

Abstract
Yersinia pestis, the bacterial agent of plague, secretes several proteins important for pathogenesis or host protection. The F1 protein forms a capsule on the bacterial cell surface and is a well-characterized protective antigen but is not essential for virulence. A type III secretion system that is essential for virulence exports Yop proteins, which function as antiphagocytic or anti-inflammatory factors. Yop effectors (e.g., YopE) are delivered across the host cell plasma membrane by a translocon, composed of YopB and YopD. Complexes of YopB, YopD, and YopE (BDE) secreted by Yersinia pseudotuberculosis were purified by affinity chromatography and used as immunogens to determine if antibodies to the translocon could provide protection against Y. pestis in mice. Mice vaccinated with BDE generated high-titer immunoglobulin G antibodies specific for BDE, as shown by enzyme-linked immunosorbent assay and immunoblotting, and were protected against lethal intravenous challenge with F1− but not F1+ Y. pestis. Mice passively immunized with anti-BDE serum were protected from lethal challenge with F1− Y. pestis. The YopB protein or a complex of YopB and YopD (BD) was purified and determined by vaccination to be immunogenic in mice. Mice actively vaccinated with BD or passively vaccinated with anti-BD serum were protected against lethal challenge with F1− Y. pestis. These results indicate that anti-translocon antibodies can be used as immunotherapy to treat infections by F1− Y. pestis.
Yersinia pestis is a gram-negative bacterium and the agent of plague, an acute, often fatal infection that can manifest in three forms: bubonic, pneumonic, and septicemic (38, 41). Y. pestis has several characteristics that would facilitate its development as a biological weapon, resulting in its classification as a category A select agent. These characteristics include the capacity for aerosol dissemination and the high fatality rate of pneumonic plague (24). There is no safe and effective vaccine currently available that can prevent the pneumonic form of the disease (46, 49). Antibiotics, such as aminoglycosides, can significantly reduce the mortality of pneumonic plague if treatment is initiated within 20 h of the onset of disease; however, antibiotic-resistant strains of Y. pestis have been identified (reviewed in reference 41). It is therefore important to develop new vaccines or immunotherapeutics for the prevention or treatment of pneumonic plague.
Y. pestis secretes several proteins that have been studied as experimental subunit vaccines (41, 46, 49). The F1 protein is encoded on the plasmid pMT1 and is assembled into an antiphagocytic capsule by a chaperone-usher pathway (38, 43). Mice actively vaccinated with recombinant F1 or passively immunized with an F1-specific monoclonal antibody (MAb) were protected against bubonic or pneumonic Y. pestis infection (46, 49). However, F1− mutants of Y. pestis have been shown to retain full virulence in animal infection models, and therefore, F1 may not be an ideal vaccine candidate (reviewed in references 38 and 46). LcrV is a multifunctional virulence protein that is encoded by plasmid pCD1 and is exported to the bacterial surface by a type III secretion system (T3SS) (6, 9). LcrV localizes to the tip of the T3SS needle structure and is secreted into the extracellular milieu (6, 9, 31). Recombinant LcrV has been extensively studied as a subunit vaccine, either alone or in combination with F1 (46, 49). Mice actively immunized with recombinant LcrV are protected from lethal infection by pneumonic or bubonic plague (46, 49). Active immunization with LcrV protects mice from lethal infection with capsulated or F1− variants of Y. pestis (2, 46, 49). In addition, anti-LcrV MAbs can passively protect mice from lethal bubonic or pneumonic Y. pestis disease (21, 22). An improved version of the recombinant LcrV vaccine has been developed by deletion of a region of the protein involved in immunosuppressive activity (11, 36). Nevertheless, an important limitation of an LcrV vaccine is the fact that enteropathogenic Yersinia species (Yersinia enterocolitica and Yersinia pseudotuberculosis) express variants of the LcrV protein that do not provide cross-protective immunity (42). Therefore, an F1− Y. pestis strain engineered to express a variant LcrV protein might be resistant to current LcrV-F1-based vaccines.
Recently, several additional protective antigens in Y. pestis were identified that could be developed into subunit vaccines or immunotherapeutics. They include the ABC transporter protein OppA (48) and the YadC outer membrane protein (32). Components of the T3SS other than LcrV have been investigated as protective antigens. Vaccination of mice with recombinant YscF, the subunit of the polymeric T3SS needle, has been shown to confer protection against Y. pestis infection (28, 47). Several of the T3SS substrates, known as Yops, have been investigated as candidate protective antigens by vaccinating mice with recombinant forms of the proteins (3, 25, 33). Vaccination with YopH, -E, -N, -K, or -M resulted in no significant protection against Y. pestis infection (3, 25, 33). Vaccination with YopD was found to provide significant protection against F1− Y. pestis, but not wild-type F1+ bacteria (3). Andrews et al. hypothesized that the failure of YopD vaccination to protect against encapsulated bacteria may result from the F1 protein blocking access of antibodies to secreted YopD at the bacterial surface (3).
YopD, along with YopB and LcrV, is required for translocation of effector Yops across the host cell plasma membrane (9, 13). YopB and YopD contain hydrophobic domains indicative of transmembrane proteins (17), and both proteins bind to LcrV (45). These observations have led to the suggestion that YopB and YopD assemble on the tip of the T3SS needle with the aid of LcrV and form a translocation pore that inserts in the host membrane (5). Anti-LcrV antibodies opsonize Yersinia by binding LcrV at the needle tip (12, 31). In the presence of anti-LcrV antibody, increased phagocytosis of Y. pestis and reduced Yop translocation into macrophages have been reported (10, 52). If YopB and YopD are also exposed at the needle tip prior to or during translocation, it is reasonable to expect that antibodies against these proteins would also neutralize Yop translocation and afford protection against Y. pestis infection. In addition, it is less likely that protection mediated by anti-translocon antibodies could be bypassed by genetic modification of Y. pestis, because YopB and YopD are essential for virulence and both proteins are highly conserved among Yersinia species (17, 19).
Shigella flexneri secretes homologs of YopB and YopD (IpaB and IpaC, respectively) via a T3SS (17, 29). Previous work has demonstrated that a complex containing IpaB, IpaC, and other factors isolated from S. flexneri can function as a subunit vaccine against shigellae in animal infection models (50). In addition, a MAb specific for IpaB was shown to neutralize S. flexneri T3SS function in an in vitro invasion assay (30). Building upon these observations, as well as the finding that mice vaccinated with recombinant YopD are protected against F1− Y. pestis (3), we have purified secreted translocon complexes and investigated their potentials as protective immunogens. Our results show for the first time that antibodies specific for the translocon can be used as immunotherapy to treat infections caused by F1− Y. pestis.
MATERIALS AND METHODS
Bacterial strains.
Escherichia coli Tuner (DE3) (lacIp) (Novagen) containing the expression vector pET28a lcrV was used for overexpression of recombinant His-LcrV. The fragment of DNA including the lcrV gene used to construct the pET28a lcrV vector was obtained using PCR. Oligonucleotides LCRV3 (5′-AGGATCCCATATGATTAGAGCCTACGAACAAAACC-3′) and LCRV4 (5′-GAATTCATTTACCAGACGTGTCATCTAGC-3′) were used as primers, and pCD1Ap was used as a template. The resulting DNA segment was inserted into pET28a (Novagen) using NdeI and EcoRI restriction sites (underlined in the oligonucleotide sequences), placing lcrV under the control of the T7 promoter and in frame with a sequence coding for six His residues (His tag) at the N terminus.
The Y. pseudotuberculosis strains used in this study are derived from the multi-yop mutant strain YP44 (yopEHJOKMB) (44). The yopE::kan allele in YP44 encodes a stable truncated form of the YopE protein (residues 1 to 131) that is secreted by the T3SS (37). YP46 [yopE(R144A)HJOKMB] was constructed by replacing the yopE::kan allele in YP44 with the full-length yopE(R144A) gene using the plasmid pSB890 and a standard allelic-recombination strategy (37). The yopE(R144A) gene sequence used for insertion into pSB890 was obtained from pYopER144A (4). The presence of the mutation was confirmed by DNA sequencing. A Yop secretion assay (37) was used to confirm the secretion of full-length YopE(R144A). YP56 (ΔyopE yopEHJOKMB) was constructed from YP44 by allelic replacement of the yopE::kan allele with a complete deletion of the yopE open reading frame. The DNA fragment containing the ΔyopE allele that was inserted into pSB890 was constructed using recombinant PCR and the oligonucleotide primers YOPE-UP-F1 (5′-CCCCCCAACCTCGTCCCAGGATAAGATGG-3′), YOPE-UP-R1 (5′-GGAAACACTATCCCCTTGTTGACTATTTATTACCTTGGCTATTA-3′), YOPE-DOWN-F2 (5′-TAATAGCCAAGGTAATAAATAGTCACAAGGGGATAGTGTTTCC-3′), and YOPE-DOWN-R2 (5′-TTCGGTGTGACTCAGGCTAAG-3′).
YP60 (ΔyopE yopEHJOKMBD) was constructed from YP56 by allelic replacement, using pSB890-ΔYopD to delete the yopD open reading frame. The DNA segment containing the ΔyopD allele that was inserted into pSB890 was constructed by recombinant PCR using the primers YopDΔ1A (5′-TGAAGGATCCAGACATGGCAGCGTTAC-3′), YopDΔ1B (5′-GGTTATTCCTCCTTAAACTTAAACAG-3′), YopDΔ1C (5′-GGAGGAATAACCCCATTGATGACCTTGGT-3′), and YopDΔ1D (5′-CGGTGGATCCCAGCTCAGGTCTTGA-3′).
The Y. pestis Δpgm mutant KIM5 (KIM6/pCD1Ap) has been described previously (27). KIM5caf1A (KIM6 caf1A::kan/pCD1Ap) was constructed from KIM6 plus caf1A::kan (39) using a two-step procedure to delete the pgm locus and transform it with pCD1Ap as described previously (14). To confirm that the KIM5 and KIM5caf1A strains constructed as described above were virulent, they were injected into mice (see below), and isolates were recovered from infected spleens after necropsy. Cultures of the isolates passed through mice were used to prepare frozen stocks (50% Luria-Bertani broth [LB], 50% glycerol), which were stored at −80°C.
To prepare inocula for injection into mice, samples of bacteria from the frozen stocks were thawed, inoculated on heart infusion (HI) agar plates, and allowed to form isolated colonies at 26°C. The isolated colonies were used to inoculate HI broth containing 25 μg/ml ampicillin (Ap) (for KIM5) or 25 μg/ml Ap and 25 μg/ml kanamycin (for KIM5caf1A). The cultures were incubated with shaking at 26°C overnight. The next day, the cultures were diluted 1:167 in HI broth (resulting in an optical density at 600 nm (OD600) of ∼0.02) with appropriate antibiotics and were grown at 26°C for approximately six generations. The cultures were subjected to centrifugation, and the bacterial pellet was suspended in phosphate-buffered saline (PBS) and diluted to 104 CFU per milliliter in PBS. We observed spontaneous loss of virulence for colonies of KIM5caf1A recovered from the frozen stock on two occasions. To ensure that bacteria recovered from frozen stock were virulent, a preliminary test was made by injection of bacteria into naïve mice approximately 1 week before the challenge of vaccinated mice.
Y. pestis KIM5/GFP (14) was used in phagocytosis assays. Cultures were grown overnight with aeration in HI broth supplemented with Ap (25 μg/ml) and chloramphenicol (25 μg/ml) at 28°C. The next day, the cultures were diluted 1:40 in the same medium supplemented with 2.5 mM CaCl2 and incubated for 2 h at 37°C with aeration in the presence of 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG) to induce expression of green fluorescent protein (GFP). Growth of KIM5 at 37°C for 2 h results in very low levels of F1 expression on the bacterial surface (40). The bacterial cultures were centrifuged, resuspended in PBS, and diluted in PBS to ∼1.5 × 108 CFU per ml.
Purification and preparation of immunogens. (i) Purification of BDE.
YP44, YP46, YP56, or YP60 harboring the expression vector pHis6-YopB (44) was grown in LB supplemented with 20 mM sodium oxalate and 20 mM magnesium chloride as described previously (37), except that incubation at 26°C was for 2 h and incubation at 37°C was for 4.5 h in the presence of 0.05 mM IPTG. Bacterial cultures (∼300 ml) were centrifuged (6,000 × g), and the resulting supernatant was concentrated to ∼10 ml using Centricon Plus-70A filter devices and the manufacturer's guidelines (Millipore). CHAPS {3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate} (Sigma no. C3023) was added to the concentrated supernatant to a final concentration of 1 mM, and affinity protein purification was carried out using the small-scale purification batch method with a His Bind Purification Kit (Novagen). Purifications were carried out using ∼300 μl (settled bead volume) of His·Bind beads, and elution was performed using a volume of ∼1 ml. Protein samples were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Gelcode Blue (Pierce) staining to monitor purification. Eluted protein was extensively dialyzed against PBS at 4°C and stored at 4°C before use in immunizations.
(ii) Purification of LcrV.
A culture of Tuner (DE3) lacIp (pET28a lcrV), starting at an OD600 nm of 0.1, was grown in 100 ml of LB containing 25 μg/ml kanamycin with shaking at 37°C. After 3 h, IPTG was added to a final concentration of 0.1 mM, and incubation was continued for 3 h to allow for overexpression of His6-LcrV. A cell pellet was obtained by centrifugation, and the pelleted cells were lysed with BugBuster Reagent (Novagen). After centrifugation (20,000 × g), the resulting supernatant (∼2 ml) was subjected to affinity chromatography using a His-Trap column (Amersham) and an AKTA fast protein liquid chromatography system. His6-LcrV protein was eluted as a single highly purified peak with an imidazole gradient. Fractions containing His6-LcrV were dialyzed against PBS and stored at −80°C. Samples of the starting supernatant, chromatography fractions, and dialyzed His6-LcrV were analyzed by SDS-PAGE with GelCode Blue staining.
To prepare immunogens for injection, protein samples (corresponding to 15 μg of LcrV per injection and 30 μg of BDE per injection) were mixed with adjuvants (MPL plus TDM; Sigma no. M6536) or alum (Imject Alum no. 77161; Pierce) according to the manufacturers’ guidelines. Adjuvant controls were prepared by mixing 100 μl of adjuvant with 100 μl of PBS. The final volumes used for injection were approximately 200 μl (100 μl MPL plus TDM, 100 μl BDE), 200 μl (100 μl MPL plus TDM, 100 μl LcrV), 250 to 290 μl (100 μl alum, 150 to 190 μl BDE), 250 to 290 μl (100 μl alum,150 to 190 μl LcrV), 200 to 250 μl (100 μl alum, 100 to 150 μl BD), or 250 μl (100 μl alum, 150 μl B).
Immunization and infection of mice.
All experiments were carried out with the approval of the Stony Brook University IACUC. Female BALB/c mice were purchased from Taconic Laboratories. Blood samples obtained before (prebleed) or after active immunization were collected using a retro-orbital procedure. Serum was isolated from blood samples using Serum-Gel serum separator tubes (Sarstedt) following the manufacturer's guidelines. For active immunization, 9-week-old mice were given a subcutaneous injection in the nape of the neck with immunogen/adjuvant mixture. Primary immunization was followed by boosts 2 weeks and 4 weeks later. The mice were bled 10 days after the final boost to obtain serum for immunoblotting, enzyme-linked immunosorbent assay (ELISA), or passive immunization. Challenge with Y. pestis was performed 30 days after the last boost.
Passive immunization was performed using 14- to 15-week-old mice. Serum for passive immunization was obtained by cardiac puncture of euthanized mice. Serum samples used for passive immunization were tested by ELISA before use to confirm that they contained the expected titers of immunoglobulin G (IgG) antibodies to BDE, BD, or LcrV (Table 1). The serum samples were pooled and used neat or mixed 1:1 with PBS. Mice were given intraperitoneal injections of 200 μl (100 μl serum plus 100 μl PBS or 200 μl of serum) on day +1 or days −1, 0, and +1 relative to infection.
TABLE 1.
Total IgG titers determined by ELISA
| Expta (adjuvant) | Mice immunized with: | ELISA antigen | Titerb |
|---|---|---|---|
| 1 (MPL + TDM) | LcrV | LcrV | 1 × 10−5 |
| BDE | BDE | 1 × 10−5 | |
| LcrV | 1 × 10−3 | ||
| 2 (alum) | LcrV | LcrV | 1 × 10−5 |
| BDE | BDE | 1 × 10−5 | |
| LcrV | 1 × 10−3 | ||
| 3 (alum) | LcrV | LcrV | 1 × 10−5 |
| BDE | BDE | 1 × 10−5 | |
| LcrV | 1 × 10−3 | ||
| 4 (alum) | BDE | BDE | 1 × 10−5 |
| BD | BDE | 1 × 10−5 | |
| B | BDE | 1 × 10−5 | |
| BDE | LcrV | <1 × 10−3 | |
| BD | LcrV | <1 × 10−3 | |
| B | LcrV | <1 × 10−3 |
The mice were injected via the lateral tail vein with 100 μl of Y. pestis suspension in PBS (∼1,000 CFU or 100 50% lethal dose [LD50]). The mice were monitored three times a day for severe signs of disease. Mice exhibiting severe signs of disease (hunched posture, ruffled fur, and immobility upon stimulation) were considered moribund and were humanely euthanized.
Depletion of anti-LcrV antibody from serum.
Beads coated with purified LcrV were used to deplete trace amounts of anti-LcrV antibodies from BDE or BD serum. His·Bind beads (His Bind Purification Kit; Novagen) in a volume of ∼50 μl (settled bead volume) were incubated at room temperature with an excess of purified His-LcrV (500 μg) for 30 min. Control beads were prepared in parallel by incubation in PBS. The beads were washed twice with 1 ml PBS, followed by incubation in 300 μl of normal mouse serum for 10 min at room temperature with occasional agitation. The beads were then incubated with 1,650 μl of BDE serum, 2,400 μl of BD serum, or 2,400 μl of control alum serum for 30 min. The beads were pelleted by centrifugation, and the serum supernatant was subjected to a second round of incubation with LcrV-coated beads as described above. Samples of the serum before and after depletion were analyzed by immunoblotting as described below.
SDS-PAGE and immunoblotting.
Protein samples were boiled for 5 min in Laemmli sample buffer containing 0.1 M dithiothreitol prior to electrophoresis. The proteins were separated on an SDS-10% polyacrylamide gel via electrophoresis and either transferred to nitrocellulose membranes for immunoblot analysis or stained with GelCode Blue for direct visualization. For immunoblot analysis, the membranes were blocked in Tris-buffered saline containing 0.05% Tween 20 (TBST) and 1% bovine serum albumin. Rabbit anti-LcrV serum (provided by Matt Nilles, University of North Dakota School of Medicine) was used at a dilution of 1:50,000 in TBST. Mouse anti-BDE, anti-LcrV, or control (alum) serum was used for immunoblotting at a dilution of 1:1,000 in TBST. Secondary horseradish peroxidase-conjugated anti-rabbit antibody (Jackson) was diluted 1:10,000 in TBST, and secondary horseradish peroxidase-conjugated anti-mouse antibody (Jackson) was diluted 1:50,000 in TBST. The immunoblots were developed by chemiluminescense (Perkin Elmer).
Measurement of antibody response by ELISA.
The presence of antibodies in immune sera was detected using antigen excess ELISA. Wells of 96-well plates were coated with antigen (0.5 μg antigen in 100 μl of 0.3 N sodium carbonate buffer, pH 9.6) by incubation overnight at 4°C. The antigen solution was removed, and the wells were washed and then blocked by incubation with 200 μl of PBS containing 0.05% Tween 20 and 1% bovine serum albumin (PBSTB) for 1 h at room temperature. The blocking solution was removed and replaced with immune sera serially 10-fold diluted in PBSTB (100 μl per well). After incubation at 37°C for 1 h, the wells were washed. Secondary horseradish peroxidase-conjugated anti-mouse antibody (Jackson) diluted 1:50,000 in PBSTB was added to each well (100 μl per well). After a 1-h incubation at 37°C, the secondary reagent was removed, the wells were washed, and 50 μl of chromogenic substrate 1-Step Ultra TMB (tetramentylbenzidine)-ELISA reagent (Pierce) was added to each well. After incubation for 5 to 30 min, 50 μl of 2 M sulfuric acid was added to each well to stop the reaction. The absorbance was read at 450 nm.
An ImmunoPure Monoclonal Antibody Isotyping kit (Pierce no. 37501) was used following the recommended guidelines to measure antibody isotypes in immune sera by ELISA.
Phagocytosis assay.
The mouse macrophage-like cell line RAW 264.7 (ATCC TIB-71) was grown in cell culture medium (Dulbecco's modified Eagle's medium plus Glutamax [Gibco BRL] supplemented with 10% heat-inactivated fetal calf serum [HyClone] and 1 mM sodium pyruvate) in a 5% CO2 humidified incubator at 37°C. Macrophages were seeded into wells of 24-well plates containing glass coverslips (1.5 × 105 cells in 1 ml of culture medium) and allowed to incubate overnight. Ten microliters of KIM5/GFP suspended in PBS (∼1.5 × 106 CFU) was mixed with 10 μl of pooled serum (BDE, LcrV, or alum control) or 10 μl of PBS (no-serum control) and incubated at 37°C for 10 min. Serum samples used for opsonization were tested by ELISA before use to confirm that they contained the expected titers of IgG antibodies to BDE or LcrV (Table 1). The bacteria were then diluted in 1 ml of cell culture medium, which was used to replace the medium overlying the macrophages. Infection was performed at a multiplicity of infection of 10 bacteria per macrophage. The plates were centrifuged for 5 min at 95 × g to induce contact between the bacteria and the macrophages. After incubation at 37°C with 5% CO2 for 25 min, the macrophages were washed once with prewarmed PBS and then fixed in 2.5% paraformaldehyde in PBS at room temperature for 30 min. Extracellular bacteria were then immunolabeled with rabbit anti-Yersinia antiserum SB349, followed by goat anti-rabbit antibody conjugated to Alexa Fluor 594 (Molecular Probes), as described previously (15), except that the permeabilization step was omitted. The coverslips were mounted on slides and examined by epifluorescence microscopy using a Zeiss Axioplan2 microscope equipped with a 40× objective. A Spot camera (Diagnostic Instruments) was used to capture sequentially Alexa Fluor 594 (red), GFP (green), and phase-contrast images in three random fields from each coverslip. The images were overlaid using Adobe Photoshop 6.0, and the percentage of internalized bacteria was quantified by counting the number of macrophage-associated intracellular bacteria (green) and extracellular bacteria (overlay of red and green). The number of intracellular bacteria was divided by the sum of internalized and extracellular bacteria and multiplied by 100.
Sequence comparison of YopB and YopD proteins from Y. pestis and Y. pseudotuberculosis.
The percent amino acid identity between YopB and YopD proteins from Y. pestis KIM (39), Y. enterocolitica W22703, and Y. pseudotuberculosis YPIII (17) was determined using BLASTP 2.2.17, available through the NCBI website (1).
Statistical analysis of data.
Mouse survival curves were plotted using Prism (GraphPad) and analyzed for significant differences using the Mantel-Haenszel log rank test. The results of phagocytosis assays were analyzed by analysis of variance and the Tukey posttest (Prism).
RESULTS
Purification of secreted BDE translocon complexes.
YopB and YopD contain hydrophobic regions predicted to function as transmembrane domains (9, 13, 44). Due to this hydrophobic character, recombinant forms of YopB and YopD are poorly expressed and largely insoluble in E. coli in the absence of their chaperone, SycD (34, 44). These characteristics present a barrier to the purification of native forms of these proteins in sufficient quantities for immunization studies. Previously, we created YP44, a multi-yop mutant of Y. pseudotuberculosis (YopEHJOKMB−) that harbors an expression vector coding for YopB with an N-terminal His tag (pHisYopB) (44). We found that His-YopB and YopD secreted from YP44(pHisYopB) under T3SS-inducing growth conditions (low calcium; 37°C) accumulated as soluble proteins (44). Furthermore, we were able to purify complexes of His-YopB and YopD from supernatants of bacterial growth media using Ni2+ chelation chromatography (44). We took advantage of these observations to purify secreted His-YopB and YopD as a complex for immunization studies. Experiments undertaken to characterize the complex purified from Y. pseudotuberculosis strain YP44(pHisYopB) showed that a truncated form of YopE (YopE1-131) that is expressed by this strain copurified with His-YopB and YopD (see Materials and Methods) (data not shown). It is not surprising that YopE copurified with the complexes, because YopE has been shown to bind to YopD in vitro (20), although the importance of this interaction has not been determined. In preliminary studies, the translocon complexes that were purified from YP44(pHisYopB) and used for immunization studies contained YopE1-131. In subsequent experiments, Y. pseudotuberculosis strain YP46 [YopE(R144A)HJOKMB−] was used to purify translocon complexes (see Materials and Methods). The complexes that were purified from YP46 and used for immunization contained the full-length catalytically inactive YopE protein YopE(R144A). Results obtained with the latter complexes are the focus of the work presented here. The presence of YopE polypeptides in the complexes is likely not important for the protective immunity we observed, since recombinant YopE showed no activity as a vaccine candidate (25) and a complex containing only YopB and YopD elicited protective immunity (see below).
To purify secreted translocon complexes, YP46(pHisYopB) was grown under T3SS-inducing conditions, culture supernatant was concentrated and subjected to affinity purification using Ni2+ chelation resin (His·Bind beads), and the results were analyzed by SDS-PAGE (see Materials and Methods). The starting supernatant contained four major bands corresponding to His-YopB, LcrV, YopD, and YopE (Fig. 1, lane 2). After incubation with His·Bind beads, the majority of His-YopB was depleted from the supernatant (lane 3) and became associated with the beads (lane 4). A small percentage of the YopD and YopE that was present in the supernatant became associated with the beads (lane 4) and, along with His-YopB, could be eluted from the beads with imidazole (lane 5). We found that inclusion of CHAPS (1 mM final concentration) in the supernatant and elution buffer was important to obtain elution of the complex from the beads. LcrV did not bind to the beads and therefore was not present at significant levels in the eluted fraction (lanes 4 and 5). Densitometric scans of gels indicated a ratio of 1:2:1 for His-YopB/YopD/YopE, although it was not determined if all three proteins are in a single complex. For simplicity, we hereafter refer to complexes copurifying with His-YopB as BDE.
FIG. 1.
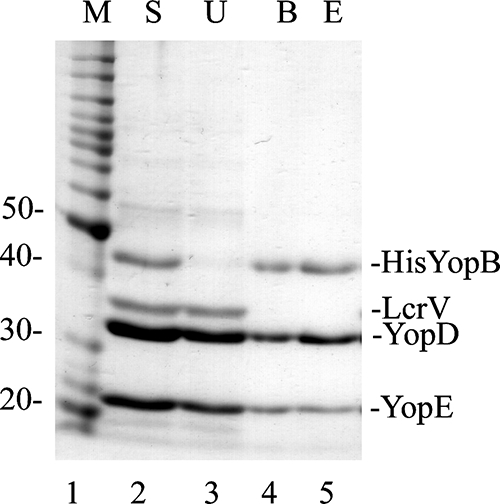
Purification of BDE translocation complex. A culture of Y. pseudotuberculosis YP46(pHis6-YopB) was grown under T3SS-inducing conditions in the presence of IPTG, and a culture supernatant containing secreted proteins was obtained following centrifugation. The culture supernatant was concentrated, supplemented with CHAPS (to 1 mM), and subjected to Ni2+ chelation affinity purification using His·Bind beads. Samples of the concentrated supernatant (S; lane 2), unbound proteins (U; lane 3), proteins bound to the beads (B; lane 4), and proteins eluted from the beads with imidazole (E; lane 5) were analyzed by SDS-PAGE and GelCode Blue staining. Lane 1 contains a protein molecular weight (MW) standard (M), and corresponding MWs are shown on the left. The positions of His-YopB, LcrV, YopD, and YopE are indicated on the right.
Antibody responses to BDE in immunized mice.
Recombinant LcrV was overexpressed in E. coli and purified from lysates (see Materials and Methods) for use as a positive control protective antigen. Samples of purified BDE and LcrV were resolved by SDS-PAGE and visualized by GelCode Blue staining (Fig. 2, lanes 1 and 2). BDE and LcrV were used to immunize BALB/c mice following a schedule of one primary injection and two booster injections at 14-day intervals. Control mice received injections of adjuvant alone. Two different adjuvants were used, either MPL plus TDM or alum (see Materials and Methods). Ten days after the second boost, serum was isolated, pooled, and used in an immunoblotting assay to detect IgG responses to BDE and LcrV. IgG antibodies in anti-BDE serum reacted strongly with three antigens corresponding to His-YopB, YopD, and YopE (Fig. 2B, lane 4). There was also weaker reaction with unknown antigens in the BDE preparation that migrated between 60 and 100 kDa (lane 4). Sera from LcrV-immunized mice contained IgG antibodies that recognized purified LcrV (lane 7), but not BDE (lane 6). Using a more sensitive immunoblotting assay performed with rabbit anti-LcrV hyperimmune serum (see Materials and Methods), trace amounts of LcrV were detected in preparations of BDE (data not shown). The trace amount of LcrV contaminating BDE generated an extremely weak antibody response because IgG antibodies in mouse anti-BDE serum that react with LcrV were difficult to detect by immunoblotting (note the absence of signal in Fig. 2, lane 5; also see Fig. 6). Thus, in mice immunized with BDE, the primary IgG response was to the three major proteins in the preparation (His-YopB, YopD, and YopE).
FIG. 2.
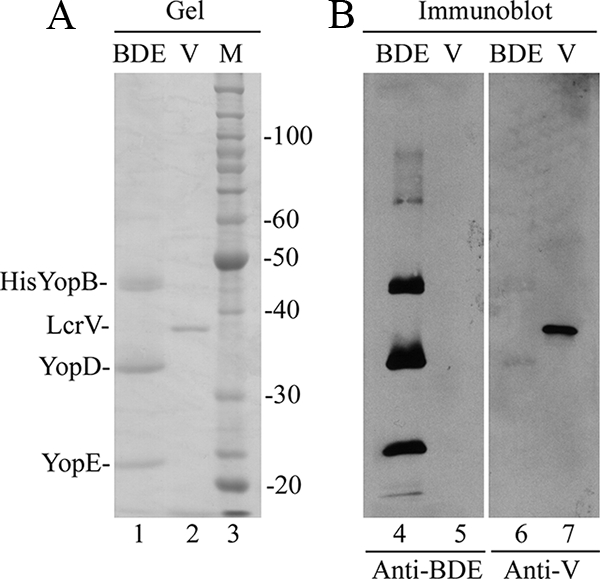
SDS-PAGE and immunoblot analyses of BDE and LcrV. (A) Samples of purified BDE (lane 1) and His-LcrV (V; lane 2) were resolved by SDS-PAGE and visualized in the gel by staining them with GelCode Blue. The positions of His-YopB, LcrV, YopD, and YopE are shown on the left. A molecular weight (MW) standard (M; lane 3) and corresponding MWs are shown on the right. (B) Proteins in gels that duplicate those shown in panel A were transferred to membranes. The membranes were processed by immunoblotting to detect antigens recognized by IgG antibodies in sera from mice immunized with BDE (anti-BDE; lanes 4 and 5) or LcrV (anti-V; lanes 6 and 7).
FIG. 6.

Evaluation of sera for protective antibodies by macrophage phagocytosis assay. KIM5/GFP was incubated with no serum or the indicated sera for 10 min. The bacteria were then used to infect (multiplicity of infection, 10) RAW 264.7 macrophages seeded onto coverslips at 1.5 × 105 cells per well in a 24-well plate. After incubation at 37°C for 30 min, the cells were washed, fixed, and processed for immunofluorescence microscopy to distinguish intracellular and extracellular bacteria. The samples were viewed with a fluorescence microscope, and the numbers of macrophage-associated intracellular and extracellular bacteria were quantified in three independent fields. Percent internalization was calculated as the number of intracellular bacteria divided by the total number of cell-associated bacteria. The results were compiled from three independent experiments. *, significantly different compared to adjuvant serum (P < 0.05; analysis of variance). The error bars indicate standard deviation.
ELISA was performed to obtain a more quantitative measurement of antibody responses to BDE and LcrV in immunized mice. Pooled sera from mice immunized with BDE or LcrV were reacted with the corresponding purified antigen, and the results of ELISA for total IgG showed the presence of specific antibody at similar titers (10−5) (Table 1). ELISA with BDE serum reacted against LcrV revealed a 100-fold-lower titer for total IgG (10−3) (Table 1). Similar results were obtained using the MPL-plus-TDM adjuvant (experiment 1) or the alum adjuvant (experiments 2 and 3). These results indicate that vaccinations with BDE or LcrV elicit similar levels of IgG antibodies in mice.
Antibody subclass titers for BDE were also determined by ELISA. As shown in Table 2, the highest titers were for IgG1, IgG2a, and IgG2b when MPL plus TDM was used as the adjuvant (experiment 1). When alum was used as the adjuvant, IgG1 had the highest titers (experiments 2 and 3), consistent with the known property of this adjuvant to skew antibody development toward a TH2 phenotype.
TABLE 2.
Anti-BDE subclass titers determined by ELISA
| Subclass | Titera for expt:
|
||
|---|---|---|---|
| 1 | 2 | 3 | |
| IgA | 1 × 10−3 | 1 × 10−3 | 1 × 10−3 |
| IgM | 1 × 10−3 | 1 × 10−3 | 1 × 10−3 |
| IgG1 | 1 × 10−5 | 1 × 10−6 | 1 × 10−5 |
| IgG2a | 1 × 10−5 | 1 × 10−4 | 1 × 10−4 |
| IgG2b | 1 × 10−5 | 1 × 10−4 | 1 × 10−4 |
| IgG3 | 1 × 10−4 | 1 × 10−3 | 1 × 10−4 |
The titer is the lowest dilution that gives OD value above 0.1.
Active vaccination of mice with BDE against Y. pestis infection.
Mice vaccinated with BDE or with LcrV or injected with adjuvant as a control were challenged with Y. pestis to test for protection. Experiments used a conditionally virulent Δpgm strain (KIM5) as the infection model under biological safety level 2 conditions. Y. pestis KIM5 delivered intravenously (i.v.) has an LD50 of ∼10 CFU in BALB/c mice (23, 33). Actively vaccinated mice were infected with ∼1,000 CFU (100 LD50) of KIM5 or KIM5caf1A. The KIM5caf1A mutant lacks the Caf1A usher and is unable to secrete F1 or assemble capsule on its surface (39). This mutant was used to determine if the presence of the F1 capsule would interfere with vaccine-mediated protection (3). As shown in Fig. 3A, mice vaccinated with either BDE or LcrV formulated with MPL plus TDM were completely protected from lethal infection by KIM5caf1A, while the adjuvant control mice were not. Similar results were obtained when mice were vaccinated with the immunogens formulated with alum as the adjuvant (Fig. 3B). However, when mice were challenged with F1+ KIM5, no significant difference in protection was observed between groups immunized with BDE or alum control (Fig. 3C). Mice vaccinated with LcrV were fully protected against F1+ KIM5 (Fig. 3C). These results suggest that the presence of the F1 capsule interferes with protection afforded by active vaccination with BDE.
FIG. 3.

Protection of actively vaccinated mice against lethal Y. pestis infection. Groups of eight BALB/c mice immunized with BDE or LcrV or injected with adjuvant as a control were infected via the i.v. route with ∼1,000 CFU (∼100 LD50) of Y. pestis KIM5caf1A (A and B) or KIM5 (C). Percent survival was monitored over a 21- day period. (A) The adjuvant was MPL plus TDM. (B and C) The adjuvant was alum. Survival curves were significantly different (LcrV versus adjuvant or BDE versus adjuvant, P < 0.05) for the experiments (A and B) as determined by a log rank test. Note that the curves for LcrV and BDE are superimposed in panels A and B.
Additional results showing that the F1 capsule allowed Y. pestis to overcome vaccine-induced immunity of the BDE immunogen came from experiments carried out under biological safety level 3 conditions with wild-type Y. pestis. The challenge was carried out as described previously (8), except that the bacteria were grown at 37°C and the inoculation volume was 25 μl. The LD50 for wild-type CO92 in this infection model is 1,000 CFU (8; D. Perlin and S. Parks, unpublished data). Vaccinated or control BALB/c mice were challenged with wild-type CO92 by intranasal challenge with ∼10,000 CFU (10 LD50) of Y. pestis CO92. The adjuvant (n = 8) and BDE (n = 8) groups all died on day 3 postinfection. All mice in the LcrV group (n = 8) survived to day 5 postinfection (Perlin and Parks, unpublished). These results indicate that, unlike LcrV, the BDE immunogen did not elicit protective immunity against F1+ Y. pestis.
Passive immunization of mice against Y. pestis infection with BDE serum.
Passive-immunization studies were carried out to determine if BDE vaccination was eliciting antibodies protective against KIM5caf1A infection. Sera were isolated from mice immunized with BDE or LcrV or injected with alum as a control. Initially, naïve mice were passively immunized with a single dose of serum (100 μl) injected intraperitoneally (i.p.) 1 day after challenge (day +1) with KIM5caf1A. Mice passively vaccinated in this manner with anti-BDE serum, or control serum, were not protected against lethal infection, while mice given LcrV serum were protected (Fig. 4A). To determine if additional doses of BDE serum would result in protection, the passive-immunization protocol was changed to include i.p. injections of serum on days −1, 0, and +1 relative to infection. Significant protection of mice against lethal KIM5caf1A infection was observed when the mice were passively immunized with BDE serum, but not control serum, using this three-dose protocol (Fig. 4B). In addition, full protection of mice was observed when the mice were passively immunized by the same regime with larger doses of BDE serum (200 μl) (Fig. 4B). These results indicate that BDE-vaccinated mice are producing protective antibodies but that protective antibodies are present at lower levels in BDE serum than in LcrV serum.
FIG. 4.

Passive immunization of naïve mice with anti-LcrV or anti-BDE serum. Naïve BALB/c mice were infected i.v. with a lethal dose of F1− Y. pestis. Passive immunization was performed by i.p. injection of doses of pooled anti-LcrV, anti-BDE, or control (alum or preimmune) sera. Mouse survival was recorded for 21 days. (A) Mice (n = 8 per group) were injected with a single dose (100 μl) of sera on day +1 relative to infection. (B) Mice were injected with three doses of sera (100 μl or 200 μl in each dose) on days −1, 0, and +1. The difference in the survival curves was significant (BDE at 100 μl versus alum at 100 μl, P < 0.05) as determined by a log rank test. For BDE at 100 μl and alum at 100 μl, n = 8; for BDE at 200 μl, n = 4. (C) Mice (n = 5 per group) were injected with three doses of sera (100 μl in each dose) on days −1, 0, and +1. BDE-depleted sera were exposed to LcrV-coated beads to remove anti-LcrV antibody prior to use for passive immunization. The difference in the survival curves was significant (non-BDE depleted versus preimmune, P < 0.05; BDE depleted versus preimmune, P < 0.05) as determined by a log rank test.
To determine if trace levels of anti-LcrV antibodies present in the BDE serum could be contributing to the protection observed, a depletion experiment was carried out (see Materials and Methods). BDE serum was incubated with beads coated with purified recombinant LcrV, and after removal of the beads, the serum was used for passive immunization. As shown in Fig. 4C, the depleted BDE serum significantly increased the survival of mice infected with KIM5caf1A compared to mice given preimmune serum. Although it appeared that better protection was obtained with the depleted serum than with the control nondepleted BDE serum (Fig. 4C), the difference between these curves was not statistically significant (P = 0.0625). Immunoblots that were overexposed after incubation with the BDE serum confirmed that trace amounts of anti-LcrV IgG were removed by the depletion procedure (Fig. 5, compare lanes 2 and 4). Therefore, the protection observed following vaccination with BDE was unlikely to be mediated by anti-LcrV antibody.
FIG. 5.

Evaluation by immunoblotting of anti-LcrV depletion. Samples of BDE (lanes 1 and 3) or LcrV (lanes 2 and 4) were resolved by SDS-PAGE, transferred to filters, and incubated with BDE serum before (Pre) or after (Post) depletion of anti-LcrV antibodies. After being processed for immunoblotting with secondary antibody, the films were overexposed to allow detection of anti-LcrV signal in predepleted sera (lane 2). The positions of His-YopB, YopD, and YopE are shown on the left. The position of LcrV is shown on the right.
Evaluation of BDE sera for protective antibodies by macrophage phagocytosis assay.
Previous studies have shown that protective anti-LcrV antibodies increase phagocytosis of Y. pestis by macrophages (10, 52). An in vitro assay was used to determine if antibodies in sera of mice immunized with BDE would increase phagocytosis of Y. pestis by macrophages. KIM5/GFP bacteria grown under conditions that do not result in extensive F1 production (see Materials and Methods) were opsonized with pooled immune sera for 10 min. The opsonized bacteria were then used to infect cultured murine RAW264.7 macrophage-like cells for 30 min, and the extent of phagocytosis was measured by microscopy-based assay. As shown in Fig. 6, increased bacterial phagocytosis was observed when KIM5 was opsonized with control serum compared to no-serum treatment. This likely represents increased phagocytosis as a result of complement opsonization, as shown previously in similar experiments utilizing Y. enterocolitica (16). Opsonization with anti-LcrV serum significantly increased phagocytosis of KIM5 compared to the control serum condition (Fig. 6), showing that anti-LcrV antibodies further stimulate uptake of the bacteria beyond that seen with complement alone. Antisera from mice vaccinated with BDE reproducibly increased bacterial phagocytosis compared to the control serum condition, although this difference was not statistically significant when the results of three independent experiments were analyzed (Fig. 6). We conclude that anti-BDE antibodies are unable to significantly increase phagocytosis of Y. pestis by macrophages, possibly because the levels of protective antibodies in BDE serum are lower than those in anti-LcrV serum.
Purification and immunological characterization of secreted B and BD translocon complexes.
The feasibility of purifying secreted His-YopB alone (B) or a His-YopB/YopD complex (BD) was investigated. For this purpose, two new Y. pseudotuberculosis strains were constructed, YP56 and YP60 (see Materials and Methods). YP56 and YP60 were transformed with pHisYopB and cultured under T3SS-inducing conditions, and the culture supernatant was concentrated and subjected to affinity purification as described above. Analysis of the eluted material by SDS-PAGE showed that BD and B could be successfully purified (Fig. 7A, lanes 2 and 3, respectively). However, yields of BD were consistently higher than those of B. Because of difficulties encountered in obtaining large quantities of purified B, we carried out challenge experiments in mice vaccinated with BD only (see below). However, sufficient amounts of B were obtained to carry out a small-scale immunization trial to compare its immunogenicity with that of BD. For this purpose, mice were immunized with BD or B formulated with alum by our standard protocol, and pooled serum samples were analyzed by immunoblotting to detect IgG responses to His-YopB, YopD, or LcrV. As shown in Fig. 7B, sera from mice immunized with BD contained IgG antibodies that strongly reacted with YopD but much more weakly with His-YopB (lane 1). A signal corresponding to LcrV was detected only when the film was overexposed (lane 2 and data not shown), indicating that anti-LcrV IgG was present in this serum at trace levels. These results showed that YopD was the immunodominant antigen in BD. Sera from mice immunized with B contained antibodies that strongly reacted with His-YopB; weak cross-reaction with YopD was also seen (Fig. 7B, lane 3). Again, a signal corresponding to LcrV was detected only when the film was overexposed (lane 4 and data not shown). ELISA performed with the BD or B serum showed the presence of IgG antibodies to the respective antigens at similarly high titers (10−5) (Table 1, experiment 4). In contrast, IgG antibodies in the BD or B serum that reacted with LcrV were present at a 100-fold-lower level (10−3 titer) (Table 1, experiment 4).
FIG. 7.
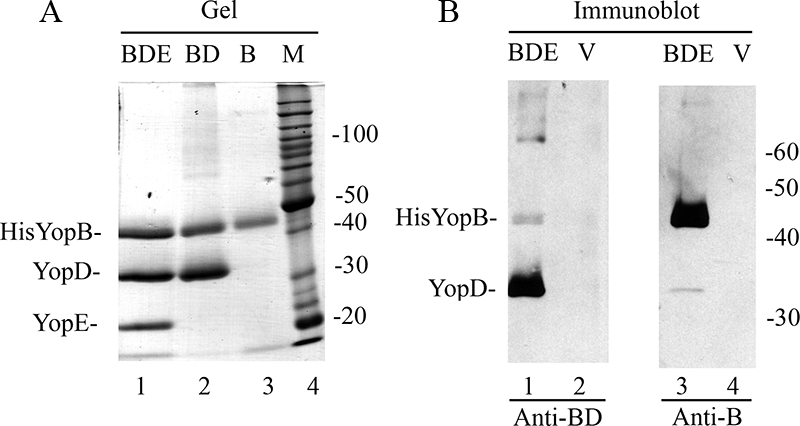
Purification and immunological characterization of BD and B. (A) Samples of purified BDE (lane 1), BD (lane 2), or B (lane 3) were resolved by SDS-PAGE and visualized by GelCode Blue staining. Lane 4 contains a molecular weight (MW) standard (M). (B) Samples of purified BDE (lanes 1 and 3) or LcrV (lanes 2 and 4) were resolved by SDS-PAGE and immunoblotted with anti-BD serum (lanes 1 and 2) or anti-B serum (lanes 3 and 4). The positions of immune-reactive antigens are indicated on the left, and the positions of MW standards are shown on the right.
Immunization of mice against Y. pestis with BD.
To determine if vaccination of mice with BD elicits protective immunity, we first challenged actively immunized mice with KIM5caf1. As shown in Fig. 8A, significant protection against lethal infection was observed in BD-immunized mice compared to the control alum-injected mice. Next, pooled sera from control alum-injected mice, or mice vaccinated with BD, were used to passively immunize mice against Y. pestis challenge. The sera (both BD and control alum) were subjected to immune depletion with LcrV-coated beads to ensure that any observed protection was not due to the presence of low levels of anti-LcrV antibodies. As shown in Fig. 8B, significant protection of mice against lethal challenge was observed when the mice were passively immunized with depleted BD serum, but not depleted alum serum. Given that YopD is the immunodominant antigen in BD (Fig. 7B, lane 1), these results suggest that anti-YopD antibodies may be sufficient for protection against KIM5caf1A infection.
FIG. 8.

Active and passive immunization of mice with BD. (A) Groups of eight BALB/c mice were immunized with BD or injected with alum adjuvant as a control. The immunized mice were infected with KIM5caf1A, and mouse survival was recorded for 21 days. The difference in the survival curves was significant (BD versus alum, P < 0.05) as determined by a log rank test. (B) Naïve BALB/c mice (n = 7 per group) were infected i.v. with KIM5caf1A. Passive immunization was performed by i.p. injection of pooled alum control sera or BD sera (100 μl in each dose) on days −1, 0, and +1. BD serum was exposed to LcrV-coated beads to deplete anti-LcrV antibody prior to use in passive immunization. Alum serum was mock depleted in parallel as a control. Mouse survival was recorded for 21 days. The difference in the survival curves was significant (BD depleted versus alum depleted, P < 0.05) as determined by a log rank test.
DISCUSSION
The goal of this research was to determine if the Yop translocon could function as a protective antigen able to elicit humoral immunity against experimental Y. pestis infection of mice. New vaccines and therapeutics are needed to prepare for the possible use of Y. pestis as a biological-weapon agent. Several scenarios involving genetic manipulation of Y. pestis can be envisioned that would limit the effectiveness of current vaccines and therapeutics. These include (i) the isolation of strains resistant to antibiotics, (ii) the generation of F1− mutants, and (iii) the generation of strains in which protective epitopes within LcrV have been altered. The last point is significant because there is evidence for natural variation of LcrV within one of the regions known to encode a protective epitope (42). To counteract these threats, additional protective antigens need to be identified and incorporated into new subunit vaccines. Alternatively, antibodies specific for newly identified protective antigens could be used to develop novel immunotherapeutics to treat infections caused by antibiotic-resistant or vaccine-resistant strains of Y. pestis.
YopD and YopB can be considered good candidates for protective antigens for several reasons. First, unlike F1, YopB and YopD are essential for virulence of yersiniae (18, 19). Second, the YopB and YopD proteins of Yersinia species are highly conserved and do not show the extensive sequence variation observed in LcrV. For example, the YopB and YopD proteins of Y. pseudotuberculosis and Y. pestis are very similar (400 out of 401 amino acids identical for YopB and 305 out of 306 amino acids identical for YopD). The YopB and YopD proteins are somewhat more divergent between Y. pestis and Y. enterocolitca. For YopD, the identity between Y. pestis and Y. enterocolitica is 301 residues out of 306 amino acids (98% identical); for YopB, the identity is 383 residues out of 401 amino acids (95% identical). The YopB and YopD proteins used in this study as protective immunogens against Y. pestis were derived from Y. pseudotuberculosis, showing direct evidence for cross-protection.
In addition to the standard BDE complex, which contains catalytically inactive YopE, we also purified a BDE translocon complex containing active YopE (unpublished data). Experiments were undertaken to determine if exposure of the latter complex to cultured cells under different conditions would result in translocation of active YopE. All results were negative, which suggests that the complex we have purified is biologically inactive with respect to translocation activity.
We do not know which of the proteins in BDE are required for eliciting protective antibodies, although we hypothesize that both YopB and YopD contribute. This hypothesis is based on the known protective activity of the recombinant YopD vaccine (3), as well as a previous study reporting that anti-YopB antibodies could decrease Y. enterocolitica colonization of intestinal lymphoid tissues in mice (7). We engineered additional Y. pseudotuberculosis strains that allowed us to purify a BD complex or B alone. The results of immunization studies with these preparations showed that both vaccines elicited high-titer antibodies. In addition, mice actively vaccinated with BD or passively immunized with BD serum were protected against lethal Y. pestis infection. Because YopD appeared to be immunodominant in the BD preparation, it is possible that anti-YopD antibodies are sufficient to provide protection against F1− Y. pestis. However, it remains possible that anti-YopB antibodies can also have protective activity. Examination of this possibility will require optimization of yields of purified B so that sufficient material is available for future vaccination studies.
LcrV has been shown to localize to the tips of T3SS needles, and recently it has been proposed to act as a folding platform for YopB and YopD as they exit the T3SS needle in an unfolded form (5). Our results showing that antibodies recognizing translocon complexes can protect mice against Y. pestis infection suggest that YopB and/or YopD is exposed, at some point, to antibody during its functions as a translocator. Andrews et al. also hypothesized that antibodies to YopD interfere with its translocation function, although it was not demonstrated that anti-YopD antibodies are protective (3). There is currently no direct evidence that YopB or YopD localizes to the T3SS needle tip in a stable fashion (31). Recent studies of T3SS function in Shigella have shown that IpaB can stably localize to the tip of the needle (35, 51). Interestingly, IpaB localization to the needle tip was induced by deoxycholate, a component of bile salts (35). It is possible that YopB and or YopD can also, in response to a signal, localize in a stable fashion to the Yersinia T3SS needle tip. Serum proteins, such as albumin, have been shown to induce secretion of YopB and YopD from Y. enterocolitica into tissue culture media (26). We hypothesize that in response to serum, YopB and/or YopD can localize to the tip of the T3SS needle and can be recognized by antibody, leading to neutralization of translocon function.
Our results show that a caf1A mutant of KIM5, unable to secrete F1 or assemble capsule (39), retains virulence in naïve mice challenged by i.v. infection. These results are consistent with earlier studies reporting that defined caf1 mutants of Y. pestis retain virulence in bubonic and pneumonic mouse infection models (38, 46). However, a role for F1 in virulence was revealed in mice vaccinated with BDE (this study) or recombinant YopD (3). Thus, the presence of F1 appears to allow Y. pestis to overcome protection mediated by antibodies against translocon proteins (3). How does F1 counteract antibody-mediated protection? The previous suggestion, that F1 masks YopD at the bacterial surface, blocking the antibody-antigen interaction (3), seems difficult to reconcile with (i) the fact that F1 does not overcome protection mediated by anti-LcrV and (ii) the concept that LcrV functions as an assembly platform for YopB and YopD (5). Thus, if antibodies are able to bind LcrV in the presence of F1, it would seem reasonable that antibodies would also bind YopB or YopD in the presence of F1. It is possible that F1 does not block antibody recognition of specific antigens (e.g., it blocks antibody access to YopB or YopD, but not LcrV) but rather plays a limited role in promoting resistance of Y. pestis to protective antibodies in general. In this scenario, it is the quality of the antibody-mediated protection that is important to reveal a function for F1. A prediction based on this hypothesis is that F1 would not allow Y. pestis to overcome protection afforded by anti-YopB or -YopD antibodies if antibodies with stronger protective activity could be raised.
In summary, we present the first evidence that anti-translocon antibodies can be used as immunotherapeutics against F1− Y. pestis. Future studies will be aimed at the development of highly protective MAbs specific for YopB and YopD. Such MAbs could be combined into a cocktail with protective anti-LcrV MAb (21), which could afford protection against wild-type, as well as vaccine-resistant, Y. pestis. In addition, MAbs specific for YopB and YopD could be used experimentally to address questions raised above concerning the locations of YopB and YopD with respect to the T3SS needle tip or the role of F1 in promoting resistance to protective antibodies.
Acknowledgments
We thank Steve Parks and David Perlin of the Public Health Research Institute for assistance with the mouse challenge assay performed at the Northeast Biodefense Center Small Animal Core. We also thank Jean Rooney and additional staff of the Department of Laboratory Animal Resources at Stony Brook University for assistance with experiments. We acknowledge the assistance of Gloria Viboud for construction of pSB890-ΔYopD. Matt Nilles at the University of North Dakota generously provided rabbit anti-LcrV serum.
This work was supported by the Northeast Biodefense Center (U54-AI057158-Lipkin and PO1-AI055621).
Editor: B. A. McCormick
Footnotes
Published ahead of print on 2 September 2008.
REFERENCES
- 1.Altschul, S. F., T. L. Madden, A. A. Schaffer, J. Zhang, Z. Zhang, W. Miller, and D. J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 253389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson, G. W., Jr., S. E. Leary, E. D. Williamson, R. W. Titball, S. L. Welkos, P. L. Worsham, and A. M. Friedlander. 1996. Recombinant V antigen protects mice against pneumonic and bubonic plague caused by F1-capsule-positive and -negative strains of Yersinia pestis. Infect. Immun. 644580-4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andrews, G. P., S. T. Strachan, G. E. Benner, A. K. Sample, G. W. Anderson, Jr., J. J. Adamovicz, S. L. Welkos, J. K. Pullen, and A. M. Friedlander. 1999. Protective efficacy of recombinant Yersinia outer proteins against bubonic plague caused by encapsulated and nonencapsulated Yersinia pestis. Infect. Immun. 671533-1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Black, D. S., and J. B. Bliska. 2000. The RhoGAP activity of the Yersinia pseudotuberculosis cytotoxin YopE is required for antiphagocytic function and virulence. Mol. Microbiol. 37515-527. [DOI] [PubMed] [Google Scholar]
- 5.Broz, P., C. A. Mueller, S. A. Muller, A. Philippsen, I. Sorg, A. Engel, and G. R. Cornelis. 2007. Function and molecular architecture of the Yersinia injectisome tip complex. Mol. Microbiol. 651311-1320. [DOI] [PubMed] [Google Scholar]
- 6.Brubaker, R. R. 2003. Interleukin-10 and inhibition of innate immunity to yersiniae: roles of Yops and LcrV (V antigen). Infect. Immun. 713673-3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burdack, S., A. Schmidt, E. Knieschies, M. Rollinghoff, and H. U. Beuscher. 1997. Tumor necrosis factor-alpha expression induced by anti-YopB antibodies coincides with protection against Yersinia enterocolitica infection in mice. Med. Microbiol. Immunol. 185223-229. [DOI] [PubMed] [Google Scholar]
- 8.Chiuchiolo, M. J., J. L. Boyer, A. Krause, S. Senina, N. R. Hackett, and R. G. Crystal. 2006. Protective immunity against respiratory tract challenge with Yersinia pestis in mice immunized with an adenovirus-based vaccine vector expressing V antigen. J. Infect. Dis. 1941249-1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cornelis, G. R. 2006. The type III secretion injectisome. Nat. Rev. Microbiol. 4811-825. [DOI] [PubMed] [Google Scholar]
- 10.Cowan, C., A. V. Philipovskiy, C. R. Wulff-Strobel, Z. Ye, and S. C. Straley. 2005. Anti-LcrV antibody inhibits delivery of Yops by Yersinia pestis KIM5 by directly promoting phagocytosis. Infect. Immun. 736127-6137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DeBord, K. L., D. M. Anderson, M. M. Marketon, K. A. Overheim, R. W. DePaolo, N. A. Ciletti, B. Jabri, and O. Schneewind. 2006. Immunogenicity and protective immunity against bubonic plague and pneumonic plague by immunization of mice with the recombinant V10 antigen, a variant of LcrV. Infect. Immun. 744910-4914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fields, K. A., M. L. Nilles, C. Cowan, and S. C. Straley. 1999. Virulence role of V antigen of Yersinia pestis at the bacterial surface. Infect. Immun. 675395-5408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Galan, J. E., and H. Wolf-Watz. 2006. Protein delivery into eukaryotic cells by type III secretion machines. Nature 444567-573. [DOI] [PubMed] [Google Scholar]
- 14.Grabenstein, J. P., H. S. Fukuto, L. E. Palmer, and J. B. Bliska. 2006. Characterization of phagosome trafficking and identification of PhoP-regulated genes important for survival of Yersinia pestis in macrophages. Infect. Immun. 743727-3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grabenstein, J. P., M. Marceau, C. Pujol, M. Simonet, and J. B. Bliska. 2004. The response regulator PhoP of Yersinia pseudotuberculosis is important for replication in macrophages and for virulence. Infect. Immun. 724973-4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grosdent, N., I. Maridonneau-Parini, M. P. Sory, and G. R. Cornelis. 2002. Role of Yops and adhesins in resistance of Yersinia enterocolitica to phagocytosis. Infect. Immun. 704165-4176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hakansson, S., T. Bergman, J. Vanooteghem, G. Cornelis, and H. Wolf-Watz. 1993. YopB and YopD constitute a novel class of Yersinia Yop proteins. Infect. Immun. 6171-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hartland, E. L., A. M. Bordun, and R. M. Robins-Browne. 1996. Contribution of YopB to virulence of Yersinia enterocolitica. Infect. Immun. 642308-2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hartland, E. L., S. P. Green, W. A. Phillips, and R. M. Robins-Browne. 1995. Role of YopD in the virulence of Yersinia enterocolitica. Contrib. Microbiol. Immunol. 13235-238. [PubMed] [Google Scholar]
- 20.Hartland, E. L., and R. M. Robins-Browne. 1998. In vitro association between the virulence proteins, YopD and YopE, of Yersinia enterocolitica. FEMS Microbiol. Lett. 162207-213. [DOI] [PubMed] [Google Scholar]
- 21.Hill, J., J. E. Eyles, S. J. Elvin, G. D. Healey, R. A. Lukaszewski, and R. W. Titball. 2006. Administration of antibody to the lung protects mice against pneumonic plague. Infect. Immun. 743068-3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hill, J., S. E. Leary, K. F. Griffin, E. D. Williamson, and R. W. Titball. 1997. Regions of Yersinia pestis V antigen that contribute to protection against plague identified by passive and active immunization. Infect. Immun. 654476-4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hines, J., E. Skrzypek, A. V. Kajava, and S. C. Straley. 2001. Structure-function analysis of Yersinia pestis YopM's interaction with alpha-thrombin to rule on its significance in systemic plague and to model YopM's mechanism of binding host proteins. Microb. Pathog. 30193-209. [DOI] [PubMed] [Google Scholar]
- 24.Inglesby, T. V., D. T. Dennis, D. A. Henderson, J. G. Bartlett, M. S. Ascher, E. Eitzen, A. D. Fine, A. M. Friedlander, J. Hauer, J. F. Koerner, M. Layton, J. McDade, M. T. Osterholm, T. O'Toole, G. Parker, T. M. Perl, P. K. Russell, M. Schoch-Spana, K. Tonat, et al. 2000. Plague as a biological weapon: medical and public health management. JAMA 2832281-2290. [DOI] [PubMed] [Google Scholar]
- 25.Leary, S. E., K. F. Griffin, E. E. Galyov, J. Hewer, E. D. Williamson, A. Holmstrom, A. Forsberg, and R. W. Titball. 1999. Yersinia outer proteins (YOPS) E, K and N are antigenic but non-protective compared to V antigen, in a murine model of bubonic plague. Microb. Pathog. 26159-169. [DOI] [PubMed] [Google Scholar]
- 26.Lee, V. T., and O. Schneewind. 1999. Type III machines of pathogenic yersiniae secrete virulence factors into the extracellular milieu. Mol. Microbiol. 311619-1629. [DOI] [PubMed] [Google Scholar]
- 27.Lilo, S., Y. Zheng, and J. B. Bliska. 2008. Caspase-1 activation in macrophages infected with Yersinia pestis KIM requires the type III secretion system effector YopJ. Infect. Immun. 763911-3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matson, J. S., K. A. Durick, D. S. Bradley, and M. L. Nilles. 2005. Immunization of mice with YscF provides protection from Yersinia pestis infections. BMC Microbiol. 538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Menard, R., P. Sansonetti, C. Parsot, and T. Vasselon. 1994. Extracellular association and cytoplasmic partitioning of the IpaB and IpaC invasins of S. flexneri. Cell 79515-525. [DOI] [PubMed] [Google Scholar]
- 30.Mills, J. A., J. M. Buysse, and E. V. Oaks. 1988. Shigella flexneri invasion plasmid antigens B and C: epitope location and characterization with monoclonal antibodies. Infect. Immun. 562933-2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mueller, C. A., P. Broz, S. A. Muller, P. Ringler, F. Erne-Brand, I. Sorg, M. Kuhn, A. Engel, and G. R. Cornelis. 2005. The V-antigen of Yersinia forms a distinct structure at the tip of injectisome needles. Science 310674-676. [DOI] [PubMed] [Google Scholar]
- 32.Murphy, B. S., C. R. Wulff, B. A. Garvy, and S. C. Straley. 2007. Yersinia pestis YadC: a novel vaccine candidate against plague. Adv. Exp. Med. Biol. 603400-414. [DOI] [PubMed] [Google Scholar]
- 33.Nemeth, J., and S. C. Straley. 1997. Effect of Yersinia pestis YopM on experimental plague. Infect. Immun. 65924-930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Neyt, C., and G. Cornelis. 1999. Role of SycD, the chaperone of the Yersinia Yop translocators YopB and YopD. Mol. Microbiol. 31143-156. [DOI] [PubMed] [Google Scholar]
- 35.Olive, A. J., R. Kenjale, M. Espina, D. S. Moore, W. L. Picking, and W. D. Picking. 2007. Bile salts stimulate recruitment of IpaB to the Shigella flexneri surface, where it colocalizes with IpaD at the tip of the type III secretion needle. Infect. Immun. 752626-2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Overheim, K. A., R. W. Depaolo, K. L. Debord, E. M. Morrin, D. M. Anderson, N. M. Green, R. R. Brubaker, B. Jabri, and O. Schneewind. 2005. LcrV plague vaccine with altered immunomodulatory properties. Infect. Immun. 735152-5159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Palmer, L. E., S. Hobbie, J. E. Galan, and J. B. Bliska. 1998. YopJ of Yersinia pseudotuberculosis is required for the inhibition of macrophage TNFα production and the downregulation of the MAP kinases p38 and JNK. Mol. Microbiol. 27953-965. [DOI] [PubMed] [Google Scholar]
- 38.Perry, R. D., and J. D. Fetherston. 1997. Yersinia pestis—etiologic agent of plague. Clin. Microbiol. Rev. 1035-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perry, R. D., S. C. Straley, J. D. Fetherston, D. J. Rose, J. Gregor, and F. R. Blattner. 1998. DNA sequencing and analysis of the low-Ca2+-response plasmid pCD1 of Yersinia pestis KIM5. Infect. Immun. 664611-4623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Philipovskiy, A. V., C. Cowan, C. R. Wulff-Strobel, S. H. Burnett, E. J. Kerschen, D. A. Cohen, A. M. Kaplan, and S. C. Straley. 2005. Antibody against V antigen prevents Yop-dependent growth of Yersinia pestis. Infect. Immun. 731532-1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prentice, M. B., and L. Rahalison. 2007. Plague. Lancet 3691196-1207. [DOI] [PubMed] [Google Scholar]
- 42.Roggenkamp, A., A. M. Geiger, L. Leitritz, A. Kessler, and J. Heesemann. 1997. Passive immunity to infection with Yersinia spp. mediated by anti- recombinant V antigen is dependent on polymorphism of V antigen. Infect. Immun. 65446-451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Runco, L. M., S. Myrczek, J. B. Bliska, and D. G. Thanassi. 2008. Biogenesis of the F1 capsule and analysis of the ultrastructure of Yersinia pestis. J. Bacteriol. 1903381-3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ryndak, M. B., H. Chung, E. London, and J. B. Bliska. 2005. Role of predicted transmembrane domains for type III translocation, pore formation, and signaling by the Yersinia pseudotuberculosis YopB protein. Infect. Immun. 732433-2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sarker, M. R., C. Neyt, I. Stainier, and G. Cornelis. 1998. The Yersinia Yop virulon: LcrV is required for extrusion of the translocators YopB and YopD. J. Bacteriol. 1801207-1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smiley, S. T. 2008. Current challenges in the development of vaccines for pneumonic plague. Exp. Rev. Vaccines 7209-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Swietnicki, W., B. S. Powell, and J. Goodin. 2005. Yersinia pestis Yop secretion protein F: purification, characterization, and protective efficacy against bubonic plague. Protein Expr. Purif. 42166-172. [DOI] [PubMed] [Google Scholar]
- 48.Tanabe, M., H. S. Atkins, D. N. Harland, S. J. Elvin, A. J. Stagg, O. Mirza, R. W. Titball, B. Byrne, and K. A. Brown. 2006. The ABC transporter protein OppA provides protection against experimental Yersinia pestis infection. Infect. Immun. 743687-3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Titball, R. W., and E. D. Williamson. 2004. Yersinia pestis (plague) vaccines. Exp. Opin. Biol. Ther. 4965-973. [DOI] [PubMed] [Google Scholar]
- 50.Turbyfill, K. R., A. B. Hartman, and E. V. Oaks. 2000. Isolation and characterization of a Shigella flexneri invasin complex subunit vaccine. Infect. Immun. 686624-6632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Veenendaal, A. K., J. L. Hodgkinson, L. Schwarzer, D. Stabat, S. F. Zenk, and A. J. Blocker. 2007. The type III secretion system needle tip complex mediates host cell sensing and translocon insertion. Mol. Microbiol. 631719-1730. [DOI] [PubMed] [Google Scholar]
- 52.Weeks, S., J. Hill, A. Friedlander, and S. Welkos. 2002. Anti-V antigen antibody protects macrophages from Yersinia pestis-induced cell death and promotes phagocytosis. Microb. Pathog. 32227-237. [DOI] [PubMed] [Google Scholar]