

Abstract
Macromolecular drugs such as proteins and gene products are presumably the most desirable therapeutic agents due to their unmatched substrate specificity and reaction efficiency. Yet, clinical use of these drugs has met with limited success, primarily due to the impermeable nature of the cell membrane that restricts cellular drug uptake to only small (<600 Da) and hydrophobic molecules. The recent discovery of the protein transduction domain (PTD) membrane-penetrating peptides, such as HIV-TAT, has finally offered the possibility of resolving this cell-membrane barrier for macromolecular drug delivery. Via covalent linkages, these PTD peptides have been shown to ferry the attached macromolecular species across membranes of all cell types, both in vitro and in vivo. Nevertheless, the lack of selectivity for PTD-mediated internalization restricts the application of this cell uptake method in clinical practice, due to concerns of inducing systemic toxicity caused by the carried drugs.
Presented herein is a modified version of our previously established “ATTEMPTS” approach in delivery of macromolecular drugs, which integrates the cell-penetrating PTDs into a heparin/protamine-regulated delivery system. In vitro findings using asparaginase (ASNase) as a model macromolecular anti-tumor agent were able to validate the feasibility of this delivery system. The chemically constructed TAT-ASNase conjugates not only were able to translocate into the MOLT-4 cells and elicit the cytotoxic effects, but also this PTD-mediated intracellular drug uptake could be regulated (with on/off control) by the addition of heparin and protamine. This modified ATTEMPTS system therefore presents a new avenue of treatment of various types of cancers and other diseases with macromolecular drugs. In vitro characterization and a preliminary proof-of-concept animal investigation that demonstrates the feasibility of this PTD-mediated ASNase therapeutic system is subsequently described.
Keywords: ATTEMPTS Approach, Asparaginase Therapy, Prodrug, Targeting, Protein Transduction Domain (PTD) Peptide
INTRODUCTION
Anticancer drug therapies are generally beset by two bottleneck limitations. The first obstacle is the absence of a preferential action of the drug on tumor cells as opposed to normal tissues. While drug interactions with the tumor target would result in desirable therapeutic efficacy, inadvertent exposure to normal cells and tissues can lead to toxic side effects. The second limitation arises from the lack of ability of most drugs, macromolecular compounds in particular, to cross the cell membrane. Currently, natural delivery of therapeutic compounds across cell membrane can only be achieved when the drug molecules are small (typically less than 700 Da) and hydrophobic. Yet, many macromolecular drugs such as therapeutic enzymes and gene products are considered to be far more desirable anti-tumor agents, due to their unmatched substrate specificity and reaction efficiency [1–3].
The first limitation of drug reaction selectivity can be circumvented by combining both targeting and prodrug features into a single delivery system, such that the drug will remain inactive following administration and also during the targeting process. Upon reaching the site of interest, the prodrug can then be converted in situ to its active form at the tumor target. Based on this principle, the “ADEPT” approach [4], which permits a specific enzymatic conversion of a prodrug into active form at the target site, has attained reasonable success in delivering small therapeutic agents to the tumor without drug-induced toxic effects. Recently, we have developed another approach termed “ATTEMPTS” [1,5], which directly introduces the prodrug feature onto the protease drug, t-PA, by temporarily blocking the catalytic domain of the drug with an appended macromolecule. After the completion of the targeting process, the original proteolytic activity of the drug is restored in situ by releasing the blockage with a second triggering agent. This ATTEMPTS approach has further advanced the utility of the targeting and prodrug type strategies to include macromolecular drugs. The lack of means to facilitate effective intracellular drug uptake by both approaches, however, has limited application of the “ADEPT” approach to only existing small and hydrophobic drugs (e.g. doxorubicin) [4), whereas the “ATTEMPTS” approach to solely enzyme drugs that exert their activities either in the circulation or in an extracellular environment.
Recent discovery of the cell-penetrating, protein transduction domain (PTD) peptides [6–15] such as HIV-TAT [7, 10, 15] has provided insight into finally resolving the membrane barrier problem for intracellular drug delivery. Both cell culture and animal studies have shown that by covalently linking TAT to nearly any drug class including hydrophilic compounds and large protein molecules (MW >150 kDa), TAT was able to transduce the attached cargos into cells of all organ types including the brain [9,10]. Although the mechanism of TAT-mediated cell transduction remains unclear, this cell entry event nevertheless appears to proceed via a first step of surface adsorption, with the binding of the cationic TAT to the anionic heparan sulfate on the cell surface, because TAT-mediated cell translocation of the attached cargos was completely inhibited in the presence of heparin, heparan or dextran sulfate [11, 12]. However, despite this unparalleled potency of TAT to transduce nearly all classes of compounds across cell membrane, the lack of selectivity of TAT has nevertheless rendered this cellular drug delivery method an unacceptable practice, due to concerns of causing drug-induced toxic effects towards normal tissues.
Because of the structural similarity between TAT and the cationic peptides employed in our previous ATTEMPTS system [1,5], we have developed a modified version of this system in an effort to achieve effective yet safe intracellular delivery of macromolecular drugs. As shown in Figure 1, the system consists of a complex composed of both targeting and drug components. The targeting component contains a specific targeting moiety, which could be an antibody (Ab) or a peptide ligand, coupled to a heparin (Hep) molecule. The drug component consists of the macromolecular drug that is covalently linked with a PTD peptide such as TAT via a disulfide bond. When combined, these two components spontaneously associate with each other through charge-charge interactions between the anionic heparin and the cationic TAT. Following administration, the prodrug feature (i.e. due to inhibition of the cell-penetrating function by heparin) and targeting moieties can prevent interaction of the drug complex with normal tissues during tumor-targeting, thereby alleviating drug-associated toxic effects. After the TAT-Drug/ Ab-Hep complexes reach the target through functions of the targeting component, protamine sulfate, a clinical heparin antidote that binds to heparin with greater affinity than TAT, can then be administered as a competing agent to dissociate the TAT-Drug conjugate from its Hep-Ab counterpart. Once relieved from heparin inhibition, the potent membrane-penetrating activity of TAT can be restored and the TAT-Drug conjugates can be taken up by the targeted tumor cells. Within tumor cells, the drug molecule can, by design, be detached from PTD through reduction of S-S linkage by the elevated level of glutathione in the cytosol, inducing tumor-specific apoptosis (see Figure 1).
Figure 1.
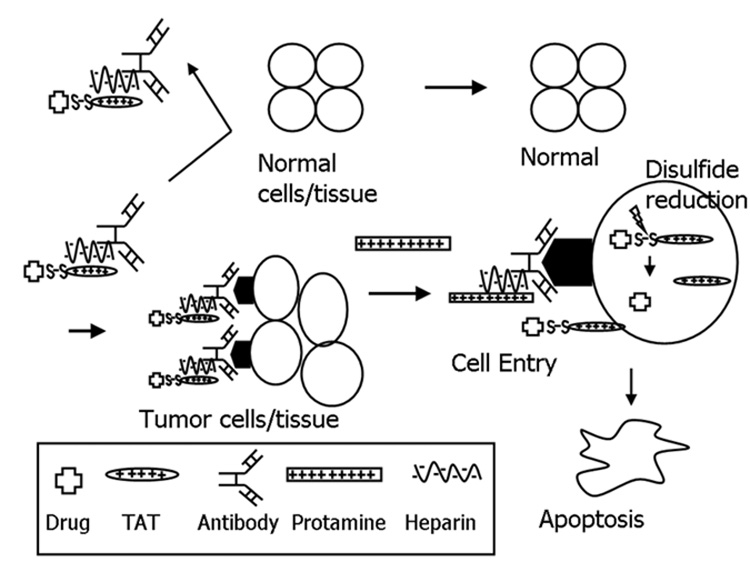
Schematic of the heparin/protamine delivery system in modulating TAT-mediated intracellular delivery of macromolecular drug.
In the present study, we selected ASNase as a model macromolecular drug to demonstrate the in vitro feasibility of this modified ATTEMPTS drug delivery system. ASNase is an enzyme drug that has been approved by the Food and Drug Administration for induction of remission in patients with acute lymphoblastic leukemia [16–25]. The mechanism of ASNase action is attributed to a systemic depletion of the non-essential amino acid asparagine (ASN). Unlike normal cells which have the ability to synthesize asparagine, some leukemic cells lack this ability and thus rely on the extracellular supply of asparagine for survival. A shortage in asparagine supply therefore leads to inhibition of DNA and RNA syntheses in leukemic cells thereby impairing cellular functions and subsequent their death. In this study, TAT was chemically linked to ASNase using 3-(2-pyridyldithio)-propionic acid N-hydroxysuccinimide ester (SPDP) cross-linking method. The cell-penetrating functions and cytotoxic effects of the TAT-ASNase conjugates were examined in vitro using the HeLa and MOLT-4 cell lines, respectively. In addition, the inhibitory effects of heparin on TAT-mediated cell internalization and the reversal of this effect by protamine were also investigated.
MATERIALS AND METHODS
Conjugation of Asparaginase (ASNase) to TAT Peptide
A modified TAT cell transduction peptide CGGGYGRKKRRQRRR was synthesized at the University of Michigan Protein Structure Core Facility. This peptide contained the 11 amino acid transduction peptide sequence (underlined) known to be effective in inducing cell internalization. Asparaginase (ASNase) was conjugated to TAT using the heterobifunctional cross-linker 3-(2-pyridyldithio)-propionic acid N-hydroxysuccinimide ester (SPDP, Sigma, USA), according to a previously established method [5].
Purification and Characterization of the TAT-ASNase Conjugates
The TAT-ASNase conjugates were purified from un-reacted ASNase and TAT with a HiTrap Heparin column (Supelco, Bellefonte, PA) connected to a HPLC system (Alltech 526 HPLC pump, Deerfield, IL) through a gradient elution (0.15–2.0 M NaCl; flow rate: 1 mL/min). Successful conjugation and purification of the TAT-ASNase conjugates were confirmed by SDS-PAGE on ready-made 12% Tris-HCl mini gels (Bio-Rad Laboratories, Hercules, CA). For cell uptake studies, free ASNase in and the TAT-ASNase conjugates were labeled with FITC according to the previously described procedures [26].
Measurement of the Enzymatic Activity of ASNase and TAT-ASNase Conjugates
Protein concentrations were determined by measuring UV absorbance at 280 nm. Enzymatic activities of ASNase and the TAT-ASNase conjugates were determined by Nesslerization [27]. Cell cytotoxicity of ASNase and the TAT-ASNase conjugates were conducted against an ASN-sensitive human leukemia cell line, MOLT-4 (ATCC, Manassas, VA), using the XTT assay [28].
Assessment of Cell Penetration and Uptake of Asp-TAT Conjugates
The cell-penetrating ability of Asp-TAT was measured by fluorescence microscopy. HeLa cells were seeded into glass chamber slides (Nunc; Naperville, IL) and cultured overnight. Cells were rinsed three times with modified Earle’s balanced salt solution (MEBSS) and then incubated with FITC-labeled ASNase or TAT-ASNase for 2 hours (70 ug/ml enzyme per 1 million cells/ml). For heparin making of TAT, approximately 5-fold molar excess with respect to ASNase was added, and 10-fold molar excess of protamine sulfate was added to unblock heparin. After washing, cells were fixed with 4% paraformaldehyde and then analyzed by fluorescence microscopy (Carl Zeiss, Gottingen, Germany).
Cell internalization of TAT-ASNase was also assessed by flow cytometry. Human leukemia MOLT-4 cells were incubated with FITC-labeled ASNase or TAT-ASNase of similar fluorescence intensity at 37 °C for 2 hours in the same manner as the confocal sample were prepared. After washing, the cell uptake of the TAT-ASNase conjugates was analyzed by flow cytometry (Facscalibur, BD Biosciences, San Jose, CA).
Leukemia Cell-Killing Ability of Internalized TAT-ASNase
Cellular uptake of the TAT-ASNase conjugates was confirmed by direct cytotoxicity studies. Human leukemia MOLT-4 cells was seeded at a density of 1 × 106 cells/well and incubated with varying concentrations of either free ASNase or TAT-ASNase conjugates for 2 hours and then washed 3 times with 50% fetal bovine serum (FBS). The treated cells were re-suspended in the original culture medium and further incubated at 37°C, 5% CO2 for 48 hours. Cell viability in the ASNase- or TAT-ASNase-treated samples was determined by the XTT cytotoxicity assay (Sigma, USA) [28,29].
Encapsulation of Asparaginase into L5178Y Cells
L5178Y, a murine T-lymphoma cell line derived from methylcholanthrene-induced lymphoma in DBA/2 mice, was purchased from ATCC (Manassas, VA). The cells were maintained and propagated in vitro using RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) and antibiotics. L5178Y cells were suspended to a concentration of 106 cells/mL. The cells were then incubated for 2 hrs with the TAT-ASNase conjugates in a 37°C, 5% CO2 atmosphere incubator at various doses ranging from 0.025 to 10 IU of equivalent ASNase activity per mL. Following three washings with RPMI-1640 containing 50% FBS, the cells were re-suspended to a concentration of 106 cells/200 µL.
Animal Preparations
Five-week-old female DBA/2 mice were purchased from Charles River Laboratories (Raleigh, NC). The mice were housed in animal facilities and were fed standard mouse chow and provided water ad libitum. They were 6 to 7 weeks old at the beginning of each experiment and the average mouse weight ranged from 14 to 19 grams. Animal experiments were conducted according to the protocol approved by the University of Michigan Committee on Use and Care of Animals (UCUCA) that conforms to the standards in the “Guide for the Care and Use of Laboratory Animals”, DHEW Pub. No (NIH) 80-23, revised 1985.
Tumor implantation and survival
Six-week-old DBA/2 mice were injected intraperitoneally with L5178Y cells (0.7×106 cells/mouse) pre-treated with either PBS solution (i.e. positive control), free ASNase (another control, see reasons below), or TAT-ASNase (i.e. the experimental group). Six mice were used in each testing group and 3 additional mice were used as the negative control (where the mice were injected with the RPMI-1640 solution only, without tumor cells). Animals were weighed to monitor weight change and the survival times were recorded.
RESULTS AND DISCUSSION
Synthesis, Purification, and Characterization of the TAT-ASNase Conjugates
Asparaginase (ASNase) was successfully conjugated to TAT via a disulfide linkage using the SPDP coupling method [5]. The TAT-ASNase conjugates were then purified using a heparin affinity HPLC system. Native asparaginase did not show any heparin binding ability, and rapidly eluted (retention time: 2.3min) from the heparin column at a low ionic strength of 0.05M NaCl (Figure 2). In contrast, following conjugation with TAT, a second peak possessing a much stronger heparin affinity (eluted at 1.22–1.68 M NaCl) with a significantly longer retention time (24–32 min) was observed in addition to the normal ASNase peak. This second major peak corresponded to the elution the TAT-ASNase conjugates, as it not only exhibited a strong ASNase activity but was also eluted at an ionic strength that was similar to free TAT (1.3M NaCl; data was not shown). This TAT-ASNase peak was relatively broad and asymmetric, indicating the presence of a heterogeneous collection of TAT-ASNase conjugates.
Figure 2.
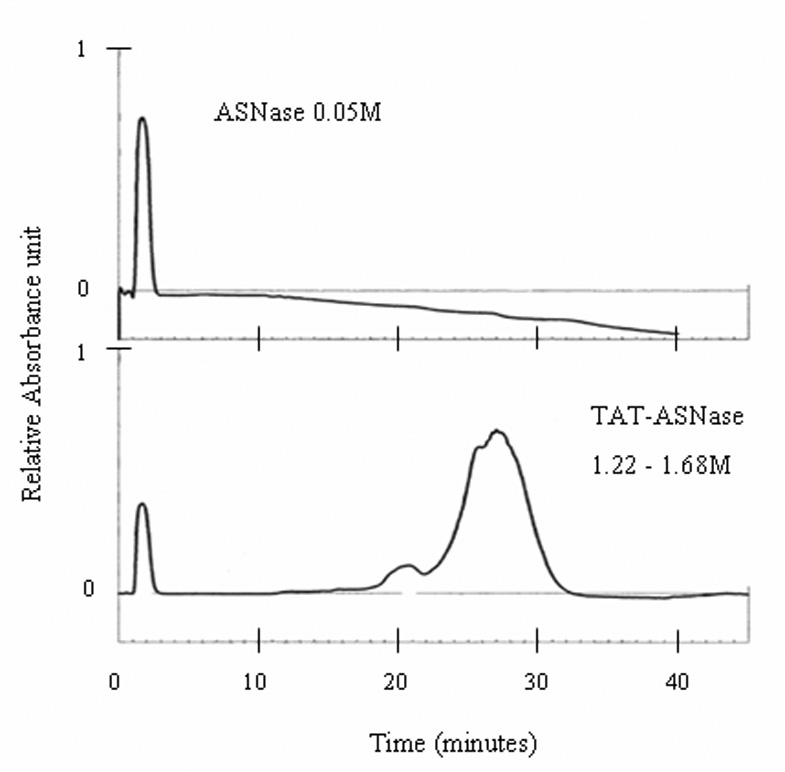
Elution of ASNase and TAT-ASNase from a heparin column. A gradient of 0.15 – 2.00 M NaCl solution was used to separate ASNase and TAT-ASNase of various heparin binding affinity.
Successful synthesis of the TAT-ASNase conjugates was further confirmed by SDS-PAGE results. Whereas native ASNase displayed a major band on the SDS gel with a molecular weight of approximately 36 kDa, the TAT-ASNase conjugates showed the presence of several bands with molecular weight slightly higher than that of native ASNase (Figure 3). Assessment based on these higher molecular weight bands suggested a heterogeneous molar ratio between TAT and ASNase in the conjugates, ranging from 1 to 5 TAT peptides per single ASNase molecule, supported by MALDI spectrum of the conjugate (data not shown).
Figure 3.
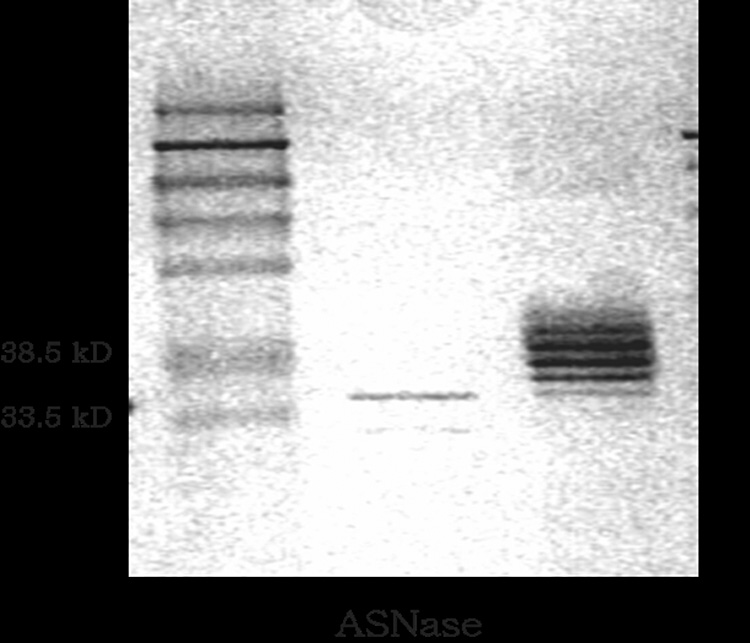
SDS-PAGE of ASNase and TAT-ASNase. ASNase migrated at its expected MW of 36KD whereas TAT-ASNase migrated at higher MW; suggesting approximately 1 to 5 TAT molecules were conjugated to each ASNase.
The concentration and enzymatic activity of the TAT-ASNase mixtures were determined by using UV spectrophotometry and Nesslerization assay, respectively. The specific activity of the TAT-ASNase was 124±20 IU/mg, compared to the original value of 228±15 IU/mg obtained for native ASNase. Based on these results, a reasonable retention of the original ASNase activity (approximately 60%) was obtained following SPDP conjugation of TAT to ASNase.
The leukemia cell-killing potency of both ASNase and TAT-ASNase was examined in vitro. Cell suspensions of 1 × 106 MOLT-4 cells/mL were incubated with various concentrations of ASNase and TAT-ASNase with enzymatic activities ranging from 10−4 IU/mL to 10 IU/mL for 48 hours, and the inhibitory effects on cell growth were measured using the XTT assay. Interestingly, both native ASNase and the TAT-ASNase conjugates displayed nearly identical IC50 values (0.0102 and 0.0100 IU/mL for ASNase and TAT-ASNase, respectively) when equal doses of ASNase activity were used (data were not shown). The results suggested that conjugation of TAT to ASNase did not impart any negative effect on the cytotoxic functions of the native enzyme.
Heparin and Protamine Induced Regulation on Cell Uptake of the TAT-ASNase Conjugates
To examine TAT-dependent cell translocation, the cellular uptake event of the TAT-ASNase conjugates was confirmed by using confocal technique with the adherent HeLa cell lines first, followed by the cytometry technique with the MOLT-4 tumor cell lines. HeLa cells were treated with FITC-labeled ASNase and TAT-ASNase. Whereas the FITC-labeled ASNase sample displayed virtually no fluorescence inside the cells (Figure 4A), there was a significant intracellular uptake of the FITC-labeled TAT-ASNase conjugates (Figure 4B). From these results, it appeared that ASNase conjugates possessed translocation abilities due to chemical conjugation with the PTD domains. Being two different cell types, TAT on the conjugate was able to ferry TAT-ASNase conjugates across either cell lines with comparable efficiency. These results were in agreement with findings by other investigators that TAT was capable of translocating the attached protein species across all the cell membrane of various cell types [9,14].
Figure 4.
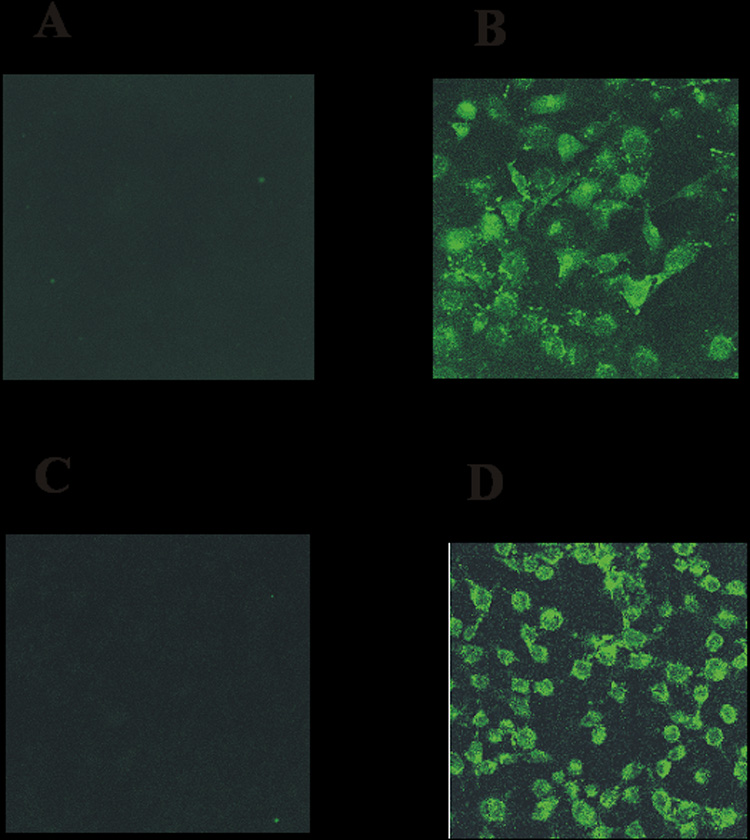
Fluorescence microscopy of: (A) FITC-ASNase; (B) FITC-TAT-ASNase; (C) FITC-TAT-ASNase with heparin; and (D) FITC-TAT-ASNase with heparin and protamine. HeLa cells were incubated with the different treatment groups.
To examine if heparin could inhibit TAT-mediated intracellular delivery of ASNase and if protamine could trigger the reversal of this inhibition, the TAT-ASNase conjugates were reacted with heparin prior to their incubation with the HeLa cells. After the addition of heparin to the chemical conjugates, no fluorescence was visible through the microscope, indicating that heparin was indeed able to inhibit the TAT-ASNase conjugates from entering the cells. These findings were in agreement with previous observations made by Mann and Frankel [6], which reported that cellular uptake of TAT was abolished in the presence of heparan or dextran sulfate. To reverse the heparin-based inhibition of TAT-mediated cell internalization of ASNase, protamine, a clinical antidote to heparin, was then added. Upon treatment with protamine, heparin-induced inhibition was completely abolished and a strong intensity of FITC fluorescence from the labeled TAT-ASNase conjugates was observed (Figure 4D).
The heparin/protamine-based control of cellular internalization by the TAT-ASNase conjugates was further validated by testing samples against the human cancer cell line MOLT-4. Significant fluorescence intensity was observed for the TAT-ASNase-treated MOLT-4 cells whereas cells treated with FITC-labeled ASNase displayed the same fluorescence intensity as untreated control cells (Figure 5A). Based on these results, it was clear that TAT conferred cell penetration ability onto ASNase and, therefore, carried the attached ASNase into the cells. To further examine whether heparin could inhibit the TAT-mediated cell translocation of ASNase and if protamine could reverse this inhibition, heparin was added to the TAT-ASNase conjugates prior to their incubation with the MOLT-4 cells. In separate experiments, protamine was subsequently added to the heparin-inhibited TAT-ASNase conjugates prior to incubation with the MOLT-4 cells. Upon the addition of heparin, the fluorescence intensity of MOLT-4 cells was reduced to a level that was equivalent to the untreated control cells (Figure 5B). This finding indicated that heparin-masked FITC-TAT-ASNase was no longer capable of internalizing cells, confirming the previous finding that heparin could effectively inhibit the TAT-mediated cell translocation of ASNase. In contrast, upon the addition of protamine to the heparin-inhibited TAT-ASNase, the observed fluorescence intensity was recovered and was comparable to that of the TAT-ASNase incubated MOLT-4 cells. This data also confirmed the previous finding that protamine was able to successfully reverse the heparin-induced inhibition of TAT-mediated cell internalization.
Figure 5.
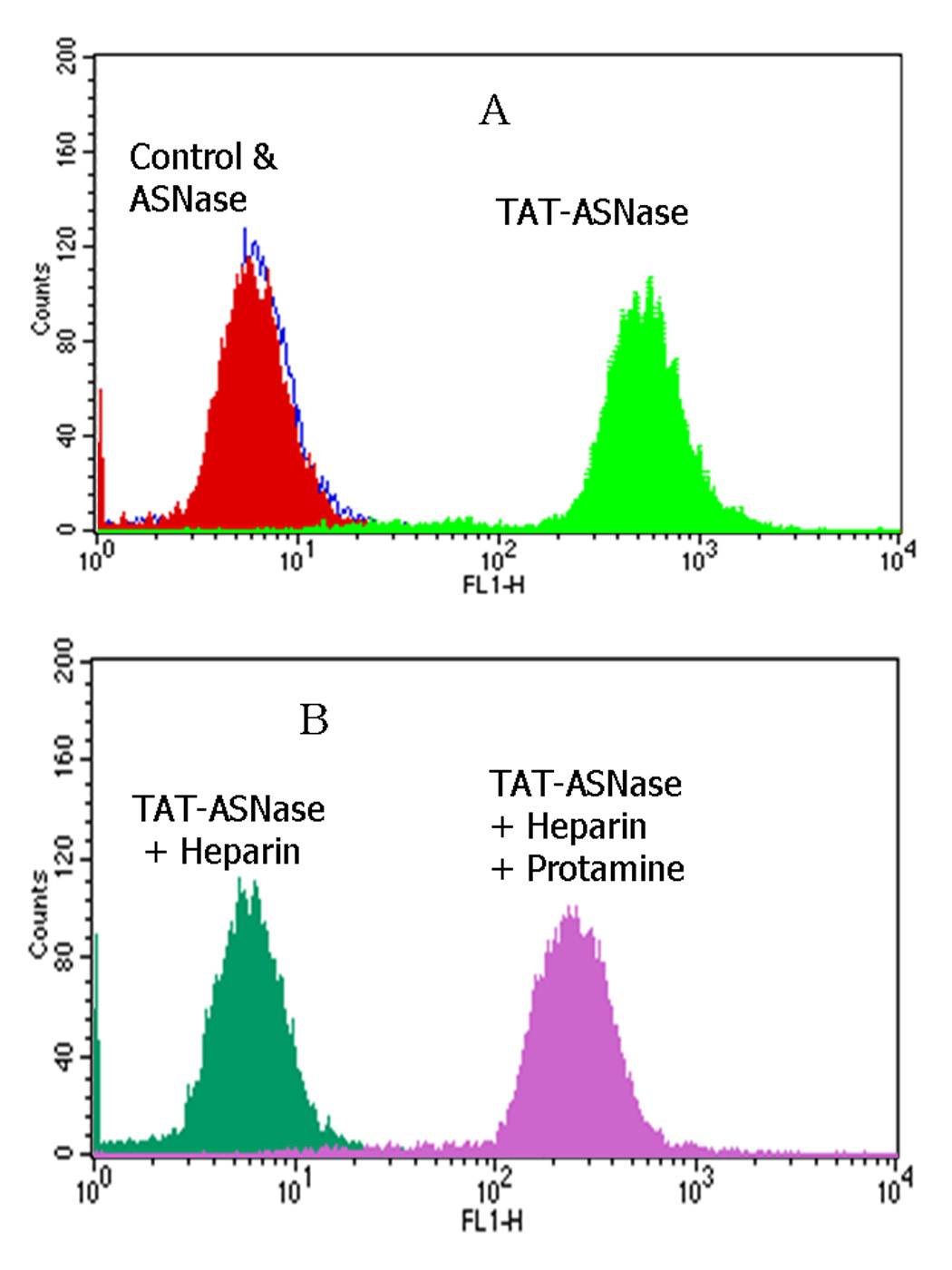
Flow cytometry analysis of FITC-TAT-ASNase conjugate. (A) ASNase and TAT-ASNase were FITC-labeled and incubated with MOLT-4 cells. (B) FITC-TAT-ASNase was incubated with heparin or heparin and protamine in MOLT-4 cells.
Leukemia Cell Killing Ability of Cell Encapsulated TAT-ASNase
To determine if TAT-ASNase could exert intracellular cytotoxic effects after cellular translocation, MOLT-4 cells were incubated with TAT-ASNase for 2 hours; a time period that has been demonstrated to allow for a complete membrane translocation of TAT-ASNase into various cell types. As a control, MOLT-4 cells were also incubated with free ASNase for the identical time period of 2 hours (Figure 4 & 5). The ASNase- or TAT-ASNase-treated cells were then thoroughly washed and further incubated in fresh culture medium for another 2 hours, followed by measurements of cell viability. As shown in the left-hand columns in Figure 6, for MOLT-4 cells incubated with either free ASNase or TAT-ASNase, cell viability of the two groups was nearly identical. This finding was due to the fact that both extracellular (free) and intracellular (internalized) ASNase could elicit cytotoxic effects on tumor cells by depleting the asparagine substrate in the culture medium that is essential for leukemic cell growth. To confirm that the internalized TAT-ASNase conjugates could induce cytotoxic effects, the treated cells were incubated with 50% FBS to neutralize the extracellular ASNase activity, prior to the 2 hr incubation for the cell viability studies. After treatment with 50% FBS, a significantly reduced viability was observed for cells treated with TAT-ASNase, compared to those treated with free ASNase. These findings indicated that TAT-ASNase was able to translocate into the cells and thereby avoid inactivation by washing with FBS, whereas the free ASNase activity employed in the control experiments which remained present in the culture medium was completely removed by the FBS washing step (Figure 6). It was hypothesized that due to the presence of a high level of reducing agents such as glutathione in the cytosol, the disulfide linkages between TAT and ASNase of the TAT-ASNase conjugates would be dissociated, therefore leaving the intracellularly delivered, cell-impermeable ASNase securely entrapped within the cells. Accordingly, the continued cytotoxic effect experienced by MOLT-4 cells was the result of asparagine depletion induced by the cell-entrapped ASNase.
Figure 6.
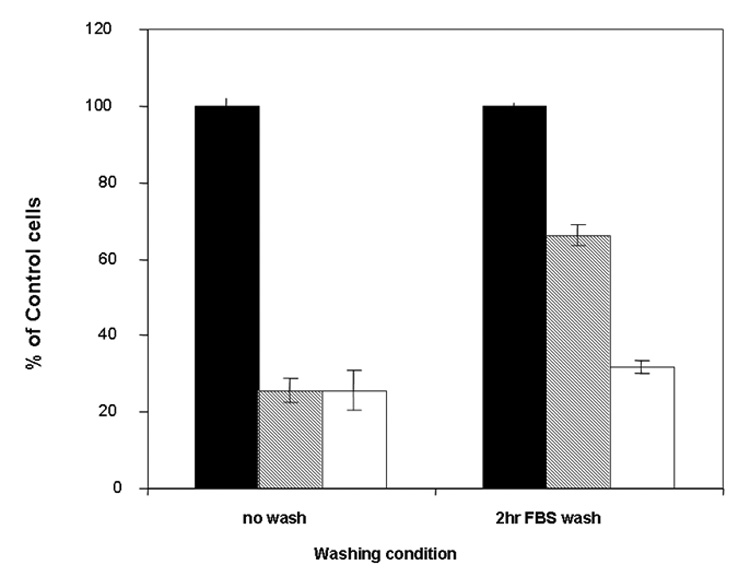
Effect of washing on cell viability. Cell viability was determined with or without washing with 50% FBS after 2 hours incubation with either control medium (black), ASNase (gray) or TAT-ASNase (no color) at 2 IU per 106 MOLT-4 cells. After washing, TAT-ASNase exhibited greater cytotoxicity than ASNase, indicating TAT-ASNase had transduced into MOLT-4 cells and was capable of maintaining its tumor killing ability.
Survival of DBA/2 Mice after L5178Y Treated with TAT-ASNase
The underlying principle of utilizing the modified ATTEMPTS approach in intracellular ASNase delivery was that elimination of nutrient asparagine (followed by apoptosis) would presumably occur only within the tumor cells, thereby aborting the toxic side effects on the high protein production organs (e.g. liver, pancreas) resulting from systemic depletion of asparagine. To provide a quick proof-of-concept demonstration of the feasibility of this drug delivery system, while no suitable murine antibody was available at the time of experiments, we chose to take an alternative approach in mimicking the tumor uptake of ASNase, by direct injection of the ASNase-encapsulated tumors cells into the testing animals. The L5178Y tumor cell line was selected for ASNase encapsulation, because it had been well documented in the literature to be highly sensitive to ASNase therapy [30] and also it is commercially available. The DBA/2 mouse was specifically chosen to be the host for the implanted tumor cells because L5178Y cells had been demonstrated to be tumorigenic in this mouse strain [31]. In addition, previous investigations had used this cell line and mouse combination to examine the success of various asparaginase therapy, therefore, a history of success using this combination has already been demonstrated and documented [32]. Moreover, another advantage of using DBA/2 mouse strain was that it did not require any special food or housing conditions associated with immune deficient mice such as NOD/SCID.
Six days after the tumor implantation, abdominal distension was visible and tumor-injected mice displayed significant increase in weight relative to negative control mice. By day 8, the differences in mouse ascites size between the control and the TAT-ASNase-treated group was clearly visible. Figure 7 illustrates the survival rates of these treated animals. As seen, the mean survival time for the positive control and TAT-ASNase-treated group were 13.9 and 15.6 days, respectively. ANOVA analysis was performed on all test groups using SPSS, and the results showed that there was a statistically significant difference in the mean survival rate between the treatment groups at p = 0.05 (F2,12 = 6.478, p = 0.012). By performing a multiple pair-wise comparison using the Tukey and Dunnett test, it was found that a statistically significant difference in the mean survival rate existed between the positive control and the TAT-ASNase-treated group, with Tukey’s and Dunnett’s statistics of p = 0.009 and p = 0.007, respectively.
Figure 7.
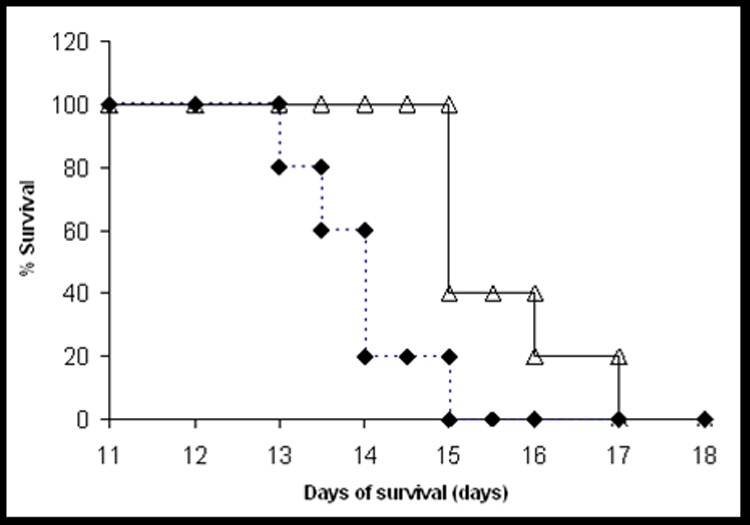
Survival curve for DBA/2 mouse bearing L5178Y mouse lymphoma cells. Each pool of 700,000 L5178Y cells was incubated with 6 IU of either TAT-ASNase (open triangle) or RPMI-1640 solution (positive control; diamond) for 2 hours, followed by washing 3 times with 50% FBS in RPMI. The DBA/2 mice of 6 weeks old were then injected intraperitoneally with one of these three treated tumor cell samples (700,000 cells per sample). Mouse survival times were recorded.
CONCLUSIONS
Macromolecular drugs such as proteins and nucleic acids are presumably the most desirable anti-cancer agents due to their unparalleled substrate specificity and reaction efficiency. Yet, clinical use of macromolecular drugs has yielded limited success, primarily due to the inability of these compounds to cross the cell membrane and exhibit their therapeutic activities. In this regard, the recent discovery of the cell-penetrating PTD peptides such as TAT has garnered significant attention with the hope that, by integrating this unique PTD-mediated cell transduction function into a delivery system, effective intracellular delivery of these macromolecular drugs could finally be achieved. However, none of the delivery systems under development to date have been able to accomplish this goal, simply because, without a tightly regulated mechanism, PTD-mediated cell internalization remains excessively potent and non-selective for cell transduction into all types of tissues, which could thereby yield massive and untoward toxic side effects. Therefore, the most outstanding novelty of our modified-ATTEMPTS approach lies in its ability to safely incorporate the supreme potency and universal applicability of the PTD-mediated cell uptake mechanism into a single system to achieve effective yet less toxic protein, gene or virtually any type of drug therapies. The approach utilizes heparin-induced inhibition on PTD to acquire the prodrug features for the PTD-mediated delivery system, followed by applying the clinically approved protamine-induced reversal on heparin inhibition to reactivate the trans-membrane activity of the PTD in achieving a selective and potent intracellular delivery of the attached drug. Using asparaginase (ASNase) as a model enzyme drug, the present results firmly demonstrate the in vitro feasibility of the proposed approach of this on/off system. Fluorescence microscopy, flow cytometry, and cell viability studies all confirmed that while ASNase by itself could not translocate into leukemic cells, the TAT-ASNase conjugates were nevertheless capable of entering the cells to exert strong cytotoxic effects. More importantly, heparin was able to completely inhibit the TAT-mediated intracellular delivery of ASNase, whereas protamine was capable of reversing the heparin-induced inhibition to allow for the recovery of TAT-mediated cell uptake of ASNase. From the preliminary in vivo survival study, TAT-ASNase treated group has shown improved survival with a statistically significant difference. To further confirm the in vivo feasibility of this modified-ATTEMPTS approach on asparaginase delivery, extensive animal studies are currently underway in our laboratory.
It should be pointed out that although ASNase has been selected as the model enzyme drug, it actually is an ideal example to reflect the benefits of the modified-ATTEMTS delivery system. As known, ASNase-associated toxicity comes mainly from systemic depletion of asparagine (ASN). This is simply because that although normal cells can survive without ASN, most organs, particularly for organs like liver that requires a tremendous amount of ASN for its high rate of protein syntheses, will suffer by this systemic ASN depletion, because they will need exogenous ASN to maintain a sufficient level of this nutrient for their normal protein biosynthesis. This is the reason why liver toxicity is one of the major concerns for ASNase therapy. Making it worse is that the short half-life of ASNase (i.e. 8–30 hrs) demands frequent injections of high doses of ASNase for therapy, further magnifying ASNase-induced toxicity and allergic responses, particularly because ASNase is obtained from bacterial resources. Therefore, a primary significance of our approach in ASNase delivery lies in its capability of allowing only tumor cells to carry their self-destructing agents (i.e. the delivered, cell-internalized ASNase) and be deprived of ASN (because any ASN substrate diffused into tumor cells would be degraded) while maintaining a nearly normal ASN concentration in the circulation. In other words, unlike current ASNase therapy that induces systemic depletion of ASN, the proposed ASNase delivery system would not affect ASN concentration in the circulation, simply because all of the administered ASNase drug will presumably be retained inside the tumor cells and not present in the circulation. Therefore, this modified-ATTEMPTS approach will not only will alleviate the toxic effects resulted from systemic depletion of ASN but also, in principle, allow for a single injection of ASNase (since tumor entrapped ASNase will be protected from proteolytic degradation and metabolic clearance), thereby drastically reducing the dose for ASNase therapy as well as its triggered immunological responses.
Lastly, another noteworthy point regarding the presented approach is that since intracellular drug uptake mediated by PTD peptides has been shown to proceed in a receptor-/transporter-independent fashion that directly targets the lipid bilayers (10–11). Hence, in principle and practice, nearly all cell types are transducible. Therefore, the approach possess not only a universal utility in delivering drugs of all types, but also a universal applicability to treating cancers of all types as long as specific targeting moieties are present. The generic nature and flexibility in applying the proposed delivery system could potentially lead to the development of an arena of new drugs that were initially considered improbable for therapeutic use due to either poor cell uptake or acute toxic side effects. Understandably, such problems have forced biotechnology and pharmaceutical companies to abandon or discard a large number of drugs that show promising therapeutic effects in vitro but limited functions or severe toxicities in vivo.
ACKNOWLEDGEMENT
This study was partially supported by NIH R01 Grants HL55461 and CA114612. Dr. Victor C. Yang is currently a Cheung Kong Scholar, appointed by the Chinese Ministry of Education, at the School of Chemical Engineering, Tianjin University, Tianjin, China. The authors would like to thank Dr. Hongfang Wang at the University of Utah for his technical advice and support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Naik SS, Liang J-F, Park YJ, Lee WK, Yang VC. Application of “ATTEMPTS” for drug delivery. J Control Release. 2005;101:35–45. doi: 10.1016/j.jconrel.2004.07.020. [DOI] [PubMed] [Google Scholar]
- 2.Stuart FD, Ponzoni M, Allen TM. Targeted delivery of antisense oligonucleotides in cancer. J Control Release. 2001;74:69–75. doi: 10.1016/s0168-3659(01)00312-1. [DOI] [PubMed] [Google Scholar]
- 3.Jain RK. Delivery of molecular medicine to solid tumors: lessons from in vivo imageing of gene expression and function. J Control Release. 2001;74:7–25. doi: 10.1016/s0168-3659(01)00306-6. [DOI] [PubMed] [Google Scholar]
- 4.Bagshawe KD. Antibody-directed enzyme prodrug therapy (Adept) J Control Release. 1994;28:187–193. [Google Scholar]
- 5.Liang JF, Li YT, Connell ME, Yang VC. Synthesis and characterization of positively charged tPA as a prodrug using heparin/protamine-based drug delivery system. AAPS PharmSci. 2000;2:1–9. doi: 10.1208/ps020107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Green M, Loewenstein PM. Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell. 1988;55:1179–1188. doi: 10.1016/0092-8674(88)90262-0. [DOI] [PubMed] [Google Scholar]
- 7.Frankel AD, Pabo CO. Cellular uptake of the tat protein from human immunodeficiency virus. Cell. 1988;55:1189–1193. doi: 10.1016/0092-8674(88)90263-2. [DOI] [PubMed] [Google Scholar]
- 8.Mann DA, Frankel AD. Endocytosis and targeting of exogenous HIV-1 Tat protein. Embo J. 1991;10:1733–1739. doi: 10.1002/j.1460-2075.1991.tb07697.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Phelan A, Elliott G, O'Hare P. Intercellular delivery of functional p53 by the herpesvirus protein VP22. Nat Biotechnol. 1998;16:440–443. doi: 10.1038/nbt0598-440. [DOI] [PubMed] [Google Scholar]
- 10.Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science. 1999;285:1569–1572. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- 11.Derossi D, Calvet S, Trembleau A, Brunissen A, Chassaing G, Prochiantz A. Cell internalization of the third helix of the Antennapedia homeodomain is receptor-independent. J Biol Chem. 1996;271:18188–18193. doi: 10.1074/jbc.271.30.18188. [DOI] [PubMed] [Google Scholar]
- 12.Ezhevsky SA, Nagahara H, Vocero-Akbani AM, Gius DR, Wei MC, Dowdy SF. Hypo-phosphorylation of the retinoblastoma protein (pRb) by cyclin D:Cdk4/6 complexes results in active pRb. Proc Natl. Acad. Sci. USA. 1997;94:10699–10704. doi: 10.1073/pnas.94.20.10699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Snyder EL, Meade BR, Dowdy SF. Anti-cancer protein transduction strategies: reconstitution of p27 tumor suppressor function. J Control Release. 2003;91:45–51. doi: 10.1016/s0168-3659(03)00212-8. [DOI] [PubMed] [Google Scholar]
- 14.Nori A, Jensen KD, Tijerina M, Kopečková P, Kopeček J. Subcellular trafficking of HPMA copolymer-Tat conjugates in human ovarian carcinoma cells. J Control Release. 2003;91:53–59. doi: 10.1016/s0168-3659(03)00213-x. [DOI] [PubMed] [Google Scholar]
- 15.Dinauer N, Lochmann D, Demirhan I, Bouazzaoui A, Zimmer A, Chandra A, Kreuter J, von Briesen H. Intracellular tracking of protamine/antisense oligonucleotide nanoparticles and their inhibitory effect on HIV-1 transactivation. J Control Release. 2004;96:497–507. doi: 10.1016/j.jconrel.2004.02.020. [DOI] [PubMed] [Google Scholar]
- 16.Mashburn LT, Wriston JC., Jr Tumor inhibitory effect of L-asparaginase from Escherichia coli. Arch Biochem Biophys. 1964;105:450–452. doi: 10.1016/0003-9861(64)90032-3. [DOI] [PubMed] [Google Scholar]
- 17.Gugliotta L, D'Angelo A, Mattioli-Belmonte M, Vigano-D'Angelo S, Colombo G, Catani L, Gianni L, Lauria F, Tura S. Hypercoagulability during L-asparaginase treatment: the effect of antithrombin III supplementation in vivo. Br J Haematol. 1990;74:465–470. doi: 10.1111/j.1365-2141.1990.tb06336.x. [DOI] [PubMed] [Google Scholar]
- 18.Gugliotta L, Mazzucconi MG, Leone G, Mattioli-Belmonte M, Defazio D, Annino L, Tura S, Mandelli F The GIMEMA Group. Incidence of thrombotic complications in adult patients with acute lymphoblastic leukaemia receiving L-asparaginase during induction therapy: a retrospective study. Eur J Haematol. 1992;49:63–66. doi: 10.1111/j.1600-0609.1992.tb00032.x. [DOI] [PubMed] [Google Scholar]
- 19.Homans AC, Rybak ME, Baglini RL, Tiarks C, Steiner ME, Forman EN. Effect of L-asparaginase administration on coagulation and platelet function in children with leukemia. J Clil Oncol. 1987;5:811–817. doi: 10.1200/JCO.1987.5.5.811. [DOI] [PubMed] [Google Scholar]
- 20.Barbui T, Finazzi G, Vigano S, Mannucci PM. L-asparaginase lowers protein C antigen. Thromb Haemost. 1984;52:216. [PubMed] [Google Scholar]
- 21.Priest JR, Ramsay NK, Bennett AJ, Krivit W, Edson JR. The effect of L-asparaginase on antithrombin, plasminogen, and plasma coagulation during therapy for acute lymphoblastic leukemia. J Pediatr. 1982;100:990–995. doi: 10.1016/s0022-3476(82)80536-2. [DOI] [PubMed] [Google Scholar]
- 22.Peterson RG, Handschumacher RE, Mitchell MS. Immunological responses to L-asparaginase. J Clin Invest. 1971;50:1080–1090. doi: 10.1172/JCI106579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dellinger CT, Miale TD. Comparison of anaphylactic reactions to asparaginase derived from Escherichia coli and from Erwinia cultures. Cancer. 1976;38:1843–1846. doi: 10.1002/1097-0142(197610)38:4<1843::aid-cncr2820380463>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 24.Fabry U, Korholz D, Jurgens H, Gobel U, Wahn V. Anaphylaxis to L-asparaginase during treatment for acute lymphoblastic leukemia in children--evidence of a complementmediated mechanism. Pediatr Res. 1985;19:400–408. doi: 10.1203/00006450-198519040-00017. [DOI] [PubMed] [Google Scholar]
- 25.Gallagher MP, Marshall RD, Wilson R. Asparaginase as a drug for treatment of acute lymphoblastic leukaemia. Essays Biochem. 1989;24:1–40. [PubMed] [Google Scholar]
- 26.Maeda H, Ishida N, Kawauchi H, Tsujimura K. Reaction of fluorescein-isothiocyanate with proteins and amino acids. I. Covalent and non-covalent binding of fluoresceinisothiocyanate and fluorescein to proteins. J Biochem (Tokyo) 1969;65:777–783. doi: 10.1093/oxfordjournals.jbchem.a129077. [DOI] [PubMed] [Google Scholar]
- 27.Shifrin S, Parrott CL, Luborsky SW. Substrate binding and intersubunit interactions in L-asparaginase. J Biol Chem. 1974;249:1335–1340. [PubMed] [Google Scholar]
- 28.Scudiero DA, Shoemaker RH, Paull KD, Monks A, Tierney S, Nofziger TH, Currens MJ, Seniff D, Boyd MR. Evaluation of a soluble tetrazolium/formazan assay for cell growth and drug sensitivity in culture using human and other tumor cell lines. Cancer Res. 1988;48:4827–4833. [PubMed] [Google Scholar]
- 29.Roehm NW, Rodgers GH, Hatfield SM, Glasebrook AL. An improved colorimetric assay for cell proliferation and viability utilizing the tetrazolium salt XTT. J Immunol Methods. 1991;142:257–265. doi: 10.1016/0022-1759(91)90114-u. [DOI] [PubMed] [Google Scholar]
- 30.Horowitz B, Madras BK, Meister A, Old LJ, Boyes EA, Stockert E. Asparagine synthetase activity of mouse leukemias. Science. 1968;160:533–535. doi: 10.1126/science.160.3827.533. [DOI] [PubMed] [Google Scholar]
- 31.Zhang ZY, Chow DA. Differential CD45 isoform expression accompanies reduced natural antibody binding in L5178Y-F9 tumor progression. J Immunol. 1997;159:344–350. [PubMed] [Google Scholar]
- 32.Veronese FM, Monfardini C, Caliceti P, Schiavon O, Scrawen MD, Beer D. Improvement of pharmacokinetic, immunological and stability properties of asparaginase by conjugation to linear and branched monomethoxy poly (ethylene glycol) J Control Release. 1996;40:199–209. [Google Scholar]