

Abstract
Acute diarrheal illness is a global health problem that may be exacerbated by concurrent infection. Using Citrobacter rodentium, a murine model of attaching and effacing diarrheagenic Escherichia coli, we demonstrate that persistent Helicobacter hepaticus infection modulates host responses to diarrheal disease, resulting in delayed recovery from weight loss and from tissue damage. Chronic colitis in concurrently infected mice is characterized by macrophage and Foxp3+ regulatory T-cell accumulation. Prolonged disease is also associated with increased interleukin-17 expression, which may be due to suppression of gamma interferon during the acute phase of diarrheal infection. This new model of polymicrobial infection provides insight into the mechanism by which subclinical infection can exacerbate morbidity due to an unrelated self-limiting infection.
Acute diarrheal illness is a major health problem worldwide. In the developed world most cases are self-limiting and can be treated with supportive care; however, in the developing world, diarrheal illness is a major cause of morbidity and mortality, particularly in children. Infection with enteropathogenic Escherichia coli is estimated to account for approximately 7% of pediatric acute diarrheal illness (24, 29). Malnutrition, immunosuppression, and concurrent disease influence the severity and outcome of diarrheal illness, but the effect of heterologous infection has not been characterized.
Citrobacter rodentium infection of laboratory mice has been studied as a model of enteropathogenic E. coli infection in children (3). In C57BL/6 mice C. rodentium infection causes loose stool progressing to diarrhea in severe cases, poor overall body condition, and weight loss (28, 37). Colonic lesions consist of epithelial hyperplasia, submucosal edema, and mucosal inflammation that ranges from mild to severe with erosions, ulcerations, and transmural serositis (20, 28). Adult C57BL/6 mice clear C. rodentium infection and recover from disease approximately 4 weeks postinoculation (wpi), with full resolution of colonic lesions by 6 wpi (20, 28). Young mice and adults of certain inbred strains develop fatal infection with C. rodentium (4, 38). Additionally, comorbidity with helminth infection alters disease severity by inducing interleukin-10 (IL-10)-expressing dendritic cells (8).
With approximately 50% of the world's population infected with Helicobacter pylori, subclincial infections in humans are common. Helicobacter hepaticus infection in laboratory mice, like H. pylori in humans, is highly prevalent and subclinical in otherwise healthy (wild-type) animals (35). Both in cultured cell systems and in vivo, H. hepaticus elicits a proinflammatory response from innate and adaptive immune cells including IL-23, gamma interferon (IFN-γ), and tumor necrosis factor alpha (TNF-α). However, persistent infection with this bacterium is balanced by regulatory responses, including IL-10 production by regulatory T (Treg) cells that prevent clinical disease (15-18, 22). Subclinical disease develops in susceptible strains, such as male A/J mice (10, 42), yet the role of IL-10 and Treg cells in suppressing clinical disease is revealed with infection of IL-10 or T-cell-deficient mice (6, 16-18, 40). Superimposition of a second, unrelated infection on the dynamic homeostasis of proinflammatory and regulatory cell populations could tip the balance and alter the outcome of the subsequent infection. We tested the hypothesis that heterologous infection can enhance morbidity by challenging mice with C. rodentium with or without concurrent H. hepaticus infection.
MATERIALS AND METHODS
Mice.
Male and female 5- to 12-week-old C57BL/6J (The Jackson Laboratory, Bar Harbor, ME) mice were used for all studies. All experiments were approved by the Massachusetts Institute of Technology Committee on Animal Care. Mice were fed a rodent diet and water ad libitum and housed in microisolator cages that were maintained in facilities approved by the Association for Assessment and Accreditation of Laboratory Animal Care, International. Until experimentally inoculated, the mice were maintained specific pathogen free of known murine bacterial, viral, and parasitic infections including all known Helicobacter spp. For each experiment, the mice were divided into four treatment groups: uninoculated (n = 10), inoculated with H. hepaticus (n = 10), inoculated with C. rodentium (n = 10), and inoculated with H. hepaticus followed by C. rodentium (n = 10). Five independent experiments were conducted: two with necropsy at 1 week after C. rodentium inoculation, one with necropsy at 2 wpi, one with necropsy at 3 wpi, and one with necropsy at 4 wpi.
Bacterial infections.
H. hepaticus 3B1 (ATCC 51449) was grown on tryptic soy agar supplemented with 5% sheep red blood cells or in tryptic soy broth (TSB) supplemented with 5% fetal calf serum at 37°C in a microaerobic environment (80% N2, 10% H2, and 10% CO2). H. hepaticus inocula were prepared from 3-day liquid cultures from which ∼2 × 108 bacteria (estimated from the optical density at 600 nm) were administered in 200 μl of TSB via intragastric gavage to individual mice. Uninoculated mice were gavaged with 200 μl of sterile TSB. For C. rodentium infections, mice were gavaged with ∼2 × 109 bacteria from an overnight culture of Kanr C. rodentium (DBS120) in 100 μl of Luria-Bertani broth, 7 to 8 weeks after H. hepaticus infection. Helicobacter status was confirmed with an all-Helicobacter PCR of fecal DNA as previously described (48). C. rodentium fecal shedding was determined by plating serial dilutions of fecal slurries (10% [wt/vol] in phosphate-buffered saline) on Luria-Bertani agar with selection for kanamycin.
Body weight measurements.
Body weights were monitored every 3 to 4 days. Mice were euthanized and excluded from the study if they lost >20% of their body weight.
Tissue collection and histology.
At necropsy fecal and tissue samples were collected. Distal colon (∼0.5 cm) was snap-frozen in liquid nitrogen and stored at −80°C until it was used for RNA isolation. The remaining colon was fixed in 10% formalin, paraffin embedded, sectioned at 5 μm, and stained with hematoxylin and eosin for histologic evaluation. Colonic tissue sections were scored on a scale of 0 to 4 (0, no lesion; 1, minimal; 2, mild; 3, moderate; and 4, severe) for inflammation, edema, hyperplasia, dysplasia, and epithelial defects by a board-certified blinded pathologist. Lesion scores are presented as histologic colitis indices that are a sum of all five categorical scores (maximum of 20). Foxp3 immunohistochemistry was performed as previously described (32), using Foxp3 antibody (FJK-16S; eBiosciences, San Diego, CA). Cells expressing Foxp3+ were counted in the distal to mid-colon at a magnification of ×20 (1 field is 1.00 mm2), excluding gut-associated lymphoid tissue. Ten fields were counted per mouse, and results are presented as the average number of Foxp3+ cells/mm2 of colon. F4/80 immunohistochemistry was performed as described for Foxp3 but using F4/80 antibody (CI:A3-1; Abcam, Cambridge, MA). Cells expressing F4/80 were counted in 20 fields of distal colon at ×40 magnification (1 field is 0.26 mm2), also excluding gut-associated lymphoid tissue. Results are presented as the average number of F4/80+ cells/mm2 of colon.
Quantitative real-time PCR.
Total RNA was isolated from distal colon using TRIzol reagent (Invitrogen, Foster City, CA), cleaned up with an RNeasy Kit (Qiagen Sciences, MD), and reverse transcribed (Invitrogen) following the manufacturers' protocols. Quantitative real-time PCR was performed on cDNA using TaqMan Gene Expression Assays (Applied Biosystems, Foster City, CA) specific for glyceraldehyde-3-phosphate dehydrogenase (GAPDH; assay no. Mm99999915_g1), IL-6 (Mm00446190_m1), MCP-1 (Mm00441242_m1), TNF-α (Mm99999068_m1), IFN-γ (Mm99999071_m1), IL-10 (Mm00439616_m1), transforming growth facter β (TGF-β; Mm00498234_m1), IL-1β (Mm00434228_m1), IL-12/IL-23p40 (Mm99999067_m1), IL-12p35 (Mm01208555_m1), IL-23p19 (Mm00518984_m1), and IL-17 (Mm00439619_m1). Each sample was calibrated to internal GAPDH levels and normalized to the average value of control (uninoculated mice) samples at the same time point.
Statistics.
Statistical significance in bacterial counts, weight change, disease indices, mRNA expression, and F4/80+ and Foxp3+ cell numbers was determined by two-way analysis of variance (ANOVA) followed by Bonferroni posttests. A Spearman correlation was used to evaluate the correlation between disease indices and cytokine mRNA expression levels. All analyses were done with GraphPad Prism software, version 4.0. P values of <0.05 were considered significant.
RESULTS
Delayed recovery from acute diarrheal illness as a result of persistent H. hepaticus infection.
In agreement with previous reports where the Helicobacter spp. status of mice was not specified (21, 43, 44), fecal shedding of C. rodentium in Helicobacter-free C57BL/6J mice reached a maximum of 109 CFU/g of feces between 1 and 2 wpi (Fig. 1a). Over the next week C. rodentium was cleared, and fecal shedding was undetectable (<103 CFU/g of feces) by 3 wpi. Because the severity of C. rodentium-induced disease can be decreased by as little as a 1 log reduction in peak bacterial load (19), we carefully compared fecal shedding with and without H. hepaticus infection. Persistent infection with H. hepaticus, confirmed by PCR on DNA isolated from fecal pellets, did not affect fecal shedding of C. rodentium (Fig. 1a). Body weight change in mice infected with H. hepaticus alone was not different from uninoculated mice, confirming the subclinical nature of the infection. In contrast, body weight loss was apparent in Helicobacter-free C. rodentium-inoculated mice by 1 wpi. These mice lost an average of 4% of their initial body weight by 12 days postinoculation (Fig. 1b) (P < 0.001). By 3 wpi these mice recovered, and their body weights were not significantly different from those of uninoculated mice. The extent of weight loss in C. rodentium-inoculated mice with a persistent subclinical H. hepaticus infection was comparable to that of Helicobacter-free C. rodentium-inoculated mice (Fig. 1b). However, concurrently infected mice did not regain body weight at 3 wpi (P < 0.001 compared to Helicobacter-free C. rodentium-inoculated mice) and remained underweight compared to control mice through 4 wpi (P < 0.001). Given that persistent subclinical H. hepaticus infection caused delayed recovery from weight loss, colonic lesions were evaluated to ascertain whether this morbidity was associated with an enhanced chronicity of mucosal disease.
FIG. 1.

Concurrent H. hepaticus infection impaired recovery of weight loss without altering the fecal shedding of C. rodentium. (a) C. rodentium fecal shedding at 4 wpi in Helicobacter-free mice or mice persistently infected with H. hepaticus. (b) Percent change in body weight from day 0 postinoculation with C. rodentium through 4 wpi in uninoculated mice and mice inoculated with H. hepaticus, C. rodentium, or H. hepaticus plus C. rodentium. Data are represented as means ± standard errors of the means. **, P < 0.01; ***, P < 0.001 (for mice inoculated with C. rodentium versus H. hepaticus plus C. rodentium); #, P < 0.05; ##, P < 0.01; ###, P < 0.001 (for uninoculated mice versus mice inoculated with H. hepaticus plus C. rodentium); ξξ, P < 0.01; ξξξ, P < 0.001 (for uninoculated mice versus mice inoculated with C. rodentium). Two-way ANOVA with Bonferroni posttests was used for statistical analysis. Cr, C. rodentium; Hh, H. hepaticus.
Prolonged colitis in mice concurrently infected with H. hepaticus.
H. hepaticus infection alone did not cause intestinal lesions, and histologic colitis in these animals was not significantly different from that of uninoculated mice (Fig. 2a, d, and g). By 1 wpi Helicobacter-free C. rodentium-inoculated mice developed colonic lesions consisting of inflammation, edema, epithelial destruction, and hyperplasia. Categorical lesion scores for each of these parameters were summed to form a histologic colitis index. At 1 wpi the median histologic colitis index in Helicobacter-free C. rodentium-inoculated mice was 4.0 (range, 0.0 to 6.0) (Fig. 2g) (P < 0.001 compared to uninoculated mice). Over the following week, histologic colitis reached maximal severity, characterized by gland elongation, goblet cell depletion, mucosal and submucosal inflammation, and epithelial defects, including erosions, crypt atrophy, and mild dysplasia (Fig. 2b). At peak severity, Helicobacter-free C. rodentium-inoculated mice had an index of 9.0 (range, 7.0 to 12.0) (Fig. 2b and g). Disease resolved in these mice during 3 and 4 wpi (at 4 wpi, index of 2.0; range, 0.5 to 4.5) (Fig. 2e and g). Concurrent infection did not significantly alter the maximal severity of histologic colitis at 2 wpi (index of 8.5; range, 5.5 to 10.0) (Fig. 2c and g), but concurrent H. hepaticus infection did delay the resolution of C. rodentium-induced lesions. At 4 wpi the index of concurrently infected mice was 4.5 (range, 2.5 to 7.5) compared with 2.0 (range, 0.5 to 4.0) in Helicobacter-free C. rodentium-inoculated mice (Fig. 2e to g) (P < 0.001). Although lesions in both Helicobacter-free and concurrently infected mice were resolving at 4 wpi, inflammation and hyperplasia were more severe in concurrently infected mice (Fig. 2 h) (P < 0.001 and P < 0.05, respectively). Thus, delayed recovery from weight loss was associated with colitis of a longer duration. Morphometrics were used to determine if the chronicity of disease in concurrently infected mice was associated with an increase in the number of macrophages, which are a hallmark of chronic inflammation (26).
FIG. 2.

C. rodentium infection causes marked colon disease that is prolonged by persistent subclinical H. hepaticus infection. (a to f) Hematoxylin- and eosin-stained colon corresponding to median histologic colitis index of the following treatment groups at the indicated time points: uninoculated (a), C. rodentium at 2 wpi (b), H. hepaticus and C. rodentium at 2 wpi (c), H. hepaticus at 3 months post-H. hepaticus inoculation (d), C. rodentium at 4 wpi (e), and H. hepaticus and C. rodentium at 4 wpi (f). (g) Histologic colitis index comprised of inflammation, edema, hyperplasia, dysplasia, and epithelial defects, each assessed on a scale of 0 to 4. Boxes represent first to third quartiles, and median values are indicated by a horizontal line. Bars represent ranges. (h) Inflammation and hyperplasia lesion scores for individual mice inoculated with C. rodentium (□) and with H. hepaticus and C. rodentium (▵) at 4 wpi. *, P < 0.05; ***, P < 0.001. Two-way ANOVA on the histologic colitis index or lesion scores from all groups and time points with Bonferroni posttests was used. Scale bar, 160 μm.
Chronic colitis is associated with an increased number of F4/80+ macrophages.
Macrophages expressing F4/80 (a macrophage-specific marker) were present in low numbers in the colon of uninoculated mice (Fig. 3a and b) (average of all time points, 20 ± 18 cells/mm2). No significant change in macrophage numbers was seen in colons of H. hepaticus-infected animals, consistent with a lack of disease (Fig. 3a and c) (average of all time points, 16 ± 20 cells/mm2). Macrophages did accumulate in the colon during C. rodentium infection (Fig. 3a) (P < 0.001). Macrophage numbers were maximal in Helicobacter-free C. rodentium-inoculated mice at 1 wpi (175 ± 36 cells/mm2), decreased by 2 wpi (72 ± 23 cells/mm2), and remained constant at 3 and 4 wpi (98 ± 34 and 125 ± 28 cells/mm2, respectively) (Fig. 3a and d). Macrophage numbers in concurrently infected mice were not significantly different from those in mice infected with C. rodentium alone at 1, 2, or 3 wpi (Fig. 3a). However, at 4 wpi, instead of staying constant at 2 and 3 wpi, as in Helicobacter-free C. rodentium-infected mice, the number of colonic macrophages significantly increased to 256 ± 40 cells/mm2 in concurrently infected mice (Fig. 3a, d, and e) (P < 0.01), indicating chronic mucosal inflammation. Natural Foxp3+ Treg cells play a key role in limiting immunopathology during infection and chronic inflammatory diseases as well as in contributing to peripheral tolerance (1). Morphometrics were used to enumerate Foxp3+ cells to determine if greater chronicity of histologic colitis was associated with accumulation of natural Treg cells.
FIG. 3.

Concurrent H. hepaticus infection increases macrophage infiltration and maintains elevated numbers of natural Treg cells in colonic tissue 4 weeks after C. rodentium inoculation. (a) Numbers of F4/80+ cells in colons enumerated in 20 fields (magnification, ×400; 0.26 mm2) per mouse distal colon (n = 3 to 4 per group) at 1, 2, 3, and 4 wpi. (b to e) Representative photomicrographs of F4/80+ macrophages in the colon of uninoculated mice (b) and mice inoculated with H. hepaticus (c) or C. rodentium (d) at 4 wpi and with H. hepaticus and C. rodentium at 4 wpi (e). (f) Numbers of Foxp3+ cells in the colon assessed from 10 fields (magnification, ×200; 1.00 mm2) per mouse distal colon (n = 3 to 4 per group) at 1, 2, 3, and 4 wpi. ***, P < 0.001 (two-way ANOVA with Bonferroni posttests). (g to j) Representative photomicrographs of Foxp3+ cells in colon from mice inoculated with C. rodentium at 2 wpi (g), H. hepaticus and C. rodentium at 2 wpi (h), C. rodentium at 4 wpi (i), and H. hepaticus and C. rodentium at 4 wpi (j). **, P < 0.01; ***, P < 0.001. Two-way ANOVA on the numbers of F4/80+ or Foxp3+ cells/mm2 of colon from all groups and time points with Bonferroni posttests was used. Error bars in panels a and f represent standard errors of the means. Scale bars, 80 μm (b to e) and 40 μm (g to j).
Foxp3+ cells accumulate in response to infectious colitis and persist during chronic disease.
As expected in the absence of histologic colitis, H. hepaticus infection alone caused no colonic accumulation of Foxp3+ Treg cells (Fig. 3f) compared to uninoculated mice (1 ± 1 Foxp3+ cells/mm2). Early after C. rodentium inoculation (1 wpi) there was no significant increase in the number of colonic Foxp3+ cells in Helicobacter-free mice, suggesting that natural Treg cells accumulate in response to colitis, not infection per se. With the development of histologic colitis at 2 and 3 wpi, there were increased numbers of Foxp3+ Treg cells in the colon of Helicobacter-free C. rodentium-inoculated mice (10 ± 6 and 21 ± 5 Foxp3+ cells/mm2, respectively) (Fig. 3f and g) (P < 0.001). By 4 wpi, colonic numbers of Foxp3+ Treg cells in Helicobacter-free C. rodentium-inoculated mice had declined and were not significantly different from basal numbers in uninoculated or H. hepaticus-infected mice (Fig. 3f and i). Colonic numbers of Foxp3+ Treg cells in concurrently infected mice began to accumulate earlier (1 wpi) than in mice infected with C. rodentium alone. At the peak of histologic colitis at 2 and 3 wpi, accumulation of Foxp3+ Treg cells in concurrently infected mice was comparable to the level in Helicobacter-free C. rodentium-inoculated mice (Fig. 3f to h). However, at 4 wpi Foxp3+ Treg-cell numbers remained elevated in concurrently infected mice, in contrast to the return to basal levels in mice infected with C. rodentium alone (Fig. 3f and j) (P < 0.001). The continued presence of natural Treg cells is consistent with a delayed recovery from histologic colitis, resulting in greater chronicity of disease.
C. rodentium colitis is associated with a proinflammatory cytokine expression profile dominated by IFN-γ.
Colonic cytokine expression levels were measured to further characterize the mechanism by which persistent subclinical infection modulated the host response to acute diarrheal illness. Adaptive immunity is required for the clearance of C. rodentium but also contributes to histologic colitis (5, 13). IL-17-producing T cells have also been recently implicated in disease pathogenesis (23). Elevated expression of the Th1 cytokines IFN-γ, TNF-α, and IL-12 coincided with increasing histologic colitis, reaching maximal expression at 2 wpi (Fig. 4a and Fig. 5) (P < 0.01 compared with uninoculated or H. hepaticus-infected mice). Expression levels of these proinflammatory cytokines declined at 3 and 4 wpi as lesions resolved. The type 2 cytokines IL-4 and IL-13 were not consistently detected in distal colons of control or infected mice. Additionally, the IL-10 expression profile mimicked that of inflammatory cytokines, possibly as a feedback mechanism to limit collateral inflammatory damage (Fig. 5). No significant changes in TGF-β expression occurred although expression trended higher at 2 wpi (Fig. 5). IL-23p19 expression was unaltered by C. rodentium infection, indicating either constitutive expression or no transcriptional induction of IL-23, a Th17 maintenance (46, 49) and Foxp3+ T-cell inhibitory factor (14). IL-17 expression increased during C. rodentium infection, reaching a plateau at 3 wpi (Fig. 4a). The association between C. rodentium disease pathogenesis and Th17 described previously was based on observations made at 8 days postinoculation (23). We also found greater colonic expression of IL-17 than IFN-γ at 1 wpi; however, later in the progression of disease IFN-γ and MCP-1 were found to predominate. Over the course of 4 weeks of infection, the dominant, IFN-γ nature of C. rodentium disease was apparent.
FIG. 4.
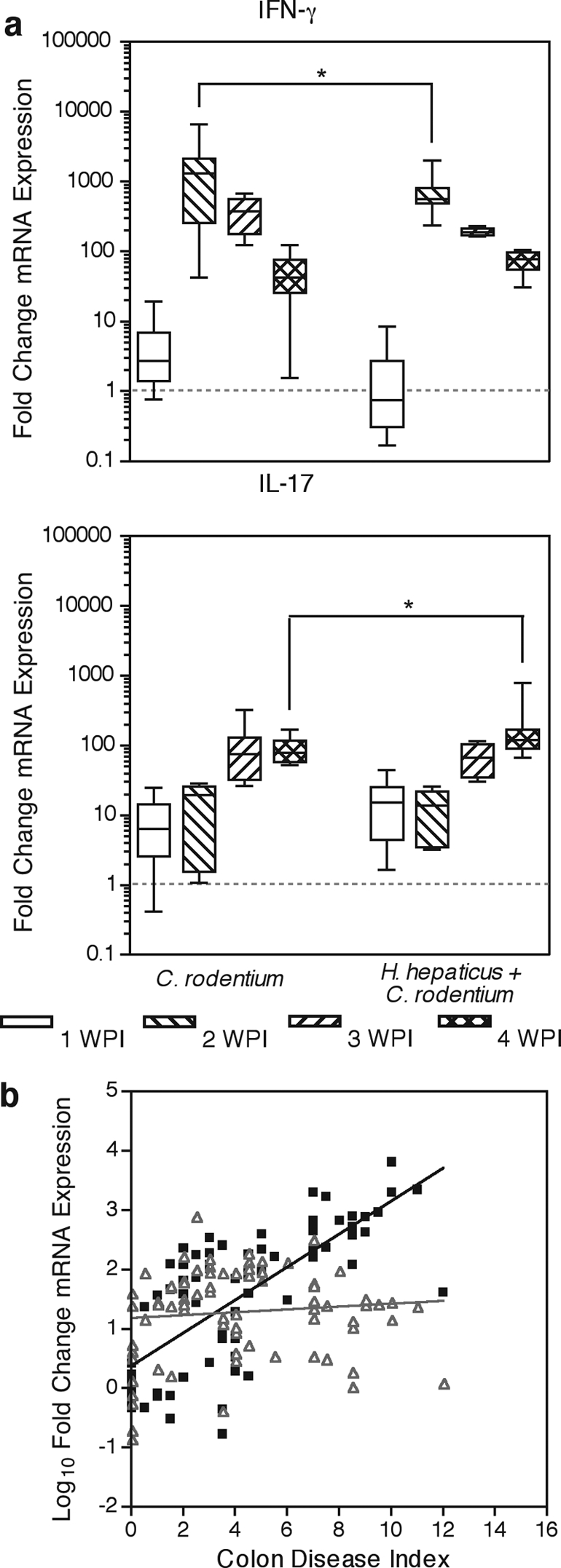
Persistent H. hepaticus infection suppresses IFN-γ expression and increases IL-17 expression in C. rodentium-infected mice at 4 wpi. (a) Colonic mRNA expression levels of IFN-γ and IL-17 were measured by quantitative real-time PCR. mRNA expression normalized to uninoculated mice is presented as box-whisker plots, where boxes represent the first to third quartiles; the median is indicated by a horizontal line. Bars represent ranges. *, P < 0.05 by two-way ANOVA with Bonferroni posttests. (b) Spearman correlation of the histologic colitis index and corresponding IFN-γ (▪) or IL-17 (▵) mRNA expression demonstrated a positive correlation between colonic disease severity and IFN-γ expression (Spearman r = 0.763; P = 0.0032) but no correlation between colonic disease severity and IL-17 (Spearman r = 0.097; P > 0.05). Solid lines are linear regressions of colon disease and cytokine expression, not Spearman correlations, to aid visualization of correlation.
FIG. 5.

C. rodentium infection induces a Th1 cytokine response that peaks at 2 weeks postinoculation, and MCP-1 expression is suppressed by concurrent H. hepaticus infection. Colonic mRNA expression levels of IL-6, TNF-α, MCP-1, IL-1β, TGF-β, IL-10, IL-12/23p40, IL-12p35, and IL-23p19 were measured by quantitative real-time PCR. mRNA expression normalized to uninoculated mice is presented in box-whisker plots indicating minimum and maximum, first and third quartiles, and median expression levels. ***, P < 0.001 by two-way ANOVA with Bonferroni posttests.
Cytokine expression and histologic colitis were correlated for individual mice in all infection groups at each time point. C. rodentium colitis correlated with colonic expression of IFN-γ but not with IL-17 (Fig. 4b). Analysis revealed a strong positive correlation between histologic colitis severity and levels of the proinflammatory cytokines IL-6, MCP-1, TNF-α, IL-1β, IL-12p35, IL-12/23p40, and in particular IFN-γ (Spearman r for IFN-γ of 0.763; P = 0.0032). Anti-inflammatory IL-10, TGF-β, and transcription factor Foxp3 expression levels had weak (Spearman r values of 0.557, 0.333, and 0.362, respectively) but significant (P < 0.01) correlation with histologic colitis severity. These weak correlations are likely due to an anti-inflammatory response to counteract the inflammation in the tissue. In contrast to other proinflammatory cytokines, such as IFN-γ, neither IL-23p19 nor IL-17 was correlated with histologic colitis (Spearman r for IL-17 of 0.097; P > 0.05).
Concurrent H. hepaticus infection suppresses IFN-γ during peak disease and enhances IL-17 expression during chronic colitis.
Persistent concurrent H. hepaticus infection did not affect colonic cytokine expression at 1 wpi (Fig. 4a). However, at 2 wpi, concurrently infected mice had significantly lower levels of IFN-γ (773-fold over uninoculated controls) and MCP-1 (45-fold) message than Helicobacter-free C. rodentium-inoculated mice (1,756-fold [P < 0.05] and 160-fold [P < 0.001], respectively) (Fig. 4a and 5). No significant differences in colonic cytokine expression were noted at 3 wpi between concurrently infected mice and mice inoculated with C. rodentium alone. By 4 wpi, when other inflammatory cytokines were returning to basal expression, IL-17 message continued to increase in concurrently infected mice, reaching a 202-fold increase over uninoculated mice. This elevated expression of IL-17 was significantly increased compared with Helicobacter-free C. rodentium-inoculated mice (92-fold over uninoculated mice) at 4 wpi (Fig. 4a) (P < 0.05). Increased IL-17 expression in concurrently infected mice was not accompanied by a change in IL-23p19 or IL-12/23p40. Overall, persistent H. hepaticus infection modulated C. rodentium-induced cytokine expression by slightly suppressing peak levels of both IFN-γ and MCP-1, as well as by modestly enhancing IL-17 expression later in the course of the disease. Decreased IFN-γ and MCP-1 expression at 2 wpi was not associated with reduced severity of colitis or body weight loss. However, the increase in IL-17 was associated with delayed recovery from weight loss, chronic colitis, and macrophage and natural Treg-cell accumulation in the colon. Given that IL-17 contributes to chronic inflammation (47) and inflammatory bowel disease (11), the association of increased IL-17 expression with increased morbidity suggests that it may prevent resolution of acute disease during self-limiting infection and may promote chronic disease progression.
DISCUSSION
To our knowledge, this is the first demonstration that a persistent subclinical bacterial infection causes delayed recovery from a self-limiting bacterial infection. Elevated IL-17 expression during chronic disease observed in concurrently infected mice is possibly due to lower IFN-γ at an earlier stage in disease progression. In this study, eradication of C. rodentium infection by the host's immune system was not altered in concurrently infected mice. Rather, H. hepaticus altered expression of a key chemokine (MCP-1) and type 1 cytokine (IFN-γ), resulting in delayed resolution of disease and greater chronicity of colitis. Persistent H. hepaticus infection did not alter mortality due to C. rodentium infection, one indicator of impaired development of adaptive immunity. By enhancing morbidity rather than mortality, as observed during concurrent helminth infection (7), concurrent infection with H. hepaticus and C. rodentium provides a useful model for evaluating the effects of subclinical infections on the outcome of a self-limiting infection.
Th1 cytokines, particularly IFN-γ, are essential for a proper host response to C. rodentium (5, 13, 34); however, the importance of IL-17-producing cells is not well defined in this disease. Our data indicate that expression of many proinflammatory cytokines is highly induced in response to C. rodentium infection and that the pattern of transient expression is not affected by persistent infection with H. hepaticus. However, IL-17 expression does not decline when mice have delayed disease resolution due to concurrent H. hepaticus infection. IL-17 has been associated with T-cell-mediated colitis (47) as well as with inflammatory bowel disease (11) although the role of this cytokine in disease is not well understood. IL-17 has been shown to recruit neutrophils to mucosal infection sites (25, 45). However, during chronic disease in concurrently infected mice, few neutrophils were observed (data not shown). The increased IL-17 expression during chronic disease may be a consequence of the decreased IFN-γ at the peak of disease, as IFN-γ has been shown to inhibit generation of IL-17-producing cells (12). Additionally, MCP-1 is an important chemokine for monocyte recruitment to mucosal tissue (33). Decreased MCP-1 in concert with IFN-γ changes cell recruitment and subsequent cytokine production by immune cells in the colon, including epithelial cells. These alterations in cellular responses may have downstream effects on disease resolution, perhaps requiring a longer recovery period or exposing the host's immune response to intestinal microbiota for an extended period of time.
The source of IL-17 in chronic colitis is unknown; however, Th17 cells are the most likely candidates. Th17 cells have been implicated in many inflammatory diseases as well as in protection from infection by extracellular pathogens, yet the role of IL-17 in disease pathogenesis versus control of infectious agents or a balance between the two has not been fully clarified (31). Recently, an increased presence of IL-17-producing T cells in the colon due to the presence of commensal microbiota was demonstrated. This increase was particularly evident during T-cell-mediated colitis (30). Therefore, suppression of IFN-γ and MCP-1 during peak colitis leading to a delayed period of recovery could act, at least in part, through enhanced exposure to intestinal microbiota. Mucosal damage and subsequent microbial exposure could directly increase the amount of IL-17 in the colon. Additional studies will be needed to evaluate the contribution of specific cell populations and cytokines to the outcome of concurrent infection.
Although the mechanism by which concurrent H. hepaticus infection causes chronic C. rodentium-induced colitis is not fully understood, we have demonstrated alterations in mucosal cytokine production and immune cell recruitment to the colon. The paucity of neutrophils at 4 wpi (data not shown) is consistent with this being a phase of chronicity or resolution. Increased numbers of macrophages were observed in the colon throughout the course of C. rodentium disease and were still present in Helicobacter-free C. rodentium-infected mice at 4 wpi. However, the number of macrophages in the colon at 4 wpi was significantly greater in mice with concurrent H. hepaticus infection, consistent with chronic mucosal inflammation.
Beyond their essential role in controlling autoimmunity (2, 39), the role of natural Treg cells in the gastrointestinal tract remains somewhat unclear (1, 27). Accumulation of natural Treg cells has been demonstrated at sites of active disease in tuberculosis, hepatitis C virus, and colitis (9, 36, 41), indicating the importance of these cells in controlling collateral damage during pathogen-directed immune and inflammatory responses. Here, we demonstrate that infection with C. rodentium causes a significant increase in the number of natural Treg cells in the colon during active disease. These natural Treg cells are likely recruited to limit host damage since they accumulate coincident with and serve as a marker for active inflammation. Indeed, the kinetics of natural Treg-cell accumulation following initial infiltration of macrophages at 1 wpi, as well as the significant increase in natural Treg-cell abundance in concurrently infected mice at 4 wpi, confirms their association with active disease in this model.
In humans, delayed disease resolution from a self-limiting infection with chronic inflammation as a consequence of persistent subclinical infection is likely to lead to increased morbidity and mortality. Multiple concurrent infections could act in concert to produce more deleterious outcomes of disease. The true prevalence of persistent subclinical infection and the sequelae of concurrent infection in people remain to be determined. Persistent subclinical infections may at least in part account for different responses to acute infections, comparable to the contribution of genetic polymorphisms. The high prevalence of persistent subclinical infection with H. pylori or Mycobacterium tuberculosis worldwide suggests that persistent infections may impact disease pathogenesis or treatment outcome of acute infections, particularly in the developing world.
Acknowledgments
This work was supported by PHS grant R01 DK052413 to D.B.S.
We thank Elizabeth Groff and Katherine Schlieper for technical assistance, Kathy Cormier for excellent histopathology, and the Division of Comparative Medicine staff for animal care.
Editor: B. A. McCormick
Footnotes
Published ahead of print on 18 August 2008.
REFERENCES
- 1.Belkaid, Y., and B. T. Rouse. 2005. Natural regulatory T cells in infectious disease. Nat. Immunol. 6353-360. [DOI] [PubMed] [Google Scholar]
- 2.Bennett, C. L., J. Christie, F. Ramsdell, M. E. Brunkow, P. J. Ferguson, L. Whitesell, T. E. Kelly, F. T. Saulsbury, P. F. Chance, and H. D. Ochs. 2001. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat. Genet. 2720-21. [DOI] [PubMed] [Google Scholar]
- 3.Borenshtein, D., McBee, M. E., and D. B. Schauer. 2008. Utility of the Citrobacter rodentium infection model in laboratory mice. Curr. Opin. Gastroenterol. 2432-37. [DOI] [PubMed] [Google Scholar]
- 4.Borenshtein, D., P. R. Nambiar, E. B. Groff, J. G. Fox, and D. B. Schauer. 2007. Development of fatal colitis in FVB mice infected with Citrobacter rodentium. Infect. Immun. 753271-3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bry, L., M. Brigl, and M. B. Brenner. 2006. CD4+-T-cell effector functions and costimulatory requirements essential for surviving mucosal infection with Citrobacter rodentium. Infect. Immun. 74673-681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cahill, R. J., C. J. Foltz, J. G. Fox, C. A. Dangler, F. Powrie, and D. B. Schauer. 1997. Inflammatory bowel disease: an immunity-mediated condition triggered by bacterial infection with Helicobacter hepaticus. Infect. Immun. 653126-3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen, C. C., S. Louie, B. McCormick, W. A. Walker, and H. N. Shi. 2005. Concurrent infection with an intestinal helminth parasite impairs host resistance to enteric Citrobacter rodentium and enhances Citrobacter-induced colitis in mice. Infect. Immun. 735468-5481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen, C. C., S. Louie, B. A. McCormick, W. A. Walker, and H. N. Shi. 2006. Helminth-primed dendritic cells alter the host response to enteric bacterial infection. J. Immunol. 176472-483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen, X., B. Zhou, M. Li, Q. Deng, X. Wu, X. Le, C. Wu, N. Larmonier, W. Zhang, H. Zhang, H. Wang, and E. Katsanis. 2007. CD4+ CD25+ FoxP3+ regulatory T cells suppress Mycobacterium tuberculosis immunity in patients with active disease. Clin. Immunol. 12350-59. [DOI] [PubMed] [Google Scholar]
- 10.Fox, J. G., X. Li, L. Yan, R. J. Cahill, R. Hurley, R. Lewis, and J. C. Murphy. 1996. Chronic proliferative hepatitis in A/JCr mice associated with persistent Helicobacter hepaticus infection: a model of helicobacter-induced carcinogenesis. Infect. Immun. 641548-1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fujino, S., A. Andoh, S. Bamba, A. Ogawa, K. Hata, Y. Araki, T. Bamba, and Y. Fujiyama. 2003. Increased expression of interleukin 17 in inflammatory bowel disease. Gut 5265-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harrington, L. E., R. D. Hatton, P. R. Mangan, H. Turner, T. L. Murphy, K. M. Murphy, and C. T. Weaver. 2005. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 61123-1132. [DOI] [PubMed] [Google Scholar]
- 13.Higgins, L. M., G. Frankel, G. Douce, G. Dougan, and T. T. MacDonald. 1999. Citrobacter rodentium infection in mice elicits a mucosal Th1 cytokine response and lesions similar to those in murine inflammatory bowel disease. Infect. Immun. 673031-3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Izcue, A., S. Hue, S. Buonocore, C. V. Arancibia-Carcamo, P. P. Ahern, Y. Iwakura, K. J. Maloy, and F. Powrie. 2008. Interleukin-23 restrains regulatory T cell activity to drive T cell-dependent colitis. Immunity 28559-570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kullberg, M. C., D. Jankovic, C. G. Feng, S. Hue, P. L. Gorelick, B. S. McKenzie, D. J. Cua, F. Powrie, A. W. Cheever, K. J. Maloy, and A. Sher. 2006. IL-23 plays a key role in Helicobacter hepaticus-induced T cell-dependent colitis. J. Exp. Med. 2032485-2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kullberg, M. C., D. Jankovic, P. L. Gorelick, P. Caspar, J. J. Letterio, A. W. Cheever, and A. Sher. 2002. Bacteria-triggered CD4+ T regulatory cells suppress Helicobacter hepaticus-induced colitis. J. Exp. Med. 196505-515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kullberg, M. C., A. G. Rothfuchs, D. Jankovic, P. Caspar, T. A. Wynn, P. L. Gorelick, A. W. Cheever, and A. Sher. 2001. Helicobacter hepaticus-induced colitis in interleukin-10-deficient mice: cytokine requirements for the induction and maintenance of intestinal inflammation. Infect. Immun. 694232-4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kullberg, M. C., J. M. Ward, P. L. Gorelick, P. Caspar, S. Hieny, A. Cheever, D. Jankovic, and A. Sher. 1998. Helicobacter hepaticus triggers colitis in specific-pathogen-free interleukin-10 (IL-10)-deficient mice through an IL-12- and gamma interferon-dependent mechanism. Infect. Immun. 665157-5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luperchio, S. A., J. V. Newman, C. A. Dangler, M. D. Schrenzel, D. J. Brenner, A. G. Steigerwalt, and D. B. Schauer. 2000. Citrobacter rodentium, the causative agent of transmissible murine colonic hyperplasia, exhibits clonality: synonymy of C. rodentium and mouse-pathogenic Escherichia coli. J. Clin. Microbiol. 384343-4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luperchio, S. A., and D. B. Schauer. 2001. Molecular pathogenesis of Citrobacter rodentium and transmissible murine colonic hyperplasia. Microbes Infect. 3333-340. [DOI] [PubMed] [Google Scholar]
- 21.Maaser, C., M. P. Housley, M. Iimura, J. R. Smith, B. A. Vallance, B. B. Finlay, J. R. Schreiber, N. M. Varki, M. F. Kagnoff, and L. Eckmann. 2004. Clearance of Citrobacter rodentium requires B cells but not secretory immunoglobulin A (IgA) or IgM antibodies. Infect. Immun. 723315-3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maloy, K. J., L. R. Antonelli, M. Lefevre, and F. Powrie. 2005. Cure of innate intestinal immune pathology by CD4+ CD25+ regulatory T cells. Immunol. Lett. 97189-192. [DOI] [PubMed] [Google Scholar]
- 23.Mangan, P. R., L. E. Harrington, D. B. O'Quinn, W. S. Helms, D. C. Bullard, C. O. Elson, R. D. Hatton, S. M. Wahl, T. R. Schoeb, and C. T. Weaver. 2006. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 441231-234. [DOI] [PubMed] [Google Scholar]
- 24.Mani, R., U. Udgaonkar, and S. Pawar. 2003. Study of enteropathogenic Escherichia coli (EPEC) diarrhoea in children. Indian J. Pathol. Microbiol. 46118-120. [PubMed] [Google Scholar]
- 25.McKenzie, B. S., R. A. Kastelein, and D. J. Cua. 2006. Understanding the IL-23-IL-17 immune pathway. Trends Immunol. 2717-23. [DOI] [PubMed] [Google Scholar]
- 26.Medzhitov, R. 2008. Origin and physiological roles of inflammation. Nature 454428-435. [DOI] [PubMed] [Google Scholar]
- 27.Mowat, A. M. 2003. Anatomical basis of tolerance and immunity to intestinal antigens. Nat. Rev. Immunol. 3331-341. [DOI] [PubMed] [Google Scholar]
- 28.Mundy, R., T. T. MacDonald, G. Dougan, G. Frankel, and S. Wiles. 2005. Citrobacter rodentium of mice and man. Cell Microbiol. 71697-1706. [DOI] [PubMed] [Google Scholar]
- 29.Nguyen, T. V., P. Le Van, C. Le Huy, K. N. Gia, and A. Weintraub. 2005. Detection and characterization of diarrheagenic Escherichia coli from young children in Hanoi, Vietnam. J. Clin. Microbiol. 43755-760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Niess, J. H., F. Leithauser, G. Adler, and J. Reimann. 2008. Commensal gut flora drives the expansion of proinflammatory CD4 T cells in the colonic lamina propria under normal and inflammatory conditions. J. Immunol. 180559-568. [DOI] [PubMed] [Google Scholar]
- 31.Ouyang, W., J. K. Kolls, and Y. Zheng. 2008. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity 28454-467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rogers, A. B., K. S. Cormier, and J. G. Fox. 2006. Thiol-reactive compounds prevent nonspecific antibody binding in immunohistochemistry. Lab. Investig. 86526-533. [DOI] [PubMed] [Google Scholar]
- 33.Serbina, N. V., T. Jia, T. M. Hohl, and E. G. Pamer. 2008. Monocyte-mediated defense against microbial pathogens. Annu. Rev. Immunol. 26421-452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Simmons, C. P., N. S. Goncalves, M. Ghaem-Maghami, M. Bajaj-Elliott, S. Clare, B. Neves, G. Frankel, G. Dougan, and T. T. MacDonald. 2002. Impaired resistance and enhanced pathology during infection with a noninvasive, attaching-effacing enteric bacterial pathogen, Citrobacter rodentium, in mice lacking IL-12 or IFN-gamma. J. Immunol. 1681804-1812. [DOI] [PubMed] [Google Scholar]
- 35.Taylor, N. S., S. Xu, P. Nambiar, F. E. Dewhirst, and J. G. Fox. 2007. Enterohepatic Helicobacter species are prevalent in mice from commercial and academic institutions in Asia, Europe, and North America. J. Clin. Microbiol. 452166-2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Uhlig, H. H., J. Coombes, C. Mottet, A. Izcue, C. Thompson, A. Fanger, A. Tannapfel, J. D. Fontenot, F. Ramsdell, and F. Powrie. 2006. Characterization of Foxp3+ CD4+ CD25+ and IL-10-secreting CD4+ CD25+ T cells during cure of colitis. J. Immunol. 1775852-5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vallance, B. A., W. Deng, M. De Grado, C. Chan, K. Jacobson, and B. B. Finlay. 2002. Modulation of inducible nitric oxide synthase expression by the attaching and effacing bacterial pathogen Citrobacter rodentium in infected mice. Infect. Immun. 706424-6435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vallance, B. A., W. Deng, K. Jacobson, and B. B. Finlay. 2003. Host susceptibility to the attaching and effacing bacterial pathogen Citrobacter rodentium. Infect. Immun. 713443-3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wan, Y. Y., and R. A. Flavell. 2007. Regulatory T-cell functions are subverted and converted owing to attenuated Foxp3 expression. Nature 445766-770. [DOI] [PubMed] [Google Scholar]
- 40.Ward, J. M., M. R. Anver, D. C. Haines, J. M. Melhorn, P. Gorelick, L. Yan, and J. G. Fox. 1996. Inflammatory large bowel disease in immunodeficient mice naturally infected with Helicobacter hepaticus. Lab. Anim. Sci. 4615-20. [PubMed] [Google Scholar]
- 41.Ward, S. M., B. C. Fox, P. J. Brown, J. Worthington, S. B. Fox, R. W. Chapman, K. A. Fleming, A. H. Banham, and P. Klenerman. 2007. Quantification and localisation of FOXP3+ T lymphocytes and relation to hepatic inflammation during chronic HCV infection. J. Hepatol. 47316-324. [DOI] [PubMed] [Google Scholar]
- 42.Whary, M. T., T. J. Morgan, C. A. Dangler, K. J. Gaudes, N. S. Taylor, and J. G. Fox. 1998. Chronic active hepatitis induced by Helicobacter hepaticus in the A/JCr mouse is associated with a Th1 cell-mediated immune response. Infect. Immun. 663142-3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wiles, S., S. Clare, J. Harker, A. Huett, D. Young, G. Dougan, and G. Frankel. 2004. Organ specificity, colonization and clearance dynamics in vivo following oral challenges with the murine pathogen Citrobacter rodentium. Cell Microbiol. 6963-972. [DOI] [PubMed] [Google Scholar]
- 44.Wiles, S., K. M. Pickard, K. Peng, T. T. MacDonald, and G. Frankel. 2006. In vivo bioluminescence imaging of the murine pathogen Citrobacter rodentium. Infect. Immun. 745391-5396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu, Q., R. J. Martin, J. G. Rino, R. Breed, R. M. Torres, and H. W. Chu. 2007. IL-23-dependent IL-17 production is essential in neutrophil recruitment and activity in mouse lung defense against respiratory Mycoplasma pneumoniae infection. Microbes Infect. 978-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang, X. O., A. D. Panopoulos, R. Nurieva, S. H. Chang, D. Wang, S. S. Watowich, and C. Dong. 2007. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J. Biol. Chem. 2829358-9363. [DOI] [PubMed] [Google Scholar]
- 47.Yen, D., J. Cheung, H. Scheerens, F. Poulet, T. McClanahan, B. McKenzie, M. A. Kleinschek, A. Owyang, J. Mattson, W. Blumenschein, E. Murphy, M. Sathe, D. J. Cua, R. A. Kastelein, and D. Rennick. 2006. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J. Clin. Investig. 1161310-1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Young, V. B., K. A. Knox, J. S. Pratt, J. S. Cortez, L. S. Mansfield, A. B. Rogers, J. G. Fox, and D. B. Schauer. 2004. In vitro and in vivo characterization of Helicobacter hepaticus cytolethal distending toxin mutants. Infect. Immun. 722521-2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou, L., I. I. Ivanov, R. Spolski, R. Min, K. Shenderov, T. Egawa, D. E. Levy, W. J. Leonard, and D. R. Littman. 2007. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat. Immunol. 8967-974. [DOI] [PubMed] [Google Scholar]