

Abstract
The transcription factor GATA4 is essential for heart morphogenesis. Heterozygous mutation of GATA4 causes familial septal defects. However, the phenotypic spectrum of heterozygous GATA4 mutation is not known. In this study, we defined the cardiac phenotypes that result from heterozygous mutation of murine Gata4. We then asked if GATA4 mutation occurs in humans with these forms of congenital heart disease (CHD). In mice, heterozygous Gata4 mutation was associated with atrial and ventricular septal defect (ASD, VSD), endocardial cushion defect (ECD), RV hypoplasia, and cardiomyopathy. Genetic background strongly influenced the expression of ECD and cardiomyopathy, indicating the presence of important genetic modifiers. In humans, non-synonymous GATA4 sequence variants were associated with ECD (2/43), ASD (1/8), and RV hypoplasia in the context of double inlet left ventricle (1/9), forms of CHD that overlapped with abnormalities seen in the mouse model. These variants were not found in at least 500 control chromosomes, and encode proteins with non-conservative amino acid substitutions at phylogenetically conserved positions, suggesting that they are disease-causing mutations. Cardiomyopathy was not associated with GATA4 mutation in humans. These data establish the phenotypic spectrum of heterozygous Gata4 mutation in mice, and suggest that heterozygous GATA4 mutation leads to partially overlapping phenotypes in humans. Additional studies will be required to determine the degree to which GATA4 mutation contributes to human CHD characterized by ECD or RV hypoplasia.
Keywords: animal models, congenital heart defects, cardiac development, genetics ofcongenital heart disease
Introduction
The morphogenetic complexity of fashioning a four-chambered heart from a straight tube mandates a precisely orchestrated interplay of multiple transcription factors, adhesion molecules, ion channels, signaling molecules and structural proteins [1]. Errors in this process result in congenital heart disease (CHD), the most common form of birth defect. Mutation of a small but growing number of genes has been shown to cause CHD [2]. Recently, mutation of the zinc finger transcription factor GATA4 was shown to cause atrial and ventricular septal defects in several unrelated extended pedigrees (Table 1) [3–6]. Among CHD patients without a family history (“sporadic” CHD), GATA4 mutations appear to be infrequent. In published studies of sporadic CHD, GATA4 mutations were found in only 2 probands out of 376 patients examined (Table 1) [7–10]. However, without prior knowledge of the phenotypic spectrum of GATA4 mutation, it was not possible to target these studies to forms of CHD most likely to be caused by GATA4 mutation. Therefore GATA4 mutation may be more frequently associated with specific forms of CHD that were not well represented in current literature.
Table 1.
Non-synonymous GATA4 Mutations Associated with Congenital Heart Disease
| Study | Study Population | Nucleotide Change | Amino acid Change | Phenotype |
|---|---|---|---|---|
| Garg et al., 2003 [3] | Familial septal defects (2 families) | 886G>A 1075delG |
G296S E359RfsX44 |
ASD ± VSD, PS (1 family; 1 case of ECD) ASD (1 family) |
| Hiaryama-Yamada et al., 2005 [4] | Familial ASD (16 families) | 1075delG 155C>T |
E359RfsX44 S52F |
ASD (1 family) ASD (1 family) |
| Okubo et al, 2004 [5] | Familial ASD (1 family) | 1074delC | S358RfsX45 | ASD ± PS (1 family) |
| Sarkozy et al, 2005 [6] | ASD (16 families; 13 sporadic) | 886G>A | G296S | ASD ± PS (2 families) |
| Sarkozy et al, 2005 [7] | ECD (9 families; 26 sporadic) | None | None | N/A |
| Nemer et al, 2006 [8] | Largely sporadic CHD (94 probands: 26 TOF; 30 VSD; 18 PS; 15 PDA; 12 ASD, 8 TA, 6 TGA, 5 CoA) | 648C>G | E216D | TOF (2 sporadic cases) |
| Zhang et al., 2006 [9] | Largely sporadic CHD (99 probands: 36 VSD, 4 ASD, 11 TOF, ECD 1, 47 other) | None | None | N/A |
| Schluterman et al., 2007 [10] | Largely sporadic CHD (157 probands: 14 ASD, 18 VSD, 7 ECD, 18 TOF, 45 LVOTO, 55 other) | None | None | |
| This study | Largely sporadic CHD (237 probands; see Table 4) | 487C>T 1037C>T 886G>T 1207C>A |
P163S A346V 296C L403M |
ECD (1 sporadic case) ECD (1 sporadic case) ASD + PS (1 family) Hypoplastic RV (1 sporadic case) |
ASD, atrial septal defect; CoA, coarctation of the aorta; ECD, endocardial cushion defect; LVOTO, LV outflow tract obstruction; PS, pulmonary stenosis; PA, pulmonary atresia; PDA, patent ductus arteriosus; TA, tricuspid atresia; TGA, transposition of the great arteries; TOF, tetralogy of Fallot;.
We studied the spectrum of cardiac abnormalities found in mice with mutation of one copy of Gata4. We found that heterozygous Gata4 mutation in mice caused endocardial cushion defect (ECD), atrial or ventricular septal defect (ASD or VSD), hypoplastic right ventricle, and cardiomyopathy. Reasoning that this might give insight into the types of heart defects that might be caused by GATA4 mutation in humans, we then looked for GATA4 mutations in patients with similar forms of CHD. We found non-synonomous GATA4 sequence variants in association with ECD, hypoplastic RV in the context of double inlet left ventricle (DILV), and ASD. These results refine our understanding of the phenotypic spectrum of GATA4 mutation.
Methods
Mice
The Gata4Δex2 allele has been described [11]. Mice were backcrossed for more than 7 generations into either the C57BL6/J (abbreviated C57; Jackson Labs) or the FVB/NCrl (FVB; Charles River Labs) genetic backgrounds. Structural abnormalities were diagnosed on H&E stained serial paraffin sections. Echocardiography was performed on unsedated 8 week old mice. Mice were held in a supine position and imaged with a 15 MHz probe. All analyses were performed blinded to genotype. Animal use was according to protocols approved by the Institutional Animal Care and Use Committee, and conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996).
For the genetic modifier screen, mice were genotyped using the Illumina MD linkage panel. A whole genome linkage scan was performed with endocardial cushion defect (ECD) as a binary trait, using the model of Xu and Atchley [12]. Genome scans were performed using the R/qtl package [13].
Patients and GATA4 Genotyping
After obtaining informed consent, blood and/or saliva were obtained from probands, and when possible from members of their nuclear family. We studied 107 probands with cardiac abnormalities consistent with those seen in the G4D mouse model (septal defect, ECD, hypoplastic RV, and cardiomyopathy). 50 of these patients were obtained from the Children’s Hospital Boston Cardiovascular Disease Registry. 33 patients with ECD were obtained from registries at Cincinnati Children’s Hospital Medical Center and Children’s Hospital of Philadelphia. Sequencing results for another 24 patients with cardiomyopathy were obtained via personal communications with M. Sarkar, C. Seidman, and J. Seidman. Genetic testing for specific molecular diagnoses was performed when clinically indicated. Individuals with known genetic abnormalities were excluded from this study. All studies were performed under protocols monitored by the respective Institutional Review Boards, and conformed with the principles outlined in the Declaration of Helsinki.
PCR-amplified genomic DNA was sequenced on both strands using primers designed to span the GATA4 coding exons and exon/intron boundaries (Supplementary Table 1). 13 patients with ECDs were genotyped by denaturing high performance liquid chromatography rather than by direct sequencing. Non-synonymous sequence variants were confirmed by re-sequencing or by allele-specific genotyping assays. Sequence positions are relative to GATA4 cDNA (NM_002052) and protein (NP_002043). We predicted the functional effect of amino acid substitutions based phylogenetic conservation and physiochemical properties of amino acid residues, using the MAPP algorithm [14].
GATA4 was sequenced in 250 control individuals. In some cases, allele specific genotyping assays were performed in additional controls. Some of these controls were previously reported [10].
Results
Murine model of Gata4 mutation
The mutant Gata4 allele used in this study, Gata4Δex2, contains a deletion of the start codon and 46% of the coding region [11, 15]. This allele does not express full length protein. In fetal heart, but not in adult heart, the allele does express a truncated protein lacking the essential N-terminal transcriptional activation domain (Figure 1)[15]. In vitro reporter assays showed loss of transcriptional activity and did not suggest dominant negative activity [15]. Prior in vivo characterization suggested that Gata4Δex2 is a loss of function allele [11, 15, 16]. although partial residual function or weak dominant negative activity cannot be excluded. We studied Gata4Δex2/WT mice (abbreviated G4D), which express 50% reduced levels of full length Gata4 protein (Figure 1)[17].
Figure 1. Gata4 protein expression in WT and G4D fetal hearts.
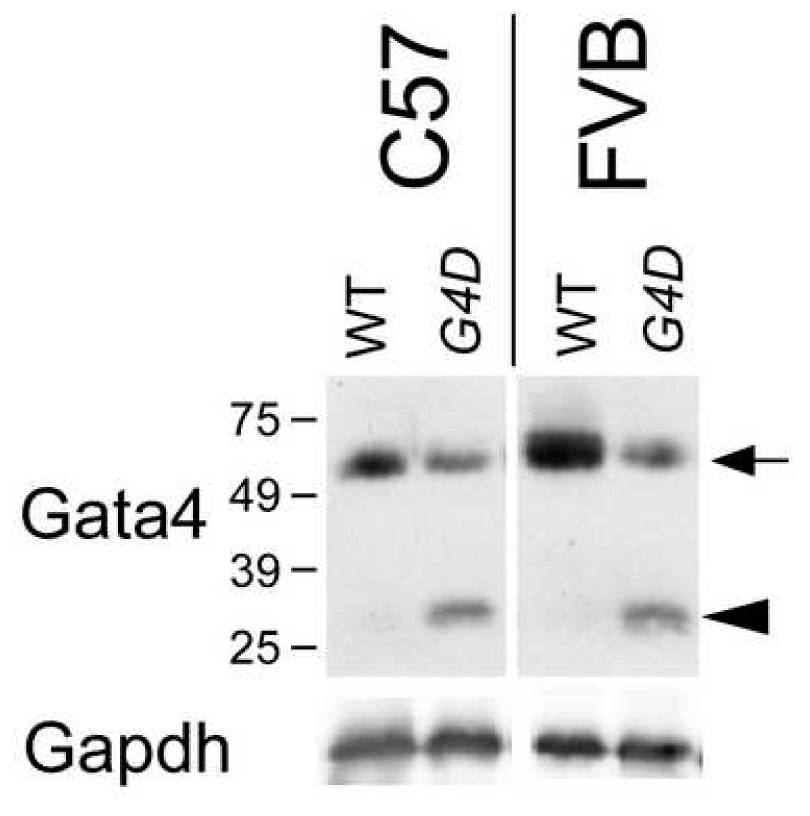
Gata4 protein in E14.5 WT and G4D hearts was measured by Western blotting. In G4D hearts, expression of full length Gata4 protein (arrow) was reduced. A truncated protein lacking the N-terminal activation domain was expressed in G4D fetal hearts (arrowhead). Expression was not different in C57 and FVB strain backgrounds.
We previously reported that in a mixed genetic background G4D hearts are structurally normal [11, 17]. However, we found that G4D mice in an inbred C57 genetic background (G4D-C57) showed decreased survival (Figure 2a). G4D-C57 mice were born at the expected Mendelian frequency, but suffered excess mortality in the perinatal period (52% mortality; Wilcoxon P< 0.0001 vs. WT; Figure 2b). Histological examination of a subset of the deceased G4D-C57 mice (20/56) demonstrated cardiac malformations in 85% (Figure 2c). The distribution of malformations was similar to that observed in G4D-C57 fetuses (Table 2; see below). In some intriguing hearts, the long axis of the RV appeared divergent from the long axis of the LV (Figure 2c). In addition to cardiac malformations, G4D-C57 mice have lung and diaphragm abnormalities, which may contribute to perinatal lethality [18].
Figure 2. Perinatal death of Gata4 mutant mice.
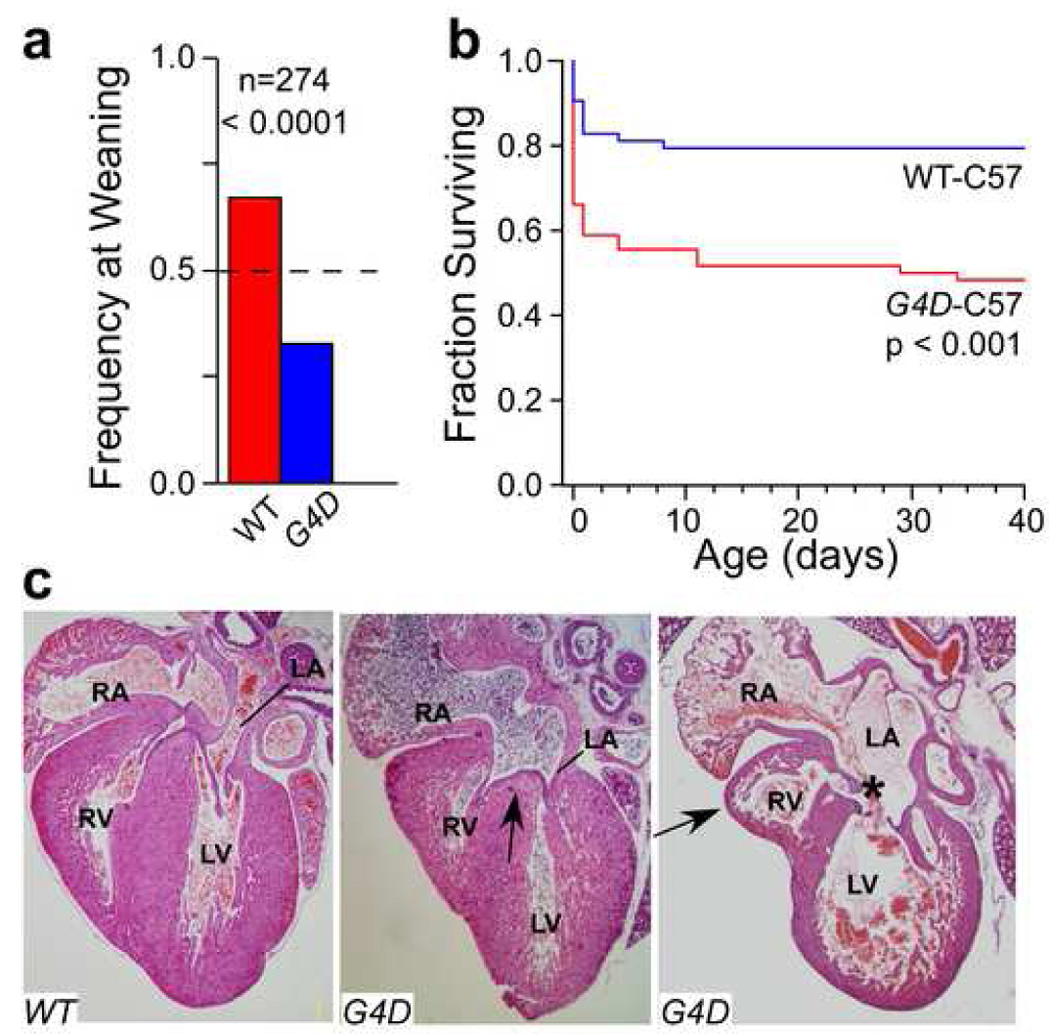
a. Frequency of G4D or WT genotypes at weaning in the C57 strain background. The expected Mendelian frequency was 50% (dashed line). b. Perinatal attrition of G4D mice in the C57 background. 141 births produced 56 G4D mice, of which 52% died, and 63 WT, of which 18% died. 22 pups were cannabilized as neonates and could not be genotyped. c. Hearts from deceased WT and G4D-C57 neonates. The middle panel shows an ASD primum defect (arrow). The right panel shows a markedly hypoplastic RV. The RV apex (arrow) is distinct from the LV apex. There is also a CAVC defect (asterisk) that is malaligned so that it opens mostly into the LV.
Table 2.
Cardiac Malformations in Unselected G4D Late Gestation Embryos
| Malformation | G4D-C57 (n=46) | G4D-FVB (n=23) | G4D-F1 (n=29) | G4D-back (n=172) |
|---|---|---|---|---|
| Normal | 24% (11) | 70% (16)* | 88% (23)* | 58% (99)* |
| ASD secundum | 24% (11) | 7% (2) | 12% (20) | |
| Isolated | 4% (2) | 7% (2) | 10% (17) | |
| With other defects | 20% (9) | 2% (3) | ||
| Endocardial Cushion Defect | 59% (27) | 4% (1)* | 3% (1)* | 24% (41)* |
| ASD primum | 7% (3) | 4% (7) | ||
| CAVC | 38% (17) | 3% (1)* | 13% (22) | |
| Inlet VSD | 18% (8) | 4% (1) | 7% (12) | |
| Ventricles | 26% (12) | |||
| Membranous VSD | 20% (9) | 22% (5) | 7% (10) | |
| Muscular VSD | 9% (4) | 4% (1) | 4% (6) | |
| Small RV sinus | 9% (4) | 9% (2) | 3% (5) |
P < 0.0001, Chi-squared test compared to G4D-C57.
Cardiac Malformations in G4D-C57 mice
To systematically determine the spectrum of structural heart defects in G4D-C57 mice, we collected an unselected group of late gestation embryos (post-coital days 15–19) and identified cardiac abnormalities on serial histological sections (Table 2). Out of 46 unselected late gestation G4D-C57 hearts examined, only 24% (11/46) appeared normal. Most malformations were severe (33/46, 72%), consisting of ECDs (27/46, 59%), VSDs (12/46, 26%), and hypoplasia of the RV (4/46, 9%). Isolated ASD secundum was observed in 2/46 (2%).
We observed a range of ECDs that included balanced complete atrioventricular canal (CAVC), LV-dominant CAVC, inlet VSD, and ASD primum (Figure 3a–e). RV hypoplasia involved the inflow portion of the right ventricle and spared the outflow portion (Figure 4). We observed 15 instances of severe RV hypoplasia; 5 occurred in the absence of ECD, and in 4 the apex of the RV was distinct from the apex of the LV (Figure 2c). These findings show that heterozygous Gata4 mutation causes aberrant endocardial cushion development and RV morphogenesis.
Figure 3. ECDs in G4D-C57 late gestation embryos.
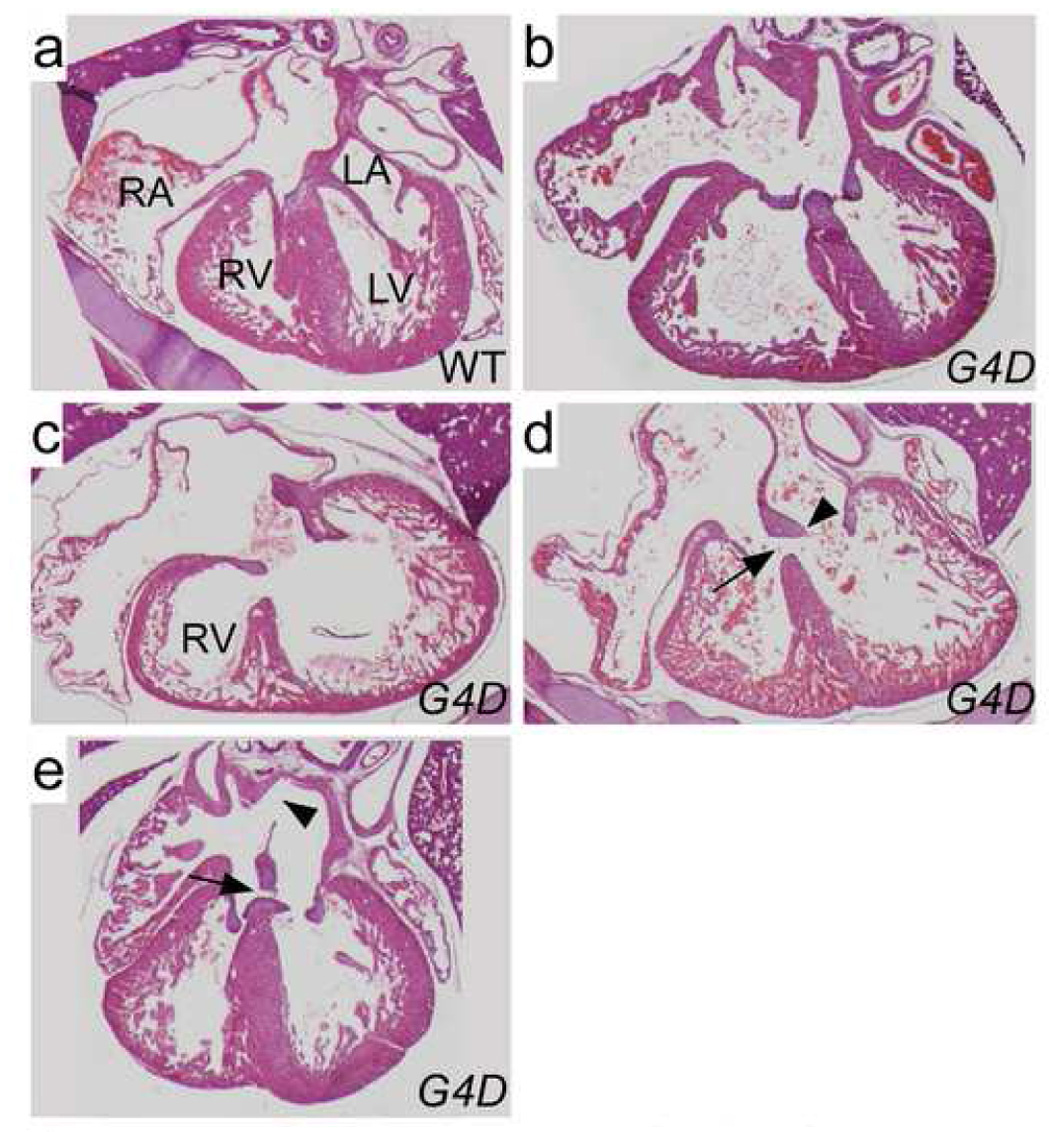
a–e. Hematoxylin and eosin stained sections demonstrating a spectrum of ECDs. a. WT control. b. Well-balanced CAVC canal defect. c. CAVC defect opening mainly into the left ventricle. The RV is moderately hypoplastic. d. Inlet VSD. The atrial septum is intact, and there are two AV valves, albeit with highly primitive leaflets (arrowhead). The ventricular portion of the AV canal is not septated, resulting in an inlet VSD (arrow). e. ASD primum (arrow) and ASD secundum (arrowhead). The ventricular portion of the AV canal is septated.
Figure 4. RV hypoplasia in G4D-C57 late gestation embryo.
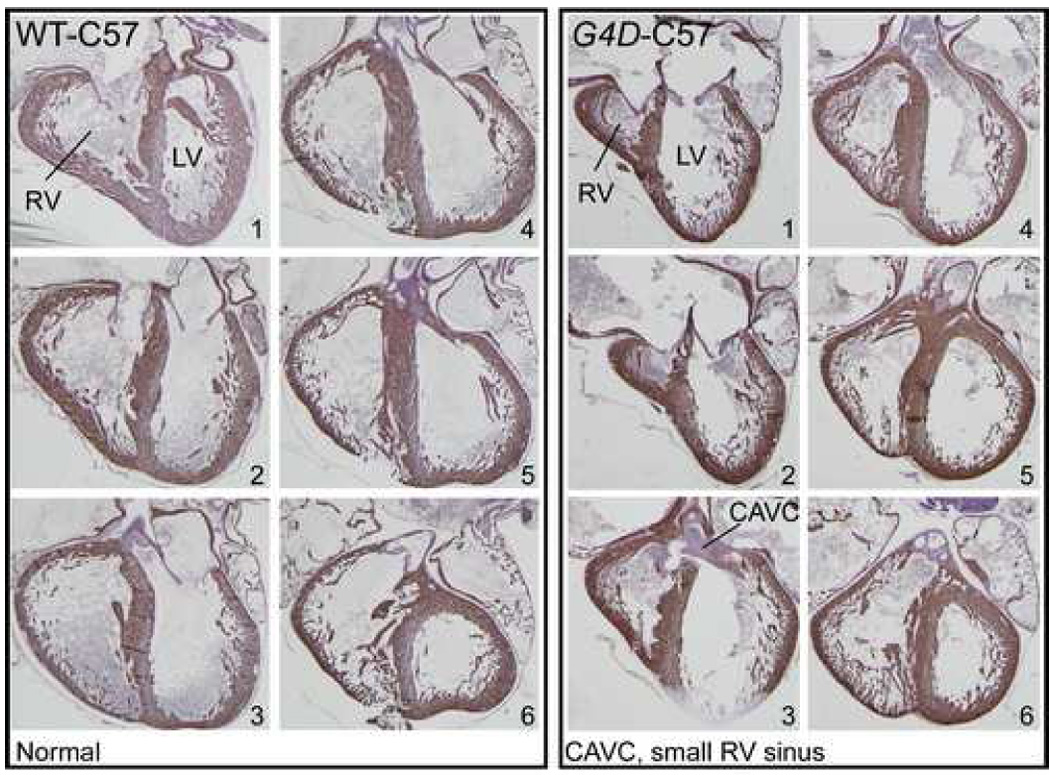
Adjacent sections from an E18.5 WT-C57 (left box of six images) or G4D-C57 littermate (right box of six images). Myocardium was visualized by desmin immunostaining, and counterstained with hematoxylin. Numbers indicate order of sections. The hypoplastic inflow portion of the RV is shown in sections 1 and 2. The outflow portion of the RV was normal in size (sections 5 and 6). In this heart, RV hypoplasia occurred in association with CAVC (section 3).
Influence of Genetic Background on G4D Phenotype
Genetic background modifies the phenotypic expression of single gene mutations in mice and in humans [19–21]. To study the effect of genetic background on phenotypic expression of heterozygous Gata4 mutation, we bred G4D into the pure FVB genetic background. In G4D-FVB fetuses, 15/21 (71%) hearts were normal (Table 2). The frequency of VSDs (5/23; 22%) and RV hypoplasia (2/23, 9%) was similar to that found in the C57 background. However, the frequency of ECDs was substantially lower (1/23, 4%; χ2 P < 0.0001). These data indicate that genetic modifiers increase the frequency of ECDs by 20-fold in G4D-C57 compared to G4D-FVB (59% in G4D-C57 vs 3% in G4D-FVB).
To further characterize the strain-specific genetic modifiers, we crossed G4D-C57 mice to FVB obtain G4D-F1 mice. 88% of G4D-F1 hearts were normal, and only 1/33 (3%; χ2 P < 0.0001) had an ECD (Table 2). This was not due to differences between the Gata4WT allele in C57 versus FVB, because the strain contributing the wild-type allele did not influence survival (data not shown). These data suggest that genetic modifier(s) in the C57 strain that increase risk of ECD are recessive.
To map the modifier(s), we crossed G4D-F1 mice to C57 to obtain G4D-backcross (G4D-back) embryos. We sectioned 172 such embryos in late gestation, and found cardiac malformations in 73 (42%; Table 2). ECDs occurred at the highest frequency (41/172, 24%), again consistent with recessive modifier(s) in C57. The ratio of males versus females was the same in affected versus unaffected embryos, suggesting that the modifier(s) were located on autosomes. We performed a whole genome linkage scan, genotyping 25 affected and 13 unaffected G4D-back embryos at 691 single nucleotide polymorphisms (SNPs) informative between C57 and FVB. We did not identify any SNP where the C57 homozygous genotype was significantly enriched amongst affected mice (data not shown). The study had an 80% likelihood of identifying a suggestive linkage (LOD ≥ 2.46) between ECD and a single strong modifier (relative risk of 23). Thus, the absence of a significant association in this study suggests multiple modifiers, each with a lower relative risk.
Abnormal Cardiac Function in G4D mice
We previously reported that depressed ventricular function is a fully penetrant phenotype of G4D mice in a C57/FVB F1 genetic background [17]. We asked if ventricular function in adult G4D mice varied with strain background. Compared to WT, G4D-FVB mice had mild ventricular dysfunction while G4D-C57 mice exhibited moderate-severe ventricular dysfunction (Table 3). This was not due to cardiac malformations, because postmortem analysis included serial histological sectioning of each heart in this study. This showed only one ASD secundum in a G4D-C57 heart.
Table 3.
Gravimetric and Echocardiographic Assessment of Ventricular Function in G4D Mice
| C57BL6/J | FVB/N | |||
|---|---|---|---|---|
| WT (n=7) | G4D (n=7) | WT (n=6) | G4D (n=5) | |
| LVEDD (mm) | 3.50 ± 0.06 | 3.92 ± 0.23 | 3.52 ± 0.06 | 3.64 ± 0.10 |
| LVESD (mm) | 1.72 ± 0.12 | 2.65 ± 0.23A | 1.54 ± 0.09 | 1.93 ± 0.05A,C |
| PWth (mm) | 0.49 ± 0.02 | 0.57 ± 0.06 | 0.52 ± 0.03 | 0.50 ± 0.02 |
| FS (%) | 51 ± 1 | 33 ± 2A | 56 ± 2 | 47 ± 2B,C |
| HR (bpm) | 680 ± 17 | 639 ± 46 | 727 ± 8 | 733 ± 26 |
| BW (g) | 30.0 ± 0.9 | 30.3 ± 0.5 | 29.1 ± 0.7 | 28.2 ± 1.7 |
| TL (mm) | 17.1 ± 0.1 | 17.1 ± 0.1 | 171 ± 0.1 | 17.2 ± 0.1 |
| HW (mg) | 158 ± 7 | 167 ± 8 | 133 ± 4 | 134 ± 8C |
| LuW (mg) | 148 ± 9 | 148 ± 7 | 145 ± 8 | 137 ± 6 |
| HW/BW (mg/g) | 5.3 ± 0.2 | 5.5 ± 0.3 | 4.6 ± 0.1 | 4.8 ± 0.1 |
| HW/TL (mg/mm) | 9.2 ± 0.4 | 9.8 ± 0.5 | 7.8 ± 0.3 | 7.8 ± 0.5C |
p < 0.01 compared to WT control of the same strain
p < 0.05 compared to WT control of the same strain
p < 0.05 for G4D-C57 compared to G4D-FVB
Results are shown as mean ± SEM. Groups were compared by ANOVA with Tukey-Kramer HSD post-hoc test. LVEDD, left ventricular end diastolic diameter; LVESD, left ventricular end systolic diamter; PWth, posterior wall thickness; FS, fractional shortening; HR, heart rate; bpm, beats per minute. BW, body weight; TL, tibial length; HW, heart weight; LuW, lung weight.
GATA4 Mutations in Human Heart Disease
To extend our study to human CHD, we analyzed the coding regions and splice donor/acceptor sites of GATA4 in genomic DNA samples from 107 patients with cardiac abnormalities in the phenotypic spectrum of the G4D mouse model (septal defect, n=8; ECD, n=43; RV hypoplasia, in the context of DILV, n=9; or cardiomyopathy, n=48) (Table 4).
Table 4.
Patient Characteristics
| Patients | GATA4 Alteration* | Probands with Family History | |
|---|---|---|---|
| Cardiac Lesion | # (%) | # (%) | History |
| Endocardial Cushion Defects | 42 (39) | 2 (4.8) | 0 |
| Double Inlet LV | 9 (8) | 1 (11.1) | 0 |
| ASD/VSD | 8 (7) | 1 (12.5) | 1 |
| Cardiomyopathy* | 48 (45) | 0 (0) | 6 |
| TOTAL | 107 | 4 (3.7) | |
24 of these patients were via personal communications from M. Sarkar, C. Seidman, and J. Seidman
GATA4 alteration is defined as a nonsynonomous sequence alteration not found in control individuals.
We identified several non-synonymous GATA4 sequence variants. Those found in control only, or in probands and controls, are listed in Supplementary Table 2. Four non-synonymous GATA4 sequence variants occurred in probands but not in controls (Table 5):
Table 5.
Non-synonymous GATA4 sequence variants associated with CHD found in this study
| Controls |
||||||||
|---|---|---|---|---|---|---|---|---|
| Proband | Sequence Variant | AA Change | Conservation | Affected Domain | All # (est allelic freq) | Ethnically Matched # (est allelic freq) | Cardiac Phenotype | Note |
| 1 | 1207C>A | L403M | XMRPH | C Terminal Domain | 0/500 (0–0.007) | 0/62 (0–0.058) | Hypoplastic RV in the context of [S,D,D] DILV with sinus venosus atrial septal defect | |
| 2 | 886G>T | G296C | XMRPH | C terminal Zn Finger /NLS junction | 0/500 (0–0.007) | 0/246 (0–0.015) | Secundum ASD, valvar PS | Sister: ASD 2, geno unknown; Father: LSVC to CS, G296C. |
| 3 | 487C>T | P163S | XMRPH | TAD2 | 0/600 (0–0.006) | 0/346 (0–0.011) | ECD (Primum ASD, cleft MV) | Father P163S reportedly unaffected. |
| 4 | 1037C>T | A346V | MPH (Ser in XR) | C Terminal Domain | 0/600 (0–0.006) | 0/346 (0–0.011) | ECD (Primum ASD, cleft MV) | Mother A346V reportedly unaffected |
Proband 1 was Libyan Jewish. Probands 2–4 were Caucasian. “Conservation” indicates identical residue in Xenopus laevis (X), Mus musculis (M), Rattus norvegicus (R), Sus scrofa (pig, P), and Homo sapiens (H). “Controls” indicates the number of control alleles with the sequence variant, and the number of control alleles examined. This is reported for all controls and for ethnically matched controls. “Est Allelic Freq” indicates the 95% confidence estimate of the allelic frequency in controls. The upper bound of this estimate was calculated from observing zero sequence variants in n control chromosomes, using a binomial distribution [32]. ASD 2, secundum ASD; LSVC, left superior vena cava; CS, coronary sinus; NLS, nuclear localization sequence; MV, mitral valve.
1. G296C occurred in a proband with secundum ASD and pulmonary stenosis, and is similar to the previously described G296S mutation, which co-segregated with secundum ASDs and PS in two unrelated pedigrees (Table 1) [3, 6]. The proband’s father had the same G296C substitution and had a persistent LSVC to coronary sinus. A sibling also had a secundum ASD, but DNA was not available for genetic analysis. This sequence variant was not found in 500 control chromosomes (246 ethnically matched). G296 is invariant from Xenopus through human, and occurs in the DNA-binding domain. The related G296S mutation reduced GATA4 DNA-binding activity as well as binding to the transcription factor Tbx5 [3].
2. L403M occurred in a proband with hypoplastic RV in the context of DILV. This patient also had a sinus venosus ASD. There was no family history of CHD. Parental DNA was not available for genotyping. This sequence variant was not found in 500 control chromosomes (62 ethnically matched). L403 is invariant from Xenopus through human, and occurs in the C-terminal domain that is required for transcriptional activation [22].
3 and 4. P163S and A346V occurred in probands with ECDs. In each case, the family history was negative for CHD, but one parent was a carrier of the sequence variant. These carrier individuals did not have clinically apparent heart disease (direct echocardiographic studies were not available). These sequence variants were not found in 600 control chromosomes (346 ethnically matched). The proline at position 163 is invariant from Xenopus through human, and occurs in a transactivation domain that is required for GATA4 activity and is conserved in GATA4, GATA5, and GATA6 [22]. The residue at position 346 is either alanine or serine (a conservative substitution in the BLOSUM62 matrix [23]) in Xenopus through human. This residue occurs in the C-terminal domain required for transcriptional activation [22]. The A346V substitution is non-conservative in the BLOSUM62 matrix [23].
We modeled the effect of these four substitutions on protein function using the MAPP algorithm [14]. Each of the four substitutions was predicted to be deleterious to protein function.
Although cardiomyopathy was a highly penetrant phenotype in G4D mice, we did not find GATA4 mutations among 48 patients with cardiomyopathy (Table 4). Conversely, none of the 22 patients with GATA4 sequence alteration reported in this study or a prior study [3] had ventricular dysfunction attributable to GATA4 mutation. These data suggest that heterozygous GATA4 mutation is not a frequent cause of cardiomyopathy in humans.
Discussion
Spectrum of Phenotypes Associated with Murine Gata4 Mutation
Using tissue-restricted gene inactivation approaches, we previously showed that Gata4 is required in the myocardial compartment for normal myocardial growth and RV morphogenesis [24], and in endocardially derived structures for normal atrioventricular valve development [15]. These processes were also disrupted by a hypomorphic mutation of Gata4, which reduced Gata4 protein by 70% and resulted in embryonic lethality due to defects in myocardial growth, endocardial cushion development, and outflow tract alignment. In this work, we extend these findings to show for the first time that heterozygous mutation of Gata4 in mice is sufficient to disrupt myocardial growth and endocardial cushion development. We did not observe conotruncal abnormalities or abnormalities of outflow tract alignment in heterozygous Gata4 mutant mice, suggesting that these abnormalities occur only after a greater perturbation of Gata4 activity. Collectively, these findings emphasize the importance of maintaining precise levels of GATA4 activity for normal development of these structures. Reduction of GATA4 activity through genetic mutation of GATA4 or interacting factors, or environmental influences (e.g., retinoic acid deficiency [25]), might lead to a similar spectrum of cardiac phenotypes.
Although human mutation of one copy of GATA4 causes familial septal defects, mice with mutation of one copy of GATA4 were previously reported to be normal [26–28]. This apparent difference between mice and humans appeared to prohibit using the heterozygous mutant mouse model to gain insights into the spectrum of abnormalities that might occur in humans with heterozygous GATA4 mutation. However, by studying the phenotype of heterozygous Gata4 mutant mice in different inbred strain backgrounds, we show that murine mutation of one copy of Gata4 is sufficient to lead to septal defect, ECD, RV hypoplasia, and cardiomyopathy, albeit with reduced penetrance and variable expressivity.
Modifiers of Cardiovascular Phenotype Due to Murine Gata4 Mutation
While reduced penetrance and variable expressivity have been frequently observed in studies of single gene mutations associated with CHD [19–21, 29], the cause of this variability is not well understood. In the case of heterozygous Gata4 mutation in mice, we show that genetic modifiers strongly influence phenotypic expression, as the C57 background increased the frequency of ECDs by 20-fold over the FVB background. LV dysfunction was also more severe in the C57 compared to the FVB background. Our screen for genetic modifiers of the ECD phenotype did not identify a single strong genetic modifier, suggesting that two or more weaker modifiers are responsible.
An inference from our experiments is that epigenetic factors must also have an important influence on the expression of cardiovascular phenotypes. Despite extensive inbreeding (now backcrossed 13 generations), G4D mutant mice display considerable phenotypic variation and partial penetrance that cannot be accounted for by genetic factors. The controlled breeding environment argues against a significant contribution of environmental factors to the observed phenotypic variation. By exclusion, this suggests that stochastic events contribute to the phenotypic heterogeneity. Similar phenotypic heterogeneity was previously noted in a careful study of Hey2 null mice in highly inbred strain backgrounds [29], indicating that the important role of epigenetic factors in modulating phenotypic expression extends beyond Gata4 mutations.
Human GATA4 Mutations
In a panel of 107 patients with largely sporadic CHD and cardiac phenotypes that overlapped those observed in the G4D mouse model, we found four GATA4 non-synonymous sequence variants (G296C, L403M, P163S, and A346V) that did not occur in control individuals. These are likely disease-causing mutations, as each is a sequence variant that alters a highly conserved residue and that was not found in controls. A strength of our study was the large number of control chromosomes analyzed (500–600 total; 62–346 ethnically matched; Table 5). Computation modeling suggested that each substitution was deleterious to protein function. Further, the related G296S mutation has been previously reported to be disease causing [3, 6]. One parent of each of the probands with P163S and A346V mutation carried the mutation but did not have overt clinical disease. This likely reflects reduced penetrance, which might be expected based on the mouse model.
We found GATA4 mutations in two patients with ECD. An additional patient with ECD and GATA4 mutation has previously been reported as a member of an extended pedigree with GATA4 mutation and predominantly ASD or VSD [3]. These data indicate that GATA4 mutation can cause ECD in humans as well as in mice. The estimated frequency of GATA4 mutation among sporadic ECD cases (2/43 in this study plus 0/34 from the literature (Table 1) = 2.6%) is significant for a single gene.
No single gene mutation has been previously associated with DILV in humans. This cardiac malformation has been proposed to result from severe RV hypoplasia with consequent malpositioning of the atrioventricular septum [30], or from abnormal endocardial cushion development [31]. Since GATA4 is a crucial regulator of both RV chamber morphogenesis and endocardial cushion development [15, 24], an association of DILV with GATA4 mutation is consistent with its known roles in heart development. In G4D mice, we observed two malformations that may represent forme frustes of DILV: malaligned CAVC in which the common AV valve opened predominantly into the LV (Figure 3c), and severe hypoplasia of the RV sinus (Figure 4).
Use of heterozygous mouse models to model human disease
While constitutive and conditional knockout studies illuminate the essential function of genes, the complete ablation of gene activity in these models is often not representative of human disease. In many cases, heterozygous mouse mutants more closely model the perturbations in gene activity that occur in human disease as a result of heterozygous mutation or environmental influences. In this study, we carefully examined the phenotype of mice with heterozygous Gata4 mutation, in order to generate hypotheses regarding potential cardiac phenotypes that may be associated with GATA4 mutation in humans. Of the 107 patients in our study with cardiac phenotypes overlapping those seen in the mouse model, 4 patients had GATA4 mutation. In contrast, we did not find GATA4 mutation in 126 patients with cardiac phenotypes not seen in the mouse model (conotruncal anomalies, n=34; heterotaxy, n=7; Ebstein’s anomaly, n=5; left-sided obstruction, n=81; data not shown). Out of the five types of phenotypes that were present in the mouse model (ECD, RV hypoplasia, ASD, VSD, cardiomyopathy), four types are associated with GATA4 mutation in humans (ECD, RV hypoplasia in the context of DILV, ASD, VSD). These results suggest that careful study of heterozygous mutant mouse models is a productive experimental strategy for developing hypotheses regarding potential human phenotypes.
Given the high penetrance of cardiomyopathy in the G4D mouse model, it was notable that we did not find GATA4 mutations among patients with cardiomyopathy. Moreover, 22 patients with GATA4 mutation did not exhibit cardiomyopathy. This might reflect a difference between the specific mutations studied in mice and humans, or a difference in phenotypic expression between mouse and humans. Thus, while mouse models are useful for generation of hypotheses regarding human phenotypes, the hypotheses require testing in humans.
In this study, we showed that heterozygous Gata4 mutation in a mouse model caused ECD, RV hypoplasia, septal defects, and cardiomyopathy. Among patients with CHD, we found GATA4 mutations associated with overlapping cardiac phenotypes, namely ECD, RV hypoplasia in the context of DILV, and septal defects. These data support the importance of fine regulation of GATA4-dependent pathways in the development of these structures, and identify GATA4 as a cause of sporadic ECD and DILV in humans. Additional targeted studies of more patients with ECD and particularly RV hypoplasia are needed to determine the degree to which GATA4 mutation contributes to these forms of CHD.
Supplementary Material
Acknowledgements
This work was supported by grants from the NIH (SRK, NIH training grant T32HLO07572; WTP, SCCOR P50HLO74734; DWB SCCOR P50HL74728 and HL69712; and EG SCCOR P50HL74731). WTP was also supported by charitable donations from Edward Marran and Karen Carpenter. Jie Shen was supported as a Bang Bao Research Scholar by a grant from Proctor and Gamble. We thank M. Sarkar, C. Seidman, and J. Seidman for unpublished data.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest: none.
References
- 1.Srivastava D, Olson EN. A genetic blueprint for cardiac development. Nature. 2000;407:221–226. doi: 10.1038/35025190. [DOI] [PubMed] [Google Scholar]
- 2.Ransom J, Srivastava D. The genetics of cardiac birth defects. Semin Cell Dev Biol. 2007;18:132–139. doi: 10.1016/j.semcdb.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 3.Garg V, Kathiriya IS, Barnes R, Schluterman MK, King IN, Butler CA, et al. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature. 2003;424:443–447. doi: 10.1038/nature01827. [DOI] [PubMed] [Google Scholar]
- 4.Hirayama-Yamada K, Kamisago M, Akimoto K, Aotsuka H, Nakamura Y, Tomita H, et al. Phenotypes with GATA4 or NKX2.5 mutations in familial atrial septal defect. Am J Med Genet A. 2005;135:47–52. doi: 10.1002/ajmg.a.30684. [DOI] [PubMed] [Google Scholar]
- 5.Okubo A, Miyoshi O, Baba K, Takagi M, Tsukamoto K, Kinoshita A, et al. A novel GATA4 mutation completely segregated with atrial septal defect in a large Japanese family. J Med Genet. 2004;41:e97. doi: 10.1136/jmg.2004.018895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sarkozy A, Conti E, Neri C, D'Agostino R, Digilio MC, Esposito G, et al. Spectrum of atrial septal defects associated with mutations of NKX2.5 and GATA4 transcription factors. J Med Genet. 2005;42:e16. doi: 10.1136/jmg.2004.026740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sarkozy A, Esposito G, Conti E, Digilio MC, Marino B, Calabro R, et al. CRELD1 and GATA4 gene analysis in patients with nonsyndromic atrioventricular canal defects.[letter] Am J Med Genet A. 2005;139(3):236–238. doi: 10.1002/ajmg.a.31018. [DOI] [PubMed] [Google Scholar]
- 8.Nemer G, Fadlalah F, Usta J, Nemer M, Dbaibo G, Obeid M, et al. A novel mutation in the GATA4 gene in patients with Tetralogy of Fallot. Hum Mutat. 2006;27:293–294. doi: 10.1002/humu.9410. [DOI] [PubMed] [Google Scholar]
- 9.Zhang L, Tumer Z, Jacobsen JR, Andersen PS, Tommerup N, Larsen LA. Screening of 99 Danish patients with congenital heart disease for GATA4 mutations. Genet Test. 2006;10:277–280. doi: 10.1089/gte.2006.10.277. [DOI] [PubMed] [Google Scholar]
- 10.Schluterman MK, krysiak AE, Kathiriya IS, Abate N, Chandalia M, Srivastava D, et al. Screening and biochemical analysis of GATA4 sequence variations identified in patients with congenital heart disease. Am J Med Genet A. 2007;143A:817–823. doi: 10.1002/ajmg.a.31652. [DOI] [PubMed] [Google Scholar]
- 11.Pu WT, Ishiwata T, Juraszek AL, Ma Q, Izumo S. GATA4 is a dosage-sensitive regulator of cardiac morphogenesis. Developmental Biology. 2004;275:235–244. doi: 10.1016/j.ydbio.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 12.Xu S, Atchley WR. Mapping quantitative trait loci for complex binary diseases using line crosses. Genetics. 1996;143:1417–1424. doi: 10.1093/genetics/143.3.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Broman KW, Wu H, Sen S, Churchill GA. R/qtl: QTL mapping in experimental crosses. Bioinformatics. 2003;19:889–890. doi: 10.1093/bioinformatics/btg112. [DOI] [PubMed] [Google Scholar]
- 14.Stone EA, Sidow A. Physicochemical constraint violation by missense substitutions mediates impairment of protein function and disease severity. Genome Res. 2005;15:978–986. doi: 10.1101/gr.3804205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rivera-Feliciano J, Lee KH, Kong SW, Rajagopal S, Ma Q, Springer Z, et al. Development of heart valves requires Gata4 expression in endothelial-derived cells. Development. 2006;133:3607–3618. doi: 10.1242/dev.02519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bosse T, Piaseckyj CM, Burghard E, Fialkovich JJ, Rajagopal S, Pu WT, et al. Gata4 Is Essential for the Maintenance of Jejunal-Ileal Identities in the Adult Mouse Small Intestine. Mol Cell Biol. 2006;26:9060–9070. doi: 10.1128/MCB.00124-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bisping E, Ikeda S, Kong SW, Tarnavski O, Bodyak N, McMullen JR, et al. Gata4 is required for maintenance of postnatal cardiac function and protection from pressure overload-induced heart failure. Proc Natl Acad Sci U S A. 2006;103:14471–14476. doi: 10.1073/pnas.0602543103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jay PY, Bielinska M, Erlich JM, Mannisto S, Pu WT, Heikinheimo M, et al. Impaired mesenchymal cell function in Gata4 mutant mice leads to diaphragmatic hernias and primary lung defects. Dev Biol. 2007;301:602–614. doi: 10.1016/j.ydbio.2006.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benson DW, Silberbach GM, Kavanaugh-McHugh A, Cottrill C, Zhang Y, Riggs S, et al. Mutations in the cardiac transcription factor NKX2.5 affect diverse cardiac developmental pathways. J Clin Invest. 1999;104:1567–1573. doi: 10.1172/JCI8154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goldmuntz E, Geiger E, Benson DW. NKX2.5 mutations in patients with tetralogy of fallot. Circulation. 2001;104:2565–2568. doi: 10.1161/hc4601.098427. [DOI] [PubMed] [Google Scholar]
- 21.McElhinney DB, Geiger E, Blinder J, Benson DW, Goldmuntz E. NKX2.5 mutations in patients with congenital heart disease. J Am Coll Cardiol. 2003;42:1650–1655. doi: 10.1016/j.jacc.2003.05.004. [DOI] [PubMed] [Google Scholar]
- 22.Morrisey EE, Ip HS, Tang Z, Parmacek MS. GATA-4 activates transcription via two novel domains that are conserved within the GATA-4/5/6 subfamily. J Biol Chem. 1997;272:8515–8524. doi: 10.1074/jbc.272.13.8515. [DOI] [PubMed] [Google Scholar]
- 23.Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. Proc Natl Acad Sci U S A. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zeisberg EM, Ma Q, Juraszek AL, Moses K, Schwartz RJ, Izumo S, et al. Morphogenesis of the right ventricle requires myocardial expression of Gata4. J Clin Invest. 2005;115:1522–1531. doi: 10.1172/JCI23769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghatpande S, Ghatpande A, Zile M, Evans T. Anterior endoderm is sufficient to rescue foregut apoptosis and heart tube morphogenesis in an embryo lacking retinoic acid. Dev Biol. 2000;219:59–70. doi: 10.1006/dbio.1999.9601. [DOI] [PubMed] [Google Scholar]
- 26.Molkentin JD, Lin Q, Duncan SA, Olson EN. Requirement of the transcription factor GATA4 for heart tube formation and ventral morphogenesis. Genes Dev. 1997;11:1061–1072. doi: 10.1101/gad.11.8.1061. [DOI] [PubMed] [Google Scholar]
- 27.Kuo CT, Morrisey EE, Anandappa R, Sigrist K, Lu MM, Parmacek MS, et al. GATA4 transcription factor is required for ventral morphogenesis and heart tube formation. Genes Dev. 1997;11:1048–1060. doi: 10.1101/gad.11.8.1048. [DOI] [PubMed] [Google Scholar]
- 28.Xin M, Davis CA, Molkentin JD, Lien CL, Duncan SA, Richardson JA, et al. A threshold of GATA4 and GATA6 expression is required for cardiovascular development. Proc Natl Acad Sci U S A. 2006;103:11189–11194. doi: 10.1073/pnas.0604604103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sakata Y, Koibuchi N, Xiang F, Youngblood JM, Kamei CN, Chin MT. The spectrum of cardiovascular anomalies in CHF1/Hey2 deficient mice reveals roles in endocardial cushion, myocardial and vascular maturation. J Mol Cell Cardiol. 2006;40:267–273. doi: 10.1016/j.yjmcc.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 30.van Praagh R, Plett JA, van Praagh S. Single ventricle. Pathology, embryology, terminology and classification. Herz. 1979;4:113–150. [PubMed] [Google Scholar]
- 31.Jiao K, Langworthy M, Batts L, Brown CB, Moses HL, Baldwin HS. Tgf{beta} signaling is required for atrioventricular cushion mesenchyme remodeling during in vivo cardiac development. Development. 2006;133:4585–4593. doi: 10.1242/dev.02597. [DOI] [PubMed] [Google Scholar]
- 32.Clopper CJ, Pearson ES. The use of confidence or fiducial limits illustrated in the case of the binomial. Biometrika. 1934;26:404–413. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.