

Abstract
An enigmatic feature of age-related neurodegenerative diseases is that they seldom, if ever, are fully manifested in nonhuman species under natural conditions. The neurodegenerative tauopathies are typified by the intracellular aggregation of hyperphosphorylated, microtubule-associated protein tau (MAPT) and the dysfunction and death of affected neurons. We document the first case of tauopathy with paired helical filaments in an aged chimpanzee (Pan troglodytes). Pathologic forms of tau in neuronal somata, neuropil threads, and plaque-like clusters of neurites were histologically identified throughout the neocortex and, to a lesser degree, in allocortical and subcortical structures. Ultrastructurally, the neurofibrillary tangles consisted of tau-immunoreactive paired helical filaments with a diameter and helical periodicity indistinguishable from those seen in Alzheimer’s disease. A moderate degree of Aβ deposition was present in the cerebral vasculature and, less frequently, in senile plaques. Sequencing of the exons and associated introns in the genomic MAPT locus disclosed no mutations that are associated with the known human hereditary tauopathies, nor any polymorphisms of obvious functional significance. Although the lesion profile in this chimpanzee differed somewhat from that of Alzheimer’s disease, the co-presence of paired helical filaments and Aβ-amyloidosis indicates that the molecular mechanisms for the pathogenesis of the two canonical Alzheimer lesions - neurofibrillary tangles and senile plaques - are present in aged chimpanzees.
Keywords: Aβ, aging, amyloid, Alzheimer’s disease, cerebral amyloid angiopathy, neurodegeneration, neurofibrillary tangles, Pan troglodytes, proteopathy, senile plaques, tau
The tauopathies are a class of human neurodegenerative disorders characterized by the intracellular aggregation of abnormally phosphorylated tau protein and selective neuronal loss (Lee et al., 2001). Tau dysfunction likely plays a primary role in disease pathogenesis, as mutations in the MAPT locus are associated with several of these tauopathies (Goedert and Jakes, 2005). In the mammalian nervous system, tau normally interacts with tubulin to stabilize microtubules and promote their assembly (Mandelkow et al., 2007). In the neurodegenerative tauopathies, tau is hyperphosphorylated by intracellular kinases, which impedes the normal binding of the protein to cytoskeletal elements and thereby augments its tendency to polymerize into higher molecular weight assemblies such as neurofibrillary tangles (Ballatore et al., 2007; Mandelkow et al., 2007). More than 20 human tauopathies are known, each with a disease-specific clinical presentation and pattern of atypical tau aggregation in neurons and/or glial cells (Goedert, 2004; Lee et al., 2001; Williams, 2006).
The most frequently occurring tauopathy is Alzheimer’s disease (AD), a dementing disorder in which specific regions of the brain are beset by neurofibrillary tangles as well as deposits of the Aβ peptide in senile plaques in the walls of cerebral blood vessels (Duyckaerts et al., 1998; Hardy and Orr, 2006). The neurofibrillary tangles in AD consist mostly of paired helical filaments, twisting ribbons of tau that have a helical half-periodicity of ∼80nm and an apparent diameter that alternates between ∼8 and 20nm (Crowther, 1991). Although genetic and biochemical evidence indicates that the pathogenic cascade of AD is initiated by aberrant Aβ, particularly the 42-amino acid form (Aβ42) (Hardy and Selkoe, 2002), the degree of tau pathology correlates strongly with cognitive decline (Arriagada et al., 1992; Crystal et al., 1988; Giannakopoulos et al., 2007; Wilcock and Esiri, 1982).
Humans are particularly, and perhaps uniquely, susceptible to Alzheimer’s disease and other tauopathies (Nelson et al., 1996; Walker and Cork, 1999). However, amino acid sequence similarities in extant species indicate that both the Aβ-precursor protein (APP) and tau protein are highly conserved evolutionarily. The amino acid sequence of APP695 is >99% identical in humans and chimpanzees (Cervenakova et al., 1994) and the tau sequence is 100% identical in the two species (Holzer et al., 2004). Additionally, all six tau isoforms that are found in the human brain also have been identified in nonhuman primates (Holzer et al., 2004; Nelson et al., 1996).
In the normal course of senescence, many mammalian species exhibit cerebral Aβ-amyloidosis (Cork et al., 1988; Elfenbein et al., 2007; Erwin et al., 2000; Gearing et al., 1996; Gearing et al., 1997; Geula et al., 2002; Hartig et al., 1997; Kimura et al., 2003; Mestre-Frances et al., 2000; Nakamura et al., 1996; Nakamura et al., 1997; Poduri et al., 1994; Roertgen et al., 1996; Walker and Cork, 1999; Walker et al., 1990), and Aβ accumulation in the brains of aged nonhuman primates can sometimes reach levels comparable to those in AD (Elfenbein et al., 2007)(R.F. Rosen, unpublished data). Aggregated tau has been histologically identified in glia and neurons of a number of aged mammals (Braak and Braak, 1994; Cork et al., 1988; Hartig et al., 1997; Kiatipattanasakul et al., 2000; Nelson et al., 1994; Nelson et al., 1993; Nelson and Saper, 1995; Roertgen et al., 1996; Schultz et al., 2001), but no documented case of nonhuman tau pathology fully recapitulates the degree and localization of intraneuronal tau accumulation seen in the human tauopathies. Furthermore, neuronal tau pathology with AD-like paired helical filaments has never been identified in a nonhuman primate (Erwin et al., 2000; Walker and Cork, 1999). Here we present the first evidence of tauopathy with human-like paired helical filaments in an aged chimpanzee.
MATERIALS AND METHODS
Subjects
A 41-year old, socially housed female chimpanzee (Pan troglodytes)(CO494) at the Yerkes National Primate Research Center spontaneously developed acute lethargy and rapidly progressive motor dysfunction suggestive of stroke. A T2-weighted magnetic resonance (MR) scan revealed a massive, left-hemispheric lesion involving mainly the temporal, parietal and occipital lobes (Fig. 1B). At necropsy, gross examination of the brain confirmed a substantial region of ischemic (non-hemorrhagic) necrosis in the left hemisphere. The right hemisphere was grossly normal. The total brain weight was 287.5 grams, less than the average weight of an adult chimpanzee brain (Herndon et al., 1999), possibly due to necrosis in the left hemisphere. The previous medical history was unremarkable except for a chronic systolic heart murmur first diagnosed at 15 years of age, moderate obesity (weight at death 61.5 kg) and high serum cholesterol (total cholesterol levels in 1995, 2000 and 2005 were 262, 244 and 359mg/dl, respectively). A T1-weighted MR scan performed in 1995 showed no obvious abnormalities of the brain (Fig. 1A).
Figure 1.

MR images of the chimpanzee brain before and during the stroke. A-D (Top row): An anterior-posterior series of coronal, T1-weighted MR images made 10 years prior to death. E-H (Bottom row): T2-weighted MR images captured after the symptoms had emerged, with the location of affected tissue in the left hemisphere indicated by asterisks. [A T2-weighted scan was not run prior to the stroke].
For comparison, age-related Aβ and tau lesions were examined in available archival neocortical and/or hippocampal tissue samples from eight additional chimpanzees aged 30 to 59 years (Table 1). Furthermore, as a reference group for the MAPT genotyping, frozen cerebella from 7 chimpanzees were selected from the Yerkes archives for tau gene sequence analysis. All studies were conducted in accordance with federal and local guidelines for the humane care and use of animals.
Table 1.
Supplemental chimpanzees examined for Aβ and tau pathology (archival material)
| Subject | Sex | Age (years) |
|---|---|---|
| N00-39Pt | f | 30 |
| N00-40Pt | m | 34 |
| N00-34Pt | m | 37 |
| N01-20Pt | f | 41 |
| Y06-147Pt | m | 43 |
| Y07-25Pt | f | 44 |
| Y06-108Pt | f | 47 |
| C-612 | f | 56 |
Collection and Preparation of Cerebral Tissue Samples
Fresh, unfixed tissue blocks (∼500 mg) were excised from the right (non-infarcted) superior temporal cortex for analysis by enzyme-linked immunosorbance assay (ELISA). [Unfixed tissue samples for ELISA also were taken from the superior temporal cortex of 5 end-stage AD cases for comparison; Table 2]. The brain then was coronally slabbed at ∼2cm thickness and immersion-fixed in 10% neutral-buffered formalin for at least 7 days. A set of bilateral, matched tissue blocks from the prefrontal, temporal, and occipital cortices (avoiding the infarct core), as well as samples from the diencephalon, globus pallidus, putamen, lower brainstem and cerebellum, were sectioned at 50μm thickness on a vibratome and stored at 4°C in phosphate-buffered saline (PBS) prior to immunostaining. Additional fixed brain samples from the right temporal lobe were cryoprotected in 30% sucrose, frozen, and cut at 50μm thickness on a freezing-sliding microtome. Frozen sections from the eight reference chimpanzees were sectioned and stored similarly, and all frozen sections were maintained in an antifreeze solution (30% sucrose/30% ethylene glycol in PBS) at -20°C until immunostaining. Separate, bilateral tissue blocks from the prefrontal, temporal and occipital lobes of chimpanzee CO494 were paraffin-embedded, sectioned at 8μm thickness, and mounted onto silanized slides (Newcomer Supply, Middleton, WI) for silver- and immuno-staining, as well as for additional histopathological stains (below).
Table 2.
AD cases examined by Aβ ELISA
| Subject | Sex | Age (years) | PMI (hours) |
|---|---|---|---|
| OS02-159 | m | 61 | 5.5 |
| OS03-300 | f | 75 | 12 |
| OS02-106 | f | 81 | 2 |
| E04-172 | f | 87 | 6 |
| OS01-128 | f | 91 | 2.5 |
PMI: postmortem interval
Antibodies and Reagents
The following antibodies were used for immunohistochemistry: AT8 (1:5,000) mouse IgG1 monoclonal antibody from Pierce Biotechnology (Rockford, IL), Cat#MN1020, in PBS, raised against partially purified human PHF-tau, has an epitope in the region around phosphoserine 202/phosphothreonine 205 and does not cross-react with normal tau (Goedert et al., 1995); CP13 (1:10,000) mouse IgG1 monoclonal antibody, a generous gift from Dr. Peter Davies (Albert Einstein College of Medicine, Bronx, NY), was produced from mice immunized with PHF-tau purified from Alzheimer brain tissue by the method described in detail in Jicha et al. (1999). The antibody was selected for reactivity with the phosphopeptide GYSSPG(phosphoS)PGTPGSRS, where the phosphoS is phosphoserine 202 of tau. No reactivity with any other phosphoserine of tau was detected (Jicha et al., 1999); PHF1 (1:10,000) mouse IgG1 monoclonal antibody, also from Dr. Davies, was raised against detergent-extracted PHF preparations, and has been epitope-mapped to the region around phosphoserine 396/404 (Greenberg et al., 1992; Otvos et al., 1994). Specificity was confirmed with Western blots of transfected cell lysates (Otvos et al., 1994); MC1 (1:10,000) mouse IgG1 monoclonal, also from Dr. Davies, was raised against Alz50-immunopurified PHFs and then epitope-mapped to similar conformation-specific regions as Alz50, but not FAC1 (Jicha et al., 1997) (in the same study, Western blot and dot blot showed specificity of MC1 to PHFs of tau protein); 6E10 (1:5,000) mouse IgG1 monoclonal antibody from Covance (Princeton, NJ), Cat# SIG-39320, Protein G-purified, in PBS, was raised against residues 1-16 of Aβ peptide, with an epitope at residues 3-8 (Kim, 1988); 4G8 (1:5,000) mouse IgG2b monoclonal antibody from Covance, Cat# SIG-39220, Protein G-purified, in PBS, was raised against residues 17-24 of Aβ peptide, with an epitope at residues 18-22 (Kim, 1988). The specificity of both 6E10 and 4G8 antibodies was characterized in the source reference. Rabbit polyclonal antibodies R361 and R398 (both at 1:15,000), a generous gift from Dr. Pankaj Mehta (Institute for Basic Research on Developmental Disabilities, Staten Island, NY), were raised against synthetic Aβ32-40 and Aβ33-42 (AnaSpec, San Jose, CA), respectively, conjugated to keyhole limpet hemocyanin in PBS. Specificity of these antibodies was examined by sandwich ELISA and Western blot analysis (Potempska et al., 1999). All antibodies to phosphorylated tau and Aβ were tested immunohistochemically with human AD tissue sections and specifically stained only abnormal tau accumulations and parenchymal and vascular amyloid deposits, respectively. Anti-ubiquitin (1:10,000) rabbit polyclonal antibody from Dako (Carpinteria, CA), Cat#Z-0458, was raised against ubiquitin isolated from cow erythrocytes and conjugated to chicken gamma globulins with glutaraldehyde, purified by solid-phase absorption with human plasma proteins (in Western blotting, the antibody labels bands corresponding to free ubiquitin and ubiquitin conjugates); anti-GFAP (1:5,000) purified immunoglobulin fraction of rabbit antiserum from Dako (Carpinteria, CA), Cat# Z0334, was raised against glial fibrillary acidic protein isolated from cow spinal cord, purified by solid-phase absorption with human and cow serum proteins (shows no cross reactivity with human plasma or cow serum by crossed immunoelectrophoresis and indirect ELISA). Immunohistochemistry with the antibody revealed the expected pattern of astrocytic staining in human AD and chimpanzee tissue sections. Anti-Iba1 (1:10,000) rabbit polyclonal antibody from Wako (Osaka, Japan), Cat# 019-19741, in TBS, purified by affinity antigen chromatography from rabbit antisera, was raised against a synthetic peptide (PTGPPAKKAISELP) corresponding to the C-terminus of Iba1, a 17-kDa EF hand protein that is specifically expressed in macrophages/ microglia and is upregulated during the activation of these cells. Specificity was confirmed by Western Blot (Imai, et al., 1996) and immunohistochemistry with the antibody revealed the expected pattern of microglial staining in human AD and chimpanzee tissue sections. Anti-α-synuclein (1:300) rabbit polyclonal antibody, a gift of Dr. Bernardino Ghetti (Indiana University, Indianapolis, Indiana), was raised against a peptide containing residues 119-137 (DPDNEAYEMPSEEGYODYE) of the C-terminus of α-synuclein (Piccardo et al, 1998). Specificity was confirmed by specific immunolabeling of Lewy bodies in Parkinson’s disease. Vectastain Elite kits (Vector Laboratories, Burlingame, CA) were used for ABC-based immunodetection of antigen-antibody complexes.
Histochemistry
Endogenous peroxidase in tissue sections was inactivated with 3% H2O2 in methanol for 10 minutes at room temperature. Nonspecific reagent binding was blocked with 2% normal serum in 0.2% Tween for one hour at room temperature. For Aβ-immunodetection, sections were pretreated for 10 minutes in 90% formic acid to expose antigenic sites. Sections were incubated in primary antibody (diluted in buffer with blocking serum) overnight at 4°C. After rinsing, sections were incubated for one hour at room temperature in biotinylated secondary antibody, rinsed, immersed for 30 minutes in avidin-biotin complex, and then developed with diaminobenzidine (DAB) or DAB complexed to nickel (Vector Laboratories). Tissue from human AD cases was used as positive control material, and non-immune mouse IgG or rabbit sera were used in place of the primary antibodies as negative controls. In some instances, a light hematoxylin counterstain was applied after immunostaining.
Double immunostaining was employed to localize multiple antigens in tissue sections. For fluorescence immunostaining, sections were blocked in 2% normal goat serum / 0.1% Triton X-100 in PBS for one hour at room temperature, incubated in a mouse monoclonal antibody (diluted in 2% normal goat serum) overnight at 4°C, rinsed well, and then incubated for 90 minutes in Cy2-conjugated anti-mouse secondary antibody (1:200; Jackson ImmunoResearch Laboratories, West Grove, PA). Sections were again rinsed thoroughly, incubated overnight in diluted rabbit polyclonal antibody at 4°C, rinsed, and placed for 90 minutes in Rhodamine-Red-X goat anti-rabbit secondary antibody (1:200; Jackson). To block endogenous autofluorescence, double-stained sections were mounted on gel-coated slides, rinsed for 5 minutes in 70% ethanol and immersed in 1% Sudan black (in 70% ethanol, filtered before each use) for 30 minutes at room temperature. Excess Sudan black was removed via 3 rinses in ethanol, and sections were coverslipped from PBS with Faramount aqueous mounting medium (Dako) for fluorescence microscopy. To visualize multiple antigens in the same sections by standard transillumination light microscopy, antibodies were applied as described above for single antigen/DAB immunohistochemistry, except that the first antibody-antigen complex was marked with DAB+nickel (black), and the subsequent antibody was marked with DAB only (brown).
Finally, we stained selected sections with the Congo Red, Bielschowsky and Campbell-Gallyas stains for AD-type lesions, with the Prussian Blue (Perls) stain for iron, with hematoxylin and eosin, and with a Gram stain for bacteria (there was no evidence of bacterial infection in any brain region). Light-microscopic photomicrographs were taken with a Leica DMLB microscope (Wetzlar, Germany) and SPOT XPlorer and FLEX digital cameras (Diagnostic Instruments, Sterling Heights, MI). Confocal images were captured with a Zeiss LSM 510 laser scanning confocal microscope. All images were edited in Photoshop (Adobe) without any further manipulations.
Quantitative Mapping of Tau and Aβ Lesions
Tau lesions (intrasomatic neurofibrillary tangles and plaque-like clusters of immunoreactive neurites) and Aβ lesions (cerebral Aβ-amyloid angiopathy [CAA] and parenchymal [senile] Aβ plaques) were mapped and quantitated bilaterally in matched CP13-, R361- and R398-immunostained sections from the prefrontal, temporal, and occipital cortices using the Neurolucida image analysis system (MBF Biosciences, Williston, VT). The prefrontal cortical sections were taken at the level of the rostral end of the middle frontal gyrus (Bailey & Bonin FE/FD; Brodmann area 10/9/46) (Bailey et al., 1950; Brodmann, 1912); temporal cortical sections taken at the level anterior to the primary auditory cortex (A1) (TA/TE, Brodmann area 20/21/22); and the occipital cortical sections were from a level between the lunate sulcus and the occipital pole, containing both area V1 and extrastriate cortex (OB/OC; Brodmann area 18/17).
A single researcher used the Neurolucida system to count every tau or Aβ lesion found in each section. Every discrete cell soma or cluster of tau-immunoreactive elements was counted as a single lesion. Two different types of tau plaques were identified and counted: neuritic and punctate. Similarly, each distinct Aβ-plaque or Aβ-reactive vascular profile was indicated on the tissue map. R361- or R398-positive vessels that were spatially continuous within the section were counted as a single lesion; otherwise, discrete vascular profiles were counted separately. The numeric densities of immunoreactive lesions were calculated from the total planar area of each section (Elfenbein et al., 2007). Lesion densities in the two hemispheres were compared statistically using paired t-tests, with a set limit of p <0.05 for significance. Neuropil threads (tau-positive processes that were not organized into plaques) were profuse and widely distributed in brain (see Fig. 2) and were not quantitated histologically.
Figure 2.

Tau- and Aβ-pathology in the aged chimpanzee. A: Tau-immunoreactive somata (2 indicated by arrowheads) and neuritic tau plaques (1 indicated by arrow) in the left prefrontal cortex. Numerous tau-immunoreactive processes (threads) occupy the intervening neuropil. Antibody AT8. B: Tau-immunoreactive neuron (arrowhead) in area CA1 of the right hippocampus; note the integrity of the nearby pyramidal cells. Antibody AT8, Hematoxylin counterstain. C: Double-immunostained section showing tau- (CP13/Nickel-DAB, black, developed first) and Aβ- (R398/DAB, brown) immunoreactivity in the left prefrontal cortex. A tau plaque (black arrow) and a tau-positive pyramidal neuron (arrowhead) are present. A focus of Aβ-immunoreactive CAA is indicated by the grey arrow. Note the absence of an Aβ-core in the tau plaques, which was typical of focal accumulations of tau neurites in this animal. D: Cerebral amyloid angiopathy (arrowheads denote two blood vessels) and a senile plaque (arrow) in the right temporal neocortex. Antibody 6E10. Bars = 100 μm (A, C), 50 μm (B), 200 μm (D).
Electron Microscopy
For ultrastructural analysis, tissue samples from the left prefrontal cortex were postfixed in 4% paraformaldehyde and 0.5% glutaraldehyde, washed in phosphate buffer (0.1M, pH 7.4) and immersed in osmium tetroxide (1% in phosphate buffer) for 20 minutes. They were then rinsed in phosphate buffer and dehydrated in a graded series of ethanol and propylene oxide. Uranyl acetate (1%) was added to the 70% ethanol (35 minute immersion) to improve contrast in the electron microscope. The sections were then embedded in epoxy resin (Durcupan ACM; Fluka, Ft. Washington, PA) on microscope slides and heated for 48 hours at 60°C. Areas of interest were selected, excised from the slide and glued onto resin blocks. Ultrathin sections were cut with a Leica Ultracut T2 (Nussloch, Germany), collected onto single-slot copper grids, and stained with lead citrate.
For immunogold EM, non-postfixed sections were preincubated in PBS containing 5% nonfat dry milk and then washed in Tris-buffered saline (TBS)-gelatin buffer (0.02 M Tris, 0.15 M NaCl, 1 μl/ml fish gelatin, pH 7.6) to block nonspecific sites. Sections were then incubated for 48 hours at 4°C in CP13 antibody (1:10,000) diluted in PBS-BSA, rinsed in TBS-gelatin, and incubated for 2 hours at room temperature in gold-conjugated goat anti-mouse Fab’ fragments (dilution 1:100; Nanogold [Nanoprobes Inc., Yaphank, NY]). Gold particles (1.4 nm) were silver-enhanced with the HQ Silver kit (Nanoprobes). As a control for the specificity of immunolabeling, omission of the primary antibody from incubation solutions completely abolished immunostaining for the corresponding antigens. The tissue was then embedded and cut as described above.
All thin sections were examined with a Zeiss EM10-C electron microscope (Oberkochen, Germany) and digital images were captured using the Dual View camera (Gatan Inc., Pleasanton, CA).
Aβ ELISA
Unfixed, right temporal cortical tissue was Dounce-homogenized in 5 volumes of homogenization buffer (50mM Tris-HCl, 150mM NaCl, and protease inhibitor tablets [Santa Cruz Biochemicals, Santa Cruz, CA]) and then centrifuged at 100,000g for 60 minutes at 4°C to generate the “soluble” supernatant. The pellet was probe-sonicated in 70% formic acid and centrifuged at 14,000rpm for 60 minutes at 4°C to generate the “insoluble” supernatant. Aβ40 and Aβ42 were measured in each extract by ELISA (The Genetics Company, Schlieren, Switzerland) according to the manufacturer’s instructions. Soluble extracts were diluted in sample buffer at a 1:50 dilution, and insoluble extracts were neutralized in 1.0M Tris base, pH 11 (1:20 dilution) and diluted in sample buffer to a 1:1,000 total dilution. All samples were run in duplicate. Plates were read at 450 nm and the average optical density values for each extract were interpolated on a 4-parameter standard curve to determine Aβ concentrations.
DNA Extraction and MAPT Genotyping
Genomic DNA from unfixed liver of the 41-year old subject (and, as a reference group, from unfixed cerebellar tissue from 7 additional chimpanzees) was purified using large-scale, automated template purification systems employing solid-phase reversible immobilization (Agencourt Bioscience Corporation, Beverly, MA). The purified DNA samples were then sequenced using ABI dye-terminator chemistry. All subsequent steps were based on sequencing by automated DNA sequencing methods. The ABI dye terminator sequence reads were run on ABI 3700/3730 (Applied Biosystems) machines and the data were transferred to Linux machines. Base calls and quality scores were determined using the program PHRED (Ewing and Green, 1998; Ewing et al., 1998).
RESULTS
Tau Histopathology
Postmortem immunohistochemical analysis using antibodies to pathologic forms of tau revealed abnormal intracellular tau immunoreactivity in multiple cortical regions of both hemispheres and, to a lesser extent, in subcortical structures. Throughout the neocortex (the most severely affected brain structure), the lesions included tau-laden neurons, neuropil threads, and diverse, plaque-like clusters of neurites (‘tau plaques’) (Fig. 2A-C). The regional density of tau lesions varied among and within cortical areas, and there were foci of particularly intense pathology (Fig. 2A). The lesions were most abundant in the prefrontal cortical samples, followed by the temporal cortex and the occipital cortex, which had the least tauopathy of the neocortical regions examined (Table 3). In the hippocampus, occasional tau-positive neurons (Fig. 2B), neuropil threads and tau plaques were present, but these were much less numerous than in most regions of neocortex. The tau plaques generally were not associated with a core of aggregated Aβ (Fig 2C). Subcortically, tau-immunoreactivity, mostly in thread-like processes, was present to varying degrees in the globus pallidus, neostriatum and diencephalon, and occasionally in white matter pathways. Scattered immunoreactive profiles also were present in the lower brainstem and, very infrequently, in the cerebellum. Overall, subcortical tauopathy was sparse relative to that in neocortex.
Table 3.
Quantitative mapping data of intrasomatic neurofibrillary tangles and tau plaque-like clusters of immunoreactive neurites in location-matched tissue blocks from the bilateral prefrontal, temporal, and occipital cortices of chimpanzee CO494. Lesion densities are expressed as number of lesions per unit area of tissue analyzed. Antibody CP13
| Region | # N-Plaque | # P-Plaque | # Cell Bodies | N-Plaque density (#/mm2) | # P-Plaque density (#/mm2) | Cell Body Density (#/mm2) |
|---|---|---|---|---|---|---|
| LFC | 158 | 79 | 349 | 0.6132 | 0.3066 | 1.3546 |
| LOC | 5 | 8 | 22 | 0.0182 | 0.0291 | 0.0802 |
| LTC | 22 | 43 | 138 | 0.0672 | 0.1314 | 0.4215 |
| RFC | 39 | 46 | 81 | 0.1246 | 0.1469 | 0.2587 |
| ROC | 0 | 0 | 0 | 0.0000 | 0.0000 | 0.0000 |
| RTC | 1 | 20 | 25 | 0.0072 | 0.1436 | 0.1795 |
L: left; R: right; FC: prefrontal cortex; OC: occipital cortex; TC: temporal cortex; N-plaque: tau neuritic plaque; P-Plaque: tau punctate plaque
In nearby frontal, temporal, and occipital cortical sections, antibodies AT8 and CP13 consistently detected the most tau lesions. Antibody PHF1 detected somewhat fewer lesions, and antibody MC1, which recognizes AD-type conformational epitopes on tau, detected the fewest tangles, tau plaques, and neuropil threads of the four anti-tau antibodies. A subset of neurons, plaque neurites and neuropil threads were immunoreactive for ubiquitin (not shown). Neuritic plaques and neurofibrillary tangles also were evident by Campbell-Gallyas and Bielschowsky silver stains (Fig. 3). Granulovacuolar degeneration was not seen. The presence of tau-only plaques in this animal prompted us to determine whether the tau-neurites might originate from astrocytes, as in the case of ‘astrocytic plaques’ in corticobasal degeneration (CBD) (Komori et al., 1998). Double-immunostaining for GFAP and CP13 in left and right frontal cortex showed that the neurites are not astrocytic processes (Fig. 4A). The neurites also were negative for the microglial marker Iba-1 (Fig. 4B) and α-synuclein (not shown).
Figure 3.

Silver-stained lesions in the left prefrontal neocortex of the aged chimpanzee. A: Campbell-Gallyas-stained neuritic plaque (tortuous black neurites, arrow). This plaque was positive with the anti-tau antibody CP-13 in an adjacent section (not shown). B: Two cortical cells stained with the Bielschowsky silver method. Bar = 50 μm for both A and B.
Figure 4.

Double-fluorescence immunostaining of tau-neurites and reactive glia in the left prefrontal cortex of the aged chimpanzee. By confocal microscopy, CP13-positive neurites (green) in tau plaques do not colocalize with GFAP-stained astrocytes (magenta) (A), nor do they colocalize with Iba1-immunoreactive microglia (magenta) (B). Neuropil threads also are negative for these glial markers. Bar = 20 μm for A and B.
Ultrastructurally, the neurofibrillary tangles consisted of dense, intraneuronal bundles of paired helical filaments that were identical in size and helical periodicity to those in humans with Alzheimer’s disease (Crowther, 1991; Metuzals, 1986) (Fig. 5, Fig. 6B). Immunogold labeling with antibody CP13 confirmed the presence of phospho-tau epitopes on the paired helical filaments (Fig. 5B,C).
Figure 5.
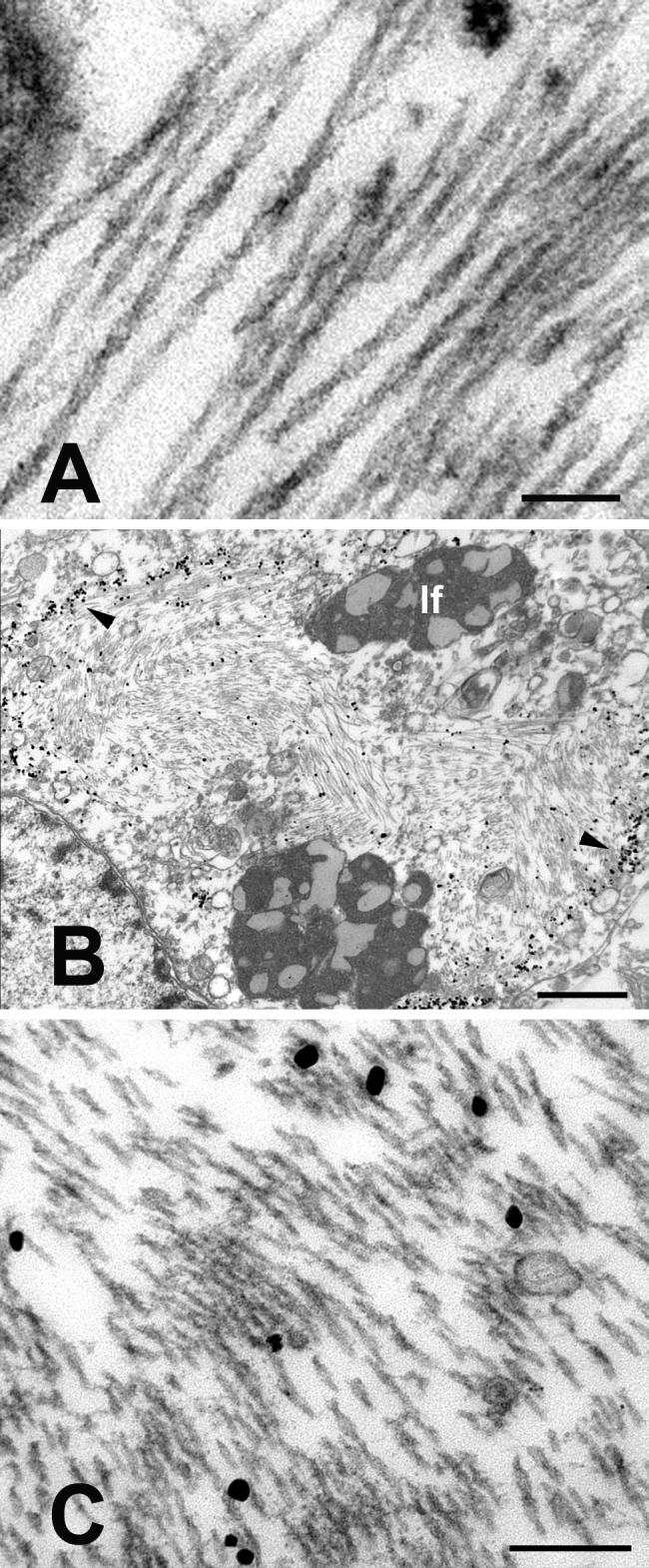
Ultrastructure of paired helical filaments in neurons from the left prefrontal cortex of the aged chimpanzee. A: High magnification view of intraneuronal paired helical filaments. The twisting, filamentous ribbons have a mean half-periodicity of ∼79nm, a maximum width of ∼20nm and a minimum width of ∼10nm. B-C: CP13/immunogold-labeled, intraneuronal paired helical filaments. B is a low magnification view of a CP13-immunoreactive neuron; the nucleus is to the lower left. Because the thick section was immunostained prior to sectioning, the immunogold preferentially decorates the periphery of the mass of PHFs (arrowheads). Note the heavy bundles of paired helical filaments in the cytoplasm. C shows a higher magnification view of CP13/immunogold-labeled PHFs from the cell in B. lf: lipofuscin. Bars = 100nm (A), 1μm (B) and 200nm (C).
Figure 6.

Tauopathy in humans with Alzheimer’s disease. A: Light micrograph showing tau-immunoreactive neurons (two indicated by arrowheads) and clusters of neurites (one indicated by arrow) in Alzheimer’s disease; antibody AT8. B: Electron micrograph of intraneuronal paired helical filaments in Alzheimer’s disease. The size and helical periodicity of the filaments are highly similar to those in chimpanzee CO494 (Figure 5). Bars = 100um (A) and 200nm (B).
Aβ Histopathology
Immunohistochemistry with antibodies to Aβ disclosed a moderate degree of CAA (Figs. 2C, D) in all of the neocortical regions examined, as well as mild, focal Aβ-plaque pathology (Fig. 2D) (Table 4). As in humans, Aβ deposits were sparse in the hippocampus compared to temporal neocortex, and were essentially absent in the basal ganglia, diencephalon, and lower brainstem. The cerebellum manifested very mild CAA. In affected regions, most Aβ-immunopositive senile plaques were diffuse in nature, as confirmed by a paucity of Congo Red staining/birefringence in parenchymal deposits. The number of Aβ42-positive plaques did not differ significantly from the number of Aβ40-positive plaques, but CAA was marginally more likely to be immunoreactive for Aβ42 than for Aβ40 (t=2.560; p= .0506). Surprisingly, the degree of total CAA was significantly greater in the right, non-stroke hemisphere than in the left hemisphere (t=4.668; p=.043) (Table 4).
Table 4.
Quantitative mapping data of Aβ parenchymal plaques and CAA in location-matched tissue blocks from the bilateral prefrontal, temporal, and occipital cortices of chimpanzee CO494. Lesion densities are expressed as number of lesions per unit area of tissue analyzed Antibodies R361 and R398 to Aβ40 and Aβ42, respectively
| Region | Total Aβ40-CAA | Total Aβ42-CAA | Total Aβ40-Plaques | Total Aβ42-Plaques | Aβ40-CAA density (#/mm2) | Aβ42-CAA density (#/mm2) | Aβ40-Plaque density (#/mm2) | Aβ42-Plaque density (#/mm2) |
|---|---|---|---|---|---|---|---|---|
| LFC | 162 | 298 | 2 | 0 | 0.5512 | 1.0147 | 0.0068 | 0.0000 |
| LOC | 45 | 78 | 0 | 0 | 0.1723 | 0.2957 | 0.0000 | 0.0000 |
| LTC | 67 | 112 | 12 | 10 | 0.2505 | 0.3329 | 0.0449 | 0.0297 |
| RFC | 358 | 666 | 57 | 26 | 1.2227 | 2.1810 | 0.1947 | 0.0851 |
| ROC | 289 | 966 | 3 | 0 | 0.9947 | 3.1774 | 0.0103 | 0.0000 |
| RTC | 118 | 284 | 43 | 50 | 0.8592 | 2.0315 | 0.3131 | 0.3577 |
L: left: R: right; FC: prefrontal cortex; OC: occipital cortex; TC: temporal cortex
Aβ ELISA
Insoluble Aβ40 and Aβ42 levels in a sample of temporal neocortex from the non-infarcted hemisphere of the chimpanzee were lower than in the 5 reference AD brains when assessed by ELISA. Soluble Aβ levels, however, were higher in the chimpanzee than in the AD cases (Fig. 7).
Figure 7.

Aβ levels in the right temporal neocortex of the chimpanzee (CO494), relative to the levels in a comparison group of 5 confirmed AD patients, measured by ELISA. A: Insoluble Aβ40 (gray, top) and Aβ42 (black, bottom) levels. B: Soluble Aβ40 and Aβ42 levels. Note the different scales on the y axes for insoluble and soluble Aβ. Bars = standard deviations for the AD group.
MAPT Genotyping
Sequence analysis of genomic DNA revealed no mutations of functional significance at the tau locus (including saitohin). Two synonymous base pair changes in exons 7 and 9 were identified, while all other polymorphisms were located in noncoding regions at distances from the nearest splicing sites ranging from 40 to 145 base pairs. One unusual, 4-base pair deletion was identified in intron 3 only in this chimpanzee, but this deletion was deemed unlikely to be of pathogenic significance because it lies 67 base pairs from the nearest predicted splicing site.
DISCUSSION
This is the first report of cerebral tauopathy with paired helical filaments in an aged chimpanzee. The tau lesions included neurofibrillary tangles, neuropil threads, and neuritic tau plaques; ultrastructurally, the neurofibrillary tangles consisted of paired helical filaments that were indistinguishable from those occurring in humans with Alzheimer’s disease (Fig. 2, Fig. 5, Fig. 6) (Crowther, 1991; Metuzals, 1986). This aged chimpanzee also exhibited a species-typical profile of cerebral β-amyloid angiopathy and infrequent Aβ-immunoreactive senile plaques (Gearing et al., 1994; Gearing et al., 1996).
The impetus for human-like tau pathology in this chimpanzee is uncertain. Any influence on the expression or splicing of tau, whether environmental or genetic, could alter the probability of developing tauopathy (Hardy et al., 2006). Several mutations in the tau gene are associated with human primary tauopathies (Goedert and Jakes, 2005; Lee et al., 2001). However, sequencing of the exons and associated introns in the genomic MAPT locus of this subject disclosed no known tauopathy-associated mutations, nor any other genetic changes of obvious functional significance. The two extended MAPT haplotypes that occur in human populations (H1 and H2) are thought to differentially influence the probability of developing neurofibrillary tangles (Hardy et al., 2006). The tau haplotype in chimpanzees, however, is a mixture of the human H1 and H2 haplotypes (Conrad et al., 2004; Holzer et al., 2004; Pittman et al., 2005), and at present it is unclear whether this gene structure affects the likelihood that chimpanzees will manifest tau lesions. While genetic or epigenetic changes might yet be discovered that regulate the pathogenicity of tau, our analysis shows that this animal did not harbor a sequence modification in the tau gene that would be expected to precipitate a human-like tauopathy.
Old age is the most important risk factor for AD and other neurodegenerative diseases in humans (Katzman and Kawas, 1999), and age probably contributed to the emergence of tau pathology in this chimpanzee. The documented maximum lifespan of Pan troglodytes is 59 years (Herndon et al., 1999). Chimpanzees in their 40’s and 50’s have been shown previously to exhibit cerebral Aβ-amyloidosis, primarily in the form of CAA (Cork and Walker, 1993; Erwin et al., 2000; Gearing et al., 1994; Gearing et al., 1996; Walker and Cork, 1999), but significant intraneuronal tau pathology has not been documented previously in chimpanzees (Gearing et al., 1994; Gearing et al., 1996; Walker and Cork, 1999). Although it is not unusual to encounter occasional tau-immunoreactive neurons and processes in older animals, immunohistochemical examination of archival, postmortem tissue samples from eight additional chimpanzees ranging from 30 to 59 years of age revealed little abnormal tau immunoreactivity (R.F. Rosen, unpublished). Thus, while age probably played a role in the ontogeny of tauopathy in this 41-year old animal, the paucity of tau lesions in other chimpanzees of similar or greater age suggests that additional factors are involved.
Tau pathology also could be related to conditions that engendered the ischemic stroke, which appears to be a rare occurrence in nonhuman primates (Borkowski et al., 2000; Fish et al., 2004). The chimpanzee in this study had high levels of cholesterol throughout her adult life, and hypercholesterolemia has been implicated as a risk factor both for stroke and tauopathy (Ohm and Meske, 2006). Furthermore, the ischemic lesion itself may have directly initiated AD-like tau pathology in this chimpanzee. In humans, epidemiological evidence suggests a relationship between cerebral ischemia and dementia, and nearly one-third of AD patients exhibit postmortem evidence of cerebral infarct (Kalaria, 2000). Experimental evidence for a relationship between ischemia and tau pathology is ambiguous, however. Total tau has been found to increase in CSF after stroke in humans, although phospho-tau in brain appears not to be elevated (Hesse et al., 2001). In rats, tau is rapidly hyperphosphorylated, via a cdk5-associated mechanism, in neurons at the site of experimental ischemia (Wen et al., 2007). However, the hyperphosphorylation occurs only in neurons in the infarcted region, and paired helical filaments and neuritic tau-plaques were not observed. Tauopathy in the chimpanzee in this investigation was advanced and widespread in both hemispheres, and was not increased in the infarcted region proper, or in the adjacent neocortex, suggesting that the lesions probably were present prior to the acute ischemic incident. Although the Perls iron stain did not reveal evidence of prior microinfarcts in the tissue sections that we sampled, the possibility that clinically silent microinfarcts may have precipitated tau pathology in the years before the chimpanzee’s death cannot be eliminated.
The amyloid cascade hypothesis implicates the multimerization of Aβ as the critical upstream effector in the pathogenic process that ultimately results in the generation of neurofibrillary tangles in AD (Hardy and Selkoe, 2002). In vivo and in vitro evidence indicates that aberrantly folded Aβ can induce tau hyperphosphorylation and polymerization in neurons (Bloom et al., 2005; Gotz et al., 2001). The Aβ42:Aβ40 ratio, in particular, has been hypothesized to govern the risk of developing AD (Kim et al., 2007; McGowan et al., 2005), and soluble Aβ oligomers are increasingly thought to play a crucial role (Catalano et al., 2006; Walsh and Selkoe, 2007). ELISA of (non-stroke) temporal neocortex from this chimpanzee revealed a relatively high ratio of Aβ42:Aβ40, similar to that in humans with AD. Furthermore, levels of buffer-soluble Aβ (low molecular weight monomers and oligomers) in the temporal cortex were well above the mean levels of soluble Aβ in AD (Fig. 7). This Aβ profile, co-present with neurofibrillary tangles, therefore suggests the possibility that excess soluble cerebral Aβ42 may have contributed to the emergence of tauopathy. The soluble Aβ quantities in this animal should be interpreted with caution, however, as the Aβ-precursor protein can be upregulated by the conditions associated with brain injury (Raby et al., 1998), including stroke (Popa-Wagner et al., 1998).
In summary, we report an unprecedented case of tauopathy with intraneuronal paired helical filaments, neuropil threads and neuritic tau plaques in an aged chimpanzee. The subject also exhibited a moderate degree of cerebral Aβ deposition, mainly in the brain vasculature. Despite tantalizing similarities, there are also important pathologic differences between this chimpanzee and humans with AD, notably in the rarity of neurofibrillary tangles in the hippocampus, the presence of unusual, tau-only neuritic plaques lacking β-amyloid cores, and the paucity of parenchymal Aβ- (senile) plaques. However, the occurrence of both tau- and Aβ-pathology indicates that the cellular and molecular machinery for generating two key hallmarks of AD - neurofibrillary tangles and Aβ-amyloidosis - is fully present in aged chimpanzees. In addition to providing evidence for biological similarities between humans and chimpanzees even late in the lifespan, these findings compel us to reconsider the assumption that humans are the only primates to manifest Alzheimer-like tauopathy with age.
Supplementary Material
ACKNOWLEDGMENTS
We thank C. Suwyn, X. Zhang, T. Nagaoka, S. Maxson, L. Strickland, H. Rees, S.N. Dodson, D. Cooper, R. Baul, and C. Allen for assistance, P. Mehta and P. Davies for antibodies, and M. Jucker, Y. Smith and S. Zola for helpful discussions. The Yerkes National Primate Research Center is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International.
Supported by NIH RR-00165, P01AG026423, P50AG025688, and the James S. McDonnell Foundation award 21002093.
LITERATURE CITED
- Arriagada PV, Marzloff K, Hyman BT. Distribution of Alzheimer-type pathologic changes in nondemented elderly individuals matches the pattern in Alzheimer’s disease. Neurology. 1992;42(9):1681–1688. doi: 10.1212/wnl.42.9.1681. [DOI] [PubMed] [Google Scholar]
- Bailey P, von Bonin G, McCulloch W. The Isocortex of the Chimpanzee. Univ. of Illinois Press; Urbana, Illinois: 1950. [Google Scholar]
- Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci. 2007;8(9):663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- Bloom GS, Ren K, Glabe CG. Cultured cell and transgenic mouse models for tau pathology linked to beta-amyloid. Biochim Biophys Acta. 2005;1739(2-3):116–124. doi: 10.1016/j.bbadis.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Borkowski R, Taylor TG, Rush J. Cerebral infarction and myocardial fibrosis in a white-handed gibbon (Hylobates lar) J Zoo Wildl Med. 2000;31(1):65–70. doi: 10.1638/1042-7260(2000)031[0065:CIAMFI]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Morphological criteria for the recognition of Alzheimer’s disease and the distribution pattern of cortical changes related to this disorder. Neurobiol Aging. 1994;15(3):355–356. doi: 10.1016/0197-4580(94)90032-9. [DOI] [PubMed] [Google Scholar]
- Brodmann K. Neue Ergebnisse uber die vergleichende histologische Lokalisation der Grosshirnrinde mit besonderer Berucksichtigung des Stirnhirns. Anat. Anz. Suppl. 1912;41:157–216. [Google Scholar]
- Catalano SM, Dodson EC, Henze DA, Joyce JG, Krafft GA, Kinney GG. The role of amyloid-beta derived diffusible ligands (ADDLs) in Alzheimer’s disease. Curr Top Med Chem. 2006;6(6):597–608. doi: 10.2174/156802606776743066. [DOI] [PubMed] [Google Scholar]
- Cervenakova L, Brown P, Goldfarb LG, Nagle J, Pettrone K, Rubenstein R, Dubnick M, Gibbs CJ, Jr., Gajdusek DC. Infectious amyloid precursor gene sequences in primates used for experimental transmission of human spongiform encephalopathy. Proc Natl Acad Sci U S A. 1994;91(25):12159–12162. doi: 10.1073/pnas.91.25.12159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad C, Vianna C, Schultz C, Thal DR, Ghebremedhin E, Lenz J, Braak H, Davies P. Molecular evolution and genetics of the Saitohin gene and tau haplotype in Alzheimer’s disease and argyrophilic grain disease. J Neurochem. 2004;89(1):179–188. doi: 10.1046/j.1471-4159.2004.02320.x. [DOI] [PubMed] [Google Scholar]
- Cork LC, Powers RE, Selkoe DJ, Davies P, Geyer JJ, Price DL. Neurofibrillary tangles and senile plaques in aged bears. J Neuropathol Exp Neurol. 1988;47(6):629–641. doi: 10.1097/00005072-198811000-00006. [DOI] [PubMed] [Google Scholar]
- Cork LC, Walker LC. Age-related lesions, nervous system. In: Jones TC, Mohr U, Hunt RD, editors. Monographs on Pathology of Laboratory Animals Nonhuman Primates II. Springer-Verlag; Berlin: 1993. pp. 173–183. [Google Scholar]
- Crowther RA. Straight and paired helical filaments in Alzheimer disease have a common structural unit. Proc Natl Acad Sci U S A. 1991;88(6):2288–2292. doi: 10.1073/pnas.88.6.2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crystal H, Dickson D, Fuld P, Masur D, Scott R, Mehler M, Masdeu J, Kawas C, Aronson M, Wolfson L. Clinico-pathologic studies in dementia: nondemented subjects with pathologically confirmed Alzheimer’s disease. Neurology. 1988;38(11):1682–1687. doi: 10.1212/wnl.38.11.1682. [DOI] [PubMed] [Google Scholar]
- Duff K, Knight H, Refolo LM, Sanders S, Yu X, Picciano M, Malester B, Hutton M, Adamson J, Goedert M, Burki K, Davies P. Characterization of pathology in transgenic mice over-expressing human genomic and cDNA tau transgenes. Neurobiol Dis. 2000;7(2):87–98. doi: 10.1006/nbdi.1999.0279. [DOI] [PubMed] [Google Scholar]
- Duyckaerts C, Colle MA, Dessi F, Piette F, Hauw JJ. Progression of Alzheimer histopathological changes. Acta Neurol Belg. 1998;98(2):180–185. [PubMed] [Google Scholar]
- Elfenbein HA, Rosen RF, Stephens SL, Switzer RC, Smith Y, Pare J, Mehta PD, Warzok R, Walker LC. Cerebral beta-amyloid angiopathy in aged squirrel monkeys. Histol Histopathol. 2007;22(2):155–167. doi: 10.14670/HH-22.155. [DOI] [PubMed] [Google Scholar]
- Erwin JM, Nimchinsky EA, Gannon PJ, Perl DP, Hof PR. The Study of Brain Aging in Great Apes. In: Hof PR, Mobbs CV, editors. Functional Neurobiology of Aging. Academic Press; 2000. pp. 447–456. [Google Scholar]
- Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1998;8(3):186–194. [PubMed] [Google Scholar]
- Ewing B, Hillier L, Wendl MC, Green P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998;8(3):175–185. doi: 10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- Fish PH, Carpenter JW, Kraft S. Diagnosis and treatment of a cerebral infarct in a chimpanzee (Pan troglodytes) J Zoo Wildl Med. 2004;35(2):203–207. doi: 10.1638/01-078. [DOI] [PubMed] [Google Scholar]
- Gearing M, Rebeck GW, Hyman BT, Tigges J, Mirra SS. Neuropathology and apolipoprotein E profile of aged chimpanzees: implications for Alzheimer disease. Proc Natl Acad Sci U S A. 1994;91(20):9382–9386. doi: 10.1073/pnas.91.20.9382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gearing M, Tigges J, Mori H, Mirra SS. Abeta40 is a major form of beta-amyloid in nonhuman primates. Neurobiol Aging. 1996;17(6):903–908. doi: 10.1016/s0197-4580(96)00164-9. [DOI] [PubMed] [Google Scholar]
- Gearing M, Tigges J, Mori H, Mirra SS. beta-Amyloid (Abeta) deposition in the brains of aged orangutans. Neurobiol Aging. 1997;18(2):139–146. doi: 10.1016/s0197-4580(97)00012-2. [DOI] [PubMed] [Google Scholar]
- Geula C, Nagykery N, Wu CK. Amyloid-beta deposits in the cerebral cortex of the aged common marmoset (Callithrix jacchus): incidence and chemical composition. Acta Neuropathol (Berl) 2002;103(1):48–58. doi: 10.1007/s004010100429. [DOI] [PubMed] [Google Scholar]
- Giannakopoulos P, Gold G, Kovari E, von Gunten A, Imhof A, Bouras C, Hof PR. Assessing the cognitive impact of Alzheimer disease pathology and vascular burden in the aging brain: the Geneva experience. Acta Neuropathol (Berl) 2007;113(1):1–12. doi: 10.1007/s00401-006-0144-y. [DOI] [PubMed] [Google Scholar]
- Goedert M. Tau protein and neurodegeneration. Semin Cell Dev Biol. 2004;15(1):45–49. doi: 10.1016/j.semcdb.2003.12.015. [DOI] [PubMed] [Google Scholar]
- Goedert M, Jakes R. Mutations causing neurodegenerative tauopathies. Biochim Biophys Acta. 2005;1739(2-3):240–250. doi: 10.1016/j.bbadis.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Goedert M, Jakes R, Vanmechelen E. Monoclonal antibody AT8 recognises tau protein phosphorylated at both serine 202 and threonine 205. Neurosci Lett. 1995;189(3):167–169. doi: 10.1016/0304-3940(95)11484-e. [DOI] [PubMed] [Google Scholar]
- Gotz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science. 2001;293(5534):1491–1495. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- Greenberg SG, Davies P, Schein JD, Binder LI. Hydrofluoric acid-treated tau PHF proteins display the same biochemical properties as normal tau. J Biol Chem. 1992;267(1):564–569. [PubMed] [Google Scholar]
- Hardy J, Orr H. The genetics of neurodegenerative diseases. J Neurochem. 2006;97(6):1690–1699. doi: 10.1111/j.1471-4159.2006.03979.x. [DOI] [PubMed] [Google Scholar]
- Hardy J, Pittman A, Myers A, Fung HC, de Silva R, Duckworth J. Tangle diseases and the tau haplotypes. Alzheimer Dis Assoc Disord. 2006;20(1):60–62. doi: 10.1097/01.wad.0000201853.54493.d8. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hartig W, Bruckner G, Schmidt C, Brauer K, Bodewitz G, Turner JD, Bigl V. Co-localization of beta-amyloid peptides, apolipoprotein E and glial markers in senile plaques in the prefrontal cortex of old rhesus monkeys. Brain Res. 1997;751(2):315–322. doi: 10.1016/s0006-8993(96)01423-0. [DOI] [PubMed] [Google Scholar]
- Herndon JG, Tigges J, Anderson DC, Klumpp SA, McClure HM. Brain weight throughout the life span of the chimpanzee. J Comp Neurol. 1999;409(4):567–572. [PubMed] [Google Scholar]
- Hesse C, Rosengren L, Andreasen N, Davidsson P, Vanderstichele H, Vanmechelen E, Blennow K. Transient increase in total tau but not phospho-tau in human cerebrospinal fluid after acute stroke. Neurosci Lett. 2001;297(3):187–190. doi: 10.1016/s0304-3940(00)01697-9. [DOI] [PubMed] [Google Scholar]
- Holzer M, Craxton M, Jakes R, Arendt T, Goedert M. Tau gene (MAPT) sequence variation among primates. Gene. 2004;341:313–322. doi: 10.1016/j.gene.2004.07.013. [DOI] [PubMed] [Google Scholar]
- Imai Y, Ibata I, Ito D, Oshawa K, Kohsaka S. A novel gene Iba1 in the major histocompatibility complex class III region encoding and EF hand protein expressed in a monocyte lineage. Biochem and Biophys Res Comm. 224:855–862. doi: 10.1006/bbrc.1996.1112. [DOI] [PubMed] [Google Scholar]
- Jicha GA, Bowser R, Kazam IG, Davies P. Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J Neurosci Res. 1997;48(2):128–132. doi: 10.1002/(sici)1097-4547(19970415)48:2<128::aid-jnr5>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Jicha GA, Weaver C, Lane E, Vianna C, Kress Y, Rockwood J, Davies P. cAMP-dependent protein kinase phosphorylations on tau in Alzheimer’s Disease. J Neurosci. 1999;19(17):7486–7494. doi: 10.1523/JNEUROSCI.19-17-07486.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalaria RN. The role of cerebral ischemia in Alzheimer’s disease. Neurobiol Aging. 2000;21(2):321–330. doi: 10.1016/s0197-4580(00)00125-1. [DOI] [PubMed] [Google Scholar]
- Katzman R, Kawas C. The epidemiology of dementia and Alzheimer disease. In: Terry RD, Katzman R, Bick KL, Sisodia SS, editors. Alzheimer Disease. 2nd edition Lippincott Williams & Wilkins; Philadelphia: 1999. pp. 95–116. [Google Scholar]
- Kiatipattanasakul W, Nakayama H, Yongsiri S, Chotiapisitkul S, Nakamura S, Kojima H, Doi K. Abnormal neuronal and glial argyrophilic fibrillary structures in the brain of an aged albino cynomolgus monkey (Macaca fascicularis) Acta Neuropathol (Berl) 2000;100(5):580–586. doi: 10.1007/s004010000215. [DOI] [PubMed] [Google Scholar]
- Kim J, Onstead L, Randle S, Price R, Smithson L, Zwizinski C, Dickson DW, Golde T, McGowan E. Abeta40 inhibits amyloid deposition in vivo. J Neurosci. 2007;27(3):627–633. doi: 10.1523/JNEUROSCI.4849-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KS, Miller DL, Sapienza VJ, Chen CMJ, Bai C, Grundke-Iqbal I, Currie JR, Wisniewski HM. Production and characterization of monoclonal antibodies reactive to synthetic cerebrovascular amyloid peptide. Neurosci Res Commun. 1988;2:121–130. [Google Scholar]
- Kimura N, Tanemura K, Nakamura S, Takashima A, Ono F, Sakakibara I, Ishii Y, Kyuwa S, Yoshikawa Y. Age-related changes of Alzheimer’s disease-associated proteins in cynomolgus monkey brains. Biochem Biophys Res Commun. 2003;310(2):303–311. doi: 10.1016/j.bbrc.2003.09.012. [DOI] [PubMed] [Google Scholar]
- Komori T, Arai N, Oda M, Nakayama H, Mori H, Yagishita S, Takahashi T, Amano N, Murayama S, Murakami S, Shibata N, Kobayashi M, Sasaki S, Iwata M. Astrocytic plaques and tufts of abnormal fibers do not coexist in corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol (Berl) 1998;96(4):401–408. doi: 10.1007/s004010050911. [DOI] [PubMed] [Google Scholar]
- Lee VM-Y, Goedert M, Trojanowski JQ. Neurodegenerative Tauopathies. Ann Rev Neurosci. 2001;24(1):1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- Mandelkow E, von Bergen M, Biernat J, Mandelkow EM. Structural principles of tau and the paired helical filaments of Alzheimer’s disease. Brain Pathol. 2007;17(1):83–90. doi: 10.1111/j.1750-3639.2007.00053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan E, Pickford F, Kim J, Onstead L, Eriksen J, Yu C, Skipper L, Murphy MP, Beard J, Das P, Jansen K, Delucia M, Lin WL, Dolios G, Wang R, Eckman CB, Dickson DW, Hutton M, Hardy J, Golde T. Abeta42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron. 2005;47(2):191–199. doi: 10.1016/j.neuron.2005.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mestre-Frances N, Keller E, Calenda A, Barelli H, Checler F, Bons N. Immunohistochemical analysis of cerebral cortical and vascular lesions in the primate Microcebus murinus reveal distinct amyloid beta1-42 and beta1-40 immunoreactivity profiles. Neurobiol Dis. 2000;7(1):1–8. doi: 10.1006/nbdi.1999.0270. [DOI] [PubMed] [Google Scholar]
- Metuzals J. Neurofilaments and paired helical filaments. In: Metuzals J, editor. Electron Microscopy and Alzheimer’s Disease. San Francisco Press, Inc.; San Francisco: 1986. pp. 34–37. [Google Scholar]
- Nakamura S, Kiatipattanasakul W, Nakayama H, Ono F, Sakakibara I, Yoshikawa Y, Goto N, Doi K. Immunohistochemical characteristics of the constituents of senile plaques and amyloid angiopathy in aged cynomolgus monkeys. J Med Primatol. 1996;25(4):294–300. doi: 10.1111/j.1600-0684.1996.tb00213.x. [DOI] [PubMed] [Google Scholar]
- Nakamura S, Tamaoka A, Sawamura N, Kiatipattanasakul W, Nakayama H, Shoji S, Yoshikawa Y, Doi K. Deposition of amyloid beta protein (Abeta) subtypes [A beta 40 and A beta 42(43)] in canine senile plaques and cerebral amyloid angiopathy. Acta Neuropathol (Berl) 1997;94(4):323–328. doi: 10.1007/s004010050714. [DOI] [PubMed] [Google Scholar]
- Nelson PT, Greenberg SG, Saper CB. Neurofibrillary tangles in the cerebral cortex of sheep. Neurosci Lett. 1994;170(1):187–190. doi: 10.1016/0304-3940(94)90270-4. [DOI] [PubMed] [Google Scholar]
- Nelson PT, Marton L, Saper CB. Alz-50 immunohistochemistry in the normal sheep striatum: a light and electron microscope study. Brain Res. 1993;600(2):285–297. doi: 10.1016/0006-8993(93)91385-6. [DOI] [PubMed] [Google Scholar]
- Nelson PT, Saper CB. Ultrastructure of neurofibrillary tangles in the cerebral cortex of sheep. Neurobiol Aging. 1995;16(3):315–323. doi: 10.1016/0197-4580(94)00175-z. [DOI] [PubMed] [Google Scholar]
- Nelson PT, Stefansson K, Gulcher J, Saper CB. Molecular evolution of tau protein: implications for Alzheimer’s disease. J Neurochem. 1996;67(4):1622–1632. doi: 10.1046/j.1471-4159.1996.67041622.x. [DOI] [PubMed] [Google Scholar]
- Ohm TG, Meske V. Cholesterol, statins and tau. Acta Neurol Scand Suppl. 2006;185:93–101. doi: 10.1111/j.1600-0404.2006.00692.x. [DOI] [PubMed] [Google Scholar]
- Otvos L, Jr., Feiner L, Lang E, Szendrei GI, Goedert M, Lee VM. Monoclonal antibody PHF-1 recognizes tau protein phosphorylated at serine residues 396 and 404. J Neurosci Res. 1994;39(6):669–673. doi: 10.1002/jnr.490390607. [DOI] [PubMed] [Google Scholar]
- Piccardo P, Mirra SS, Young K, Gearing M, Dlouhy SR, Ghetti B. [alpha]-Synuclein accumulation in Gerstmann-Straussler-Schenker disease (GSS) with prion protein gene (PRNP) F198S. Neurobiol Aging. 1998;19:S724. [Google Scholar]
- Pittman AM, Myers AJ, Abou-Sleiman P, Fung HC, Kaleem M, Marlowe L, Duckworth J, Leung D, Williams D, Kilford L, Thomas N, Morris CM, Dickson D, Wood NW, Hardy J, Lees AJ, de Silva R. Linkage disequilibrium fine mapping and haplotype association analysis of the tau gene in progressive supranuclear palsy and corticobasal degeneration. J Med Genet. 2005;42(11):837–846. doi: 10.1136/jmg.2005.031377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poduri A, Gearing M, Rebeck GW, Mirra SS, Tigges J, Hyman BT. Apolipoprotein E4 and beta amyloid in senile plaques and cerebral blood vessels of aged rhesus monkeys. Am J Pathol. 1994;144(6):1183–1187. [PMC free article] [PubMed] [Google Scholar]
- Popa-Wagner A, Schroder E, Walker LC, Kessler C. beta-Amyloid precursor protein and beta-amyloid peptide immunoreactivity in the rat brain after middle cerebral artery occlusion: effect of age. Stroke. 1998;29(10):2196–2202. doi: 10.1161/01.str.29.10.2196. [DOI] [PubMed] [Google Scholar]
- Potempska A, Mack K, Mehta P, Kim KS, Miller DL. Quantification of sub-femtomole amounts of Alzheimer amyloid beta peptides. Amyloid. 1999;6(1):14–21. doi: 10.3109/13506129908993283. [DOI] [PubMed] [Google Scholar]
- Raby CA, Morganti-Kossmann MC, Kossmann T, Stahel PF, Watson MD, Evans LM, Mehta PD, Spiegel K, Kuo YM, Roher AE, Emmerling MR. Traumatic brain injury increases beta-amyloid peptide 1-42 in cerebrospinal fluid. J Neurochem. 1998;71(6):2505–2509. doi: 10.1046/j.1471-4159.1998.71062505.x. [DOI] [PubMed] [Google Scholar]
- Roertgen KE, Parisi JE, Clark HB, Barnes DL, O’Brien TD, Johnson KH. Abeta-associated cerebral angiopathy and senile plaques with neurofibrillary tangles and cerebral hemorrhage in an aged wolverine (Gulo gulo) Neurobiol Aging. 1996;17(2):243–247. doi: 10.1016/0197-4580(95)02069-1. [DOI] [PubMed] [Google Scholar]
- Schultz C, Hubbard GB, Tredici KD, Braak E, Braak H. Tau pathology in neurons and glial cells of aged baboons. Adv Exp Med Biol. 2001;487:59–69. doi: 10.1007/978-1-4615-1249-3_5. [DOI] [PubMed] [Google Scholar]
- Walker LC, Cork LC. The Neurobiology of Aging in Nonhuman Primates. In: Terry RD, Katzman R, Bick KL, Sisodia SS, editors. Alzheimer Disease. 2nd edition Lippincott Williams & Wilkins; Philadelphia: 1999. pp. 223–243. [Google Scholar]
- Walker LC, Masters C, Beyreuther K, Price DL. Amyloid in the brains of aged squirrel monkeys. Acta Neuropathol (Berl) 1990;80(4):381–387. doi: 10.1007/BF00307691. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ. Abeta oligomers - a decade of discovery. J Neurochem. 2007;101(5):1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- Wen Y, Yang SH, Liu R, Perez EJ, Brun-Zinkernagel AM, Koulen P, Simpkins JW. Cdk5 is involved in NFT-like tauopathy induced by transient cerebral ischemia in female rats. Biochim Biophys Acta. 2007;1772(4):473–483. doi: 10.1016/j.bbadis.2006.10.011. [DOI] [PubMed] [Google Scholar]
- Wilcock GK, Esiri MM. Plaques, tangles and dementia. A quantitative study. J Neurol Sci. 1982;56(2-3):343–356. doi: 10.1016/0022-510x(82)90155-1. [DOI] [PubMed] [Google Scholar]
- Williams DR. Tauopathies: classification and clinical update on neurodegenerative diseases associated with microtubule-associated protein tau. Intern Med J. 2006;36(10):652–660. doi: 10.1111/j.1445-5994.2006.01153.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.