

Abstract
North Carolina macular dystrophy (NCMD) is an autosomal dominant macular disease, was mapped to 6q14-q16.2, the disease-causing gene has yet not been identified. It shares phenotypic similarity with age-related macular degeneration including drusen and choroidal neovascularization. We collected six families with NCMD including 75 members, and conducted clinical characterization and genetic mapping for these families. Forty five patients were diagnosed as NCMD; all six NCMD families were mapped to MCDR1 locus using genetic linkage analysis. MCDR1 interval was refined to 3 cM (1.8mb) between D6S1716 to D6S1671 via fine mapping using microsatellite markers in these six families, all eleven annotated genes within the interval were analyzed by mutation screening in coding regions, no mutation was found, suggesting a potential novel gene or a new pathological mechanism causing NCMD. The refinement of MCDR1 locus will aid the disease-causing gene identification. Functional studies of NCMD genes should provide important insights into pathogenetic mechanisms of NCMD and age-related macular degeneration.
Keywords: NCMD, NCDR1, fine mapping, interval
1. Introduction
The inherited macular dystrophies are characterized by bilateral visual loss and the finding of generally symmetrical macular abnormalities, they can present as genetic heterogeneity including autosomal dominant, autosomal recessive, X linked recessive and mitochondrial inheritance. NCMD is an autosomal dominant, highly penetrant disease with congenital or infantile onset. NCMD shares many important clinical and histopathological similarities with AMD including an abnormal accumulation of drusen, atrophy of the RPE and overlying photoreceptor cells, choroidal neovascularizatioon and loss of central vision (Small, Hermsen, Gurney, Fetkenhour & Folk, 1992a). Lefler et al. (Lefler, Wadsworth & Sidbury, 1971) reported a large family with multiple generations from North Carolina that was affected with a form of drusen and macular degeneration, therefore the term North Carolina macular dystrophy. North Carolina macular dystrophy has been described under different names such as central areolar pigment epithelial dystrophy (CAPED), central pigment epithelial and choroidal degeneration, and central retinal pigment epithelial dystrophy in several branches of a large family (Frank, Landers, Williams & Sidbury, 1974; Small et al., 1992a). The disease in this family was apparently inherited from three founding Irish brothers who lived in the late eighteenth and early nineteenth centuries (Frank et al., 1974; Small et al., 1992a). It is generally non-progressive and shows a wide range of intrafamilial and interfamilial variations in clinical phenotypes. These clinical manifestations can be divided into three grades. Grade I represents drusen in the central macula. Grade II denotes subretinal neovascularization or scar. Grade III exhibits well-demarcated choreoretinal atrophy with hyperpigmentation on the edge of the lesion. A large family with North Carolina macular dystrophy inherited as an autosomal dominant, fully penetrant trait was studied. Linkage analysis of this kindred, now known to include more than 2000 individuals, localized the disease -causing gene to chromosome 6q16 between D6S275 and D6S475 in a 4.5 cM interval. This locus has been designated by Online Mendelian Inheritance of Man (OMIM) as MCDR1, for macular degeneration locus 1 (OMIM #136550) (Small et al., 1999; Small, Weber, Roses & Pericak-Vance, 1993; Small, Weber, Roses, Lennon, Vance & Pericak-Vance, 1992b). Several European and South American families with a NCMD phenotype were also localized to the MCDR1 locus, supporting its role as a significant cause of childhood macular degeneration (Rabb, Mullen, Yelchits, Udar & Small, 1998; Reichel et al., 1998; Sauer et al., 1997; Small, Garcia, Gallardo, Udar & Yelchits, 1998; Small, Puech, Mullen & Yelchits, 1997). We performed clinic study and linkage mapping in six families with NCMD and refined MCDR1 to 1.8 million base pairs.
2. Methods
2.1. Study subjects
This project was approved by the Institutional Board of the Institute of Ophthalmology and the Institutional Review Board of the University of Utah Health Science Center, USA. Informed consent was obtained from all participants. Seventy five members from six families were included; all participatants underwent ophthalmic examination including best corrected Snellen visual acuity determination and fundus examination. Fluorescein angiography was carried out on some patients. Affected patients were designated on the basis of decrease visual acuity and the macular lesions.
2.2. Establishment of cell lines and DNA and RNA isolation
Blood was collected by venepuncture, Lymphoblastoid cell lines were established by Epstein-Bar virus transformation of peripheral mononuclear cells as previously described (Chou et al., 1992). Haploid somatic hybrids cell lines were established using a GMP Conversion Technology (Yan et al., 2000b). Genomic DNA and RNA was isolated from the blood samples, EBV-transformed lymphoblastoid cells and haploid somatic hybrids using a Puregene genomic DNA purification kit or VERSAGENE™ RNA Kit (Gentra Systems, Minneapolis, Massachusetts, USA). cDNA was made using SuperScript™ III First-Strand Synthsis Sytem for RT-PCR (Invitrogen Corporation, Carlsbad, CA).
2.3. Genotyping and linkage analysis
In order to obtain a high density coverage of STR markers in the Minimum Genetic Interval (MGI), we first determined the precise physical location of each known STR marker listed on the Marshfield genetic maps (http://research.marshfieldclinic.org/genetics/GeneticResearch/compMaps.asp) by using public NCBI (http://www.ncbi.nlm.nih.gov/) and UCSC (http://genome.ucsc.edu/) databases (Table 1). To generate additional STR markers, we searched genomic DNA sequences from public NCBI and UCSC databases and identified 30 potential candidates. These potential STR markers included CA, ATA, GATA, GAAG repeats. We then tested the degree of polymorphisms of these STR markers by genotyping them on six North Carolina macular dystrophy families and 100 unrelated Caucasian individuals.
Table 1.
Fine Genetic map and list of polymorphic DNA markers
| STR Marker | Repeats | Location(mb) | Recombination | BAC ID | |||
|---|---|---|---|---|---|---|---|
| Family 663 | Family 348 | Family 324 | Family 71 | ||||
| D6S249 | AG/CT | 98.22 | U | X | U | U | AL596208 |
| D6S1284 | AAGG/CCTT | 98.82 | U | X | U | U | AL589740 |
| D6S1716 | CA/TG | 98.85 | U | X | U | U | AL589740 |
| S3-34301 | CA/TG | 99.01 | U | U | U | U | AL607071 |
| S3-201660 | AAGG/CCTT | 99.18 | U | U | U | U | AL590395 |
| S3-188835 | CA/TG | 99.27 | U | U | U | U | AL590395 |
| S3-263101 | CA/TG | 99.34 | U | U | U | U | AL590395 |
| S3-328991 | CA/TG | 99.41 | U | U | U | U | AL589826 |
| S4-674 | CA/TG | 99.48 | U | U | U | U | AL022395 |
| S4-40977 | CA/TG | 99.52 | U | U | U | U | AL078603 |
| S4126261 | CA/TG | 99.61 | U | U | U | U | AL078603 |
| S4-198781 | TA/TA | 99.68 | U | U | U | U | AL59022 |
| D6S1717 | CA/TG | 99.78 | U | U | U | U | AL591803 |
| S4-364451 | CA/TG | 99.83 | U | U | U | U | AL034371 |
| D6S1565 | CA/TG | 99.84 | U | U | U | U | AL034371 |
| S4-398191 | CA/TG | 99.87 | U | U | U | U | AL034371 |
| S4-444001 | CA/TG | 99.91 | U | U | U | U | AL513550 |
| S5-162761 | GAAA/TTTC | 100.13 | U | U | U | U | AL137784 |
| S5-277706 | CA/TG | 100.25 | U | U | U | U | AL035087 |
| S5-336600 | CA/TG | 100.31 | U | U | U | U | AL390959 |
| S5-388393 | CA/TG | 100.36 | U | U | U | U | AL390959 |
| S5-433030 | GAAA/TTTC | 100.40 | U | U | U | U | AL590725 |
| S5-452941 | AG/CT | 100.42 | U | U | U | U | AL590725 |
| S6-34593 | CA/TG | 100.50 | U | U | U | U | AL590725 |
| D6S1671 | CA/TG | 100.66 | U | U | U | U | AL080285 |
| S6-170561 | CA/TG | 100.69 | X | U | U | U | AL080285 |
| D6S475 | GATATATC | 100.71 | X | U | U | U | AL080285 |
Note: X denotes a recombination event between a marker and disease in an indicated family; U denotes no recombination between a marker and disease in an indicated family. Information of BAC ID was obtained from NCBI, assembly of March 2006 of UCSC database was used for physical location.
Twenty six of these 30 STR markers appeared to be informative and highly polymorphic and were used in genotyping analyses (Table 1). This approach allowed us to saturate the MGI with STR markers with an average coverage of 100 kb between two STR markers and determine the linear order of markers (Table 1)
Thirty STR markers were genotyped using the primers either found from Marshfield Clinic or designed by ourselves. The genotyping was performed using P32 labeling method using genomic DNA isolated from blood (Yang et al., 2002; Zhang et al., 2001). Two-point LOD scores were calculated using the subroutine MLINK of the LINKAGE program (v.5.1; http://www.hgmp.mrc.ac.uk/; Human Genome Mapping Project Resources Center, Cambridge, UK) (Lathrop, Lalouel, Julier & Ott, 1984, Lathrop, Lalouel, Julier & Ott, 1985). An autosomal-dominant mode of inheritance with full penetrance and a disease allele frequency of 0.0001 were assumed in the computations.
2.4. Sequencing analysis
Exons and cDNAs of POU3F2, FBXL4, LOC389416, C6orf168, COQ3, C6orf111, USP45, LOC401270, CCNC, PRDM13 and MCHR2 were PCR amplified and subject to sequencing analysis. For exons amplification of each gene, we use genomic DNA from blood and haploid somatic hybrids. We used RNA from EBV-transformed lymphoblastoid cell lines and haploid somatic hybrids cell lines to perform RT-PCR. All PCR products were purified using a QIAquick Gel Extraction Kit (QIAGEN Science, MD, USA) and sequenced using Big Dye Terminator v3.1 Cycle Sequencing Kit (ABI, Foster City, CA, USA).
3. Results
3.1. Clinical evaluation
Six multiple generation pedigrees (324, 348, 71, 633, 419, and 3784) with NCMD were identified for study (Fig. 1). Family 324, 348, 71, 633 and 3784 are of Caucasian origin whereas family 419 is of Chinese origin. These pedigrees revealed autosomal dominant inheritance, with affected members in each generation. Clinical evaluation and genetic studies on Family 324 were previously published (Reichel et al., 1998). Family 3784 was referred by an ophthalmologist with documented fundus photographs on affected patients. Thus careful clinical evaluations were performed in every individual to ensure an accurate diagnosis. A complete ophthalmic history and examination was performed on each patient. This included assessment of visual acuity, and detailed examination of the anterior segment and fundus. Individuals were diagnosed to be affected if they had decreased visual acuity along with macular atrophy, or if drusen were found in the posterior pole. Color fundus photographs were obtained for all affected individuals. At least four affected individuals in each kindred underwent fluorescein angiography. Forty two affected patients in NCMD families demonstrated a wide range of clinical phenotypes, including confluent drusen in the macula Fig. 2A, B, D), choroidal neovascularization (Fig. 2F, G, H), subretinal scars (Fig. 2E), or macular staphloma (Fig 2, C).
Figure 1.

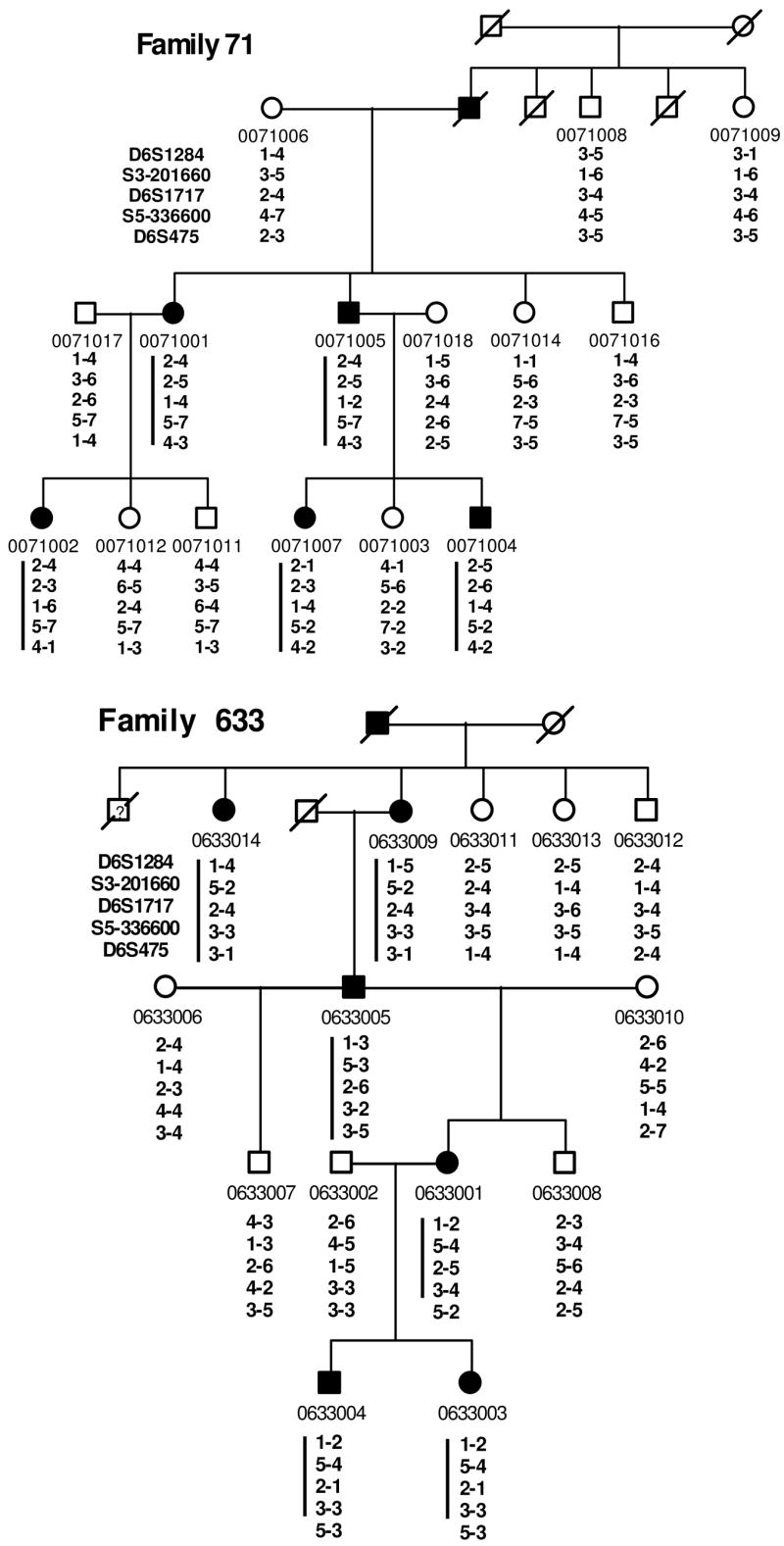

Pedigrees of six families (348, 324, 633, 71, 3784, and 419) with North Carolina macular dystrophy. The marker loci analyzed are given in the order from 6cen to 6qter (top to bottom). Square, male; Circle, female; Filled symbol represents affected with macular dystrophy; slashed symbol, deceased. A phase-known disease haplotype in each family is indicated by a vertical bar. Bottom of the 2nd page: summary of disease haplotypes shared in different families as shown in bas graphs. Black, shaded, and striped bar indicates a disease chromosome with alleles of STR markers listed in the left.
Figure 2.
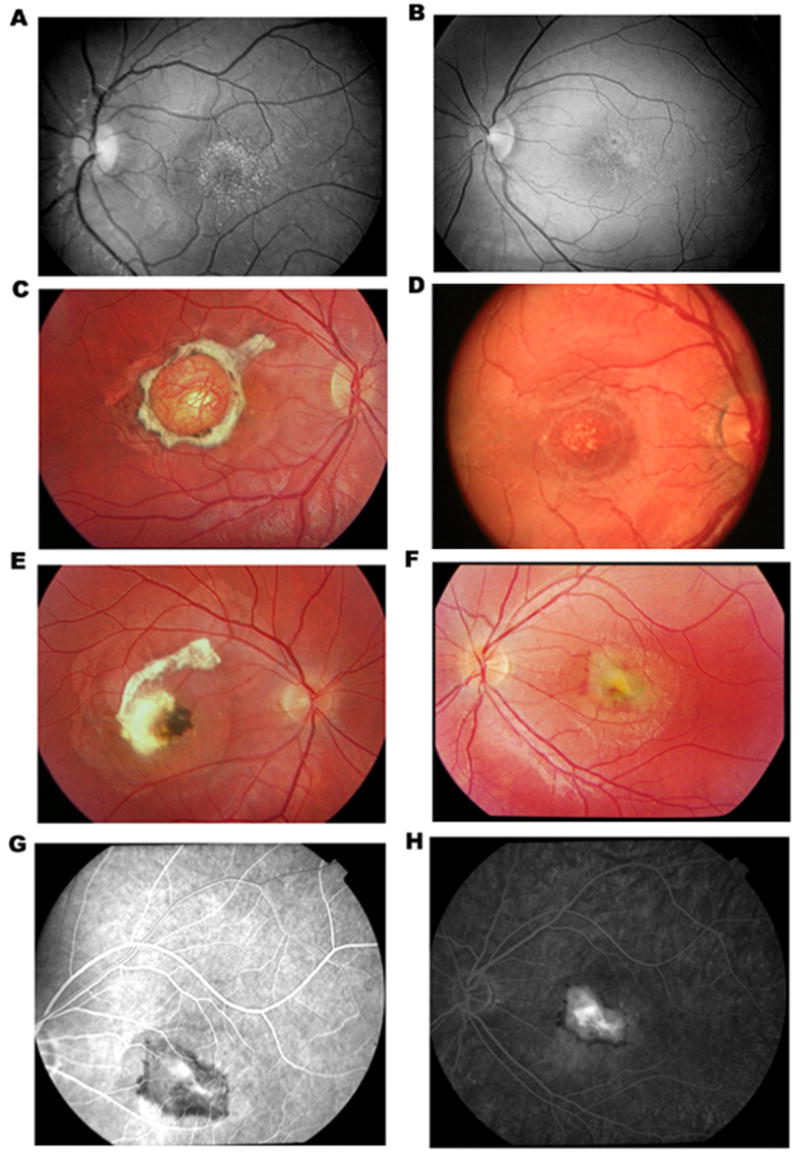
Fundus photographs of affected patients with North Carolina macular dystrophy, showing inter and intra familiar variations. A, patient #0348004 and B, patient #0348008 in family 348, demonstrating fine, confluent drusen in the macula (Grade I). C, patient #0633001 in family 633, demonstrating a macular staphloma (Grade III), D, patient #0633009 in family 633, demonstrating confluent macular drusen (Grade I). E and F, right eye and left eye of patient #0633004 in family 633, demonstrating a subretinal scar due to a prior choroidal neovasularization (CNV) in the right eye and a new CNV in the left eye. G and H, early and late frames of fluorescein angiography of the left eye of patient # 0633004, demonstrating hyperfluorescence in the macula corresponding to an active CNV.
3.2. Generation of EBV-transformed lymphoblastoid cell lines and haploid somatic hybrids
EBV transformed lymphoblastoid cell lines from three affected and two normal individuals in each kindred were established. In addition, using a GMP Conversion Technology (Yan et al., 2000b) we were able to separate normal and MCDR1 containing chromosomes and create haploid somatic hybrid cell lines (haploid samples) for one affected patient in family 348, 324, 71, 633, 419 and 3784. Since there is no normal allele to mask a disease allele, haploid samples allow one to unambiguously determine a disease haplotype, or identify a mutation (Yan, Kinzler & Vogelstein, 2000a).
3.3. Genetic linkage analysis
Because the clinical features observed in family 348, 324, 71, 633, 419, and 3784 were characteristic of NCMD, genetic linkage mapping for the responsible gene was conducted using DNA markers linked to the locus for MCDR1 on chromosomal 6q16 using established methods (Garibaldi & Zhang, 1999, Kniazeva et al., 1999; Kniazeva et al., 2000). Positive linkage to the MCDR1 locus was observed (Table 2). The likelihood (two point lod scores) of linkage between locus D6S1284, D6S1717, and D6S475 and the MCDR1 locus was determined (Table 2). The maximum lod score, 15.8, was achieved with marker D6S1717 at recombination fraction 0. To further refine the minimal genetic interval, haplotypes of individuals in each family were analyzed for recombination events. We genotyped additional 23 STR markers and identified haplotypes which segregated with disease (Table 1). Individuals with recombinant haplotypes are indicated (as an X) in Table 1. One recombination event occurred at D6S249, D6S1284, and D6S1716 in individual #0348004 of family 348 (Fig. 1 & Table 1), another occurred at D6S475 in individual #0633001 of family 663 (Fig. 1 and Table 1). Therefore, haplotype analyses suggest that the disease gene is most likely located in the interval between locus D6S1716 and D6S1671, corresponding to 1.86 million bps and containing 10 known or predicted genes (Table 1).
Table 2.
Two-point lod scores between STRs and Disease phenotype
| Recombination fractions | 0 | 0.1 | 0.2 | 0.3 | 0.4 | |
|---|---|---|---|---|---|---|
| Family348 | −3.49 | 2.83 | 2.36 | 1.67 | 0.84 | |
| D6S1284 | Family324 | 3.21 | 2.63 | 1.99 | 1.32 | .0.62 |
| Family71 | 3.01 | 2.47 | 1.87 | 1.24 | 0.58 | |
| Family633 | 3.13 | 2.59 | 1.99 | 1.34 | 0.63 | |
| Total | 5.86 | 10.5 | 8.21 | 5.57 | 2.67 | |
| Family348 | 4.12 | 3.41 | 2.65 | 1.82 | 0.93 | |
| S3-201660 | Family324 | 3.3 | 2.7 | 2.03 | 1.31 | 0.58 |
| Family71 | 3.01 | 2.46 | 1.85 | 1.19 | 0.53 | |
| Family633 | 2.11 | 1.7 | 1.26 | 0.8 | 0.34 | |
| Total | 12.5 | 10.3 | 7.79 | 5.12 | 2.38 | |
| Family348 | 4.52 | 3.78 | 2.96 | 2.04 | 1.02 | |
| D6S1717 | Family324 | 4.7 | 3.93 | 3.08 | 2.12 | 1.06 |
| Family71 | 3.3 | 2.72 | 2.06 | 1.34 | 0.61 | |
| Family633 | 3.31 | 2.76 | 2.15 | 1.46 | 0.7 | |
| Total | 15.8 | 12.7 | 10.3 | 6.96 | 3.39 | |
| Family348 | 2.71 | 2.07 | 1.37 | 0.67 | 0.15 | |
| S5-336600 | Family324 | 2.9 | 2.22 | 1.47 | 0.72 | 0.16 |
| Family71 | 0.81 | 0.63 | 0.5 | 0.38 | 0.22 | |
| Family633 | 2.38 | 1.94 | 1.47 | 0.97 | 0.46 | |
| Total | 8.78 | 6.86 | 3.34 | 2.74 | 0.99 | |
| D6S475 | Family348 | 3.51 | 2.9 | 2.24 | 1.52 | 0.77 |
| Family324 | 3.65 | 3.01 | 2.32 | 1.58 | 0.8 | |
| Family71 | 2.71 | 2.21 | 1.65 | 1.05 | 0.45 | |
| Family633 | −13 | 1.15 | 1.29 | 0.89 | 0.42 | |
| Total | −3.12 | 9.27 | 7.5 | 5.04 | 2.44 |
3.4. Completion of a high-resolution genetic linkage map of the MCDR1 locus
As the first step in positional cloning of MCDR1, we performed fine linkage mapping in order to establish a high-resolution genetic linkage map of the MCDR1 locus. This approach allows us to refine the MGI containing the MCDR1 locus. Individuals showing recombination with either D6S1716 or D6S475 were genotyped with new STR markers. The result indicated that there are only one recombination event between D6S1716 and the MCDR1 locus and another between the S6-170561 and MCDR1 locus. Therefore, the MCDR1 locus is positioned between D6S1716 (the centromeric boundary marker) and S6-170561 (the telemetric boundary marker).
To further narrow the MGI, we applied linkage disequilibrium analyses in which disease haplotypes shared in different independent families are investigated. This analysis is particularly useful to refine a MGI due to an ancestral founder mutation. We compared the disease allele of each STR marker of six North Carolina macular dystrophy families to construct extended disease haplotypes (Fig. 1). This analysis revealed that the family 348, 324, and 71 shared an identical disease haplotype between D6S1284 and S6-170561, indicating that they originated from an ancestral founder mutation. However, haplotypes of these three families diverged at marker D6S1671. Therefore the MGI has been refined to a region between marker D6S1716 (98.85 mb) and D6S1671 (100.66 mb), corresponding to approximately 1.81mb (Table 1).
3.5. The examination of candidate genes for MCDR1
A search for potential genes using public human genome databases revealed 10 known or predicted genes within the 1.8 million base pairs of interval (POU3F2, FBXL4, LOC389416, C6orf168, COQ3, C6orf111, USP45, LOC401270, CCNC, PRDM13 and MCHR2). All exons and cDNA of the 11 genes were analyzed. Unfortunately, we have not identified any DNA alteration in the coding region or splicing junctions that provide convincing evidence that they cause North Carolina macular dystrophy phenotype.
4. Discussion
Over the past decade, much progress has been made in identifying genes for juvenile macular dystrophy. Many loci for macular dystrophy have been mapped and six genes cloned (Allikmets et al., 1997; Heon et al., 1996; Kniazeva et al., 2000; Michaelides, Hunt & Moore, 2003; Nichols, Sheffield, Vandenburgh, Drack, Kimura & Stone, 1993; Petrukhin et al., 1998; Sun, Molday & Nathans, 1999; Weber, Vogt, Wolz, Ives & Ewing, 1994; Wells et al., 1993; Weng, Mata, Azarian, Tzekov, Birch & Travis, 1999; Zhang et al., 2001). Functional studies of these genes have provided important insights into pathogenetic mechanisms of macular degeneration. For example, ABCR is the protein product of the recessive Stargardt macular dystrophy gene, ABCA4, that functions as an outwardly directed flippase for removal of N-retinyliden-phosphatidylethanolamine (A2E, (Gregory et al., 1996; Stone et al., 1999)). ELOVL4 is mutated in dominant Stargardt macular dystrophy and encodes an enzyme involved in elongation of very long chain fatty acid (Cameron, 2007; Karan et al., 2005; Zhang et al., 2001).
It is important to study the genetic basis of North Carolina macular dystrophy, as it causes juvenile macular degeneration with visual impairment. Furthermore, the significance of proposed studies on North Carolina macular dystrophy lies in its relationship with AMD. North Carolina macular dystrophy shares important clinical features with age-related macular degeneration (AMD). Drusen is a hallmark of AMD and choroidal neovascularization (CNV) is one of the most important complications of AMD. Both of them are present in patients with North Carolina macular dystrophy. Therefore, these observations make North Carolina macular dystrophy a unique genetic model for AMD. NCDR1 was initially mapped to 6q16 and then refined to an interval between D6S249 and D6S1671 (Small et al., 1999, Small et al., 1992b). Through this study, we refined MCDR1 interval to 1.81mb between D6S1716 and D6S1671. No disease-causing mutation was identified in the candidate genes within the interval suggesting that MCDR1 mutations may lie in a novel gene or caused a new pathogenic mechanism. Understanding the molecular mechanisms of MCDR1 should lead to novel insights into the pathogenesis of macular degeneration. Elucidation of the function of the MCDR1 gene may increase our understanding of retinal cell biology in general and drusen formation in particular.
Acknowledgments
This research was supported by grants from Knights Templar Eye Research Foundation (ZY), and the following grants (to KZ): NIH (R01EY14428, R01EY14448); the Ruth and Milton Steinbach Fund, Ronald McDonald House Charities, the Macular Vision Research Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allikmets R, Singh N, Sun H, Shroyer NF, Hutchinson A, Chidambaram A, et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet. 1997;15(3):236–246. doi: 10.1038/ng0397-236. [DOI] [PubMed] [Google Scholar]
- Cameron DJ, Zongzhong Tong, Zhenglin Yang, Jack Kaminoh, Shin Kamiyah, Haoyu Chen, et al. Essential role of Elovl4 in very long chain fatty acid synthesis, skin permeability barrier functiona, and neonatal survival. International Journal of Biological Sciences. 2007;3:111–119. doi: 10.7150/ijbs.3.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou YH, Brown EM, Levi T, Crowe G, Atkinson AB, Arnqvist HJ, et al. The gene responsible for familial hypocalciuric hypercalcemia maps to chromosome 3q in four unrelated families. Nat Genet. 1992;1(4):295–300. doi: 10.1038/ng0792-295. [DOI] [PubMed] [Google Scholar]
- Frank HR, Landers MB, 3rd, Williams RJ, Sidbury JB. A new dominant progressive foveal dystrophy. Am J Ophthalmol. 1974;78(6):903–916. doi: 10.1016/0002-9394(74)90800-9. [DOI] [PubMed] [Google Scholar]
- Garibaldi DC, Zhang K. Molecular genetics of macular degeneration. Int Ophthalmol Clin. 1999;39(4):117–142. doi: 10.1097/00004397-199903940-00009. [DOI] [PubMed] [Google Scholar]
- Gregory CY, Evans K, Wijesuriya SD, Kermani S, Jay MR, Plant C, et al. The gene responsible for autosomal dominant Doyne’s honeycomb retinal dystrophy (DHRD) maps to chromosome 2p16. Hum Mol Genet. 1996;5(7):1055–1059. doi: 10.1093/hmg/5.7.1055. [DOI] [PubMed] [Google Scholar]
- Heon E, Piguet B, Munier F, Sneed SR, Morgan CM, Forni S, et al. Linkage of autosomal dominant radial drusen (malattia leventinese) to chromosome 2p16–21. Arch Ophthalmol. 1996;114(2):193–198. doi: 10.1001/archopht.1996.01100130187014. [DOI] [PubMed] [Google Scholar]
- Karan G, Lillo C, Yang Z, Cameron DJ, Locke KG, Zhao Y, et al. Lipofuscin accumulation, abnormal electrophysiology, and photoreceptor degeneration in mutant ELOVL4 transgenic mice: A model for macular degeneration. Proc Natl Acad Sci U S A. 2005;102(11):4164–4169. doi: 10.1073/pnas.0407698102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kniazeva M, Chiang MF, Morgan B, Anduze AL, Zack DJ, Han M, et al. A new locus for autosomal dominant stargardt-like disease maps to chromosome 4. Am J Hum Genet. 1999;64(5):1394–1399. doi: 10.1086/302377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kniazeva M, Traboulsi EI, Yu Z, Stefko ST, Gorin MB, Shugart YY, et al. A new locus for dominant drusen and macular degeneration maps to chromosome 6q14. Am J Ophthalmol. 2000;130(2):197–202. doi: 10.1016/s0002-9394(00)00585-7. [DOI] [PubMed] [Google Scholar]
- Lathrop GM, Lalouel JM, Julier C, Ott J. Strategies for multilocus linkage analysis in humans. Proc Natl Acad Sci U S A. 1984;81(11):3443–3446. doi: 10.1073/pnas.81.11.3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathrop GM, Lalouel JM, Julier C, Ott J. Multilocus linkage analysis in humans: detection of linkage and estimation of recombination. Am J Hum Genet. 1985;37(3):482–498. [PMC free article] [PubMed] [Google Scholar]
- Lefler WH, Wadsworth JA, Sidbury JB., Jr Hereditary macular degeneration and amino-aciduria. Am J Ophthalmol. 1971;1(1 Part 2):224–230. doi: 10.1016/0002-9394(71)90394-1. [DOI] [PubMed] [Google Scholar]
- Michaelides M, Hunt DM, Moore AT. The genetics of inherited macular dystrophies. J Med Genet. 2003;40(9):641–650. doi: 10.1136/jmg.40.9.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols BE, Sheffield VC, Vandenburgh K, Drack AV, Kimura AE, Stone EM. Butterfly-shaped pigment dystrophy of the fovea caused by a point mutation in codon 167 of the RDS gene. Nat Genet. 1993;3(3):202–207. doi: 10.1038/ng0393-202. [DOI] [PubMed] [Google Scholar]
- Petrukhin K, Koisti MJ, Bakall B, Li W, Xie G, Marknell T, et al. Identification of the gene responsible for Best macular dystrophy. Nat Genet. 1998;19(3):241–247. doi: 10.1038/915. [DOI] [PubMed] [Google Scholar]
- Rabb MF, Mullen L, Yelchits S, Udar N, Small KW. A North Carolina macular dystrophy phenotype in a Belizean family maps to the MCDR1 locus. Am J Ophthalmol. 1998;125(4):502–508. doi: 10.1016/s0002-9394(99)80191-3. [DOI] [PubMed] [Google Scholar]
- Reichel MB, Kelsell RE, Fan J, Gregory CY, Evans K, Moore AT, et al. Phenotype of a British North Carolina macular dystrophy family linked to chromosome 6q. Br J Ophthalmol. 1998;82(10):1162–1168. doi: 10.1136/bjo.82.10.1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer CG, Schworm HD, Ulbig M, Blankenagel A, Rohrschneider K, Pauleikhoff D, et al. An ancestral core haplotype defines the critical region harbouring the North Carolina macular dystrophy gene (MCDR1) J Med Genet. 1997;34(12):961–966. doi: 10.1136/jmg.34.12.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small KW, Garcia CA, Gallardo G, Udar N, Yelchits S. North Carolina macular dystrophy (MCDR1) in Texas. Retina. 1998;18(5):448–452. [PubMed] [Google Scholar]
- Small KW, Hermsen V, Gurney N, Fetkenhour CL, Folk JC. North Carolina macular dystrophy and central areolar pigment epithelial dystrophy. One family, one disease. Arch Ophthalmol. 1992a;110(4):515–518. doi: 10.1001/archopht.1992.01080160093040. [DOI] [PubMed] [Google Scholar]
- Small KW, Puech B, Mullen L, Yelchits S. North Carolina macular dystrophy phenotype in France maps to the MCDR1 locus. Mol Vis. 1997;3:1. [PubMed] [Google Scholar]
- Small KW, Udar N, Yelchits S, Klein R, Garcia C, Gallardo G, et al. North Carolina macular dystrophy (MCDR1) locus: a fine resolution genetic map and haplotype analysis. Mol Vis. 1999;5:38. [PubMed] [Google Scholar]
- Small KW, Weber J, Roses A, Pericak-Vance P. North Carolina macular dystrophy (MCDR1). A review and refined mapping to 6q14-q16.2. Ophthalmic Paediatr Genet. 1993;14(4):143–150. doi: 10.3109/13816819309042913. [DOI] [PubMed] [Google Scholar]
- Small KW, Weber JL, Roses A, Lennon F, Vance JM, Pericak-Vance MA. North Carolina macular dystrophy is assigned to chromosome 6. Genomics. 1992b;13(3):681–685. doi: 10.1016/0888-7543(92)90141-e. [DOI] [PubMed] [Google Scholar]
- Stone EM, Lotery AJ, Munier FL, Heon E, Piguet B, Guymer RH, et al. A single EFEMP1 mutation associated with both Malattia Leventinese and Doyne honeycomb retinal dystrophy. Nat Genet. 1999;22(2):199–202. doi: 10.1038/9722. [DOI] [PubMed] [Google Scholar]
- Sun H, Molday RS, Nathans J. Retinal stimulates ATP hydrolysis by purified and reconstituted ABCR, the photoreceptor-specific ATP-binding cassette transporter responsible for Stargardt disease. J Biol Chem. 1999;274(12):8269–8281. doi: 10.1074/jbc.274.12.8269. [DOI] [PubMed] [Google Scholar]
- Weber BH, Vogt G, Wolz W, Ives EJ, Ewing CC. Sorsby’s fundus dystrophy is genetically linked to chromosome 22q13-qter. Nat Genet. 1994;7(2):158–161. doi: 10.1038/ng0694-158. [DOI] [PubMed] [Google Scholar]
- Wells J, Wroblewski J, Keen J, Inglehearn C, Jubb C, Eckstein A, et al. Mutations in the human retinal degeneration slow (RDS) gene can cause either retinitis pigmentosa or macular dystrophy. Nat Genet. 1993;3(3):213–218. doi: 10.1038/ng0393-213. [DOI] [PubMed] [Google Scholar]
- Weng J, Mata NL, Azarian SM, Tzekov RT, Birch DG, Travis GH. Insights into the function of Rim protein in photoreceptors and etiology of Stargardt’s disease from the phenotype in abcr knockout mice. Cell. 1999;98(1):13–23. doi: 10.1016/S0092-8674(00)80602-9. [DOI] [PubMed] [Google Scholar]
- Yan H, Kinzler KW, Vogelstein B. Tech.sight. Genetic testing--present and future. Science. 2000a;289(5486):1890–1892. doi: 10.1126/science.289.5486.1890. [DOI] [PubMed] [Google Scholar]
- Yan H, Papadopoulos N, Marra G, Perrera C, Jiricny J, Boland CR, et al. Conversion of diploidy to haploidy. Nature. 2000b;403(6771):723–724. doi: 10.1038/35001659. [DOI] [PubMed] [Google Scholar]
- Yang Z, Peachey NS, Moshfeghi DM, Thirumalaichary S, Chorich L, Shugart YY, et al. Mutations in the RPGR gene cause X-linked cone dystrophy. Hum Mol Genet. 2002;11(5):605–611. doi: 10.1093/hmg/11.5.605. [DOI] [PubMed] [Google Scholar]
- Zhang K, Kniazeva M, Han M, Li W, Yu Z, Yang Z, et al. A 5-bp deletion in ELOVL4 is associated with two related forms of autosomal dominant macular dystrophy. Nat Genet. 2001;27(1):89–93. doi: 10.1038/83817. [DOI] [PubMed] [Google Scholar]