

Abstract
Francisella tularensis is a highly virulent bacterial pathogen that invades and replicates within numerous host cell types, including macrophages and epithelial cells. In an effort to better understand this process, we screened a transposon insertion library of the F. tularensis live vaccine strain (LVS) for mutant strains that invaded but failed to replicate within alveolar epithelial cell lines. One such strain isolated from this screen contained an insertion in the gene FTL_1914, which is conserved among all sequenced Francisella species yet lacks significant homology to any gene with known function. A deletion strain lacking FTL_1914 was constructed. This strain did not replicate in either epithelial or macrophage-like cells, and intracellular replication was restored by the wild-type allele in trans. Based on the deletion mutant phenotype, FTL_1914 was termed ripA (required for intracellular proliferation, factor A). Following uptake by J774.A1 cells, F. tularensis LVS ΔripA colocalized with LAMP-1 then escaped the phagosome at the same rate and frequency as wild-type LVS-infected cells. Electron micrographs of the F. tularensis LVS ΔripA mutant demonstrated the reentry of the mutant bacteria into double membrane vacuoles characteristic of autophagosomes in a process that was not dependent on replication. The F. tularensis LVS ΔripA mutant was significantly impaired in its ability to persist in the lung and in its capacity to disseminate and colonize the liver and spleen in a mouse model of pulmonary tularemia. The RipA protein was expressed during growth in laboratory media and localized to the cytoplasmic membrane. Thus, RipA is a cytoplasmic membrane protein conserved among Francisella species that is required for intracellular replication within the host cell cytoplasm as well as disease progression, dissemination, and virulence.
Francisella tularensis is a highly virulent zoonotic pathogen that is the etiologic agent of the disease tularemia. Francisella species have been isolated from over 250 animal species including mice, rabbits, and squirrels (5). Transmission to humans from infected animals occurs through arthropod bites (41), physical contact with infected animal tissues (10), consumption of contaminated water (9, 50), or inhalation of aerosolized organisms (19). Transmission by inhalation results in the most aggressive form of tularemia, where as few as 10 CFU can lead to a disease (55) that can rapidly progress (62) and result in mortality rates as high as 60% if untreated (17). Individual cases of tularemia occur throughout the Northern hemisphere (35), with clusters of outbreaks occurring in Scandinavia (63), Europe (28, 33), and the American Midwest (56). Reported cases of tularemia peaked in the United States during the 1930s and 1940s but have since declined, with only 1,368 cases reported between 1990 and 2000 (11).
F. tularensis strains infectious for humans are subdivided into two groups, type A (Francisella tularensis subsp. tularensis) and type B (Francisella tularensis subsp. holoarctica). Type A is characterized by high virulence for humans and geographic localization to North America, although recently type A was isolated in Europe as well (29). Type B is distributed throughout the Northern hemisphere, and infections with this strain have an associated mortality lower than that for type A. A type B F. tularensis strain was used to create an attenuated live vaccine strain (LVS) for use in the Soviet Union (59); however, this vaccine is not universally protective, nor is it licensed for use in the United States. F. tularensis LVS is attenuated in humans but remains pathogenic for mice and other rodents, making it an excellent model for laboratory studies on Francisella pathogenesis (21).
F. tularensis is a facultative intracellular pathogen. The ability to replicate in various cell types such as alveolar macrophages (3, 45, 48, 58), dendritic cells (8), and lung alveolar type II epithelial cells (22, 31) are all considered to be important in the pathogenesis of Francisella. Virulence factors such as mglA (4), iglA (16), iglC (38, 39, 54), tolC (24), clpB (42), type IV pili (20, 23, 30), and a recently identified 58-kDa protein (60) have all been characterized using macrophage and mouse models of infection. Yet, the actual function of many of these proteins remains to be determined. We screened a transposon insertion library to identify additional genes that contribute to the intracellular growth of F. tularensis. Herein, we describe one such gene that is required for intracellular growth and virulence in a mouse model of tularemia.
MATERIALS AND METHODS
Bacterial strains and cell culture.
Escherichia coli TOP10 and M15 strains were used for cloning and expression of six-histidine fusion proteins, respectively. E. coli cultures were propagated in Luria broth supplemented with ampicillin at 100 μg/ml or kanamycin at 20 μg/ml as necessary for maintaining antibiotic selection. Francisella tularensis LVS was obtained from the Centers for Disease Control and Prevention, Atlanta, GA. F. tularensis LVS was propagated on chocolate agar (25 g/liter brain heart infusion [BHI], 50 μg/ml hemoglobin, 15 g/liter agar) supplemented with 1% IsoVitaleX (Becton-Dickson), complete BHI broth (35 g/liter BHI, 50 μg/ml hemin, 1% IsoVitaleX), or Chamberlain's defined medium (12). All bacterial strains cultured on chocolate agar were grown at 37°C. Broth cultures were incubated in a shaking water bath at 37°C.
TC-1 (ATCC CRL-2785) is a tumor cell line derived from mouse primary lung epithelial cells that was cultured in RPMI 1640 supplemented with 2 mM l-glutamine, 1.5 g/liter sodium bicarbonate, 10 mM HEPES, 1.0 mM sodium pyruvate, 0.1 mM nonessential amino acids, and 10% fetal bovine serum. J774A.1 (ATCC TIB-67) is a reticulum cell sarcoma mouse macrophage-like cell line that was cultured in Dulbecco's minimal essential medium with 4.5 g/liter glucose, 2 mM l-glutamine, and 10% fetal bovine serum. Cell lines were cultured at 37°C in a 5% CO2 atmosphere.
Plasmids and molecular techniques.
Cloning of F. tularensis LVS DNA was conducted by PCR amplification of genomic DNA by use of Pfu turbo DNA polymerase (Stratagene) and cloned into the pCR Blunt-II TOPO vector (Invitrogen) by use of the manufacturer's protocols. Plasmids for complementation experiments were created by ligating cloned regions of the F. tularensis LVS genome into the pKK multiple cloning site (MCS), a derivative of plasmid pKK214 gfp (1) where a gfp Tetr fragment was removed and replaced with a fragment containing an MCS and a Kanr allele. Kanamycin selection was maintained at 20 μg/ml for E. coli TOP10 and 10 μg/ml for F. tularensis LVS. The kanamycin resistance gene was an F. tularensis codon-optimized version of aphA1 synthesized by Blue Heron Biotechnology that was expressed from a modified F. tularensis groEL promoter (18).
Gentamicin protection assay.
To determine the rate of intracellular invasion and replication, F. tularensis LVS strains were cultured to mid-exponential phase in Chamberlain's defined medium and then added to J774A.1 or TC-1 monolayers at a multiplicity of infection (MOI) of 100 in 200 μl of prewarmed tissue culture medium. Gentamicin protection assays were then conducted as described previously (31). Assays were done in triplicate and statistical significance was determined using unpaired t tests with unequal variance on the log transforms of recovered CFU to compare mutant strains to the wild type.
Mutagenesis and allelic exchange.
A ripA deletion construct was made by splice overlap extension (SOE) PCR (34) using primers designed to delete the ripA locus while maintaining 1-kb flanking regions. All ripA sequence between the start and stop codons was deleted while leaving the start and stop codons intact. The construct was sequenced to confirm that the deletion was in frame and to ensure the integrity of the flanking DNA sequence. A BamHI-NotI fragment containing the deleted allele was ligated into pMP590 sacB Kanr for allelic exchange (40). Kanamycin (10 μg/ml) was used to select for plasmid integration and 10% sucrose to counterselect for resolution as described by LoVullo et el. (40).
Invasion and vacuolar escape assay.
Intracellular bacteria accessible to cytoplasmically delivered antibodies were enumerated by the methods described previously (15). The F. tularensis LVS ΔripA mutant's accessibility to the cytoplasm was compared to that of the wild type at 20 min, 60 min, and 180 min postinvasion into J774A.1 cells in triplicate assays per time point. Bacteria accessible to the cytoplasm were described as cytoplasmic and bacteria inaccessible to the cytoplasm as vacuolar.
Fluorescence microscopy.
J774A.1 cells were cultured in eight-well chamber slides (Nunc). F. tularensis LVS(pKK214 gfp) and F. tularensis LVS ΔripA(pKK214 gfp) were prepared for inoculation as described above. Invasion assay synchronization, epifluorescent microscopy, and LAMP-1 colocalization quantitation were done as described in the work of Craven et al. (15) at the described times postinoculation into J774A.1 mouse macrophage-like cells. Three independent assays were conducted for quantitation, with multiple captures at each time point examined. LVS green fluorescent protein (GFP)-positive objects at 24 h contained multiple bacteria and were scored as a group. Three independent counts of over 100 GFP-positive objects were scored as positive or negative for LAMP-1 colocalization. The mean percentages of colocalization ± the standard deviations were recorded, and mutant values were compared to those for the wild type with an unpaired two-tailed t test.
Electron microscopy.
Monolayers of J774A.1 cells infected with LVS or the LVS ΔripA mutant were rinsed with phosphate-buffered saline (PBS) and fixed in 3% glutaraldehyde-0.15 M sodium phosphate, pH 7.4. Following three additional rinses with sodium phosphate buffer, the monolayers were postfixed for 1 h in 1% osmium tetroxide-1.25% potassium ferrocyanide-0.15 M sodium phosphate buffer, rinsed in deionized water, dehydrated using increasing concentrations of ethanol (30%, 50%, 75%, 100%, and 100% again for 10 min each), and embedded in Polybed 812 epoxy resin (Polysciences, Inc., Warrington, PA). The embedded samples were sectioned parallel and perpendicular to the substrate at 70 nm using a diamond knife. Ultrathin sections were collected on 200-mesh copper grids and stained with 4% aqueous uranyl acetate for 15 min, followed by Reynolds’ lead citrate for 7 min. Sections were observed using a Leo EM910 transmission electron microscope at 80 kV (LEO Electron Microscopy, Thornwood, NY) and photographed using a Gatan Bioscan digital camera (Gatan, Inc., Pleasanton, CA).
Mouse model of pulmonary infection.
Francisella strains were prepared for intranasal inoculation by culturing in Chamberlain's defined medium to mid-exponential phase prior to suspension in PBS. Anesthetized 6- to 8-week-old female C57BL/6 mice were inoculated intranasally with 105 CFU of F. tularensis LVS suspended in 50 μl of PBS. Inoculations were conducted with groups of four mice per strain for each time point. Organ burdens were evaluated by homogenization and serial dilution before plating on chocolate agar to determine recovered CFU per organ. Statistical analysis was conducted on log transforms of organ burdens by use of independent unpaired t tests to compare mutant strains to the wild type.
Membrane fractionation.
F. tularensis LVS was cultured to mid-exponential phase in Chamberlain's defined medium, pelleted by centrifugation, and resuspended in lysis buffer (10 mM Tris, pH 7.5, 150 mM NaCl). Lysozyme was added to 0.1 mg/ml and the bacterial suspension incubated on ice for 30 min. A 1:1 cell suspension with 0.1-mm silica beads was beaten for 20 min in a Disruptor Genie (Scientific Industries). Cell lysates were collected after the beads settled and the beads were washed three times with lysis buffer. Cell lysates were clarified by centrifugation at 12,000 × g for 4 min. Crude membrane fractions were collected by ultracentrifugation at 100,000 × g for 90 min, and the supernatants were saved as the cytosolic fractions. Crude membrane fractions were washed one time by resuspension in lysis buffer and ultracentrifugation. Cytoplasmic membrane fractions were solubilized with lysis buffer and extracted with 10% Sarkosyl (Sigma) added to a 0.2% final concentration. Outer membrane fractions were then pelleted by ultracentrifugation at 100,000 × g for 60 min (16, 44).
Recombinant RipA and anti-RipA antiserum production.
The F. tularensis LVS ripA gene was cloned and sequenced to confirm the integrity of the DNA sequence. The ripA allele was ligated into a pQE30 (Qiagen) vector to construct an N-terminal six-histidine tag fusion. The ripA::6His allele was induced using 0.1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for 4 h and cells were harvested by centrifugation. The cell pellet was resuspended (40 mM Tris-HCl, pH 8.0, 1 mM EDTA, 0.1 M NaCl) and lysozyme treated at 1 mg/ml for 30 min before sonication. The lysate was clarified by centrifugation. The crude membrane fraction was pelleted by ultracentrifugation at 115,000 × g for 60 min. RipA::6His was purified from the crude membrane fraction using a Qiagen native purification protocol with buffers containing 1% Triton X-100 and 10 mM β-mercaptoethanol. The purified RipA::6His was analyzed for purity by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Coomassie staining. The purified protein was methanol chloroform precipitated (61) and resuspended in PBS. Purified RipA::6His was sent to Proteintech Group, Inc., for the production of rabbit antiserum by the standard Proteintech long protocol. Preimmune bleeds were collected from each rabbit used in the protocol. Antiserum was screened for reactivity and specificity by Western blotting of the purified protein.
SDS-PAGE and Western analysis.
SDS-PAGE was conducted by the methods of Laemmli (36) utilizing a denaturing discontinuous 12.5% acrylamide gel. Amounts of total protein loaded in each sample were equivalent, as determined by a bicinchoninic acid assay (Pierce). Proteins were transferred to nitrocellulose and blocked in 5% bovine serum albumin blocking buffer (0.05% Tween 20, PBS) overnight. Antibody incubations were conducted in 0.5% bovine serum albumin wash buffer (0.05% Tween 20, PBS). Membranes were washed in 0.05% Tween 20-PBS. Primary incubations and secondary antibody incubations were for 60 min. Rabbit anti-RipA serum was used at 1:1,000, chicken anti-AtpB immunoglobulin Y (IgY) at 1:5,000 (AgriSera), mouse anti-GFP IgG at 1:1,000 (Abcam), goat anti-rabbit IgG-horseradish peroxidase at 1:10,000 (Sigma), and goat anti-chicken IgG-horseradish peroxidase at 1:10,000 (Sigma). The signal was developed using Pierce SuperSignal Pico substrate per the manufacturer's protocols and detected by autoradiography.
RESULTS
Identification of RipA.
To identify genes involved in Francisella intracellular growth, we created and screened a transposon mutant library of F. tularensis LVS for strains that entered but did not replicate within TC-1 cells, which are a transformed lung epithelial cell line. We identified one mutant strain that invaded epithelial cells at the same frequency as F. tularensis LVS but did not replicate following invasion (Fig. 1A). At 6 h postinoculation, there was no significant difference between the number of CFU recovered from mutant- and wild-type-infected TC-1 cells. However, by 24 h postinoculation there were significantly fewer (P < 0.001) mutant organisms (1.9 × 105 CFU) than wild-type LVS (1.1 × 107 CFU) recovered from the Tn5 mutant- and wild-type LVS-infected cells, respectively.
FIG. 1.
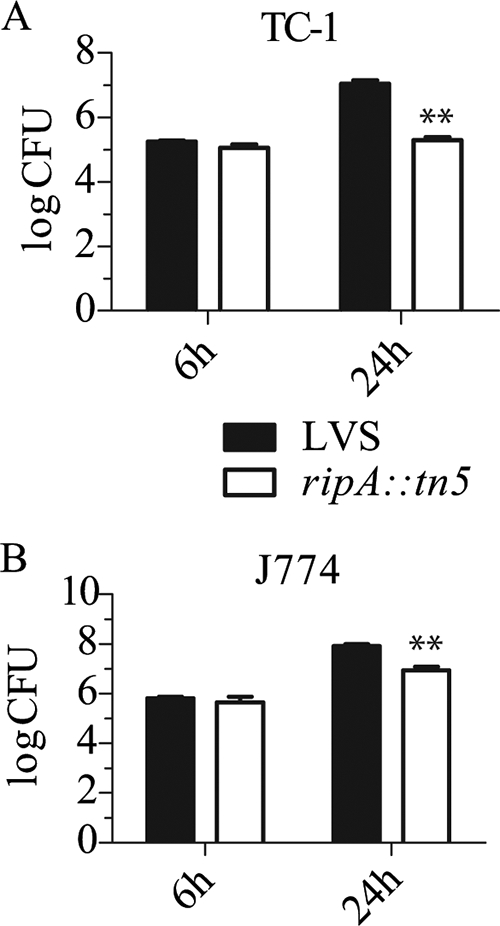
Gentamicin protection assays with the LVS ripA::Tn5 mutant. (A and B) TC-1 lung epithelial cells (A) or J774A.1 mouse macrophage-like cells (B) were infected with either LVS or the LVS ripA::Tn5 mutant at an MOI of 100. Indicated are mean CFU (n = 3) at 6 h and 24 h postinoculation with error bars ± 1 standard deviation. Student's t tests were conducted comparing LVS to the LVS ripA::Tn5 mutant at each time point (P values of <0.01 are demarcated by **).
To compare the levels of intracellular growth of the Tn5 mutant strain and wild-type LVS in epithelial and macrophage-like cell lines, identical assays were performed using the J774A.1 mouse macrophage-like cell line (Fig. 1B). As in the TC-1 epithelial cell line, the mutant and wild-type strains entered J774A.1 cells at the same frequency. At 24 h postinoculation, 8.2 × 107 and 8.7 × 106 CFU (P < 0.01) were recovered from LVS- and Tn5 mutant-infected cells, respectively. Thus, in contrast to the no-growth phenotype observed for TC-1 cells, the Tn5 mutant replicated within J774A.1 cells but to a lesser extent than wild-type LVS. Similar results were obtained with other lung epithelial cell lines (MLE-12, A549) and with bone marrow-derived mouse macrophages (data not shown). The monocyte and epithelial cell lines were propagated in modestly different medium formulations, but swapping the media had no impact on the cell-type-specific growth defect of the mutant (data not shown).
Analysis of the DNA sequence flanking the Tn5 insertion showed that the interrupted allele was FTL_1914 (Fig. 2A). This allele was termed ripA (required for intracellular proliferation, factor A). The ripA locus is predicted to encode a 178-amino-acid membrane protein conserved in all sequenced Francisella strains. There is only one copy of the locus in each sequenced genome. pBLAST (25) was used to identify similar non-F. tularensis proteins (E < 1). These were hypothetical membrane proteins of unknown function in Streptomyces coelicolor A3(2) (E = 1 × 10−10), Beggiatoa sp. strain PS (E = 2 × 10−10), Moritella sp. strain PE36 (E = 1 × 10−9), Sulfitobacter sp. strain NAS-14.1 (E = 0.006), and Clostridium perfringens type C strain JGS1495 (E = 0.044), along with a predicted membrane protein linked to a retron element in an Escherichia coli clinical isolate (E = 0.36) and a nitric oxide reductase (NorW) in Aeromonas salmonicida (E = 0.60). NorW plays a role in nitric oxide detoxification through interactions with NorR and NorV; however, there are no NorR or NorV homologs encoded in the Francisella genome and the amino acid similarity is to a NorW fragment not part of the functional oxidoreductase. Thus, at this time protein homology analyses did not reveal any obvious clues to the function of RipA.
FIG. 2.

(A) Graphical representation of the genomic organization of the LVS ripA locus. Loci in close proximity to ripA (FTL_1914) on the chromosome are FTL_1912 (30S ribosomal protein S1), FTL_1913 (Sua5/YciO/YrdC family protein), and FTL_1915 (acetyltransferase). Primers utilized for SOE PCR are marked by arrows. int, internal; ext, external. (B) DNA sequence of the ripA deletion marked with the forward and reverse overlapping internal primers used in the SOE PCR. The predicted ribosome binding site (RBS) and the remaining ripA codons (M and *) are marked.
Creation and initial characterization of a ripA deletion mutant.
The intracellular growth of the LVS Tn5::ripA mutant was not restored by in trans complementation with wild-type ripA. Thus, to circumvent potential complications caused by the transposon insertion, an F. tularensis LVS ΔripA strain lacking all but the start and stop codons was created for use in subsequent phenotypic assays. The deletion allele was generated by SOE PCR (Fig. 2B), cloned into the allelic exchange vector pMP590 (40), and introduced into LVS as described previously (27, 40). Following sucrose selection to isolate resolved plasmid integrants, strains lacking wild-type ripA and possessing the ripA deletion locus were identified by PCR. The integrity of the ΔripA locus and the flanking DNA was confirmed by sequencing. The growth of the mutant strain in Chamberlain's defined medium was not significantly different from that of wild-type LVS. After 24 h of growth in Chamberlain's defined medium, the mutant and wild-type strains reached equivalent maximum densities (data not shown).
Francisella intracellular growth requires ripA.
Mutant and wild-type strains were subjected to gentamicin protection assays conducted with J774A.1 (Fig. 3B) and TC-1 (Fig. 3A) cell lines to determine the contribution of ripA to intracellular growth. The invasion frequencies of the deletion mutant were not significantly different from those of the wild type (P > 0.4) in either cell line. However, in contrast to what was seen for the wild type, the number of recovered intracellular mutant CFU decreased after 24 h in both cell types, indicating that the ΔripA mutant had an intracellular survival defect. The mean recoveries for LVS and the LVS ΔripA mutant were 1.23 × 107 and 2.60 × 102 CFU (P < 0.001), respectively, at 24 h for TC-1 lung epithelial cells. The mean recoveries for LVS and the LVS ΔripA mutant were 1.04 × 108 and 1.07 × 105 CFU (P < 0.001), respectively, at 24 h for J774A.1 cells. The intracellular growth defect of the LVS ΔripA mutant was not due to increased sensitivity to gentamicin (data not shown). The intracellular replication of the LVS ΔripA mutant was restored by in trans complementation with ripA, demonstrating a direct cause-and-effect relationship between ripA expression and intracellular growth.
FIG. 3.
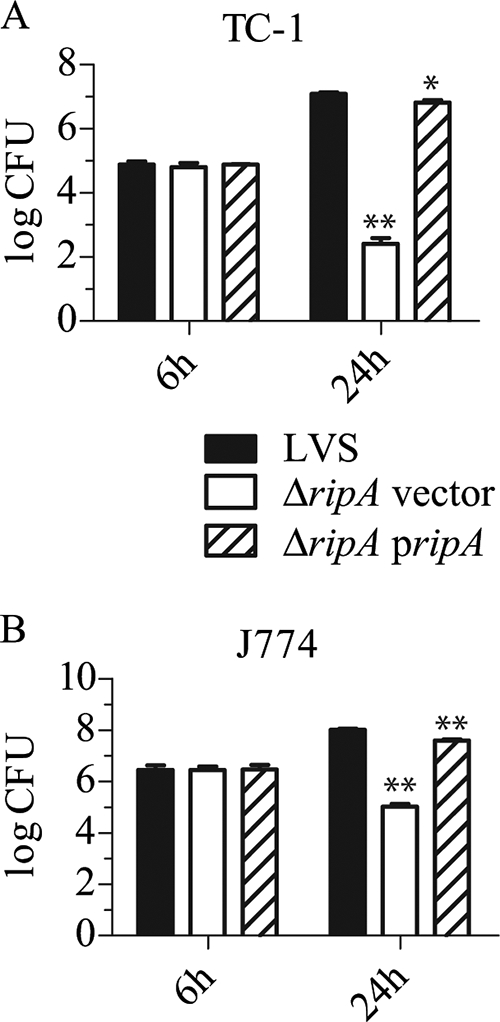
Gentamicin protection assays with the ΔripA vector and complemented (ΔripA pripA) mutants (A) TC-1 lung epithelial cells or (B) J774A.1 mouse macrophage-like cells were infected at an MOI of 100. Indicated are mean CFU (n = 3) at 6 h and 24 h postinoculation with error bars ± 1 standard deviation. Student's t tests were conducted comparing strains to wild-type LVS at each time point (P values of <0.05 are demarcated by *, and P values of <0.01 are demarcated by **).
F. tularensis ΔripA mutant is unaffected in its ability to escape the phagosome.
Following uptake by host cells, wild-type F. tularensis organisms escape the phagosome and replicate within the cytoplasm (13, 26). With the notable exception of F. novicida ΔiglD (53), F. tularensis LVS ΔpurMCD (47), and F. tularensis LVS clpB (42) mutants, most intracellular-growth-defective mutant strains of Francisella fail to escape the phagosome (39, 54). To determine the contribution of ripA to this property, we compared the phagosome escape frequencies and rates of the wild type and the ΔripA mutant in J774A.1 cells. Briefly, monolayers were inoculated with GFP-expressing strains at an MOI of 100. Invasion was synchronized as described previously (13). Infected cells were treated with digitonin to selectively permeabilize the cytoplasmic, but not the phagosomal, membrane 20, 60, and 180 min postinvasion and probed with Alexa 647-conjugated anti-F. tularensis antibody. Extracellular and cytoplasmic bacteria were antibody accessible, but bacteria contained by an intact phagosomal membrane were not. Antibody-bound and unbound organisms were quantified via fluorescence-activated cell sorting, and the contribution of extracellular bacteria was accounted for in corresponding samples not treated with digitonin. At each time point, the percentages of antibody-accessible mutant and wild-type bacteria were statistically identical (Fig. 4A). Thus, the ΔripA strain accessed the cytoplasm at the same rate as wild-type F. tularensis LVS, demonstrating that ripA is not required for phagosome escape and that failure to escape the phagosome does not account for the intracellular growth defect conferred by the ΔripA allele.
FIG. 4.

Intracellular trafficking of the LVS ΔripA mutant. (A) Cytoplasmic accessibility of LVS(pKK214 gfp) and LVS ΔripA(pKK214 gfp) in J774A.1 macrophage-like cells at 20 min, 1 h, and 3 h postinoculation (PI). Vacuolar bacteria were defined as bacteria inaccessible to the cytoplasmically delivered α-Francisella lipopolysaccharide antibodies. Mean percentages of vacuolar (n = 3) values are plotted and error bars are representative of ±1 standard deviation. The percentage of accessibility with saponin permeabilization is marked with a broken line without symbols. (B) Colocalization of LVS(pKK214 gfp) and LVS ΔripA(pKK214 gfp) with LAMP-1 in J774A.1 macrophage-like cells. The percentages of LAMP-1 colocalization were determined from three independent experiments and recorded as the mean percentages of LAMP-1 positivity, with error bars representative of ±1 standard deviation. At 24 h PI, LAMP-1 colocalization done on a per-cell basis. Individual bacteria were not distinguishable in wild-type images. (C) Representative epifluorescent images of LVS(pKK214 gfp) and LVS ΔripA(pKK214 gfp) at 20 min, 3 h, and 24 h PI in J774A.1 macrophage-like cells. The 20-min and 3-h images demonstrate similar staining patterns for LVS(pKK214 gfp) and LVS ΔripA(pKK214 gfp). The 24-h image demonstrates that LVS ΔripA(pKK214 gfp) may be visualized as individual bacteria colocalized with LAMP-1 at this time point. Extracellular bacteria were stained with 6-(7-amino-4-methylcoumarin-3-acetyl)amino hexanoic acid and colorized blue. GFP captures are demarcated by LVS (bottom panels) and colorized green in merged images (top panels). LAMP-1 is stained with Alexa 647 and colorized red. Individual channels for GFP and Alexa 647 captures are marked as LVS and LAMP-1, respectively.
Francisella ripA mutant colocalizes with LAMP-1.
Francisella-containing phagosomes associate with early and late maturation antigens EEA-1 and LAMP-1 before membrane integrity is compromised and the bacteria are released to the cytoplasm. There is evidence to suggest that early phagosome maturation steps are required for Francisella survival and subsequent intracellular replication (17, 62). We compared mutant and wild-type colocalizations with LAMP-1 in infected cells to determine if the lack of ripA affected phagosome trafficking. J774A.1 cells were inoculated with GFP-expressing wild-type and ΔripA strains as described above, permeabilized, and probed with Alexa 647-conjugated LAMP-1 antibody. The colocalization of LAMP-1 with bacterial cells was evaluated by epifluorescence. Consistent with what has been reported by other groups (13, 27), at 20 and 180 min postinoculation, 50% and 22%, respectively, of intracellular LVS colocalized with LAMP-1 (Fig. 4B). The number of wild-type organisms colocalizing with LAMP-1 reached a minimum of 1.5% at 12 h postinoculation, which was followed by an increase to 26% at 24 h (Fig. 4B). This reacquisition of LAMP-1 colocalization has been described previously as the replication-dependent formation of Francisella-containing vacuoles (13). The colocalization of the LVS ΔripA mutant with LAMP-1 was not statistically different from what was seen for the wild type at any point tested (P > 0.05; unpaired two-tailed t test), save for that at 24 h postinoculation, when 40% of the mutant organisms colocalized with LAMP-1. It should be noted that LVS ΔripA mutant-infected cells contained only one to three bacteria per cell, whereas wild-type-infected cells had what appeared to be hundreds of bacteria 24 h postinoculation. Thus, the concentration of wild-type bacteria in these cells may impact the LAMP-1 colocalization calculations. More important than the difference in the proportions of mutant and wild-type organisms that colocalized with LAMP-1 in 24-h-infected cells is the observation that LAMP-1 colocalization with the LVS ΔripA mutant increased between 12 and 24 h. This process of reassociation with LAMP-1-positive vacuoles was previously thought to require bacterial replication (13). These results demonstrate that replication per se is not required for this process and that the failure to reassociate with LAMP-1-positive vacuoles does not account for the intracellular growth deficiency of the ΔripA mutant.
Francisella ripA mutant reenters a membrane-bound vacuole after escape to the cytoplasm.
To confirm the intracellular trafficking conclusions drawn from the phagosome escape and LAMP-1 colocalization studies described above, we evaluated mutant and wild-type subcellular localization 15 min and 3 and 24 h postinoculation by electron microscopy. Mutant and wild-type bacteria were found in membrane-bound vacuoles at 15 min and were free in the cytoplasm at 3 h postinoculation (Fig. 5). By 24 h, bacteria within membrane-bound vacuoles were evident. Wild-type bacteria were present at high density and were located in either the cytosol or membrane-bound vacuoles. One to three mutant bacteria were present in the cytosol or within membrane-bound vacuoles, some of which possessed double or multiple membranes characteristic of autophagosomes (Fig. 5).
FIG. 5.

Transmission electron microscopy of intracellular wild-type and ΔripA F. tularensis LVS. Individual wild-type and mutant bacteria within membrane-bound vacuoles 20 min and 24 h postinoculation are indicated by arrows. Note the presence of multiple membranes in the 24-h samples. Wild-type and mutant bacteria were free in the cytosol 3 h postinoculation.
RipA is required for virulence in a mouse model of infection.
We used a mouse model of pulmonary tularemia to assess the role of ripA in F. tularensis virulence and pathogenesis. Anesthetized C57BL/6 mice were inoculated intranasally with 105 CFU of either LVS(pKK MCS) (wild type plus vector), LVS ΔripA(pKK MCS) (mutant plus vector), or LVS ΔripA(pKK MCS ripA) (mutant plus plasmid complement). Bacterial burdens were measured by serial dilution and plating of homogenized organs. Lung burdens were determined 2 hours postinoculation to monitor the delivery of each strain to the lung. Lung, liver, and spleen organ burdens (Fig. 6) were determined 1, 3, 7, and 14 days postinoculation.
FIG. 6.
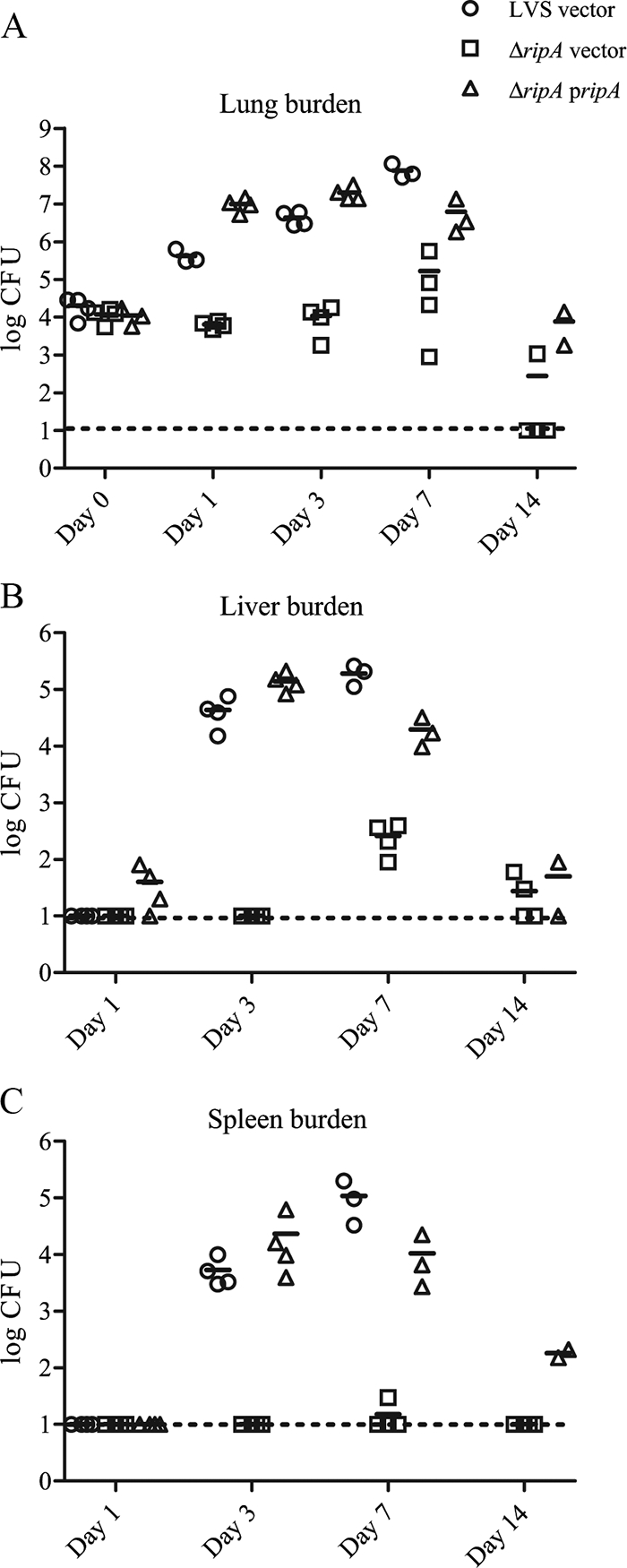
Bacterial organ burdens from infected mice (n = 4) were quantified as CFU per organ and graphed as individual data points. Mean organ burdens are demarcated by horizontal broken lines. Organ burdens in mice inoculated with LVS vector-only, LVS ΔripA vector-only, and LVS ΔripA(pJRF146 ripA) (ripA pripA) strains. Lung (A), liver (B), and spleen (C) burdens were determined at 2 h (lung only) or at 1 day, 3 days, 7 days, and 14 days [LVS ΔripA vector only and LVS ΔripA(pJRF146 ripA)] postinoculation. The limit of detection is marked as a broken line at ∼10 CFU. Burdens that were below the limit of detection are plotted at 10 CFU.
The CFU recovered from the lungs at 2 h postinoculation, 2.48 × 104, 1.18 × 104, and 1.12 × 104 for LVS, the LVS ΔripA mutant, and LVS ΔripA(pJRF146 ripA), respectively (Fig. 6A), were not statistically different (P > 0.2; independent two-tailed t test with unequal variance); thus, equivalent numbers of each strains were delivered to the lung. By day 1 the organ burdens of LVS ΔripA mutant-infected animals were significantly lower than those of wild-type and complemented mutant strains (P < 0.001). Within the first 24 h postinoculation, the number of LVS ΔripA organisms in the lung decreased, whereas the number of both wild-type and complemented strains increased by at least 1 order of magnitude over the number of organisms initially delivered (Fig. 6A). In contrast to the wild-type and complemented mutant strains, the LVS ΔripA mutant was not detected in the liver or spleen before day 7 (Fig. 6B and C). At day 7, all LVS ΔripA mutant-infected mice had detectable liver burdens but only one LVS ΔripA mutant-infected mouse had a detectable spleen burden (>30 CFU). In mice infected with the LVS ΔripA mutant or with LVS ΔripA(pJRF146 ripA), organ burdens were reduced at day 14 relative to day 7 (P < 0.01).
Mice infected with wild-type LVS were euthanized at day 7 due to morbidity. Collectively, these data demonstrate the importance of ripA in establishing a Francisella infection.
Characterization and localization of the RipA protein.
Subcellular fractions of LVS, the LVS ΔripA mutant, and LVS ΔripA(pJRF146 ripA) were analyzed by Western blotting using anti-RipA antiserum (Fig. 7A). RipA migrated at a relative molecular mass of 17 kDa. RipA protein was not detected in LVS ΔripA lysates. Complementation with the native ripA allele in a multicopy plasmid resulted in increased RipA expression relative to what was seen for the wild-type strain.
FIG. 7.
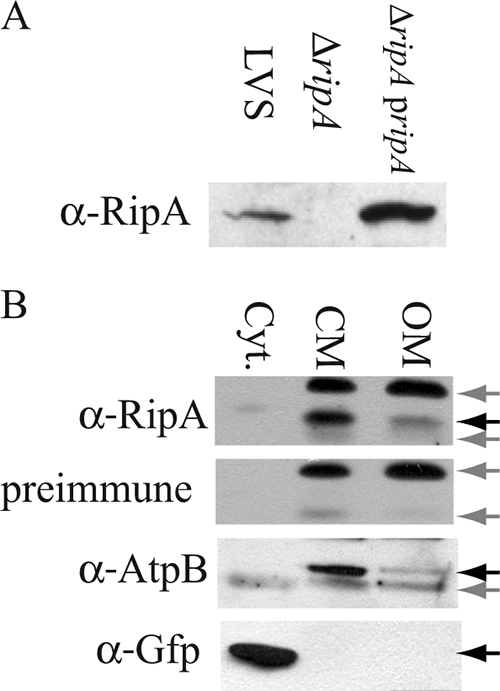
(A) Western blot analysis of cytoplasmic membrane enriched fractions of LVS, the LVS ΔripA mutant, and LVS ΔripA(pJRF146 ripA) (ripA pripA). Rabbit anti-RipA (α-RipA) (1:1,000) antiserum was used as the primary antibody. (B) Subcellular localization of RipA. Western analysis of cytosolic (Cyt.), cytoplasmic membrane (CM), and outer membrane (OM) enriched fractions of LVS(pKK214 gfp). Nonspecific bands were determined with preimmune serum from the mouse used to generate the mouse anti-RipA serum. AtpB was used as a marker for the cytoplasmic membrane and GFP for the cytoplasm. Specific bands (RipA, AtpB, and GFP) are marked with black arrows. Nonspecific bands are marked with gray arrows.
When the translated amino acid sequence was analyzed with TopPred (14), RipA was predicted to be localized to the cytoplasmic membrane with three transmembrane domains and the amino terminus in the cytoplasm. Western analysis of cytoplasmic, cytoplasmic membrane, and outer membrane enriched protein fractions (16, 44) prepared from GFP-expressing LVS revealed that RipA localized to the cytoplasmic membrane (Fig. 7B). RipA was differentiated from proteins that reacted nonspecifically with the antiserum by probing corresponding samples with preimmune serum. Antibodies recognizing the FoF1 ATP synthase β subunit (AtpB) (37) and GFP were used as markers for cytoplasmic membrane and cytoplasmic components, respectively. No GFP was detected in either membrane fraction. Low levels of AtpB and RipA were detected in the outer membrane fraction, indicating the presence of low-level contamination with cytoplasmic membrane proteins. However, the relative band intensities demonstrate that the majority of RipA was present in the cytoplasmic membrane fraction of F. tularensis.
DISCUSSION
The highly virulent nature of F. tularensis was recognized even before it was isolated and identified in the 1920s as the etiologic agent of a plague-like disease in rodents. It is evident that the ability to survive and replicate within diverse host cell types is fundamental to F. tularensis pathogenesis. While the processes of host cell entry, phagosome escape, and intracellular replication have been described, the mechanisms used by F. tularensis to achieve these have not. The recent creation and improvement of tools and procedures for the genetic manipulation of F. tularensis have led to the identification of a number of genes that are required for intracellular growth and/or virulence in animal models of tularemia.
We screened a transposon insertion mutant library of F. tularensis LVS to identify previously unstudied genes required for proliferation within host cells. One such mutant strain contained a transposon insertion in FTL_1914 (ripA), which is conserved among Francisella species. The transposon insertion mutant possessed an unusual phenotype in that it failed to replicate within cell lines of epithelial origin yet retained the ability to replicate within monocyte cell lines and bone marrow-derived macrophages. We created an F. novicida U112 Tn5::ripA mutant strain by replacing the wild-type gene with the LVS Tn5::ripA allele via homologous recombination, and this strain had the same intracellular growth phenotype as LVS Tn5::ripA (data not shown). However, F. tularensis LVS and Schu S4 (data not shown) deletion mutant strains failed to replicate in any cell type tested. We cannot account for the differences in intracellular growth conferred by the transposon insertion and deletion mutant alleles. Only the deletion mutant phenotype was complemented by wild-type ripA in trans, suggesting that the transposon insertion may have exerted cis-acting or polar effects or possibly created selective pressure for secondary unlinked mutations that compensated for the growth defect in macrophages.
Even though ripA has homology to a limited number of genes in other unrelated bacteria, this information does not provide significant insight into the function of ripA. Thus, we thoroughly characterized the expression and subcellular localization of RipA, as well as the ΔripA mutant phenotypes, to provide a basis for discerning the mechanism(s) by which this protein contributes to intracellular growth.
Intracellular pathogens must overcome the host innate immune response to successfully colonize the intracellular niche. The primary host defense is centered on the antimicrobial properties of the phagosome. Most successful intracellular pathogens either escape the phagosome or divert phagosome maturation to their own ends. Francisella quickly escapes the phagosome into the cytoplasmic environment, where it replicates (13, 26, 39). Since the recovery of the LVS ΔripA mutant from infected epithelial cells or macrophages decreased dramatically after the initial invasion, we wanted to assess if the decreased survival was due to altered intracellular trafficking of the LVS ΔripA mutant. The LVS ΔripA mutant became accessible to the cytoplasm at the same rate as wild-type LVS. The mutant also colocalized with the late endosomal/lysosomal marker LAMP-1 with the same frequency as the wild type. Thus, the intracellular survival defect was not due to failure to escape from and subsequent killing in the phagolysosome.
Autophagy is another important means by which host cells clear pathogens from the host cytoplasm (43, 51). Most cytoplasmic pathogens studied to date either evade or hijack autophagic mechanisms. For example, Shigella flexneri and Listeria monocytogenes evade autophagy in a process requiring de novo protein synthesis (46, 51), while Legionella pneumophila, Salmonella enterica, and Coxiella burnetii exploit the autophagic machinery (6, 7, 32, 52, 57). Francisella is different from other intracellular pathogens that have been examined because its evasion of autophagy is not dependent on de novo protein synthesis (13). Francisella reenters autophagosome-like LAMP-1-positive vacuoles after replication in the cytoplasm. The LVS ΔripA mutant also localizes with LAMP-1 after escape to the cytoplasm. Electron micrographs showed the F. tularensis LVS ΔripA mutant present in vacuoles with double membranes characteristic of autophagosomes by 24 h postinoculation. In contrast to what has been published regarding wild-type Francisella (13), the reentry of ΔripA bacteria into Francisella-containing vacuoles was not dependent on replication. We hypothesize that the autophagy of the intracellular F. tularensis LVS ΔripA mutant occurs in the cytoplasm, although this may or may not have a direct bactericidal effect on the bacteria. Further work needs to be done to determine if the LVS ΔripA mutant requires de novo protein synthesis to stimulate compartmentalization during the cytoplasmic phase of its life cycle and what effect the compartmentalization of the LVS ΔripA mutant has on intracellular bacterial survival.
The survival defect of the LVS ΔripA mutant in the cytoplasm is likely due to increased sensitivity to host antimicrobial factors, its inability to acquire a necessary nutrient, or failure to adapt to the cytoplasmic environment. Nutritional deprivation or auxotrophy would have a bacteriostatic effect on the bacteria (2). For example, the F. tularensis ΔpurF strain, a purine auxotroph, fails to replicate in host cell cytoplasm but persists for an extended period before declining gradually (49). This is not the phenotype demonstrated by the F. tularensis ΔripA mutant, which declines dramatically between 6 h and 24 h in the intracellular niche. This rapid loss of intracellular viability coupled with the fact that the ΔripA strain replicates at the same rate and extent as wild-type LVS in chemically defined minimal Chamberlain's medium suggests, albeit not conclusively, that the F. tularensis ΔripA mutant is not an auxotroph. We are currently working to determine if the ΔripA strain is affected in its ability respond and adapt to the host cell cytoplasmic environment and are identifying RipA-interacting proteins with the aim of inferring its function based on the properties of the interacting proteins.
Acknowledgments
We gratefully acknowledge Victoria Madden of the Microscopy Services Laboratory and Larry Arnold of the Flow Cytometry Facility at UNC—Chapel Hill for their guidance and assistance.
This work was supported by a Southeast Regional Center of Excellence in Biodefense and Emerging Infections grant (NIH/NIAID U54-AI057157) and by the National Institutes of Health (R21-AI053399).
Editor: S. R. Blanke
Footnotes
Published ahead of print on 2 September 2008.
REFERENCES
- 1.Abd, H., T. Johansson, I. Golovliov, G. Sandstrom, and M. Forsman. 2003. Survival and growth of Francisella tularensis in Acanthamoeba castellanii. Appl. Environ. Microbiol. 69600-606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Appelberg, R. 2006. Macrophage nutriprive antimicrobial mechanisms. J. Leukoc. Biol. 791117-1128. [DOI] [PubMed] [Google Scholar]
- 3.Balagopal, A., A. S. MacFarlane, N. Mohapatra, S. Soni, J. S. Gunn, and L. S. Schlesinger. 2006. Characterization of the receptor-ligand pathways important for entry and survival of Francisella tularensis in human macrophages. Infect. Immun. 745114-5125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baron, G. S., and F. E. Nano. 1998. MglA and MglB are required for the intramacrophage growth of Francisella novicida. Mol. Microbiol. 29247-259. [DOI] [PubMed] [Google Scholar]
- 5.Berdal, B. P., R. Mehl, N. K. Meidell, A. M. Lorentzen-Styr, and O. Scheel. 1996. Field investigations of tularemia in Norway. FEMS Immunol. Med. Microbiol. 13191-195. [DOI] [PubMed] [Google Scholar]
- 6.Beron, W., M. G. Gutierrez, M. Rabinovitch, and M. I. Colombo. 2002. Coxiella burnetii localizes in a Rab7-labeled compartment with autophagic characteristics. Infect. Immun. 705816-5821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Birmingham, C. L., A. C. Smith, M. A. Bakowski, T. Yoshimori, and J. H. Brumell. 2006. Autophagy controls Salmonella infection in response to damage to the Salmonella-containing vacuole. J. Biol. Chem. 28111374-11383. [DOI] [PubMed] [Google Scholar]
- 8.Bosio, C. M., and S. W. Dow. 2005. Francisella tularensis induces aberrant activation of pulmonary dendritic cells. J. Immunol. 1756792-6801. [DOI] [PubMed] [Google Scholar]
- 9.Celebi, G., F. Baruonu, F. Ayoglu, F. Cinar, A. Karadenizli, M. B. Ugur, and S. Gedikoglu. 2006. Tularemia, a reemerging disease in northwest Turkey: epidemiological investigation and evaluation of treatment responses. Jpn. J. Infect. Dis. 59229-234. [PubMed] [Google Scholar]
- 10.Centers for Disease Control and Prevention. 2005. Tularemia transmitted by insect bites—Wyoming, 2001-2003. MMWR Morb. Mortal. Wkly. Rep. 54170-173. [PubMed] [Google Scholar]
- 11.Centers for Disease Control and Prevention. 2002. Tularemia—United States, 1990-2000. MMWR Morb. Mortal. Wkly. Rep. 51181-184. [PubMed] [Google Scholar]
- 12.Chamberlain, R. E. 1965. Evaluation of live tularemia vaccine prepared in a chemically defined medium. Appl. Microbiol. 13232-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Checroun, C., T. D. Wehrly, E. R. Fischer, S. F. Hayes, and J. Celli. 2006. Autophagy-mediated reentry of Francisella tularensis into the endocytic compartment after cytoplasmic replication. Proc. Natl. Acad. Sci. USA 10314578-14583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Claros, M. G., and G. von Heijne. 1994. TopPred II: an improved software for membrane protein structure predictions. Comput. Appl. Biosci. 10685-686. [DOI] [PubMed] [Google Scholar]
- 15.Craven, R. R., J. D. Hall, J. R. Fuller, S. Taft-Benz, and T. H. Kawula. 2008. Francisella tularensis invasion and trafficking in lung epithelial cells. Infect. Immun. 762833-2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Bruin, O. M., J. S. Ludu, and F. E. Nano. 2007. The Francisella pathogenicity island protein IglA localizes to the bacterial cytoplasm and is needed for intracellular growth. BMC Microbiol. 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dennis, D. T., T. V. Inglesby, D. A. Henderson, J. G. Bartlett, M. S. Ascher, E. Eitzen, A. D. Fine, A. M. Friedlander, J. Hauer, M. Layton, S. R. Lillibridge, J. E. McDade, M. T. Osterholm, T. O'Toole, G. Parker, T. M. Perl, P. K. Russell, and K. Tonat. 2001. Tularemia as a biological weapon: medical and public health management. JAMA 2852763-2773. [DOI] [PubMed] [Google Scholar]
- 18.Ericsson, M., I. Golovliov, G. Sandstrom, A. Tarnvik, and A. Sjostedt. 1997. Characterization of the nucleotide sequence of the groE operon encoding heat shock proteins chaperone-60 and -10 of Francisella tularensis and determination of the T-cell response to the proteins in individuals vaccinated with F. tularensis. Infect. Immun. 651824-1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feldman, K. A., R. E. Enscore, S. L. Lathrop, B. T. Matyas, M. McGuill, M. E. Schriefer, D. Stiles-Enos, D. T. Dennis, L. R. Petersen, and E. B. Hayes. 2001. An outbreak of primary pneumonic tularemia on Martha's Vineyard. N. Engl. J. Med. 3451601-1606. [DOI] [PubMed] [Google Scholar]
- 20.Forslund, A. L., K. Kuoppa, K. Svensson, E. Salomonsson, A. Johansson, M. Bystrom, P. C. Oyston, S. L. Michell, R. W. Titball, L. Noppa, E. Frithz-Lindsten, M. Forsman, and A. Forsberg. 2006. Direct repeat-mediated deletion of a type IV pilin gene results in major virulence attenuation of Francisella tularensis. Mol. Microbiol. 591818-1830. [DOI] [PubMed] [Google Scholar]
- 21.Fortier, A. H., M. V. Slayter, R. Ziemba, M. S. Meltzer, and C. A. Nacy. 1991. Live vaccine strain of Francisella tularensis: infection and immunity in mice. Infect. Immun. 592922-2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gentry, M., J. Taormina, R. B. Pyles, L. Yeager, M. Kirtley, V. L. Popov, G. Klimpel, and T. Eaves-Pyles. 2007. Role of primary human alveolar epithelial cells in host defense against Francisella tularensis infection. Infect. Immun. 753969-3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gil, H., J. L. Benach, and D. G. Thanassi. 2004. Presence of pili on the surface of Francisella tularensis. Infect. Immun. 723042-3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gil, H., G. J. Platz, C. A. Forestal, M. Monfett, C. S. Bakshi, T. J. Sellati, M. B. Furie, J. L. Benach, and D. G. Thanassi. 2006. Deletion of TolC orthologs in Francisella tularensis identifies roles in multidrug resistance and virulence. Proc. Natl. Acad. Sci. USA 10312897-12902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gish, W., and D. J. States. 1993. Identification of protein coding regions by database similarity search. Nat. Genet. 3266-272. [DOI] [PubMed] [Google Scholar]
- 26.Golovliov, I., V. Baranov, Z. Krocova, H. Kovarova, and A. Sjostedt. 2003. An attenuated strain of the facultative intracellular bacterium Francisella tularensis can escape the phagosome of monocytic cells. Infect. Immun. 715940-5950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Golovliov, I., A. Sjostedt, A. Mokrievich, and V. Pavlov. 2003. A method for allelic replacement in Francisella tularensis. FEMS Microbiol. Lett. 222273-280. [DOI] [PubMed] [Google Scholar]
- 28.Gurycova, D. 2006. Epidemiologic characteristics of tularemia in Slovakia. Bratisl. Lek. Listy 107224. [PubMed] [Google Scholar]
- 29.Gurycova, D. 1998. First isolation of Francisella tularensis subsp. tularensis in Europe. Eur. J. Epidemiol. 14797-802. [DOI] [PubMed] [Google Scholar]
- 30.Hager, A. J., D. L. Bolton, M. R. Pelletier, M. J. Brittnacher, L. A. Gallagher, R. Kaul, S. J. Skerrett, S. I. Miller, and T. Guina. 2006. Type IV pili-mediated secretion modulates Francisella virulence. Mol. Microbiol. 62227-237. [DOI] [PubMed] [Google Scholar]
- 31.Hall, J. D., R. R. Craven, J. R. Fuller, R. J. Pickles, and T. H. Kawula. 2007. Francisella tularensis replicates within alveolar type II epithelial cells in vitro and in vivo following inhalation. Infect. Immun. 751034-1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hernandez, L. D., M. Pypaert, R. A. Flavell, and J. E. Galan. 2003. A Salmonella protein causes macrophage cell death by inducing autophagy. J. Cell Biol. 1631123-1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hofstetter, I., J. Eckert, W. Splettstoesser, and A. M. Hauri. 2006. Tularaemia outbreak in hare hunters in the Darmstadt-Dieburg district, Germany. Euro. Surveill. 11E060119.3. [DOI] [PubMed] [Google Scholar]
- 34.Horton, R. M., H. D. Hunt, S. N. Ho, J. K. Pullen, and L. R. Pease. 1989. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 7761-68. [DOI] [PubMed] [Google Scholar]
- 35.Johansson, A., J. Farlow, P. Larsson, M. Dukerich, E. Chambers, M. Bystrom, J. Fox, M. Chu, M. Forsman, A. Sjostedt, and P. Keim. 2004. Worldwide genetic relationships among Francisella tularensis isolates determined by multiple-locus variable-number tandem repeat analysis. J. Bacteriol. 1865808-5818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227680-685. [DOI] [PubMed] [Google Scholar]
- 37.Lai, E. M., U. Nair, N. D. Phadke, and J. R. Maddock. 2004. Proteomic screening and identification of differentially distributed membrane proteins in Escherichia coli. Mol. Microbiol. 521029-1044. [DOI] [PubMed] [Google Scholar]
- 38.Lai, X. H., I. Golovliov, and A. Sjostedt. 2004. Expression of IglC is necessary for intracellular growth and induction of apoptosis in murine macrophages by Francisella tularensis. Microb. Pathog. 37225-230. [DOI] [PubMed] [Google Scholar]
- 39.Lindgren, H., I. Golovliov, V. Baranov, R. K. Ernst, M. Telepnev, and A. Sjostedt. 2004. Factors affecting the escape of Francisella tularensis from the phagolysosome. J. Med. Microbiol. 53953-958. [DOI] [PubMed] [Google Scholar]
- 40.LoVullo, E. D., L. A. Sherrill, L. L. Perez, and M. S. Pavelka, Jr. 2006. Genetic tools for highly pathogenic Francisella tularensis subsp. tularensis. Microbiology 1523425-3435. [DOI] [PubMed] [Google Scholar]
- 41.Markowitz, L. E., N. A. Hynes, P. de la Cruz, E. Campos, J. M. Barbaree, B. D. Plikaytis, D. Mosier, and A. F. Kaufmann. 1985. Tick-borne tularemia. An outbreak of lymphadenopathy in children. JAMA 2542922-2925. [DOI] [PubMed] [Google Scholar]
- 42.Meibom, K. L., I. Dubail, M. Dupuis, M. Barel, J. Lenco, J. Stulik, I. Golovliov, A. Sjostedt, and A. Charbit. 2008. The heat-shock protein ClpB of Francisella tularensis is involved in stress tolerance and is required for multiplication in target organs of infected mice. Mol. Microbiol. 671384-1401. [DOI] [PubMed] [Google Scholar]
- 43.Nakagawa, I., A. Amano, N. Mizushima, A. Yamamoto, H. Yamaguchi, T. Kamimoto, A. Nara, J. Funao, M. Nakata, K. Tsuda, S. Hamada, and T. Yoshimori. 2004. Autophagy defends cells against invading group A Streptococcus. Science 3061037-1040. [DOI] [PubMed] [Google Scholar]
- 44.Nikaido, H. 1994. Isolation of outer membranes. Methods Enzymol. 235225-234. [DOI] [PubMed] [Google Scholar]
- 45.Nutter, J. E., and Q. N. Myrvik. 1966. In vitro interactions between rabbit alveolar macrophages and Pasteurella tularensis. J. Bacteriol. 92645-651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ogawa, M., and C. Sasakawa. 2006. Intracellular survival of Shigella. Cell. Microbiol. 8177-184. [DOI] [PubMed] [Google Scholar]
- 47.Pechous, R., J. Celli, R. Penoske, S. F. Hayes, D. W. Frank, and T. C. Zahrt. 2006. Construction and characterization of an attenuated purine auxotroph in a Francisella tularensis live vaccine strain. Infect. Immun. 744452-4461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Polsinelli, T., M. S. Meltzer, and A. H. Fortier. 1994. Nitric oxide-independent killing of Francisella tularensis by IFN-gamma-stimulated murine alveolar macrophages. J. Immunol. 1531238-1245. [PubMed] [Google Scholar]
- 49.Quarry, J. E., K. E. Isherwood, S. L. Michell, H. Diaper, R. W. Titball, and P. C. Oyston. 2007. A Francisella tularensis subspecies novicida purF mutant, but not a purA mutant, induces protective immunity to tularemia in mice. Vaccine 252011-2018. [DOI] [PubMed] [Google Scholar]
- 50.Reintjes, R., I. Dedushaj, A. Gjini, T. R. Jorgensen, B. Cotter, A. Lieftucht, F. D'Ancona, D. T. Dennis, M. A. Kosoy, G. Mulliqi-Osmani, R. Grunow, A. Kalaveshi, L. Gashi, and I. Humolli. 2002. Tularemia outbreak investigation in Kosovo: case control and environmental studies. Emerg. Infect. Dis. 869-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rich, K. A., C. Burkett, and P. Webster. 2003. Cytoplasmic bacteria can be targets for autophagy. Cell. Microbiol. 5455-468. [DOI] [PubMed] [Google Scholar]
- 52.Romano, P. S., M. G. Gutierrez, W. Beron, M. Rabinovitch, and M. I. Colombo. 2007. The autophagic pathway is actively modulated by phase II Coxiella burnetii to efficiently replicate in the host cell. Cell. Microbiol. 9891-909. [DOI] [PubMed] [Google Scholar]
- 53.Santic, M., M. Molmeret, J. R. Barker, K. E. Klose, A. Dekanic, M. Doric, and Y. Abu Kwaik. 2007. A Francisella tularensis pathogenicity island protein essential for bacterial proliferation within the host cell cytosol. Cell. Microbiol. 92391-2403. [DOI] [PubMed] [Google Scholar]
- 54.Santic, M., M. Molmeret, K. E. Klose, S. Jones, and Y. A. Kwaik. 2005. The Francisella tularensis pathogenicity island protein IglC and its regulator MglA are essential for modulating phagosome biogenesis and subsequent bacterial escape into the cytoplasm. Cell. Microbiol. 7969-979. [DOI] [PubMed] [Google Scholar]
- 55.Saslaw, S., H. T. Eigelsbach, J. A. Prior, H. E. Wilson, and S. Carhart. 1961. Tularemia vaccine study. II. Respiratory challenge. Arch. Intern. Med. 107702-714. [DOI] [PubMed] [Google Scholar]
- 56.Staples, J. E., K. A. Kubota, L. G. Chalcraft, P. S. Mead, and J. M. Petersen. 2006. Epidemiologic and molecular analysis of human tularemia, United States, 1964-2004. Emerg. Infect. Dis. 121113-1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Swanson, M. S., and R. R. Isberg. 1995. Association of Legionella pneumo-phila with the macrophage endoplasmic reticulum. Infect. Immun. 633609-3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thorpe, B. D., and S. Marcus. 1964. Phagocytosis and intracellular fate of Pasteurella tularensis. II. In vitro studies with rabbit alveolar and guinea pig alveolar and peritoneal mononuclear phagocytes. J. Immunol. 93558-565. [PubMed] [Google Scholar]
- 59.Tigertt, W. D. 1962. Soviet viable Pasteurella tularensis vaccines. A review of selected articles. Bacteriol. Rev. 26354-373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Twine, S., M. Bystrom, W. Chen, M. Forsman, I. Golovliov, A. Johansson, J. Kelly, H. Lindgren, K. Svensson, C. Zingmark, W. Conlan, and A. Sjostedt. 2005. A mutant of Francisella tularensis strain SCHU S4 lacking the ability to express a 58-kilodalton protein is attenuated for virulence and is an effective live vaccine. Infect. Immun. 738345-8352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wessel, D., and U. I. Flugge. 1984. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem. 138141-143. [DOI] [PubMed] [Google Scholar]
- 62.White, J. D., J. R. Rooney, P. A. Prickett, E. B. Derrenbacher, C. W. Beard, and W. R. Griffith. 1964. Pathogenesis of experimental respiratory tularemia in monkeys. J. Infect. Dis. 114277-283. [DOI] [PubMed] [Google Scholar]
- 63.Wik, O. 2006. Large tularaemia outbreak in Varmland, central Sweden, 2006. Euro. Surveill. 11E060921.1. [DOI] [PubMed] [Google Scholar]