

Abstract
Selection of the AUG start codon for translation in eukaryotes is governed by codon-anticodon interactions between the initiator Met-tRNAiMet and the mRNA. Translation initiation factor 2 (eIF2) binds Met-tRNAiMet to the 40S ribosomal subunit, and previous studies identified Sui− mutations in eIF2 that enhanced initiation from a noncanonical UUG codon, presumably by impairing Met-tRNAiMet binding. Consistently, an eIF2γ-N135D GTP-binding domain mutation impairs Met-tRNAiMet binding and causes a Sui− phenotype. Intragenic A208V and A382V suppressor mutations restore Met-tRNAiMet binding affinity and cell growth; however, only A208V suppresses the Sui− phenotype associated with the eIF2γ-N135D mutation. An eIF2γ-A219T mutation impairs Met-tRNAiMet binding but unexpectedly enhances the fidelity of initiation, suppressing the Sui− phenotype associated with the eIF2γ-N135D,A382V mutant. Overexpression of eIF1, which is thought to monitor codon-anticodon interactions during translation initiation, likewise suppresses the Sui− phenotype of the eIF2γ mutants. We propose that structural alterations in eIF2γ subtly alter the conformation of Met-tRNAiMet on the 40S subunit and thereby affect the fidelity of start codon recognition independent of Met-tRNAiMet binding affinity.
A key fidelity step in protein synthesis is the selection of the translation start site on an mRNA. Ribosomes bind to the mRNA near the 5′ cap and scan in a 3′ direction in search of a start codon. Selection of the AUG start site for translation initiation in eukaryotes is dependent on the base-pairing interaction between the AUG codon on the mRNA and the CAU anticodon of the initiator Met-tRNA, which is bound to the ribosome. In addition several eukaryotic translation initiation factors (eIFs), including eIF1, eIF1A, eIF2, eIF3, and eIF5, play important roles in start site selection. The binding of the Met-tRNAiMet to the P site of the 40S ribosomal subunit is facilitated by the factor eIF2 (12, 13). The heterotrimeric eIF2 complex, consisting of the core γ subunit, which contains separate binding sites for the α- and β-subunits, binds the initiator Met-tRNAiMet in a GTP-dependent manner, forming a ternary complex (TC). The eIF2γ and its archaeal homolog, aIF2γ, are structurally analogous to EF-Tu (in eukaryotes, eEF1A) and consist of three distinct domains: a GTP-binding domain (G domain), domain II, and domain III (Fig. 1A). Like the tRNA-binding pocket in EF-Tu, the β-barrel domains II and III pack against the G domain in aIF2γ to form the Met-tRNAiMet binding pocket, where the 3′ (aminoacyl) end of the Met-tRNAiMet directly contacts domain II (24, 25).
FIG. 1.

Growth and Gcd− and Sui− phenotypes associated with the N135D mutation in Switch I of eIF2γ and its intragenic suppressors. (A) Schematic of eIF2γ. eIF2γ is divided into three domains based on sequence homology with structurally characterized EF-Tu and aIF2γ. The locations of conserved G domain sequence motifs G1/P-loop, G2, G3/DXXG, and G4/NKXD are indicated along with the locations of the structurally important and nucleotide-sensitive Switch I and Switch II elements. Above the schematic are sequence alignments around the sites of the eIF2γ-N135D mutation in Switch I and the suppressor mutations T115A, A208V, A219T, and A382V. The mutated residues in yeast (S. cerevisiae) eIF2γ are depicted in bold and aligned with the corresponding sequences from archaeal (Methanococcus jannaschii and Pyrococcus abyssi) aIF2γ, human eIF2γ, and Escherichia coli translation elongation GTPase EF-Tu. (B) Growth rate analysis of yeast expressing WT and mutant forms of eIF2γ. Derivatives of yeast strain J292 expressing the indicated WT or mutant form of eIF2γ from low-copy-number plasmids and carrying either a high-copy-number (H.C.) IMT4 plasmid expressing tRNAiMet (+; H.C. tRNAiMet; pC1683 [24]), or an empty vector (-) were grown to saturation, and 4-μl volumes of serial dilutions (with optical densities at 600 nm of 1.0, 0.1, 0.01, 0.001, and 0.0001) were spotted on minimal medium with essential nutrients (SD) and incubated for 3 days at 30°C. The doubling times during exponential growth in liquid SD medium are shown in parentheses. (C) Gcd− phenotypes. A WT GCN4-lacZ reporter construct (p180) was introduced into the WT and mutant eIF2γ strains described for panel A and carrying either an empty high-copy-number vector or a high-copy-number plasmid expressing tRNAiMet . Transformants were grown to an A600 of 0.7 in SD medium, whole-cell extracts were prepared, and β-galactosidase activity (nmol of o-nitrophenyl-β-d-galactopyranoside cleaved per min per mg) was measured for three independent transformants. Means and standard errors are plotted. For the H.C. vector data (black bars), P was <0.0002 for WT versus N135D and for N135D versus N135D,A208V or N135D,A219T; P was <0.003 for N135D versus A219T or N135D,A382V. (D) Sui− phenotypes. HIS4AUG-lacZ or his4UUG-LacZ reporters were introduced into the strains described for panel C. β-Galactosidase activities were measured from one to four independent transformants and were used to calculate the mean UUG/AUG ratio ± the standard error. White and black bars denote transformants carrying a high-copy-number IMT4 plasmid and empty vector, respectively; gray bars denote transformants carrying neither the high-copy-number plasmid nor vector. For H.C. vector data (black bars), P was <0.003 for WT versus N135D, for N135D versus A219T, and for N135D versus N135D,A208V or N135D,A219T or N135D,A219T,A382V; P was ∼0.09 for N135D versus N135D,A382V.
The G domain of eIF2γ/aIF2γ, like the G domains of Ras and related small GTPases, the translational GTPases, and the heterotrimeric G proteins, contains Switch I (marked by a conserved Thr residue in the G2 sequence motif) and Switch II (containing the G3 Asp-X-X-Gly sequence motif) elements (Fig. 1A) that directly contact the bound nucleotide and undergo structural rearrangements in response to the presence of GTP versus GDP (20, 23-25, 27, 30). In EF-Tu the Switch elements undergo dramatic structural rearrangements, including the refolding of Switch I from a β-hairpin structure to a short α-helix upon GTP binding. This movement of the Switch elements in EF-Tu upon GTP binding triggers the reorientation of domains II and III from an open to a closed structure and formation of the tRNA-binding pocket (20, 23). In contrast, the three domains of aIF2γ are closely packed in the closed conformation in the presence of GDP or GTP (25, 32). As GTP binding to eIF2 is essential for Met-tRNAiMet binding, it is believed that the modest structural changes in the two Switch elements of eIF2γ upon GTP binding enable formation of the Met-tRNAiMet-binding pocket.
The eIF2 plays a key role in the early steps of translation initiation and interacts with other translation factors to form critical preinitiation complexes. The eIF2 binds Met-tRNAiMet to the P site of the 40S ribosomal subunit, and binding of the factors eIF1, eIF1A, eIF3, and eIF5 generates a 43S preinitiation complex (12, 13). This 43S complex is recruited to the 5′ end of an mRNA and then scans along the mRNA in a 3′ direction until the anticodon of the Met-tRNAiMet base pairs with an AUG codon. The factor eIF5 interacts with the G domain of eIF2γ to trigger GTP hydrolysis to GDP+Pi, and the factor eIF1 is positioned near the P site of the 48S complex and is thought to monitor the interaction between Met-tRNAiMet and the AUG codon (1, 2, 4, 17, 18, 21). Repositioning of eIF1 upon proper codon-anticodon interaction converts the 43S complex from a scanning-competent “open” state to a “closed” nonscanning state and is coupled with the release of Pi from eIF2 (4, 18, 21, 22). Following dissociation of the eIF2-GDP binary complex from the 48S complex and joining of the 60S ribosomal subunit, the resulting 80S monosome enters the elongation phase of protein synthesis. Notably, like many but not all G proteins, eIF2γ binds GDP with higher affinity than GTP (16). In order for eIF2 to function in additional rounds of translation initiation, eIF2-GDP must be recycled to eIF2-GTP in a reaction accelerated by the guanine nucleotide exchange factor eIF2B (13).
Two in vivo assays have been used to study eIF2γ function in the yeast Saccharomyces cerevisiae. The first assay monitors the translation of the GCN4 mRNA, which encodes a transcriptional activator of amino acid biosynthetic enzyme genes (14). The expression of GCN4 is controlled by the abundance of eIF2 TCs. Four short open reading frames (ORFs) in the GCN4 mRNA leader restrict the flow of scanning ribosomes to the GCN4 ORF. In wild-type (WT) cells, phosphorylation of eIF2 by the kinase GCN2 inhibits the activity of eIF2B and results in a reduction of TC levels. This lowering of TC abundance enables ribosomes scanning the GCN4 mRNA leader following translation of uORF1 to bypass the inhibitory uORFs 2 to 4 and reinitiate translation at the GCN4 AUG codon. In contrast, GCN4 expression is constitutively repressed in yeast lacking GCN2 (14). However, mutations in eIF2 that impair TC formation, for example, by lowering eIF2 abundance or decreasing the affinity of eIF2 for Met-tRNAiMet, constitutively derepress GCN4 expression in the absence of GCN2 function (Gcd− phenotype) (9, 14, 31). Thus, the eIF2γ-N135K mutation impairs Met-tRNAiMet binding (15), induces the expression of a GCN4-lacZ reporter gene, and enables gcn2Δ cells to grow on medium containing 3-aminotriazole, an inhibitor of histidine biosynthesis.
The second assay monitors the effects of eIF2γ mutations on start codon recognition at the HIS4 gene in yeast. Donahue and colleagues identified yeast mutants that bypassed the requirement for the normal HIS4 gene AUG start codon and were able to grow on medium lacking histidine. Analysis of these mutants revealed that alterations in the eIF2α (SUI2), eIF2β (SUI3), eIF1 (SUI1), eIF5 (TIF5), and eIF2γ (GCD11/SUI4) genes enabled translation to initiate at the UUG codon encoding the third amino acid in the HIS4 protein (3, 5, 7, 8, 15, 33). Biochemical analysis of the eIF2γ-N135K mutant revealed a defect in Met-tRNAiMet binding (15). This defect was not due to impaired GTP binding, as might have been expected due to the location of the mutation in the Switch I element of the G domain. Rather, the eIF2γ-N135K mutation increased the dissociation rate of Met-tRNAiMet from the active GTP-bound eIF2 complex (15). Additional biochemical analyses, including studies on the eIF2β and eIF5 mutant factors, led to the proposal that mutations that weaken Met-tRNAiMet binding to eIF2 or mutations that accelerate GTP hydrolysis by eIF2 induce premature release of Met-tRNAiMet from eIF2 on the scanning 40S subunit and enable ribosomes to initiate translation from the non-AUG codon in the HIS4 mRNA (15).
Supporting this idea, Hannig and coworkers' examination of spontaneous Gcd− mutants of GCD11 revealed a Sui− phenotype in strains expressing eIF2γ-R510H (9). Intriguingly, there was no correlation between the Gcd− and Sui− phenotypes associated with the various eIF2γ mutants. Several GCD11 mutants strongly derepressed GCN4 expression, yet they did not enhance expression of a HIS4 reporter containing a UUG start codon. In contrast, the gcd11-R510H mutant moderately derepressed GCN4 expression, and it strongly stimulated translation of the mutant his4UUG-lacZ reporter (9). As the R510H mutation maps to the tRNA-binding domain III of eIF2γ, this mutation has been proposed to weaken Met-tRNAiMet binding to eIF2. These findings reinforce the notion that impaired binding of Met-tRNAiMet to eIF2 enables release of eIF2 from Met-tRNAiMet on the scanning 40S ribosome in the absence of AUG codon recognition and thus allows translation initiation at a noncognate UUG codon.
To further examine the role of eIF2γ and its Switch elements in translation initiation and, more specifically, in start site selection, we first generated the Switch I eIF2γ-N135D mutant. This mutation significantly impaired yeast cell growth and conferred a Sui− phenotype as previously reported for the lethal eIF2γ-N135K mutation (15). In addition, biochemical and molecular analyses revealed that the eIF2γ-N135D mutation impaired Met-tRNAiMet binding. Next, we screened for intragenic suppressors of the growth defect of the eIF2γ-N135D strain. Interestingly, an A382V suppressor mutation restored wild-type Met-tRNAiMet binding but did not rescue the Sui− phenotype. This novel Sui− mutation in eIF2γ that does not impair Met-tRNAiMet binding, combined with the suppression of the Sui− phenotype by overexpression of eIF1, leads us to propose that the conformation of Met-tRNAiMet in the eIF2 TC and on the scanning ribosome plays a critical role in selecting the translation start site in a manner governed by eIF1.
MATERIALS AND METHODS
Detailed descriptions of plasmid and yeast strain constructions and details of the suppressor screening are presented in the supplemental material.
Protein purification and biochemical assays.
For purification of eIF2, yeast strain J293 was cotransformed with a high-copy-number LEU2 plasmid expressing His8-GCD11 (wild type or mutant) and high-copy-number pC2887 expressing eIF2α, eIF2β, and tRNAiMet . Six-liter cultures of the transformants were grown in synthetic complete (SC)-His-Leu medium to an A600 of 2.0, and the cells were harvested, flash frozen in liquid nitrogen, and then broken using a micromill. Protein purification using Ni2+-affinity chromatography was performed as described previously (16).
Nitrocellulose filter binding assays using [3H]GDP (11.5 Ci/mmol; 1 mCi/ml) or [3H]GTP (7.5 Ci/mmol; 0.5 mCi/ml) and purified eIF2 were performed as described previously (16). Initiator tRNAiMet was prepared by in vitro transcription, aminoacylated using [3H]Met (84.0 Ci/mmol; 5 mCi/ml), and  binding to eIF2 was assessed using filter binding assays as described previously (16). For analysis of eIF2 GTPase activity, eIF2 ternary complexes were formed in the presence of [γ-33P]GTP (3,000 Ci/mmol; 10 mCi/ml) and Met-tRNAiMet . Ternary complexes were mixed with eIF1, eIF1A, eIF5, 40S subunits, and AUG codon, and the rate of GTP hydrolysis was assessed as described elsewhere (2).
binding to eIF2 was assessed using filter binding assays as described previously (16). For analysis of eIF2 GTPase activity, eIF2 ternary complexes were formed in the presence of [γ-33P]GTP (3,000 Ci/mmol; 10 mCi/ml) and Met-tRNAiMet . Ternary complexes were mixed with eIF1, eIF1A, eIF5, 40S subunits, and AUG codon, and the rate of GTP hydrolysis was assessed as described elsewhere (2).
Assays of β-galactosidase activity in whole-cell extracts from strains containing the GCN4-lacZ reporter plasmid p180, or the HIS4AUG-lacZ (p367) or his4UUG-lacZ (p391) reporter plasmids (a kind gift from Tom Donahue, Indiana University), were performed as described previously (19).
RESULTS
The eIF2γ-N135D mutation impairs Met-tRNAiMet binding, derepresses GCN4 expression, and enhances initiation at a UUG codon.
The Thr137 residue in eIF2γ, part of the conserved G2 sequence motif (Fig. 1A), corresponds to the universally conserved Switch I Thr found in all G proteins. Via its backbone, this Thr directly contacts the γ-phosphate of GTP, and through its side chain it helps coordinate the Mg2+ ion required for nucleotide binding (27, 30). To examine the role of the Switch I element in eIF2γ, we mutated Asn135, located two residues before the Thr137 in Switch I, to Lys (N135K), Asp (N135D), and Ala (N135A). Introduction of plasmids encoding these mutant forms of eIF2γ in place of a plasmid encoding the WT gene in the gcd11Δ strain J212 resulted in growth defects. Cells expressing eIF2γ-N135D exhibited a marked slow-growth phenotype (Fig. 1B, compare rows 1 and 3), whereas the N135A mutation slightly impaired cell growth rates (data not shown). As observed previously (15), the eIF2γ-N135K (SUI4) mutant was recessive lethal (data not shown). Western analyses revealed that eIF2γ-N135D was expressed at levels comparable to WT eIF2γ (Fig. 2A, compare lanes 1 to 4). Moreover, overexpression of eIF2γ-N135D from a high-copy-number plasmid resulted in a much more severe slow-growth phenotype (see Fig. S2, right panel, in the supplemental material). These results indicate that the Switch I mutation in eIF2γ-N135D impairs cell growth by altering eIF2 function and not by lowering eIF2 protein levels in the cell.
FIG. 2.

Analysis of eIF2γ expression in vivo and purification of WT and mutant eIF2 complexes. (A) Western blot analysis of eIF2γ expression. Whole-cell extracts (20 and 40 μg) of strains described in Fig. 1B were subjected to immunoblot analysis using anti-yeast eIF2γ (upper panel, gray arrowhead) or anti-yeast eIF2Bɛ (GCD6) antiserum (lower panel, open arrowhead). Immune complexes were visualized using enhanced chemiluminescence, and the amount of eIF2γ was quantified relative to the eIF2Bɛ loading control. The amount of the eIF2γ mutants relative to WT (100%) is summarized above the lanes. (B) Purification of eIF2 complexes containing WT or mutant eIF2γ subunits. eIF2 complexes were purified from strains overexpressing eIF2α, eIF2β, eIF2γ, and His8-eIF2γ (WT or mutant) as described in Materials and Methods. Two different amounts (0.5 and 2 μg) of the eIF2 complexes were resolved by 4 to 20% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and visualized by staining with GelCode blue. The positions of the eIF2γ subunit (gray arrowhead) and eIF2α and eIF2β subunits (open arrowhead, doublet) are indicated.
The previous studies on the eIF2γ-N135K mutant revealed a defect in Met-tRNAiMet binding and suggested altered GTPase properties for the mutant factor (15). To examine the biochemical properties of the eIF2 complex containing eIF2γ-N135D, we purified WT and mutant eIF2 complexes from strains overexpressing eIF2α, eIF2β, and His-tagged versions of WT or mutant eIF2γ. As shown in Fig. 2B (lanes 1 to 4), the WT and mutant eIF2 complexes were purified to near homogeneity. As the N135D mutation is in the Switch I element, the mutation may affect guanine nucleotide binding or hydrolysis. Nitrocellulose filter binding assays were used to monitor the binding of GTP and GDP to the eIF2 complexes. As observed previously (16), WT eIF2 bound GTP with a Kd of ∼2 μM, which corresponds to an ∼100-fold-lower affinity than for GDP (Kd, ∼26 nM) (Table 1; see also Fig. S3 and S4 in the supplemental material). The eIF2γ-N135D mutation did not significantly alter the affinity for GTP (Kd, ∼1.6 μM) or GDP (Kd, ∼30 nM). To examine the impact of the N135D mutation on eIF2 GTPase activity, WT or mutant TCs were formed in the presence of [γ-33P]GTP and then incubated with 40S subunits, initiation factors eIF1 and eIF1A, and AUG codon. Addition of the factor eIF5 triggered GTP hydrolysis with similar rates for WT eIF2 (0.29 s−1) and the N135D mutant (0.22 s−1) (Table 1; see also Fig. S6 in the supplemental material). Thus, the N135D mutation in eIF2γ did not markedly affect guanine nucleotide binding or hydrolysis by eIF2, and the mutant factor was competent to bind the 40S subunit and form 43S complexes (data not shown). In contrast, the N135D mutation impaired Met-tRNAiMet binding to eIF2. Whereas WT eIF2 bound Met-tRNAiMet with a Kd of ∼5 nM, the Kd for Met-tRNAiMet binding to the N135D mutant form of eIF2 was elevated roughly fourfold (Kd, ∼21 nM) (Table 1; see also Fig. S5 in the supplemental material). Supporting this biochemical defect in Met-tRNAiMet binding, significantly less Met-tRNAiMet coprecipitated with eIF2γ-N135D than with WT eIF2γ from crude yeast extracts (see Fig. S1 in the supplemental material). Moreover, the slow-growth phenotype of yeast expressing the eIF2γ-N135D mutant was partially suppressed by overexpression of the IMT4 gene encoding tRNAiMet (Fig. 1B, row 2).
TABLE 1.
Summary of biochemical properties of eIF2 complexes containing WT or mutant eIF2γ subunitsa
| eIF2γ | GDP Kd (nM) | GTP Kd (μM) |
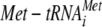 Kd (nM) Kd (nM) |
GTPase hydrolysis rate (s−1) |
|---|---|---|---|---|
| WT | 25.88 (±3.18) | 2.00 (±0.12) | 5.31 (±0.92) | 0.29 (±0.01) |
| N135D mutant | 30.35 (±6.29) | 1.60 (±0.17) | 20.62 (±6.89) | 0.22 (±0.02) |
| A208V mutant | 39.07 (±9.54) | 1.15 (±0.03) | 1.14 (±0.33) | 0.20 (±0.02) |
| N135D-A208V mutant | 36.72 (±3.50) | 1.43 (±0.05) | 3.49 (±0.66) | 0.19 (±0.02) |
| A219T mutant | >500 | >10 | 15.02 (±4.07) | 0.29 (±0.01) |
| N135D-A219T mutant | >500 | >10 | 3.91 (±0.53) | 0.52 (±0.11) |
| A382V mutant | 37.82 (±8.98) | 1.30 (±0.07) | 5.11 (±1.46) | 0.20 (±0.01) |
| N135D-A382V mutant | 33.08 (±3.44) | 1.72 (±0.01) | 4.76 (±0.67) | 0.24 (±0.01) |
Dissociation constants (Kd) for GDP, GTP, and 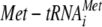 and the rate of GTP hydrolysis were determined as described in Materials and Methods (see also Fig. S3 to S6 in the supplemental material). All values are the means (± standard deviations) of at least two independent experiments.
and the rate of GTP hydrolysis were determined as described in Materials and Methods (see also Fig. S3 to S6 in the supplemental material). All values are the means (± standard deviations) of at least two independent experiments.
Given the defect in Met-tRNAiMet binding, we next examined whether the eIF2γ-N135D mutation would affect GCN4 expression in yeast cells. As shown in Fig. 1C, the eIF2γ-N135D mutation increased expression of a GCN4-lacZ reporter ∼20-fold in cells grown in amino acid-complete medium, in which GCN4 expression is normally repressed (Gcd− phenotype). This derepression of GCN4-lacZ expression was blocked in reporters containing only uORF4 (data not shown), indicating the eIF2γ-N135D mutation does not cause ribosomes to simply skip over the uORFs in the GCN4 mRNA leader. Thus, impaired assembly of the TC in the eIF2γ-N135D strain likely blocked efficient reinitiation at the inhibitory uORFs in the GCN4 mRNA leader, allowing more ribosomes to reinitiate translation further downstream at the GCN4 ORF. At odds with this simple interpretation, overexpression of tRNAiMet did not suppress the derepression of GCN4-lacZ expression (Fig. 1C), although this treatment partially suppressed the slow-growth phenotype associated with the eIF2γ-N135D mutation (Fig. 1B). This apparent paradox likely reflects the greater sensitivity of GCN4 expression than of general growth of the cells to changes in TC levels, as GCN4 is fully derepressed in WT cells by a level of eIF2α phosphorylation that does not inhibit growth. Alternatively, it is possible that the eIF2γ mutation affects eIF2 function in another way that alters GCN4 expression.
We asked next whether the N135D mutation in eIF2γ conferred a Sui− phenotype. In cells expressing WT eIF2γ, expression of a HIS4-lacZ reporter with a UUG start codon is less than 5% the level observed with a reporter containing an AUG start codon (UUG/AUG ratio, 0.04) (Fig. 1D). The eIF2γ-N135D mutation increased the UUG/AUG ratio nearly threefold (Fig. 1D), indicating that like the N135K mutation, the eIF2γ-N135D mutation confers a Sui− phenotype. Interestingly, overexpression of tRNAiMet enhanced the Sui− phenotype associated with the eIF2γ-N135D mutation (Fig. 1D). This latter finding suggests that the Sui− phenotype is not simply due to decreased Met-tRNAiMet binding affinity, but instead may reflect an altered property of the scanning 43S complex.
Isolation of intragenic suppressors of the eIF2γ-N135D mutation.
To gain further insights into the role of the Switch I element in eIF2γ and more specifically into the effect of the N135D mutation on eIF2γ function, we screened for intragenic suppressors of the eIF2γ-N135D mutation. A plasmid carrying the eIF2γ-N135D mutant gene was subjected to random mutagenesis, and then the library of mutant plasmids was screened to identify mutations that suppressed the slow-growth phenotype associated with the eIF2γ-N135D mutation. Four suppressor mutations were identified: Thr115 to Ala, Ala208 to Val, Ala219 to Thr, and Ala382 to Val (Fig. 1A). Using the structure of the EF-Tu-GDPNP-Phe-tRNAPhe complex (20) to model the locations of the suppressor mutations, the T115A suppressor is located immediately after the G1 motif (GXXXXGKTT), which is involved in α-phosphate binding, and the A208V suppressor is located near the end of Switch II at the C terminus of helix α2 (Fig. 1A; see also Fig. 4A, panel 3, below). The A219T suppressor is in β-sheet β7 at the base of the GTP-binding site, and the A382V suppressor resides in domain II in close proximity to the binding site for the aminoacyl, CCA-3′ end of Met-tRNAiMet (Fig. 1A; see also Fig. 4A, panel 2, below). As shown in Fig. 1B, the A208V mutation was slightly better than the A219T and A382V suppressors in rescuing the growth defect associated with the N135D mutation in eIF2γ. Interestingly, whereas yeast expressing the eIF2γ-A382V or eIF2γ-A208V single mutants grew like strains expressing WT eIF2γ, the eIF2γ-A219T single mutant conferred a slow-growth phenotype (Fig. 1B).
FIG. 4.

Models depicting the locations of eIF2γ mutations and their impacts on Met-tRNAiMet binding and start codon selection. (A) Model of eIF2γ mutations on the structure of the EF-Tu-GDPNP-Phe-tRNA complex. (1) Ribbon representation of the EF-Tu ternary complex (PDB ID code 1TTT), showing Phe-tRNA (orange) and GDPNP (cyan) bound to the EF-Tu. The EF-Tu G domain is depicted in brown, while domains 2 and 3 are marine blue and green, respectively. The lime sphere represents Mg2+. The side chains of key residues are depicted as sticks, with Asn135 in blue and the sites of suppressor mutations in green. The dashed box indicates the region shown in panels 2 and 3. (2) Model focusing on the locations of the N135D mutation (blue) in the Switch I element (warm pink), the A382V mutation (green) in domain 2, and the A219T mutation (green) in the G domain. (3) A different orientation of the structure in panel 2, rotated to reveal the location of the A208V suppressor mutation (green) in the Switch II element (magenta) of the G domain. (B) Hypothetical models depicting the impacts of eIF2γ mutations on Met-tRNAiMet binding and start codon selection. (1) In WT complexes, Met-tRNAiMet binds to eIF2 with high affinity (upward arrow in key below image) and in an optimum conformation that favors AUG codon recognition (base-pairing between the anticodon of the tRNA and the AUG codon of the mRNA. (2) The eIF2γ-N135D Switch I mutation impairs Met-tRNAiMet binding (pink; downward arrows in key) and lowers the stringency for AUG recognition (loss of base pair contact between the Met-tRNAiMet and the first nucleotide of the codon). The Met-tRNAiMet is bound in an altered conformation relative to its position on WT eIF2 (light gray). We propose that in this 48S complex eIF1 fails to properly monitor the codon-anticodon interaction and Met-tRNAiMet selects UUG as the start codon. (3) The A208V mutation in eIF2γ-N135D,A208V stabilizes Met-tRNAiMet binding (red; upward arrows in key) and restores the proper positioning of the Met-tRNAiMet in the 48S complex, resulting in a high-fidelity codon-anticodon interaction and suppression of the Sui− phenotype. (4) The eIF2γ-A219T mutation weakens the affinity for Met-tRNAiMet (pink); however, the Met-tRNAiMet is positioned in the WT conformation, resulting in good codon-anticodon interactions and no Sui− phenotype. (5) The A382V mutation in domain 2 of eIF2γ-N135D,A382V stabilizes Met-tRNAiMet binding (red); however, the Met-tRNAiMet is bound in an altered conformation, as observed with the original eIF2γ-N135D mutant. The improper position of Met-tRNAiMet in the 48S complex results in a failure of eIF1 to properly monitor the codon-anticodon interaction, and Met-tRNAiMet selects UUG as the start codon, resulting in a Sui− phenotype.
Western analyses revealed that the various eIF2γ single and double mutants were expressed close to the level of WT eIF2γ, although the expression of the eIF2γ-N135D,A219T mutant was significantly reduced (Fig. 2A). Importantly, overexpression of the eIF2γ mutants in yeast did not enhance the suppressor phenotype (see Fig. S2 in the supplemental material), and overexpression of eIF2γ-A219T exacerbated the growth defect, as observed for eIF2γ-N135D. These results indicate that the suppressor phenotypes are not related to eIF2γ protein levels. To further characterize the eIF2γ suppressors and gain insights into the suppression mechanism, we overexpressed and purified eIF2 complexes containing the eIF2γ suppressor proteins as described earlier. Analysis of the various eIF2 complexes by sodium dodecyl sulfate-polyacrylamide gel electrophoresis revealed comparable purities and integrities for all of the proteins (Fig. 2B). In the following sections we describe the genetic and biochemical characterization of the various mutant eIF2 complexes.
Coupling between the Switch I and Switch II elements in eIF2γ governs Met-tRNAiMet binding and the fidelity of start codon recognition.
The A208V suppressor mutation is located in the Switch II element (Fig. 1A). Introduction of the single A208V mutation in eIF2γ resulted in a WT growth phenotype, and like the eIF2γ-N135D,A208V double mutant, the growth of the eIF2γ-A208V mutant was slightly inhibited by overexpression of tRNAiMet (Fig. 1B). The GTP- and GDP-binding properties and the GTPase activity of the eIF2 complex containing eIF2γ-A208V were comparable to WT eIF2 (Table 1). Thus, despite its location in the critical G domain Switch II element, the A208V mutation does not substantially affect the interaction of eIF2 with guanine nucleotides. However, eIF2 complexes containing the eIF2γ-A208V mutation bound Met-tRNAiMet with nearly fivefold-higher affinity than did WT eIF2 complexes (Table 1). Importantly, the A208V mutation restored Met-tRNAiMet-binding affinity in the eIF2γ-N135D, A208V complex to levels comparable to WT eIF2 (Table 1) and likewise restored Met-tRNAiMet binding to eIF2 in crude cell extracts (see Fig. S1 in the supplemental material). Thus, the Switch II A208V mutation enhances Met-tRNAiMet-binding affinity in otherwise-WT eIF2 and suppresses the Met-tRNAiMet-binding defect associated with the Switch I N135D mutation. These results are consistent with the notion that physical and/or functional interactions between the Switch I (N135D) and Switch II (A208V) elements in eIF2γ influence the formation of the Met-tRNAiMet-binding pocket in eIF2.
Consistent with the biochemical findings, the eIF2γ-N135D,A208V mutation suppressed the Gcd− (Fig. 1C) and Sui− (Fig. 1D) phenotypes observed with the original eIF2γ-N135D mutation. Moreover, the single eIF2γ-A208V mutation had practically no impact on the expression of the GCN4-lacZ and the his4UUG-lacZ reporters compared to strains expressing WT eIF2 (Fig. 1C and D). The ability of the A208V mutation to enhance the Met-tRNAiMet-binding affinity of the eIF2γ-N135D mutant in vitro supports the notion that ternary complex formation governs the Gcd− phenotype, while Met-tRNAiMet release from eIF2 during scanning governs the Sui− properties associated with these eIF2 mutations. We propose that the A208V mutation alters the structure of the Switch II element and either (i) directly enhances Met-tRNAiMet binding by creating new contacts with the tRNA and/or (ii) indirectly restores Met-tRNAiMet binding by repositioning the Switch I element.
The eIF2γ-A219T mutation impairs Met-tRNAiMet binding but does not confer a Sui− phenotype.
The A219T suppressor mutation is located in the G domain at the base of the GTP-binding site (Fig. 1A; see also Fig. 4A, below). The main chain of Ala219 is in close proximity to the conserved His111 in the G-1 [GXXXHGK(S/T)] sequence motif. As the G-1 sequence motif, or P-loop, interacts with the β-phosphate of GTP and helps coordinate the Mg2+ ion required for GTP binding, the A219T mutation might indirectly affect nucleotide binding via alterations in the structure of the P-loop. Consistent with this prediction, the eIF2 complexes containing either the eIF2γ-A219T or the eIF2γ-N135D,A219T mutant exhibited severe defects in GDP and GTP binding. The Kd values were outside the range to be measured accurately in our assays, but we estimate that GDP- and GTP-binding affinities were decreased at least 20- and 5-fold, respectively (Table 1). As GTP binding is required for Met-tRNAiMet binding to eIF2, Met-tRNAiMet-binding assays were performed in the presence of excess GTP (2 mM). Under these conditions, the A219T mutation suppressed the Met-tRNAiMet-binding defect associated with the N135D mutation in eIF2γ (Table 1). Thus, despite a defect in GTP binding, these results suggest that the A219T mutation suppresses the deleterious affects of the N135D mutation by restoring Met-tRNAiMet binding. Supporting this notion, the GTP concentration in yeast cells (∼600 to 1,500 μM) (6) is sufficiently high to overcome the nucleotide-binding defect of the eIF2γ-N135D,A219T complex.
Interestingly, introducing the A219T mutation alone into eIF2γ resulted in a slow-growth phenotype that was suppressed by overexpression of tRNAiMet (Fig. 1B). Consistently, biochemical analysis of eIF2 containing this mutant subunit revealed a defect in Met-tRNAiMet binding (Kd, ∼15 nM, compared to 5 nM for WT eIF2). These results are consistent with previous studies, which showed that Met-tRNAiMet binding stabilized GTP binding to eIF2 (16) and that overexpression of tRNAiMet could rescue the yeast cell growth defects associated with certain G domain mutations in eIF2γ (10).
Further characterization of the eIF2γ-N135D,A219T and eIF2γ-A219T mutants revealed WT or nearly WT GTPase activities (Table 1), and both proteins were competent for 43S complex formation (data not shown). Interestingly, the eIF2γ-A219T single mutant showed a more dramatic Gcd− phenotype than the double mutant (Fig. 1C), which correlates with the significantly impaired Met-tRNAiMet binding by eIF2 complexes containing the eIF2γ-A219T mutant subunit (Table 1). This Gcd− phenotype is also consistent with the ability of overexpressed tRNAiMet to suppress the slow-growth phenotype in the eIF2γ-A219T mutant strain.
The mutual cosuppression by the eIF2γ-N135D and eIF2γ-A219T mutations is intriguing. Strains expressing either single mutant grew slowly, and the growth defect was suppressed by overexpression of tRNAiMet (Fig. 1B). Moreover, the eIF2γ-N135D,A219T double mutant grew faster than either single mutant (Fig. 1B). Thus, we propose that the N135D and A219T mutations impair eIF2γ function and eIF2 Met-tRNAiMet-binding activity via different but mutually complementary changes to the eIF2 G domain structure.
Given the effects of the A219T mutation on Met-tRNAiMet binding, we next examined the impact of this mutation on start codon fidelity. Whereas the eIF2γ-N135D mutation impaired Met-tRNAiMet binding and conferred a Sui− phenotype, the eIF2γ-N135D,A219T double mutant restored Met-tRNAiMet binding and suppressed the Sui− phenotype (Table 1 and Fig. 1D). Interestingly, although the single A219T mutation in eIF2γ impaired Met-tRNAiMet binding to a similar extent as the N135D mutation (Table 1), the A219T mutation did not confer a Sui− phenotype (Fig. 1D). Thus, weaker binding of Met-tRNAiMet to eIF2 does not always enhance translation initiation at a non-AUG codon and confer a Sui− phenotype. To resolve this apparent paradox, we tested whether the A219T mutation conferred an off-setting Ssu− (suppressor of Sui−) phenotype that suppressed the Sui− phenotype associated with decreased Met-tRNAiMet binding. While the gcd11-A219T mutation failed to suppress the strong Sui− phenotype (increased UUG/AUG ratio) associated with either the dominant SUI3-2 mutation in eIF2β or the dominant SUI5 mutation in eIF5 (data not shown), the A219T mutation did suppress the Sui− phenotype associated with the eIF2γ-N135D,A382V double mutant (Fig. 1D). Interestingly, as will be described in the next section, the A382V mutation suppresses the Met-tRNAiMet-binding defect but not the Sui− phenotype associated with the eIF2γ-N135D mutation. Thus, the eIF2γ-N135D,A382V double mutant confers a Sui− phenotype despite WT Met-tRNAiMet-binding activity (Fig. 1D and Table 1). The suppression of this Sui− phenotype in the eIF2γ-N135D,A219T,A382V triple mutant (Fig. 1D) reveals an underlying Ssu− (suppressor of Sui−) property of the A219T mutation and provides an explanation for the lack of a Sui− phenotype in the eIF2γ-A219T single mutant despite its poor Met-tRNAiMet-binding property. While the previously described Sui− phenotypes of eIF2γ mutants were attributed to reduced binding affinity for Met-tRNAiMet, our identification of eIF2γ mutations that enhance (A219T) or weaken (N135D, A382V) start codon fidelity, independent of effects on Met-tRNAiMet binding, indicates a more direct role for eIF2 in AUG recognition.
The A382V suppressor mutation restores the Met-tRNAiMet-binding activity but does not suppress the Sui− phenotype of the eIF2γ-N135D mutant.
The A382V suppressor mutation resides in domain II of the eIF2γ subunit, near to the proposed binding site for the 3′ end of Met-tRNAiMet (Fig. 1A; see also Fig. 4A, panel 2, below). Yeast expressing either the eIF2γ-N135D,A382V or the eIF2γ-A382V mutant subunit grew with nearly WT growth rates (Fig. 1B), and cell growth was not affected by overexpression of tRNAiMet . Biochemical analysis of eIF2 complexes containing a WT eIF2γ subunit, the suppressor mutant eIF2γ-A382V subunit, or the double mutant eIF2γ-N135D,A382V subunit revealed similar binding affinities for GDP, GTP, and Met-tRNAiMet (Table 1), and the mutant eIF2 complexes were competent to bind the ribosome and form 43S complexes (data not shown). Moreover, the three eIF2 complexes hydrolyzed GTP at similar rates (Table 1). Thus, the A382V mutation suppressed the Met-tRNAiMet-binding defect associated with the eIF2γ-N135D mutation. Despite this restoration of Met-tRNAiMet binding, the eIF2γ-N135D,A382V mutant exhibited a Gcd− phenotype (Fig. 1C). This derepression of GCN4 expression might be due to the poorer expression of the eIF2γ-N135D,A382V mutant protein (Fig. 2A, lanes 11 to 12) (80% of WT eIF2γ) or possibly a defect in the rate of 43S complex formation.
In addition to the Gcd− phenotype, the eIF2γ-N135D,A382V mutant, but not the eIF2γ-A382V mutant, enhanced translation initiation at the UUG codon in the his4UUG-lacZ reporter construct (Fig. 1D). This significant Sui− phenotype, comparable in magnitude to that observed with the original eIF2γ-N135D mutant, was not affected by overexpression of tRNAiMet (Fig. 1D). Thus, despite restoring Met-tRNAiMet-binding affinity and suppressing the slow-growth phenotype associated with the eIF2γ-N135D mutation, the A382V mutation failed to suppress the Sui− phenotype. We favor the notion that the A382V mutation in conjunction with the N135D mutation alters ribosomal scanning and AUG selection, resulting in a Sui− phenotype that is independent of Met-tRNAiMet binding. As the A382V mutation is adjacent to residues making key contacts with the CCA end of Met-tRNAiMet (see Fig. 4A, panel 2, below), one possibility is that the suppressor mutation alters the local conformation in domain II and thereby stabilizes the interaction with Met-tRNAiMet . However, this subtly altered conformation of Met-tRNAiMet in the ternary complex (and subsequently in the 48S complex) may affect start codon recognition, leading to enhanced utilization of UUG as a start codon. Finally, as the eIF2γ-A382V mutation did not confer a Sui− phenotype in the absence of the N135D mutation (Fig. 1D), the proposed altered Met-tRNAiMet-binding conformation in the eIF2γ-N135D,A382V complex likely depends on structural alterations introduced by both mutations.
Overexpression of eIF1 suppresses the Sui− phenotype of the eIF2γ-N135D and the eIF2γ-N135D,A382V mutants.
The ability of both the A219T and the A382V mutations to suppress the slow-growth phenotype associated with the eIF2γ-N135D mutation (Fig. 1B), yet their different impacts on Met-tRNAiMet-binding affinity (Table 1) and the Sui− phenotype (Fig. 1D), led us to consider alternative models for how the eIF2γ mutations affect start codon recognition. Recently it has been revealed that the factor eIF1 plays a central role in AUG codon selection. eIF1 binds in close proximity to the Met-tRNAiMet in the P site of the 40S ribosomal subunit (17), and proper base-pairing of the anticodon of the Met-tRNAiMet in the 48S complex with the AUG start codon on an mRNA induces a repositioning or release of eIF1 from the P site (18, 28). Coupled with this repositioning of eIF1, Pi is released from the eIF2-GDP-Pi complex in the 48S complex (1). The release of Pi, and the proposed accompanying release of eIF2-GDP, establishes the start site of translation initiation (1, 4, 28). Given the importance of eIF1 in monitoring the codon-anticodon interaction during translation initiation, it is likely that eIF1 plays a pivotal role in manifesting the Sui− phenotype. Supporting this hypothesis, it has previously been shown that overexpression of eIF1 can suppress the Sui− phenotype associated with mutations in eIF3, eIF5, and eIF4G (11, 29). Moreover, the Sui− phenotype of several eIF1 mutants was associated with an increased rate of dissociation of eIF1 from the 48S complex (4).
To further explore the mechanism underlying the Sui− phenotype of the eIF2γ mutants, and the involvement of eIF1 in this process, we examined the impact of eIF1 overexpression on the growth and Sui− phenotypes of the eIF2γ mutants. As shown in Fig. 3B, overexpression of eIF1 suppressed the Sui− phenotype of the eIF2γ-N135D and the eIF2γ-N135D,A382V mutants. This result suggests that premature repositioning or release of eIF1 from the 48S complex might contribute to the Sui− phenotype in these mutants. Examination of initiation complexes in extracts from formaldehyde-treated cells expressing WT eIF2γ or eIF2γ-N135D revealed no differences in the amount of eIF1 bound to 40S subunits (see Fig. S7 in the supplemental material), suggesting that the eIF2γ mutation does not induce significant release of eIF1 from the 48S complex. As an alternative, overexpression of eIF1 might by mass action drive the equilibrium governing the open and closed conformations of the scanning 40S subunit to the open state and cause the scanning 40S subunits to bypass the UUG codons in the Sui− mutants. Consistent with this latter notion, overexpression of eIF1 reduced the already low UUG/AUG ratio in cells expressing WT eIF2γ as well as those expressing the eIF2γ-N135D,A208V double mutant (Fig. 3B). In addition, overexpression of eIF1 failed to suppress the Gcd− phenotype (Fig. 3C) and growth defects (Fig. 3A) of the eIF2γ-N135D and the eIF2γ-N135D,A382V mutants. We conclude that overexpression of eIF1 specifically suppressed the Sui− phenotype associated with the eIF2γ mutations but that premature repositioning of eIF1 likely did not contribute to the general growth defect and Gcd− phenotype in the eIF2γ mutants.
FIG. 3.

Overexpression of eIF1 suppresses the Sui− phenotype of eIF2γ mutants. (A) Growth rate analysis of yeast expressing WT or mutant forms of eIF2γ and overexpressing eIF1. Derivatives of yeast strain J292 expressing WT eIF2γ, eIF2γ-N135D, or eIF2γ-N135D, A382V and carrying either an empty vector (-), a high-copy-number (H.C.) plasmid expressing eIF1 (+, pC2888), or a high-copy-number plasmid expressing tRNAiMet (bottom two rows) were serially diluted, spotted on SD medium, and incubated for 3 days at 30°C as described for Fig. 1B. The doubling times during exponential growth in liquid SD medium are shown in parentheses. (B) Suppression of the Sui− phenotype by overexpression of eIF1. Derivatives of yeast strain J292 expressing the indicated forms of eIF2γ and carrying either a high-copy-number plasmid expressing eIF1 or an empty vector were transformed with the HIS4AUG-lacZ or his4UUG-LacZ reporters, and β-galactosidase activities and UUG/AUG ratios were determined as described for Fig. 1D. For comparisons in the absence and presence of eIF1, P was ∼0.002 for WT, ∼0.006 for N135D,A208V, and <0.006 for N135D and for N135D,A382V. (C) The GCN4-lacZ reporter p180 was introduced into the strains described for panel B, and β-galactosidase activities were measured and plotted as described for Fig. 1C.
DISCUSSION
Previous work established the critical role of eIF2 in selecting the start codon for translation. Mutations that stimulate initiation at a UUG codon in the HIS4 mRNA have been identified in all three subunits of eIF2 as well as in the factor eIF5, which stimulates the GTPase activity of eIF2 (3, 7, 15). These previous studies indicated that reduced Met-tRNAiMet-binding affinity or hyperactive GTP hydrolysis by eIF2 facilitated initiation at the non-AUG codon (15). In this work we expand on these previous studies and isolate intragenic suppressor mutations that restore yeast cell growth in strains expressing the eIF2γ-N135D mutant that impairs Met-tRNAiMet binding and increases translation initiation at a UUG codon. The first and second suppressors we described (A208V and A219T) restored Met-tRNAiMet binding, yeast cell growth, and AUG codon fidelity for initiation. The third suppressor mutation (A382V) restored Met-tRNAiMet affinity and yeast cell growth but did not restore AUG codon fidelity. Interestingly, introduction of the A219T mutation into WT eIF2γ impaired Met-tRNAiMet binding but did not confer a Sui− phenotype. While the A208V suppressor supports the model linking decreased Met-tRNAiMet binding with increased initiation at a UUG codon, the A382V suppressor and the independent A219T mutation differ from this paradigm and suggest that eIF2γ influences start codon selection in multiple ways. The finding that overexpression of eIF1 suppressed initiation at the non-AUG codon in both the original eIF2γ-N135D mutant and the eIF2γ-N135D,A382V suppressor mutant suggests that eIF2γ mutations may affect eIF1 function to regulate start codon selection.
The biochemical characterization of the eIF2γ-N135D mutant indicated that the Switch I element of eIF2γ is involved in Met-tRNAiMet binding by eIF2. The mutant eIF2 complex exhibited a fourfold decrease in Met-tRNAiMet-binding affinity but no significant defect in guanine nucleotide binding or hydrolysis. Moreover, the slow-growth phenotype of the eIF2γ-N135D mutant was partially suppressed by overexpression of the IMT4 gene encoding tRNAiMet . As mutation of Asn135 either to negatively charged Asp (in this work) or to positively charged Lys (15) lowered Met-tRNAiMet-binding affinity, it is unlikely that the Met-tRNAiMet-binding defect was due to charge repulsion of the phosphate groups in the tRNAiMet . Likewise, modeling of Met-tRNAiMet bound to the eIF2 complex places Asn135 remote from the tRNA (Fig. 4A, panels 1 and 2). We favor a model in which the eIF2γ-N135D mutation alters the conformation of the Switch I region and indirectly perturbs the Met-tRNAiMet-binding pocket.
Further supporting the notion that the Switch I mutation altered the Met-tRNAiMet-binding pocket, the eIF2γ-N135D mutation conferred a Sui− phenotype. While the Sui− phenotype of the eIF2γ-N135K mutation was previously attributed to reduced Met-tRNAiMet-binding affinity (15), we propose that the conformation of Met-tRNAiMet within the eIF2 ternary complex, which dictates the geometry of codon-anticodon interaction, may also be playing a role in the Sui− phenotype. Accordingly, the altered Switch I conformation in the eIF2γ-N135D mutant restricts the Met-tRNAiMet to bind in an altered conformation and with weak affinity (Fig. 4B, panel 2). This altered geometry may enhance the selection of noncognate codon-anticodon interactions by the scanning ribosome, while the decreased Met-tRNAiMet affinity will result in the Met-tRNAiMet being released from eIF2 into the ribosomal P site in the absence of the correct codon-anticodon interaction. Thus, mutations in the components, like eIF2, of the scanning 48S complex that alter the geometry of, and bypass the requirement for perfect complementarity in, the codon-anticodon interaction cause a Sui− phenotype enabling translation to initiate at a non-AUG codon.
The intragenic suppressors of the eIF2γ-N135D mutation provide further support to the notion that the conformation of Met-tRNAiMet on eIF2 plays a critical role in start codon selection. The A208V mutation in the Switch II element suppressed both the growth defect and the Sui− phenotype associated with the eIF2γ-N135D mutation but not the lethal phenotype associated with the eIF2γ-N135K mutation (data not shown). We propose that the A208V mutation alters the structure of the Switch II element and (i) directly enhances Met-tRNAiMet binding by creating new contacts with the tRNA and/or (ii) indirectly restores Met-tRNAiMet binding by repositioning the Switch I element (Fig. 4A). Consistent with the former model, eIF2 complexes containing the eIF2γ-A208V mutation bound Met-tRNAiMet with higher affinity than did WT eIF2 (Table 1; see also Fig. S5 in the supplemental material). Moreover, overexpression of tRNAiMet exacerbated the growth defect in strains expressing eIF2γ-A208V (Fig. 1B), perhaps due to excessive Met-tRNAiMet binding. In contrast, the latter model is supported by the physical and functional interactions observed between the Switch I and Switch II elements in eIF5B (26) and other G proteins (30). The Switch I and II elements in eIF2γ are ∼4.5 Å apart (Fig. 4A, panel 3), and the mutation in Switch II may directly or indirectly cause the reorientation of Switch I into a more favorable position. Thus, the A208V mutation in Switch II may enable the Switch I element containing the N135D mutation to adopt a conformation favorable for Met-tRNAiMet binding and high-fidelity AUG codon recognition (Fig. 4B, panel 3).
Two of the intragenic suppressor mutations in eIF2γ uncouple Met-tRNAiMet-binding affinity and the Sui− phenotype. The eIF2γ-N135D,A382V suppressor mutation located in domain II restored Met-tRNAiMet-binding affinity without suppressing the Sui− phenotype, while the eIF2γ-A219T mutation (in the absence of the N135D mutation) impaired Met-tRNAiMet binding yet did not confer a Sui− phenotype (Table 1 and Fig. 2; see also Fig. S5 in the supplemental material). The A382V suppressor is located near the residues in domain II that are predicted to make polar contacts with the CCA end of Met-tRNAiMet (Fig. 4A, panel 1). We propose that the A382V mutation restores a productive Met-tRNAiMet-binding pocket, thereby rescuing the Met-tRNAiMet-binding defect and the growth defect associated with the eIF2γ-N135D mutation. However, the Met-tRNAiMet remains in an altered conformation (Fig. 4B, panel 5), thus affecting the geometry of the codon-anticodon interaction and lowering the fidelity of AUG codon recognition. It is noteworthy that the proposed repositioning of Met-tRNAiMet on the eIF2 mutants is likely to be quite subtle and beyond the resolution of most commonly available techniques. While the Sui− mutations enhance initiation at the noncognate UUG codon, they do not promote initiation at other near-cognate codons (CUG or GUG), suggesting that the Sui− mutations subtly affect base-pairing between the Met-tRNAiMet anticodon and the mRNA codon.
In contrast to the A382V mutation, the A219T mutation in the G domain (Fig. 4A) impaired Met-tRNAiMet and GTP binding; however, no Sui− phenotype was observed. Interestingly, the A219T and the original N135D mutations both impair Met-tRNAiMet binding and cause a slow-growth phenotype in yeast that can be suppressed by overexpression of tRNAiMet . When combined, the N135D and A219T mutations mutually suppress their growth and Met-tRNAiMet-binding defects. We propose that the N135D and A219T mutations alter the structure of the Met-tRNAiMet-binding pocket in compensatory manners. Both mutations weaken Met-tRNAiMet binding; however, when the mutations are combined, a high-affinity binding site for Met-tRNAiMet reforms. Moreover, while the N135D mutation impairs both the affinity and orientation of Met-tRNAiMet binding to eIF2, causing a Sui− phenotype, the A219T mutation confers a hyperaccuracy, Ssu−, phenotype (Fig. 1D). The A219T mutation suppressed the Sui− phenotype of the original eIF2γ-N135D mutant and the eIF2γ-N135D,A382V mutant (Fig. 1D) but not the dominant Sui− phenotypes of the eIF2β-S264Y (SUI3-2) and eIF5-G31R (SUI5) mutants (data not shown). Similar, though stronger, Ssu− mutations in eIF1 were found to decrease the dissociation rate of eIF1 from the 48S complex (4) and to require AUG recognition to convert the scanning-competent “open” 48S complex to the nonscanning “closed” complex (18, 21). We propose that the A219T mutation induces the Met-tRNAiMet to bind in an orientation (Fig. 4B, panel 4) that restricts the open-to-closed transition of the 48S complex to perfect codon-anticodon interactions. Taken together, our suppressor studies indicate that both the positioning and affinity of Met-tRNAiMet on eIF2 are important determinants of start codon recognition.
The factor eIF1 plays a critical role in the fidelity of start codon selection. Mutations in the SUI1 gene encoding eIF1 were first identified as conferring a Sui− phenotype by enhancing initiation at a UUG codon (3, 33). Subsequent studies revealed that eIF1 prevents initiation at non-AUG codons during scanning (1, 4, 22). This checkpoint role of eIF1 dissociation from the 48S complex in selecting the start site for translation initiation is mechanistically linked to the conversion of the open to closed form of the 48S complex (18, 21). Our data provide additional evidence supporting this checkpoint role of eIF1. Overexpression of eIF1 suppressed the Sui− phenotypes associated with the eIF2γ-N135D and eIF2γ-N135D,A382V mutations (Fig. 3B). However, overexpression of eIF1 did not suppress the growth defect or Gcd− phenotypes in these mutants (Fig. 3A and C), consistent with the notion that these phenotypes are due to the defect in Met-tRNAiMet-binding affinity. The suppression of the Sui− phenotypes of the eIF2γ mutants by overexpression of eIF1 is consistent with the previous reports that overexpression of eIF1 suppresses the Sui− phenotype due to mutations in eIF5, eIF3, and eIF2β (11, 29). Thus, our data support the findings from these other studies and demonstrate the critical role of eIF1 in governing translation start site selection.
Supplementary Material
Acknowledgments
We thank Alan Hinnebusch and Jon Lorsch for comments on the manuscript and helpful discussions; Lee Kapp, Mike Acker, Mikkel Algire, and Jon Lorsch for help in establishing the biochemical assays; members of the Hinnebusch and Dever labs for helpful suggestions; Tom Donahue for plasmids; and John Hershey for support.
This work was supported in part by the Intramural Program of the NIH, NICHD (T.E.D.).
Footnotes
Published ahead of print on 15 September 2008.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Algire, M. A., D. Maag, and J. R. Lorsch. 2005. Pi release from eIF2, not GTP hydrolysis, is the step controlled by start-site selection during eukaryotic translation initiation. Mol. Cell 20251-262. [DOI] [PubMed] [Google Scholar]
- 2.Alone, P. V., and T. E. Dever. 2006. Direct binding of translation initiation factor eIF2γ-G domain to its GTPase-activating and GDP-GTP exchange factors eIF5 and eIF2Bɛ. J. Biol. Chem. 28112636-12644. [DOI] [PubMed] [Google Scholar]
- 3.Castilho-Valavicius, B., H. Yoon, and T. F. Donahue. 1990. Genetic characterization of the Saccharomyces cerevisiae translational initiation suppressors sui1, sui2 and SUI3 and their effects on HIS4 expression. Genetics 124483-495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cheung, Y. N., D. Maag, S. F. Mitchell, C. A. Fekete, M. A. Algire, J. E. Takacs, N. Shirokikh, T. Pestova, J. R. Lorsch, and A. G. Hinnebusch. 2007. Dissociation of eIF1 from the 40S ribosomal subunit is a key step in start codon selection in vivo. Genes Dev. 211217-1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cigan, A. M., E. K. Pabich, L. Feng, and T. F. Donahue. 1989. Yeast translation initiation suppressor sui2 encodes the α subunit of eukaryotic initiation factor 2 and shares identity with the human α subunit. Proc. Natl. Acad. Sci. USA 862784-2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ditzelmuller, G., W. Wohrer, C. P. Kubicek, and M. Rohr. 1983. Nucleotide pools of growing, synchronized and stressed cultures of Saccharomyces cerevisiae. Arch. Microbiol. 13563-67. [DOI] [PubMed] [Google Scholar]
- 7.Donahue, T. 2000. Genetic approaches to translation initiation in Saccharomyces cerevisiae, p. 487-502. In N. Sonenberg, J. W. B. Hershey, and M. B. Mathews (ed.), Translational control of gene expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 8.Donahue, T. F., A. M. Cigan, E. K. Pabich, and B. Castilho-Valavicius. 1988. Mutations at a Zn(II) finger motif in the yeast eIF-2β gene alter ribosomal start-site selection during the scanning process. Cell 54621-632. [DOI] [PubMed] [Google Scholar]
- 9.Dorris, D. R., F. L. Erickson, and E. M. Hannig. 1995. Mutations in GCD11, the structural gene for eIF-2γ in yeast, alter translational regulation of GCN4 and the selection of the start site for protein synthesis. EMBO J. 142239-2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Erickson, F. L., and E. M. Hannig. 1996. Ligand interactions with eukaryotic translation initiation factor 2: role of the γ-subunit. EMBO J. 156311-6320. [PMC free article] [PubMed] [Google Scholar]
- 11.He, H., T. von der Haar, C. R. Singh, M. Ii, B. Li, A. G. Hinnebusch, J. E. McCarthy, and K. Asano. 2003. The yeast eukaryotic initiation factor 4G (eIF4G) HEAT domain interacts with eIF1 and eIF5 and is involved in stringent AUG selection. Mol. Cell. Biol. 235431-5445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hershey, J. W. B., and W. C. Merrick. 2000. Pathway and mechanism of initiation of protein synthesis, p. 33-88. In N. Sonenberg, J. W. B. Hershey, and M. B. Mathews (ed.), Translational control of gene expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 13.Hinnebusch, A. G. 2000. Mechanism and regulation of initiator methionyl-tRNA binding to ribosomes, p. 185-243. In N. Sonenberg, J. W. B. Hershey, and M. B. Mathews (ed.), Translational control of gene expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 14.Hinnebusch, A. G. 2005. Translational regulation of GCN4 and the general amino acid control of yeast. Annu. Rev. Microbiol. 59407-450. [DOI] [PubMed] [Google Scholar]
- 15.Huang, H., H. Yoon, E. M. Hannig, and T. F. Donahue. 1997. GTP hydrolysis controls stringent selection of the AUG start codon during translation initiation in Saccharomyces cerevisiae. Genes Dev. 112396-2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kapp, L. D., and J. R. Lorsch. 2004. GTP-dependent recognition of the methionine moiety on initiator tRNA by translation factor eIF2. J. Mol. Biol. 335923-936. [DOI] [PubMed] [Google Scholar]
- 17.Lomakin, I. B., V. G. Kolupaeva, A. Marintchev, G. Wagner, and T. V. Pestova. 2003. Position of eukaryotic initiation factor eIF1 on the 40S ribosomal subunit determined by directed hydroxyl radical probing. Genes Dev. 172786-2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maag, D., C. A. Fekete, Z. Gryczynski, and J. R. Lorsch. 2005. A conformational change in the eukaryotic translation preinitiation complex and release of eIF1 signal recognition of the start codon. Mol. Cell 17265-275. [DOI] [PubMed] [Google Scholar]
- 19.Moehle, C. M., and A. G. Hinnebusch. 1991. Association of RAP1 binding sites with stringent control of ribosomal protein gene transcription in Saccharomyces cerevisiae. Mol. Cell. Biol. 112723-2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nissen, P., M. Kjeldgaard, S. Thirup, G. Polekhina, L. Reshetnikova, B. F. Clark, and J. Nyborg. 1995. Crystal structure of the ternary complex of Phe-tRNAPhe, EF-Tu, and a GTP analog. Science 2701464-1472. [DOI] [PubMed] [Google Scholar]
- 21.Passmore, L. A., T. M. Schmeing, D. Maag, D. J. Applefield, M. G. Acker, M. A. Algire, J. R. Lorsch, and V. Ramakrishnan. 2007. The eukaryotic translation initiation factors eIF1 and eIF1A induce an open conformation of the 40S ribosome. Mol. Cell 2641-50. [DOI] [PubMed] [Google Scholar]
- 22.Pestova, T. V., S. I. Borukhov, and C. U. T. Hellen. 1998. Eukaryotic ribosomes require initiation factors 1 and 1A to locate initiation codons. Nature 394854-859. [DOI] [PubMed] [Google Scholar]
- 23.Polekhina, G., S. Thirup, M. Kjeldgaard, P. Nissen, C. Lippmann, and J. Nyborg. 1996. Helix unwinding in the effector region of elongation factor EF-Tu-GDP. Structure 41141-1151. [DOI] [PubMed] [Google Scholar]
- 24.Roll-Mecak, A., P. Alone, C. Cao, T. E. Dever, and S. K. Burley. 2004. X-ray structure of translation initiation factor eIF2γ: implications for tRNA and eIF2α binding. J. Biol. Chem. 27910634-10642. [DOI] [PubMed] [Google Scholar]
- 25.Schmitt, E., S. Blanquet, and Y. Mechulam. 2002. The large subunit of initiation factor aIF2 is a close structural homologue of elongation factors. EMBO J. 211821-1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shin, B. S., M. G. Acker, D. Maag, J. R. Kim, J. R. Lorsch, and T. E. Dever. 2007. Intragenic suppressor mutations restore GTPase and translation functions of a eukaryotic initiation factor 5B switch II mutant. Mol. Cell. Biol. 271677-1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sprang, S. R. 1997. G protein mechanisms: insights from structural analysis. Annu. Rev. Biochem. 66639-678. [DOI] [PubMed] [Google Scholar]
- 28.Unbehaun, A., S. I. Borukhov, C. U. Hellen, and T. V. Pestova. 2004. Release of initiation factors from 48S complexes during ribosomal subunit joining and the link between establishment of codon-anticodon base-pairing and hydrolysis of eIF2-bound GTP. Genes Dev. 183078-3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Valasek, L., K. H. Nielsen, F. Zhang, C. A. Fekete, and A. G. Hinnebusch. 2004. Interactions of eukaryotic translation initiation factor 3 (eIF3) subunit NIP1/c with eIF1 and eIF5 promote preinitiation complex assembly and regulate start codon selection. Mol. Cell. Biol. 249437-9455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vetter, I. R., and A. Wittinghofer. 2001. The guanine nucleotide-binding switch in three dimensions. Science 2941299-1304. [DOI] [PubMed] [Google Scholar]
- 31.Williams, N. P., A. G. Hinnebusch, and T. F. Donahue. 1989. Mutations in the structural genes for eukaryotic initiation factors 2α and 2β of Saccharomyces cerevisiae disrupt translational control of GCN4 mRNA. Proc. Natl. Acad. Sci. USA 867515-7519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yatime, L., Y. Mechulam, S. Blanquet, and E. Schmitt. 2007. Structure of an archaeal heterotrimeric initiation factor 2 reveals a nucleotide state between the GTP and the GDP states. Proc. Natl. Acad. Sci. USA 10418445-18450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoon, H. J., and T. F. Donahue. 1992. The sui1 suppressor locus in Saccharomyces cerevisiae encodes a translation factor that functions during tRNAiMet recognition of the start codon. Mol. Cell. Biol. 12248-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.