

Abstract
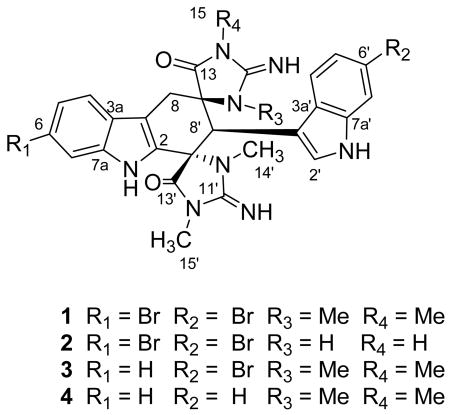
An extensive study of the secondary metabolites produced by the marine sponge Smenospongia cerebriformis (Duchassaing & Michelotti, 1864) (Order Dictyoceratida: Family Thorectidae has led to the isolation of two new bisspiroimidazolidinone derivatives, dictazoline A (1) and B (2), along with the known soft coral metabolites tubastrindole A (3) and B (4). The structures were assigned by 2D NMR spectroscopic methods.
We recently initiated a broad screening program to discover kinase and protease inhibitors from marine organisms. A cytotoxic sponge sample, subsequently identified as Smenospongia cerebriformis (Duchassaing & Michelotti, 1864) (Order Dictyoceratida: Family Thorectidae), was collected from Hospital Point on Solarte Isle, Boca del Toro, on the northwest coast of Panama. Fractionation of the extract has led to the isolation of 2 new bisspiroimidazolidinone alkaloids, dictazoline A (1) and B (2), along with the structurally-related compounds tubastrindole A (3) and B (4) previously isolated from a soft coral.1,2 We report here the isolation and structure determination of the new compounds 1 and 2.
Results and Discussions
Dictazoline A (1) was isolated as an optically active colorless powder ([α]D -4.1 (c 0.12, MeOH)). The molecular weight of 1 was obtained from the mass spectrum, which showed a cluster of pseudo-molecular ion peaks at m/z 665.0643 [M+H]+ indicative of a dibrominated compound. Based on HR-ESI-TOFMS data, the molecular formula was defined as C28H26Br2N8O2, which indicated 1 contained 19 double bond equivalents. This supposition was supported by the signals visible in the 13C NMR spectrum. In total 28 carbon resonances were observed in the 13C NMR and multiplicity-edited HSQC spectra. These could be ascribed to 15 quaternary, 8 methine, 1 methylene, and 4 methyl carbons. On the basis of the number of sp2 carbons and their chemical shifts, 1 was comprised of 2 amides (C-13, δC 175.6; C-13′, δC 173.0), 2 imines (C-11, δC 160.4; C-11′, δC 157.2), and 8 double bonds that accounted for 12 of the total 19 degrees of unsaturation implied by the molecular formula. This indicated 1 contained 7 rings.
Absorptions at 3390 and 1643 cm-1 in the IR spectrum were characteristic of carbonyl and N-H vibrations, initially suggestive of an amide. This functionality was clearly visible in the 13C NMR spectrum at δC 175.6 and 173.0. Starting with these carbon residues, HMBC and COSY experiments (Table 1) established a series of partial structures. The latter carbon (C-13′) showed a HMBC correlation from a methyl singlet at δH 2.77 (C-15′) indicative of a N-methyl amide, which also correlated to a quaternary sp2 carbon at δC 157.2 (C-11′). The chemical shift of C-11′ was suggestive of either a guanido or carbamate unit, the former seemed most likely given the 2 oxygen required by molecular formula had already been assigned to amide residues. This assignment of a guanidino unit was supported by the attachment of a second nitrogen to C-11′ based on HMBC correlations from a second N-methyl signal (C-14′). This N-methyl group showed only one other HMBC correlation, to a quaternary carbon resonating downfield at δC 70.6 (C-9′). In turn, C-9′ was placed directly adjacent to the amide carbonyl (C-13′) based on HMBC correlations from H-8′ to C-9′ and to C-13′. This defined one ring as a N-N-dimethyl-4-imidazolidinone. Close inspection of the spectroscopic data revealed an identical spiroimidazolidinone ring (C-9 to C-15) based on similar 13C NMR chemical shifts and HMBC correlations. These two rings were clearly both vicinal to C-8′ based on HMBC correlations from H-8′ (Fragment A).
Table 1.
NMR Spectroscopic Data (500 MHz), MeOH-d4 for Dictazoline A (1).
| Position | δC, mult. | δH (J in Hz) | COSY | HMBCa | ROESY |
|---|---|---|---|---|---|
| 2 | 128.3, qC | H-8, H-8′ | |||
| 3 | 114.2, qC | H-4, H-8 | |||
| 3a | 125.8, qC | H-4, H-5, H-7 | |||
| 4 | 121.3, CH | 7.52, d (8.6) | H-5 | H-8β | |
| 5 | 124.0, CH | 7.23, dd (8.6, 1.6) | H-4, H-7 | ||
| 6 | 118.1, qC | H-7 | |||
| 7 | 115.3, CH | 7.51, d (1.6) | H-5 | ||
| 7a | 139.0, qC | H-4, H-7 | |||
| 8 | 27.9, CH2 | 3.39, d (17.1)b | H-4, H-14 | ||
| 3.60, d (17.1) | H-8′ | ||||
| 9 | 71.3, qC | H-8, H-8′, H-14 | |||
| 11 | 160.4, qCc | H-14, H-15 | |||
| 13 | 175.6, qC | H-8, H-8′, H-15 | |||
| 14 | 29.8, CH3 | 3.16, s | H-2′, H-8β | ||
| 15 | 26.0, CH3 | 2.59, s | |||
| 2′ | 126.1, CH | 7.07, s | H-15′, H-14 | ||
| 3′ | 106.2, qC | H-2′, H-4′, H-8′ | |||
| 3a′ | 128.3, qC | H-2′, H-4′, H-7′, H-8′ | |||
| 4′ | 120.1, CH | 7.40, d (8.5) | H-5′ | H-8′ | |
| 5′ | 123.9, CH | 7.15, dd (8.5, 1.7) | H-4′, H-7′ | ||
| 6′ | 116.3, qC | H-4′, H-7′ | |||
| 7′ | 115.5, CH | 7.48,d (1.7) | H-5′ | ||
| 7a′ | 137.3, qC | H-2′, H-4′ | |||
| 8′ | 44.7, CH | 4.36, s | H-4′, H-8α, H-14′ | ||
| 9′ | 70.6, qC | H-8′, H-14′ | |||
| 11′ | 157.2, qCc | H-14′, H-15′ | |||
| 13′ | 173.0, qC | H-8′, H-15′ | |||
| 14′ | 26.9, CH3 | 2.89, s | H-8′ | ||
| 15′ | 24.6, CH3 | 2.77, s | H-2′ |
HMBC correlations, optimized for 7 Hz, are from proton(s) to the indicated carbon.
α-proton.
13C chemical shift determined from HMBC experiment.
Clearly, based on the UV absorptions at 220 and 291 nm, 1 contained indole chromophores. This was confirmed by analysis of HMBC and COSY spectroscopic data. Briefly, COSY correlation between two aromatic resonances, H-4′ and H-5′, established their vicinal relationship, while a meta orientation between H-5′ and H-7′ was evident based on a long range COSY correlation. The proton chemical shift and multiplicity of H-2′ (δH 7.07, s) were indicative of H-2 of an indole ring. The final HMBC connectivites needed to establish a disubstituted indole core were as follows: H-2′, H-4′, H-7′ to C-3a′ and C-7a′; H-4′, H-2′ to C-3′ (Fragment B). Based on analysis of similar HMBC and COSY correlations 1 contained a second indole moiety (Fragment C), which was tri- rather than disubstituted.
HMBC correlations provided the final carbon-carbon connectivites. Fragment B was linked to C-8′ of fragment A by a HMBC correlation from H-8′ to C-3′. Fragment D clearly was located between C-9 of fragment A and C-3 of fragment C based on HMBC correlations from H-8 to C-13 and C-3a. Finally C-9′ was connected to C-2 to form a 6-membered ring, which explained the HMBC correlation between H-8′ and C-2. Based on an accounting of the heteroatoms remaining, bromines were attached to C-6 and C-6′ to give the planar structure 1.
The relative configuration of 1 was assigned by analysis of ROESY correlations (Figure 2A). A suite of ROESY correlations between H-8′, H-8α, H-14′ established the relative configuration of C-8′ and C-9′, while crosspeaks between H-8β and H-14 established the configuration of the other spirocenter at C-9. Thus the relative configuration of 1 is 9R*, 8′R*, 9′S*.
Figure 2.

A) Key ROESY correlations used to establish the relative configuration of 1. B) Key ROESY correlations used to establish the structure and relative configuration of 2.
Detailed LC-MS analysis of the dichloromethane soluble residue led to the identification of a more polar analog. Subsequent isolation and characterization confirmed that 1 and 2 possessed similar structures. A comparison of the high-resolution ESI-TOF data indicated that 2 (C26H22Br2N8O2) was 28 amu smaller than 1. This difference was easily explained by the two missing N-methyl resonances in the 1H NMR. Analysis of the 2D NMR spectra (See Table S1 in Supporting Information) allowed the structure of 2 to be defined as depicted. A comparison of the fragments observed during the MS analysis indicated that the two N-methyl groups were in the same spiroimidazolidinone ring (Figure 3). In the MS spectrum of 1, a retro Diels-Alder reaction produced a prevalent m/z peak at 333.0360 corresponding to constitutional isomers 5 and 6. Conversely, the MS spectrum of 2 contained distinct fragments of equal intensity at m/z 333.0360 (5) and 305.0046 (7). The later ion, which was not present in the spectrum of 1, indicated both methyl groups were attached to the same spiroimidazolidinone ring.3 A direct comparison of the 1H NMR chemical shift between 1 and 2 assigned the two N-methyl groups in 2 as H-14′ and H-15′ (1 δH-14′ 2.77, δH-15′ 2.89, δH-14 3.16, δH-15 2.59; 2 δH-14′ 2.85, δH-15′ 2.95), Analysis of the HMBC spectrum provide no additional supporting evidence for this assignment though as the C-9/C-9′ and C-13/C-13′ pair of signals were accidentally isochronous at 500 MHz. In the end, analysis of the ROESY spectroscopic data provided justification for the placement of the methyl groups as depicted. Specifically, clear ROESY crosspeaks were observed between H-15′/H-2′, H-8′/H-8α, and H-8′/H-14′ (Figure 2B). These correlations also defined the configuration of all chiral centers in 2, with the exception of the C-9. Based a comparison of the carbon chemicals shifts, the configuration of C-9 in 2 was assigned as the same as for compound 1.
Figure 3.

Key Fragments observed in MS spectra of 1 and 2
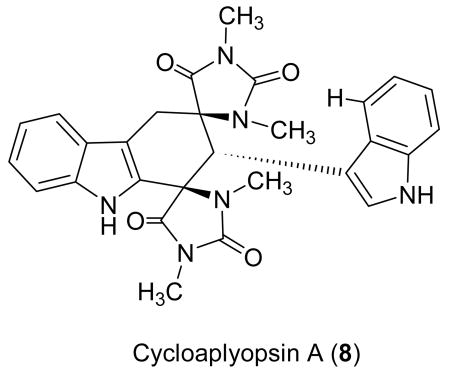
There is a report of a “dimer of 6-bromo-2′-de-N-methylaplysinopsin”, isolated from a dendrophylliid coral that would produce the same low-resolution pseudo-molecular ions given by 2.4 Unfortunately, the exact structure of this dimer is not described nor is sufficient spectroscopic data provided for a meaningful comparison with 2. Compounds 1 and 2 are structurally related to the bis-indole alkaloid cycloaplysinopsin A (8)5 and tubarstindoles1 both isolated from a Tubastraea sp. of coral. Two of the latter series of compounds, tubastrindole A (3) and B (4) were also isolated from the crude extract of this sponge. The occurrence of these metabolites in two dissimilar sources suggests that the true producer maybe an associated or symbiotic microbe. Interestingly, the cycloaplysinopsin A (8) have been reported to occur as an enantioenriched mixture (65:35), based on analysis of the proton spectrum in the presence of the chiral shift reagent Eu(fod)2. Mancini et al. propose a stereoselective Diels-Alder reaction catalyzed by a chiral environment between monomeric indole derivative 5 to explain the low optical activity of 8 ([α]D –34 (c 0.005 g/100 mL, MeOH), although the absolute configuration of the major enantiomer was not determined.5
Due to the small amount of material, only compounds 3 and 4 were evaluated for inhibition of inhibition of the serine/theronine kinase PKC. Initially, compound 4 displayed weak inhibition of PKCδ, but repeated retesting indicated these result were not statistically significant. These results are consistent with previous reports indicated this class of compounds displayed no cytotoxicity, nor any antifungal or antibacterial activity. Compounds 3 and 4 were also ineffective at reducing β-secretase proteolytic cleavage of amyloid precursor protein, an assay of relevance to possible treatments of Alzheimer's Disease.6
Experimental Section
General Experimental Procedures
Optical rotations were measured on JASCO-DIP-700 polarimeter at the sodium line (589 nm). UV spectra were obtained on a Hewlett-Packard 8453 spectrophotometer and IR bands were measured as a thin film on a NaCl disc using a Perkin Elmer 1600 series FTIR. NMR spectra were acquired on a Varian Inova 500 MHz spectrometer operating at 500 or 125 MHz using the residual solvent signals as an internal reference (CD3OD δH 3.30 ppm, δC 49.0 ppm). High-resolution mass spectral data were obtained on an Agilent MSD-TOF using the ESI mode. Gradient separations used a Shimadzu system consisting of LC-20AT Solvent Delivery Modules, a SPD-M20A VP Diode Photodiode Array Detector, and a SCL-20A VP System Controller
Collection
The sponge sample was collected from Hospital Point on Solarte Isle, Boca del Toro, on the northwest coast of Panama, from a depth of 2-3 m, on January 8, 2000. In life the sponge forms a thick encrusting pad with raised oscules and a honeycombed to conulose surface. The color in life is pinkish brown, darkening to wood brown out of the water. The texture is springy, and the sponge exudes slime. Large dark brown laminated and pithed fibers dominate the skeleton, and are concentrated at the surface. The sponge is most closely comparable to Smenospongia cerebriformis (Duchassaing & Michelotti, 1864) (Order Dictyoceratida: Family Thorectidae). A voucher specimen has been deposited in the Natural History Museum, London (BMHN 2000.12.11.6)
Extraction and Isolation of BMNH 2000.12.11.6
The freeze-dried sponge (114 g) was exhaustively extracted with 1:1 i-PrOH:CH2Cl2 (3 × 3 L) to afford 14.85 g of lipophilic extract. Partitioning using a modified Kupchan procedure yielded 4 fractions of 6.07, 1.88, 2.94 and 5.78 g from the hexanes, CH2Cl2, n-BuOH and H2O phases. The organic residue from the n-BuOH phase (2.94 g) was separated on a Sephadex LH-20 column (1300 × 30 mm) eluting with MeOH (flow rate 1.74 mL/min). The 33 fractions were analyzed by TLC and pooled into 7 fractions. Fraction 2 (582.5 mg) was chromatographed on a Si gel flash column (6.0 g) eluting with a gradient of CH2Cl2-MeOH. LC-MS analysis of the resulting fractions indicated one contained a series of halogenated compounds. Separation of this fraction by RP-HPLC [Luna C8, 250 × 10 mm, a linear gradient from 5-30% MeCN in H2O with 0.01 % formic acid in both solvents over 40 min, flow rate 3 mL/min, PDA and ELSD detection] afforded dictazoline A (1, tR 34.5 min, 0.2 mg, 1.3 × 10-3 % yield).
The residue from the CH2Cl2 partition was separated in similar manner as described above (Sephadex LH-20 and a Si Flash Column). LC-MS analysis once again indicated polyhalogenated compounds in one of the fractions. This fraction (20 mg) was separated by Sephadex LH-20 again to yield several fractions, which were further purified by RP-HPLC [Luna C8, 250 × 10 mm, a linear gradient from 5-40% MeCN in H2O w 0.01 % formic acid in both solvents over 40 min, flow rate 3 mL/min, PDA and ELSD detection] afforded dictazoline B (2, tR 27.0 min, 0.9 mg, 6.0 × 10-3 % yield).
The known compounds 3 (tR 22.0 min, 5 mg) and 4 (tR 37.0 min, 2 mg) were isolated by RP-HPLC separation of the first fraction from the Sephadex LH-20 column of the CH2Cl2 partition. Specific conditions were: Cosmosil C18-AR, 250 × 10 mm, Solvent A = 50:50 MeOH:H2O with (50 mM NH4OAc) and Solvent B = 80:20 MeOH:H2O with (50 mM NH4OAc), 35% Solvent B for 30 min then 70% solvent B, flow rate 2 mL/min, detection at 220 nm.
Dictazoline A (1)
colorless powder; [α]D22 –4.1 (c 0.12, MeOH); UV (MeOH) λmax (log ε) 224 (4.2) 291 (3.5) nm; IR (NaCl) νmax 3390, 1643, 1591, 1353 cm-1; See Table 1 for tabulated spectral data; HRESI-TOFMS m/z 665.0643 [calcd for C28H2779Br2N8O2+, 665.0618].
Dictazoline B (2)
colorless powder; [α]D22 –9.0 (c 0.2, MeOH); UV (MeOH) λmax (log ε) 224 (4.2) 289 (3.0); IR (NaCl) νmax 3340, 3244, 1699, 1665, 1590, 1385, 1262 cm-1; See Table S1 in Supporting Information for tabulated spectral data; HRESI-TOFMS m/z 637.0314 [calcd for C26H2379Br2N8O2+, 637.0305].
Figure 1.

Initial structural fragments from analysis of 2D NMR.
Acknowledgments
This work was funded by grants from the Victoria S. and Bradley L. Geist Foundation (20070461), the National Science Foundation (OCE04-32479), and the National Institute of Environmental Health Sciences (P50 ES012740). The content is solely the responsibility of the authors and does not necessarily represent the official view of the National Institute of Environmental Health Sciences, the National Institutes of Health, or the National Science Foundation. Funds for the upgrades of the NMR instrumentation were provided by the CRIF program of the Nation Science Foundation (CH E9974921) and the Elsa Pardee Foundation. The purchase of the Agilent LC-MS was funded by grant W911NF-04-1-0344 from the Department of Defense. We thank A. Sorribas for the BACE1 data, and W. Yoshida for the NMR data.
Footnotes
Supporting Information Available: Tabulate NMR data for 2 along with 1H, 13C, gCOSY, gHMBC, and gHSQC NMR Spectra for 1 and 2. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Iwagawa T, Miyazaki M, Okamura H, Nakatani M, Doe M, Takemura K. Tetrahedron Lett. 2003;44:2533–2535. doi: 10.1016/S0040-4039(03)00331-9. [DOI] [Google Scholar]
- 2.The structural depictions of 1-4 in this paper are drawn in accordance with IUPAC rules in which the lowest octant is assigned an R* configuration in molecules where the absolute configuration is unknown. See Cross LC, Klyne W. Pure & Appl Chem. 1976;45:11–30. doi: 10.1351/pac197645010011.
- 3.It should be noted that compound 5 is the well-known sponge metabolite aplysinopsin often isolated from dictyoceratid and astrophorid sponges. See. Kauzlauskas R, Murphy PT, Quinn RJ, Wells RJ. Tetrahedron Lett. 1977:61–64. doi: 10.1016/S0040-4039(01)92550-X.
- 4.Koh EGL, Sweatman H. J Exp Mar Biol. 2000;251:141–160. doi: 10.1016/S0022-0981(00)00222-7. [DOI] [PubMed] [Google Scholar]
- 5.Mancini I, Guella G, Zibrowius H, Pietra F. Tetrahedron. 2003;59:8757–8762. doi: 10.1016/j.tet.2003.09.038. [DOI] [Google Scholar]
- 6.Hardy J. Curr Alzheimer Res. 2006;3:71–73. doi: 10.2174/156720506775697098. [DOI] [PubMed] [Google Scholar]