

Abstract
Nelfinavir was once one of the most commonly used protease inhibitors (PIs). To investigate the genetic mechanisms of multidrug resistance in protease isolates with the primary nelfinavir resistance mutation D30N, we analyzed patterns of protease mutations in 582 viruses with D30N from 460 persons undergoing HIV-1 genotypic resistance testing at Stanford University Hospital from 1997 to 2005. Three patterns of mutational associations were identified. First, D30N was positively associated with N88D but negatively associated with N88S. Second, D30N and L90M were negatively associated except in the presence of N88D, which facilitated the cooccurrence of D30N and L90M. Third, D30N + N88D + L90M formed a stable genetic backbone for the accumulation of additional protease inhibitor (PI) resistance mutations. In 16 patients having isolates with more than one combination of mutations at positions 30, 88, and 90, all exhibited one of the steps in the following progression: D30N → D30N + N88D → D30N + N88D + L90M → D30N + N88D + L90M + (L33F ± I84V or M46I/L ± I54V). Although nelfinavir is now used less frequently than other PIs, the well-delineated mutational pathway we describe is likely to influence patterns of cross-resistance in viruses from persons who experience virologic failure while receiving this PI.
INTRODUCTION
Eight protease inhibitors (PIs) have been licensed in the United States and Europe for the treatment of HIV-1 infection. However, because of the high levels of cross-resistance among these PIs, patients in whom two or three PI drugs have failed often develop viruses with decreased susceptibility to all PIs. Indeed, with two exceptions—D30N, which reduces susceptibility solely to nelfinavir, and I50L, which reduces susceptibility solely to atazanavir—all PI resistance mutations are associated with decreased HIV-1 susceptibility to two or more PIs.1
Most of the drug resistance mutations responsible for resistance to multiple PIs such as V82A, I84V, and L90M rarely occur in combination with D30N.2-4 To investigate the genetic mechanisms of multiple PI resistance in protease isolates with D30N, we analyzed the patterns of protease mutations in isolates containing this mutation in a large population of individuals undergoing genotypic resistance testing from 1997 to 2005.
MATERIALS AND METHODS
Patients and samples
Subjects included all patients for whom plasma samples were submitted for genotypic resistance testing at Stanford University Hospital from July 1997 to July 2005. Genotypic resistance testing of plasma HIV-1 samples was performed using a previously described sequencing protocol.5 Briefly, RNA was ex-tracted from 0.2 to 0.5 ml of plasma using a guanidine thiocyanate lysis reagent. Reverse-strand cDNA was generated from viral RNA, and first-round polymerase chain reaction (PCR) was performed using Superscript One-Step RT-PCR (Life Technologies, Rockville, MD). Nested PCR was used to amplify a 1.3-kb product encompassing the protease gene and the first 300 residues of the reverse transcriptase (RT) gene. Direct PCR cycle sequencing was performed with AmpliTaq DNA FS polymerase and dRhodamine terminators (Applied Biosystems Inc., Foster City, CA).
To sequence molecular clones, HIV-1 RNA was extracted from four cryopreserved plasma samples that had previously undergone genotypic testing and was reverse transcribed using Superscript III RT and was amplified with Pfu DNA polymerase (Stratagene). PCR product from these samples was cloned using TOPO TA cloning reagents (Invitrogen Life Sciences). Molecular clones from each sample were then sequenced using PCR cycle sequencing as described previously. Phenotypic susceptibility tests were performed using the PhenoSense Assay of Monogram Biosciences (South San Francisco, CA).
Analysis
For more than 90% of patients, one virus sequence per patient was analyzed. However, when a patient had viruses at different times with different patterns of mutations at positions 30, 88, and 90 then each of these nonredundant virus sequences was used. The levels of statistical significance for analyses using only one virus sequence per patient were no different from those obtained using the nonredundant dataset.
PI resistance mutations were defined as the following non-polymorphic protease mutations: L23I, L24I, D30N, V32I, L33F, M46ILV, I47VA, G48VM, I50VL, F53L, I54VTLMSA, G73CSAT, V82ATFLSM, I84VAC, N88DST, and L90M. Mutations present as part of an electrophoretic mixture (i.e., more than one peak was present at a position on the sequence electropherogram) were classified as mutations.
To determine whether two mutations were associated, we used chi-squared tests to compare the proportion of sequences containing both mutations with the proportion of sequences expected to contain both mutations had the mutations been independent (i.e., the expected proportion was determined by mul-tiplying the proportions of isolates with each individual mutation). This analysis was performed using all sequences, as well as using only those sequences for which electrophoretic mixtures were not present at either of the two positions examined for cooccurrence. However, because the levels of statistical significance were similar regardless of the approach, we reported the results obtained using all sequences so that the number of isolates used in different pair-wise comparisons was consistent.
RESULTS
Prevalence of mutations at positions 30, 88, and 90
From 1997 to 2005, we sequenced HIV-1 protease sequences from 8060 plasma virus isolates from 5174 persons. One or more PI resistance mutations were present in 43.0% (3463) of viruses; and viruses with PI resistance mutations were present in 39.0% (2018) of persons. Mutations at positions 30, 88, and/or 90 were present in 31.7% (2556) of viruses; and viruses with mutations at one of these three positions were present in 30.0% (1550) of persons. D30N was present in 7.2% (582) of 8060 viruses and in viruses from 8.9% (460) of 5174 persons. N88D, N88S, N88T, and N88G were present in 5.7%, 1.3%, 0.2%, and 0.1% of viruses and in 6.5%, 1.5%, 0.2%, and 0.1% of persons. L90M was present in 24.1% (1948) of viruses and in 23.2% (1198) of persons.
Combinations of mutations at positions 30, 88, and 90
Table 1 summarizes the patterns of protease mutations at positions 30, 88, and 90 in 1623 viruses from 1550 persons. In 73 persons, more than one virus was included because different patterns of mutations at these three positions were present at different times. Table 1 reveals three significant features of the relationship among the mutations at these three positions.
Table 1.
Patterns of Protease Mutations at Positions 30, 88, and 90 in 1623 Viruses from 1550 Persons
| D30 | N88 | L90 | Number | Percentage of total with mutation at positions 30, 88, 90 | Percentage with additional PI resistance mutation |
|---|---|---|---|---|---|
| - | - | M | 1034 | 63.7 | 71 |
| N | D | - | 255 | 15.7 | 18 |
| N | - | - | 126 | 8.7 | 20 |
| - | S | - | 63 | 3.9 | 73 |
| N | D | M | 55 | 3.4 | 71 |
| - | D | M | 20 | 1.2 | 90 |
| - | S | M | 16 | 1.0 | 56 |
| - | D | - | 14 | 0.9 | 72 |
| N | - | M | 6 | 0.4 | 17 |
| - | T | - | 6 | 0.4 | 83 |
| - | T | M | 4 | 0.2 | 100 |
| - | G | M | 4 | 0.3 | 75 |
| N | S | - | 3 | 0.2 | 0 |
| N | T | - | 1 | 0.1 | 100 |
First, D30N was positively associated with N88D but negatively associated with N88S. Whereas D30N and N88D were present in the same sequence more frequently than expected (19.6% vs. 6.0%; p < 0.001), D30N and N88S were present in the same virus less frequently than expected (0.1% vs. 1.4%; p < 0.001).
Second, D30N and L90M were negatively associated with one another except in the presence of N88D. Whereas D30N and L90M occurred together less frequently than expected in all 1623 sequences (3.8% vs. 20.0% of sequences; p < 0.001), there was no negative association between these two mutations in the 333 viruses containing N88D (Table 2).
Table 2.
Interaction Between D30N and L90M and its Modulation by N88D
| Number of isolates | Percentage D30N | Percentage L90M | Percentage D30N + L90M | Expected percentage with D30N + L90M | p | |
|---|---|---|---|---|---|---|
| Interaction between D30N and L90M among all isolates with mutation at D30N and/or L90M | ||||||
| Mutant isolates | 1623 | 28.5 | 70.2 | 3.8 | 20.0 | <0.001 |
| Effect of N88D on the interaction between D30N and L90M | ||||||
| With N88D | 344 | 90.1 | 21.8 | 16.0 | 19.6 | 0.2 |
| Without N88D | 1279 | 9.9 | 78.2 | 0.5a | 7.7 | <0.001 |
Of the six viruses containing D30N and L90M, four had electrophoretic mixtures at one or both positions.
Third, only 18% and 20% of viruses with D30N and D30N + N88D, respectively, occurred in combination with additional PI resistance mutations. In contrast, viruses with most other patterns of mutations at these positions, including D30N + N88D + L90M, usually did occur in combination with additional PI resistance mutations. In particular, the mutations L33F, M46I/L, F53L, I54V/L, and I84V occurred in 16-44% of viruses with D30N + N88D + L90M (Fig. 1). In 16 patients with isolates having different combinations of D30N, N88D, and L90M, all exhibited one of the steps in the following progression: D30N → D30N + N88D → D30N + N88D + L90M → D30N + N88D + L90M + (L33F ± M46I/L ± F53L ± I54V/L ± I84V).
FIG. 1.

Patterns of drug resistance mutations in 43 isolates containing the mutations D30N, N88D, and L90M. Each of these isolates was obtained from a different person. Positions with electrophoretic mixtures are underlined.
To confirm that the D30N, N88D, and L90M can coexist in the same virus genomes, we sequenced several clones from four isolates in which D30N, N88D, and L90M had originally been detected by direct cycle sequencing. Sequences of these clones showed that all three mutations were present in two or more clones from each of the four isolates (Fig. 2).
FIG. 2.

Sequences of molecular clones of HIV protease from four isolates that contained the protease mutations D30N, N88D, and L90M by direct PCR sequencing. The one-letter amino acid code is used to indicate differences from consensus B. The residue at positions 30, 88, and 90 are shown in bold.
Phenotypic impact of viruses with mutations at positions 30, 88, and 90
Table 3 shows the phenotypic susceptibilities of clinical isolates containing the most commonly occurring patterns of mutations at positions 30, 88, and 90 obtained from the Stanford HIV Drug Resistance Database. Approximately 25% of these phenotypic results were obtained on samples from persons in this study, including eight samples containing mutations at positions 30, 88, and 90. Only one pattern—D30N + N88D + L90M—was associated with a decreased susceptibility of 3-fold or more to more than one or two PIs. These mutations alone reduced susceptibility to nelfinavir, ritonavir, atazanavir, and saquinavir. But in combination with either L33F ± I84V or M46I/L ± I54V, these mutations were associated with reduced susceptibility to each of the PIs with a range of 3- to 10-fold decreased susceptibility for Amprenavir and Loprinavir to >100-fold for Nelfinavir and Saquinavir.
Table 3.
Drug Susceptibilities of Isolates Containing Mutations at Positions 30, 88, and 90a
| Mutation pattern | APV | ATV | IDV | LVP | NFV | RTV | SQV |
|---|---|---|---|---|---|---|---|
| L90M | 1.0 (n = 12) | 1.8 (n = 4) | 1.8 (n = 16) | 1.1 (n = 10) | 4.6 (n = 15) | 3.0 (n = 16) | 1.5 (n = 16) |
| D30N | 0.5 (n = 9) | 1.8 (n = 5) | 0.9 (n = 12) | 0.7 (n = 3) | 14 (n = 12) | 0.6 (n = 12) | 0.4 (n = 12) |
| N88S | 0.1 (n = 7) | 10 (n = 2) | 2.1 (n = 7) | 0.5 (n = 1) | 11 (n = 7) | 1.2 (n = 7) | 1.2 (n = 7) |
| D30N N88D | 0.9 (n = 19) | 3.6 (n = 12) | 1.5 (n = 22) | 0.85 (n = 12) | 50 (n = 22) | 1.4 (n = 22) | 2 (n = 21) |
| D30N + N88D + L90M | 1.3 (n = 10) | 4.0 (n = 1) | 2.7 (n = 10) | 1.2 (n = 1) | 71 (n = 10) | 3.8 (n = 10) | 5.3 (n = 10) |
| D30N + N88D + L90M + L33F | 4.6 (n = 3) | 13 (n = 2) | 3.5 (n = 3) | 4.9 (n = 2) | 98 (n = 3) | 8.4 (n = 3) | 16 (n = 3) |
| D30N + N88D + L90M + I54V | 2.5 (n = 4) | 41 (n = 1) | 12 (n = 4) | N/A | 179 (n = 4) | 34 (n = 4) | 82 (n = 4) |
| D30N + N88D + L90M + I84V | 8.3 (n = 1) | 79 (n = 1) | 16 (n = 1) | 12 (n = 1) | 600 (n = 1) | 7 (n = 1) | 1000 (n = 1) |
| D30N + N88D + L90M + M46I | 2.5 (n = 1) | 6.6 (n = 1) | 6 (n = 1) | 2.4 (n = 1) | 121 (n = 1) | 2 (n = 1) | 15 (n = 1) |
| D30N + N88D + L90M + L33F + I54V | 11 (n = 1) | 5.9 (n = 1) | 2 (n = 1) | 2.9 (n = 1) | 52 (n = 1) | 5.7 (n = 1) | 20 (n = 1) |
| D30N + N88D + L90M + L33F + I84V | 42 (n = 1) | 96 (n = 1) | 21 (n = 1) | 27 (n = 1) | 200 (n = 1) | 81 (n = 1) | 200 (n = 1) |
| D30N + N88D + L90M + 46L + I54V | 1.8 (n = 1) | 34 (n = 1) | 14 (n = 1) | 7.9 (n = 1) | 600 (n = 1) | 37 (n = 1) | 87 (n = 1) |
None of the isolates included in this table had electrophoretic mixtures at any of the nonpolymorphic PI-resistance mutations.
Replication capacity (Monogram Biosciences) was available on five of eight isolates with D30N + N88D + L90M. Four had replication capacity values of 46-82%. One had a replication capacity of 10%; but this isolate also had the NNRTI resistance mutation G190Q, which is known to significantly impair virus replication.6
Treatment history and virologic outcome associated with mutations at positions 30, 88, and 90
Complete antiretroviral treatment histories were available on 27 of the 55 persons containing at least one isolate with mutations at positions 30, 88, and 90. Four of these patients received nelfinavir as their only PI for a median of 39 months (range: 30-46 months) and 10 received nelfinavir followed by ritonavir-boosted saquinavir. Twelve of the remaining 13 patients received nelfinavir followed by two or more PIs.
Of 25 persons with follow-up treatment history and plasma HIV-1 RNA levels, 16 had sustained virologic response indicated by at least two consecutive RNA levels below 50 copies/ml. Sustained responses occurred in four persons receiving efavirenz and two persons receiving lopinavir/ritonavir in combination with two or more NRTIs. Ten responses occurred in persons receiving efavirenz in combination with lopinavir/ritonavir, indinavir/ritonavir, or amprenavir/ritonavir.
DISCUSSION
There are three genetic mechanisms of nelfinavir resistance in patients receiving this drug as their first PI: D30N ± N88D, L90M ± M46I/L, and less commonly N88S.7 We identified three new and strongly significant patterns of association among mutations in viruses containing the primary nelfinavir-associated mutation, D30N. First, D30N was strongly associated with N88D but negatively associated with N88S. Second, the two highly antagonistic PI resistance mutations, D30N and L90M, frequently occurred in the same viruses, but only when a third PI resistance mutation, N88D, was also present. Third, the combination of D30N, N88D, and L90M forms a stable genetic backbone for the accumulation of additional mutations, particularly L33F, M46I/L, F53L, I54V/L, and I84V, which rarely occurred in the presence of D30N alone or D30N and N88D (without L90M).
Sequences of molecular clones from four patients confirmed that the mutations D30N + N88D + L90M did occur in thesame viral genomes. Phenotypic studies confirmed that isolates with these plus additional mutations had reduced susceptibility to multiple PIs. However, the reduction in susceptibility to amprenavir and lopinavir was generally not high and salvage therapy with one of these PIs, in combination with ritonavir and other antiretrovirals, was frequently successful at suppressing virus replication.
D30N is the only charged substrate cleft PI resistance mutation and N88D restores charge within the mutated protease enzyme, potentially stabilizing the molecule for the addition of the common PI resistance mutation L90M. The three-dimensional structures of proteases with D30N, N88D, and L90M individually have each been determined,8,9 but the structure of protease with combinations of mutations at these positions has not been studied. Figure 3A shows the superposition of three structures containing D30N alone, N88D alone, and L90M alone.8 Figure 3B shows the proximity of the van der Waals surface of these three residues in the enzyme.
FIG. 3.
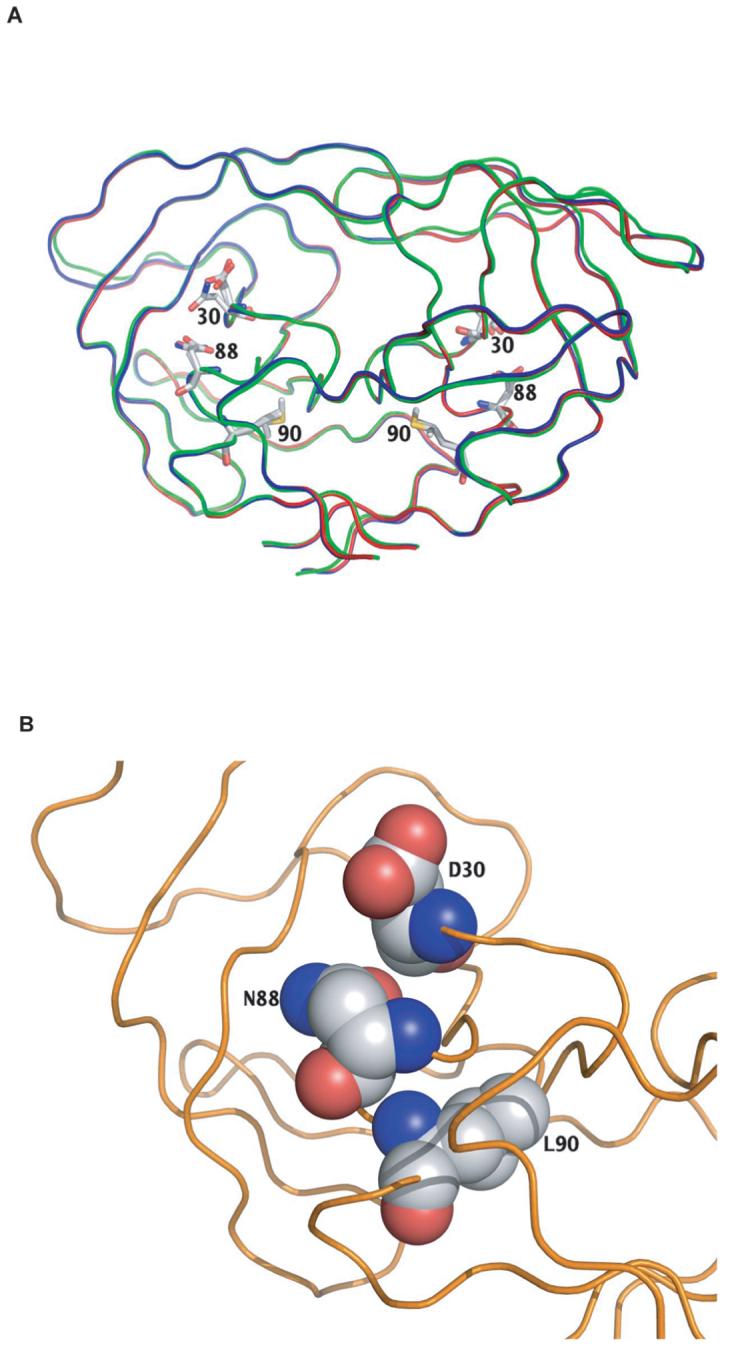
Structural relationship among protease residues 30, 88, and 90. (A) Superimposition of the three-dimensional structures of mutant viruses reported by Mahalingam et al. containing D30N alone (2F80, blue ribbon), N88D alone (1FG 8, green ribbon), and I90M alone (2F8, red ribbon).8 (B) van der Waals surfaces of wild-type residues D30, N88, and L90 deduced from wild-type protease complexed with the PI TMC114 (1T7J).9
The concept that HIV-1 might develop multidrug resistance through restricted mutational pathways was once believed to afford a strategy for treating patients with drugs specifically intended to limit HIV-1 evolution. This strategy, however, has lost much of its relevance as more potent drugs and drug combinations, which completely suppress HIV-1 replication, have become available. Nonetheless, the well-delineated mutational pathway to multiple PI resistance that we describe is relevant to the large number of persons who have been successfully maintained on a nelfinavir-containing regimen or who have developed virologic failure with the protease mutation D30N while receiving nelfinavir.
SEQUENCE DATA
Patient plasma HIV-1 protease sequences used in our analysis containing D30N + N88D + L90M mutations (114 sequences) have been submitted to GenBank under the following accession IDs: AY796625, AY030574, AY030661, AY030740, AY030814, AY030982, AY031012, AY798398, AY031117, AY031132, AY031136, AY031249, AY031271, AF544562, AY031358, AY047419, AY031475, AY031485, AY031507, AY796682, AY031648, AY031739, AY031782, AY032099, AY797073, AY032240, AY032274, AY797172, AY796689, AY797124, AY797096, AY032486, AY032495, AY032564, AY032538, AY797524, AY796881, AF544574, AY797646, AY797649, AY797671, AY796691, AY797735, AY047427, AY797453, AF544599, AY031674, AY797231, AY030418, AF085089, AY047380, AF544409, AY798344, AY031837, AY797289, AY796756, AY047437, AY796757, AY031159, AY797254, AY797255, AY032452, AY797416, AY030978, AY797672, AY796996, AY797525, AY796442, AY797736, AY797026, AY797097, AY797074, AY796758, AY796690, AY797417, AY796998, AY798183, AY798212, AY796626, AY798416, AY796692, AY796693, AY797526, AY797376, AY797737, AY796997, AY797673 DQ780897, DQ780898, DQ780899, DQ780900, DQ780901, DQ780902, DQ780903, DQ780904, DQ780905, DQ780906, DQ780907, DQ780908, DQ780909, DQ780910, DQ780911, DQ780912, DQ780913, DQ780914, DQ780915, DQ780916, DQ780917, DQ780918, DQ780919, DQ780920, DQ780921, DQ780922, DQ780923.
ACKNOWLEDGMENTS
Y.M. was supported by NIH/NIAID AI46148-01. R.W.S. and C.A.S. were supported by NIH/NIGMS GM66524. The authors also acknowledge support from Agouron Pharmaceuticals/Pfizer and from Shivender Shandilya, M.S., for assistance with the figures.
REFERENCES
- 1.Rhee SY, Liu T, Ravela J, Gonzales MJ, Shafer RW. Distribution of human immunodeficiency virus type 1 protease and reverse transcriptase mutation patterns in 4,183 persons undergoing genotypic resistance testing. Antimicrob Agents Chemother. 2004;48(8):3122–3126. doi: 10.1128/AAC.48.8.3122-3126.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sugiura W, Matsuda Z, Yokomaku Y, et al. Interference between D30N and L90M in selection and development of protease inhibitor-resistant human immunodeficiency virus type 1. Antimicrob Agents Chemother. 2002;46(3):708–715. doi: 10.1128/AAC.46.3.708-715.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kantor R, Fessel WJ, Zolopa AR, et al. Evolution of primary protease inhibitor resistance mutations during protease inhibitor salvage therapy. Antimicrob Agents Chemother. 2002;46(4):1086–1092. doi: 10.1128/AAC.46.4.1086-1092.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu TD, Schiffer CA, Gonzales MJ, et al. Mutation patterns and structural correlates in human immunodeficiency virus type 1 protease following different protease inhibitor treatments. J Virol. 2003;77(8):4836–4847. doi: 10.1128/JVI.77.8.4836-4847.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shafer RW, Hertogs K, Zolopa AR, et al. High degree of interlaboratory reproducibility of human immunodeficiency virus type 1 protease and reverse transcriptase sequencing of plasma samples from heavily treated patients. J Clin Microbiol. 2001;39(4):1522–1529. doi: 10.1128/JCM.39.4.1522-1529.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang W, Gamarnik A, Limoli K, Petropoulos CJ, Whitcomb JM. Amino acid substitutions at position 190 of human immunodeficiency virus type 1 reverse transcriptase increase susceptibility to delavirdine and impair virus replication. J Virol. 2003;77(2):1512–1523. doi: 10.1128/JVI.77.2.1512-1523.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rhee SY, Gonzales MJ, Kantor R, Betts BJ, Ravela J, Shafer RW. Human immunodeficiency virus reverse transcriptase and protease sequence database. Nucleic Acids Res. 2003;31(1):298–303. doi: 10.1093/nar/gkg100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mahalingam B, Louis JM, Hung J, Harrison RW, Weber IT. Structural implications of drug-resistant mutants of HIV-1 protease: High-resolution crystal structures of the mutant protease/substrate analogue complexes. Proteins. 2001;43(4):455–464. doi: 10.1002/prot.1057. [DOI] [PubMed] [Google Scholar]
- 9.Kovalevsky AY, Tie Y, Liu F, et al. Effectiveness of nonpeptide clinical inhibitor TMC-114 on HIV-1 protease with highly drug resistant mutations D30N, I50V, and L90M. J Med Chem. 2006;49(4):1379–1387. doi: 10.1021/jm050943c. [DOI] [PMC free article] [PubMed] [Google Scholar]