, R. Salavati, and K. Stuart. 2003. TbMP57 is a 3′ terminal uridylyl transferase (TUTase) of the Trypanosoma brucei editosome. Mol. Cell 111525-1536. [DOI] [PubMed] [Google Scholar]
- 16.Gao, G., and L. Simpson. 2003. Is the Trypanosoma brucei REL1 RNA ligase specific for U-deletion RNA editing, and is the REL2 RNA ligase specific for U-insertion editing? J. Biol. Chem. 27827570-27574. [DOI] [PubMed] [Google Scholar]
- 17.Hannaert, V., F. Bringaud, F. R. Opperdoes, and P. A. Michels. 2003. Evolution of energy metabolism and its compartmentation in Kinetoplastida. Kinetoplastid Biol. Dis. 211-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harris, M. E., D. R. Moore, and S. L. Hajduk. 1990. Addition of uridines to edited RNAs in trypanosome mitochondria occurs independently of transcription. J. Biol. Chem. 26511368-11376. [PubMed] [Google Scholar]
- 19.Huang, C. E., S. F. O'Hearn, and B. Sollner-Webb. 2002. Assembly and function of the RNA editing complex in Trypanosoma brucei requires band III protein. Mol. Cell. Biol. 22:3194-3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Igo, R. P., Jr., S. S. Palazzo, M. L. K. Burgess, A. K. Panigrahi, and K. Stuart. 2000. Uridylate addition and RNA ligation contribute to the specificity of kinteoplastid insertion RNA editing. Mol. Cell. Biol. 208447-8457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Igo, R. P., Jr., D. S. Weston, N. L. Ernst, A. K. Panigrahi, R. Salavati, and K. Stuart. 2002. Role of uridylate-specific exoribonuclease activity in Trypanosoma brucei RNA editing. Eukaryot. Cell 1112-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iuchi, S. 2001. Three classes of C2H2 zinc finger proteins. Cell Mol. Life Sci. 58625-635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jensen, B. C., C. T. Kifer, D. L. Brekken, A. C. Randall, Q. Wang, B. L. Drees, and M. Parsons. 2006. Characterization of protein kinase CK2 from Trypanosoma brucei. Mol. Biochem. Parasitol. 15128-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kable, M. L., S. D. Seiwert, S. Heidmann, and K. Stuart. 1996. RNA editing: a mechanism for gRNA-specified uridylate insertion into precursor mRNA. Science 2731189-1195. [DOI] [PubMed] [Google Scholar]
- 25.Kang, X., A. M. Falick, R. E. Nelson, G. Gao, K. Rogers, R. Aphasizhev, and L. Simpson. 2004. Disruption of the zinc finger motifs in the Leishmania tarentolae LC-4 (=TbMP63) L-complex editing protein affects the stability of the L-complex. J. Biol. Chem. 2793893-3899. [DOI] [PubMed] [Google Scholar]
- 26.Kang, X., G. Gao, K. Rogers, A. M. Falick, S. Zhou, and L. Simpson. 2006. Reconstitution of full-round uridine-deletion RNA editing with three recombinant proteins. Proc. Natl. Acad. Sci. USA 10313944-13949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kang, X., K. Rogers, G. Gao, A. M. Falick, S. Zhou, and L. Simpson. 2005. Reconstitution of uridine-deletion precleaved RNA editing with two recombinant enzymes. Proc. Natl. Acad. Sci. USA 1021017-1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Law, J. A., C. E. Huang, S. F. O'Hearn, and B. Sollner-Webb. 2005. In Trypanosoma brucei RNA editing, band II enables recognition specifically at each step of the U insertion cycle. Mol. Cell. Biol. 25:2785-2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Law, J. A., S. O'Hearn, and B. Sollner-Webb. 2007. In Trypanosoma brucei RNA editing, TbMP18 (band VII) is critical for editosome integrity and for both insertional and deletional cleavages. Mol. Cell. Biol. 27:777-787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Law, J. A., S. F. O'Hearn, and B. Sollner-Webb. 2008. Trypanosoma brucei RNA editing protein TbMP42 (band VI) is crucial for the endonucleolytic cleavages but not the subsequent steps of U-deletion and U-insertion. RNA. 14:1187-1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu, D., M. A. Searles, and A. Klug. 2003. Crystal structure of a zinc-finger-RNA complex reveals two modes of molecular recognition. Nature 42696-100. [DOI] [PubMed] [Google Scholar]
- 32.Madison-Antenucci, S., J. Grams, and S. L. Hajduk. 2002. Editing machines: the complexities of trypanosome RNA editing. Cell 108435-438. [DOI] [PubMed] [Google Scholar]
- 33.Miller, M. M., K. Halbig, J. Cruz-Reyes, and L. K. Read. 2006. RBP16 stimulates trypanosome RNA editing in vitro at an early step in the editing reaction. RNA 121292-1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muller, U. F., L. Lambert, and H. U. Goringer. 2001. Annealing of RNA editing substrates facilitated by guide RNA-binding protein gBP21. EMBO J. 201394-1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murzin, A. G. 1993. OB(oligonucleotide/oligosaccharide binding)-fold: common structural and functional solution for non-homologous sequences. EMBO J. 12861-867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Niemann, M., M. Brecht, E. Schluter, K. Weitzel, M. Zacharias, and H. U. Goringer. 2008. TbMP42 is a structure-sensitive ribonuclease that likely follows a metal ion catalysis mechanism. Nucleic Acids Res. 364465-4473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O'Hearn, S., C. E. Huang, M. Hemann, A. Zhelonkina, and B. Sollner-Webb. 2003. Trypanosoma brucei RNA editing complex: band II is structurally critical and maintains band V ligase, which is nonessential. Mol. Cell. Biol. 237909-7919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Panigrahi, A. K., N. L. Ernst, G. J. Domingo, M. Fleck, R. Salavati, and K. D. Stuart. 2006. Compositionally and functionally distinct editosomes in Trypanosoma brucei. RNA 121038-1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Panigrahi, A. K., A. Schnaufer, N. Carmean, R. P. Igo, Jr., S. P. Gygi, N. L. Ernst, S. S. Palazzo, D. S. Weston, R. Aebersold, R. Salavati, and K. D. Stuart. 2001. Four related proteins of the Trypanosoma brucei RNA editing complex. Mol. Cell. Biol. 216833-6840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Panigrahi, A. K., A. Schnaufer, N. L. Ernst, B. Wang, N. Carmean, R. Salavati, and K. Stuart. 2003. Identification of novel components of Trypanosoma brucei editosomes. RNA 9484-492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Parker, H. L., T. Hill, K. Alexander, N. B. Murphy, W. R. Fish, and M. Parsons. 1995. Three genes and two isozymes: gene conversion and the compartmentalization and expression of the phosphoglycerate kinases of Trypanosoma (Nannomonas) congolense. Mol. Biochem. Parasitol. 69269-279. [DOI] [PubMed] [Google Scholar]
- 42.Pelletier, M., and L. K. Read. 2003. RBP16 is a multifunctional gene regulatory protein involved in editing and stabilization of specific mitochondrial mRNAs in Trypanosoma brucei. RNA 9457-468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pfaffl, M. W. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ramakers, C., J. M. Ruijter, R. H. L. Deprez, and A. F. M. Moorman. 2003. Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci. Lett. 33962-66. [DOI] [PubMed] [Google Scholar]
- 45.Rogers, K., G. Gao, and L. Simpson. 2007. Uridylate-specific 3′-5′ exoribonucleases involved in uridylate-deletion RNA editing in trypanosomatid mitochondria. J. Biol. Chem. 28229073-29080. [DOI] [PubMed] [Google Scholar]
- 46.Salavati, R., N. L. Ernst, J. O'Rear, T. Gilliam, S. Tarun, Jr., and K. Stuart. 2006. KREPA4, an RNA binding protein essential for editosome integrity and survival of Trypanosoma brucei. RNA 12:819-831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schnaufer, A., N. Ernst, J. O'Rear, R. Salavati, and K. Stuart. 2003. Separate insertion and deletion sub-complexes of the Trypanosoma brucei RNA editing complex. Mol. Cell 12307-319. [DOI] [PubMed] [Google Scholar]
- 48.Schnaufer, A., A. K. Panigrahi, B. Panicucci, R. P. Igo, Jr., R. Salavati, and K. Stuart. 2001. An RNA ligase essential for RNA editing and survival of the bloodstream form of Trypanosoma brucei. Science 2912159-2162. [DOI] [PubMed] [Google Scholar]
- 49.Seiwert, S. D., S. Heidmann, and K. Stuart. 1996. Direct visualization of uridylate deletion in vitro suggests a mechanism for kinetoplastid RNA editing. Cell 84831-841. [DOI] [PubMed] [Google Scholar]
- 50.Simpson, L., R. Aphasizhev, G. Gao, and X. Kang. 2004. Mitochondrial proteins and complexes in Leishmania and Trypanosoma involved in U-insertion/deletion RNA editing. RNA. 10159-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stefl, R., L. Skrisovska, and F. H. Allain. 2005. RNA sequence- and shape-dependent recognition by proteins in the ribonucleoprotein particle. EMBO Rep. 633-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stuart, K., T. E. Allen, S. Heidmann, and S. D. Seiwert. 1997. RNA editing in kinetoplastid protozoa. Microbiol. Mol. Biol. Rev. 61105-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stuart, K. D., A. Schnaufer, N. L. Ernst, and A. K. Panigrahi. 2005. Complex management: RNA editing in trypanosomes. Trends Biochem. Sci. 3097-105. [DOI] [PubMed] [Google Scholar]
- 54.Tarun, S. Z., Jr., A. Schnaufer, N. L. Ernst, R. Proff, J. Deng, W. Hol, and K. Stuart. 2008. KREPA6 is an RNA binding protein essential for editosome integrity and survival of Trypanosoma brucei. RNA 14347-358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Theobald, D. L., R. M. Mitton-Fry, and D. S. Wuttke. 2003. Nucleic acid recognition by OB-fold proteins. Annu. Rev. Biophys. Biomol. Struct. 32115-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Trotter, J. R., N. L. Ernst, J. Carnes, B. Panicucci, and K. Stuart. 2005. A deletion site editing endonuclease in Trypanosoma brucei. Mol. Cell 20403-412. [DOI] [PubMed] [Google Scholar]
- 57.Vanhamme, L., D. Perez-Morga, C. Marchal, D. Speijer, L. Lambert, M. Geuskens, S. Alexandre, N. Ismaïli, U. Göringer, R. Benne, and E. Pays. 1998. Trypanosoma brucei TBRGG1, a mitochondrial oligo(U)-binding protein that colocalizes with an in vitro RNA editing activity. J. Biol. Chem. 273:21825-21833. [DOI] [PubMed] [Google Scholar]
- 58.Wang, B., N. L. Ernst, S. S. Palazzo, A. K. Panigrahi, R. Salavati, and K. Stuart. 2003. TbMP44 is essential for RNA editing and structural integrity of the editosome in Trypanosoma brucei. Eukaryot. Cell 2578-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang, Z., J. C. Morris, M. E. Drew, and P. T. Englund. 2000. Inhibition of Trypanosoma brucei gene expression by RNA interference using an integratable vector with opposing T7 promoters. J. Biol. Chem. 27540174-40179. [DOI] [PubMed] [Google Scholar]
- 60.Wirtz, E., S. Leal, C. Ochatt, and G. A. M. Cross. 1999. A tightly regulated inducible expression system for conditional gene knockouts and dominant-negative genetics in Trypanosoma brucei. Mol. Biochem. Parasitol. 9989-101. [DOI] [PubMed] [Google Scholar]
- 61.Wolfe, S. A., L. Nekludova, and C. O. Pabo. 2000. DNA recognition by Cys2His2 zinc finger proteins. Annu. Rev. Biophys. Biomol. Struct. 29183-212. [DOI] [PubMed] [Google Scholar]
- 62.Worthey, E. A., A. Schnaufer, I. S. Mian, K. Stuart, and R. Salavati. 2003. Comparative analysis of editosome proteins in trypanosomatids. Nucleic Acids Res. 316392-6408. [DOI] [PMC free article] [PubMed] [Google Scholar]