

Abstract
Dopamine (DA) signaling at synapses is tightly coordinated through opposing mechanisms of vesicular fusion-mediated DA release and transporter-mediated DA clearance. Altered brain DA signaling is suspected to underlie multiple brain disorders, including schizophrenia, Parkinson's disease, bipolar disorder, and attention-deficit hyperactivity disorder (ADHD). We identified a pedigree containing two male children diagnosed with ADHD who share a rare human DA transporter (DAT; SLC6A3) coding variant, Ala559Val. Among >1000 control and affected subjects, the Val559 variant has only been isolated once previously, in a female subject with bipolar disorder. Although hDAT Ala559Val supports normal DAT protein and cell surface expression, as well as normal DA uptake, the variant exhibits anomalous DA efflux from DA-loaded cells. We also demonstrate that hDAT Ala599Val exhibits increased sensitivity to intracellular Na+, but not intracellular DA, and displays exaggerated DA efflux at depolarized potentials. Remarkably, the two most common ADHD medications, amphetamine and methylphenidate, both block hDAT Ala559Val-mediated DA efflux, whereas these drugs have opposite actions at wild-type hDAT. Our findings reveal that DA efflux, typically associated with amphetamine-like psychostimulants, can be produced through a heritable change in hDAT structure. Because multiple gene products are known to coordinate to support amphetamine-mediated DA efflux, the properties of hDAT Ala559Val may have broader significance in identifying a new mechanism through which DA signaling disorders arise. Additionally, they suggest that block of inappropriate neurotransmitter efflux may be an unsuspected mechanism supporting the therapeutic actions of existing transporter-directed medications.
Keywords: dopamine, transport, attention deficit hyperactivity disorder, amphetamine, methylphenidate, mutation
Introduction
The neurotransmitter dopamine (DA) exerts powerful control over both motor and cognitive behavior (Carlsson, 1987). In the striatum, nucleus accumbens, and frontal cortex, DA is released by vesicular fusion mechanisms and cleared by cocaine-sensitive dopamine transporters (DATs; SLC6A3), which are presynaptic, 12 transmembrane domain (TM) proteins. Genetic ablation of DAT in transgenic mice greatly prolongs DA clearance rates, eliminates cocaine-induced hyperactivity, and triggers multiple changes in presynaptic and postsynaptic signaling pathways (Giros et al., 1996).
Evidence of a dependence on DAT for normal DA clearance and signaling has stimulated efforts to implicate DAT in multiple brain disorders that are associated with altered DA signaling or that are sensitive to medications targeting DA pathways, including schizophrenia, Parkinson's disease, bipolar disorder, and attention-deficit hyperactivity disorder (ADHD). In particular, data from animal models and human neuroimaging and genetic studies suggest altered availability or function of the DAT in ADHD (Mazei-Robison and Blakely, 2006). Furthermore, two prominent ADHD medications, methylphenidate (MPH; Ritalin) and amphetamine (AMPH; Adderall), both target DAT (Volkow et al., 2001). One challenge to the connection of DAT with ADHD, however, is the difference in mechanism of action of methylphenidate and amphetamine at DAT; methylphenidate blocks DAT-mediated DA uptake, whereas AMPH both blocks DA uptake and triggers DAT reversal, generating DA efflux. One view of the therapeutic action of both agents is that both agents elevate synaptic DA, overcoming deficiencies in DA signaling in cortical and basal ganglia circuits. Alternative ideas stress an “inverted U” shape for DA modulation of neuronal activity and behavior (Williams and Dayan, 2005; Arnsten, 2007), a framework that stresses less a linear relationship between synaptic DA availability and function and more the sharpening of DA output/response to achieve optimum signaling.
Given the importance of DAT in DA neurotransmission and as a site of action for ADHD medications, we examined whether ADHD subjects bear functional DAT coding variants (Mazei-Robison et al., 2005). We identified a single nonsynonymous variant, Ala559Val in hDAT (hDAT A559V) TM12, shared by two Caucasian, male siblings with ADHD. Initial characterization of the hDAT A559V variant in COS-7 and SH-SY5Y cells demonstrated normal hDAT protein expression, surface trafficking, and DA uptake, as well as unaltered potencies for AMPH, cocaine, or MPH in DA uptake inhibition. Also, hDAT A559V displays comparable sensitivity to hDAT for modulation by multiple second-messenger signaling pathways (Mazei-Robison and Blakely, 2005). With the appreciation that DAT-mediated DA efflux can be altered independently of inward DA transport (Khoshbouei et al., 2003, 2004; Fog et al., 2006), and that DAT-mediated DA efflux can be modulated to impact neuronal responses (Falkenburger et al., 2001), we examined the impact of the hDAT A559V mutation on DA efflux. Here, we report the discovery that hDAT A559V is a “reverse of function” variant, conferring anomalous, transporter-mediated DA efflux that is exacerbated when cells are depolarized. Moreover, we find that the outward DA transport mediated by hDAT A559V is attenuated by both AMPH and MPH.
Materials and Methods
Cell culture and transport assays.
HEK-293 cells for transport assays were plated, cultured, and transfected as reported previously (Mazei-Robison and Blakely, 2005). Cell surface biotinylation using sulfosuccinimidyl-2-(biotinamido)ethyl-1,3-dithiopropionate-biotin (sulfo-NHS-SS-biotin) (1.0 mg/ml; Pierce) and Western blotting methods using an N-terminal antibody against hDAT (1:2500) (Millipore Bioscience Research Reagents) were performed as described previously (Mazei-Robison and Blakely, 2005). DAT activity was assayed with 3,4-[7-3H]-dihydroxyphenylethylamine ([3H]DA; ∼20.5 Ci/mmol; PerkinElmer) for 10 min at 37°C as described previously (Apparsundaram et al., 1998) with nonspecific activity determined by 10 min pretreatment with 1 μm GBR-12909 [1-{2-[bis-(4-fluorophenyl)methoxy]ethyl}-4-(3-phenylpropyl)piperazine] (Sigma-Aldrich). For superfusion assays of DA efflux, coverslips containing transfected or control cells prelabeled with [3H]DA were transferred to superfusion chambers (0.2 ml) and superfused with buffer at 25°C and a perfusion rate of 0.7 ml/min. After a washout period of 45 min to establish stable efflux of radioactivity, 2 min fractions were collected. After 6 min of baseline values, buffers remained under control conditions or were switched to buffer including monensin (10 μm). Cells were then lysed in 1% SDS, and [3H]DA retained or released was estimated by scintillation counting. For electrophysiological experiments, hDAT, hDAT A559V, or hDAT R237Q DNAs were cotransfected with pEGFP-C1 (Invitrogen). Only fluorescent cells were then used in electrophysiological assays. Assays were conducted ∼24 h after transfection.
Cell surface biotinylation.
For cell surface biotinylation assays, HEK-293 cells were cultured in 24-well plates, transfected as described above and labeled with sulfo-NHS-SS-biotin (1.0 mg/ml; Pierce) before purification and analysis via SDS-PAGE/Western blots as described previously (Mazei-Robison and Blakely, 2005). DAT was detected using a rat monoclonal primary antibody to the N terminus of hDAT (1:2500) (Millipore Bioscience Research Reagents) and a goat-anti-rat-HRP-conjugated secondary antibody (1:5000; Jackson ImmunoResearch).
Measurement of DAT currents and DA efflux.
Electrophysiological experiments to monitor DAT charge and DA flux were performed as described previously (Khoshbouei et al., 2003, 2004; Fog et al., 2006). Briefly, HEK-293 cells were cultured in serum-containing medium on poly-d-lysine-coated 35 mm MaTek plates and transfected as described above with recordings obtained ∼24 h later. For nonclamped amperometric experiments, cells were washed twice with Krebs'–Ringer's–HEPES assay buffer and were then actively loaded with 1 μm DA in assay buffer for 45 min at 37°C. Fluorescent cells were selected for analysis, and a carbon fiber electrode was opposed to the plasma membrane and held at +700 mV to oxidize DA. Total charge flux and amperometric currents were recorded at 25°C using Axopatch 200B with a low-pass Bessel filter set at 100 Hz. Current–voltage relationships were generated by stepping membrane voltage from a holding potential of −20 mV to voltages between −100 and +100 mV in 20 mV increments for 1 s. Current was calculated as average current during the final 100 ms of the voltage step. To elicit maximum DA efflux, as described by Khoshbouei et al. (2003), 90 mm Na+ and 100 μm to 2 mm DA was used in the recording pipette (pipette solution: 30 mm KCl, 90 mm NaCl, 0.1 mm CaCl2, 2 mm MgCl2, 1.1 mm EGTA, 10 mm HEPES, 30 mm dextrose, and 2 mm DA, adjusted to pH 7.35). For NaCl titrations, KCl was substituted to maintain osmolarity. Data were recorded and analyzed using pClamp8 software.
Results
hDAT A559V increases outward DA transport without affecting inward transport
The identification and pedigree of two male ADHD probands carrying the hDAT A559V variant have been described previously (Mazei-Robison and Blakely, 2005). To evaluate the DA influx/efflux characteristics of wild-type and hDAT A559V in a system devoid of vesicular DA release, we expressed hDAT and hDAT mutants in HEK-293T cells. First, we established levels of hDAT protein expression and hDAT-mediated DA influx (Fig. 1 A,B). As in the COS-7 and SH-SY5Y cell lines (Mazei-Robison and Blakely, 2005), HEK-293T cells transiently transfected with hDAT A559V cDNA exhibit comparable DA uptake at both a low (50 nm) and high (3 μm) concentration of DA (Fig. 1 A), supported by similar levels of total and surface protein (Fig. 1 B). The hDAT coding variant Arg237Gln (R237Q), like the hDAT A559V substitution, is predicted to lie near the extracellular end of a TM domain (Cargill et al., 1999; Goldberg et al., 2003; Yamashita et al., 2005). We analyzed this variant in parallel with hDAT A559V and found it to exhibit levels of total DAT protein, surface expression, and DA influx similar to hDAT. Next, we preloaded either hDAT or variant hDAT-transfected cells with DA to examine outward DA efflux using an extracellular carbon fiber electrode (Fig. 1 C,D). Recordings from single, unclamped cells revealed that whereas amperometric currents were unaffected by application of cocaine (for raw currents, see Fig. 1 C) or MPH (for raw currents, see Fig. 1 D) with hDAT cells, indicating minimal spontaneous DA efflux, amperometric signals from hDAT A559V cells were significantly reduced by these antagonists. Comparative analyses indicate that hDAT A559V-transfected cells displayed an ∼300% increase in basal DA efflux currents compared with hDAT-transfected cells (p < 0.01, unpaired t test). This effect is not a common property of hDAT mutations, because hDAT R237Q-transfected cells displayed basal DA efflux comparable with hDAT (Fig. 1 C,D).
Figure 1.

hDAT A559V mutation produces cocaine- and MPH-sensitive DA efflux. A, Inward transport of 50 nm (n = 3) or 3 μm [3H]DA (n = 3) by hDAT or hDAT variants. DA transport activity is expressed as a percentage of that obtained with hDAT and reported as a mean ± SEM. Mean 50 nm DA hDAT transport activity was 8.89 × 10−18 ± 5.9 × 10−19 mol/cell/min and mean 3 μm DA hDAT transport activity was 2.85 × 10−16 ± 2.1 × 10−17 mol/cell/min. B, Top, Representative Western blot, using 40 μg of biotinylated protein or 4 μg of total cellular lysate. Bottom, Quantitation of total and surface hDAT protein expression (n = 3). C, hDAT A559V mediates enhanced outward DA leak. DA outward leak from hDAT (n = 4), hDAT A559V (n = 5), and hDAT R237Q (n = 3) cells is reported as mean amperometric current ± SEM. Cells were actively loaded with 1 μm DA, and DA leak was measured by amperometry as described in Materials and Methods; DAT specificity was defined by 10 μm cocaine. *p < 0.05, one-way ANOVA followed by Dunnett's multiple-comparison test. D, DA outward leak from hDAT (n = 6), hDAT A559V (n = 7), and hDAT R237Q (n = 3) cells as defined by 10 μm MPH. *p < 0.05, one-way ANOVA followed by Dunnett's multiple-comparison test. Traces provide examples of recordings under each condition.
hDAT A559V demonstrates an increased sensitivity for voltage- and intracellular sodium-triggered DA efflux
To gain insights into the mechanisms supporting altered DA efflux by A559V, amperometry was combined with whole-cell patch-clamp recordings. This technique allows for the control of intracellular ionic and neurotransmitter concentrations as well as membrane potential. Whole-cell hDAT currents, reported after antagonist subtraction, reflect the net movements of charged species (e.g., Na+, Cl−, DA+) across the transporter. Under conditions that normally elicit DA efflux in hDAT-transfected cells (high intracellular Na+ and DA, depolarization) (Khoshbouei et al., 2003), we monitored a significant potentiation of DAT-mediated outward currents for hDAT A559V (Fig. 2 A) (p < 0.01, two-way ANOVA).
Figure 2.

Electrophysiogical evidence for hDAT A559V-mediated DA efflux. A, hDAT A559V cells exhibit significantly increased outward DA current in whole-cell patch-clamp experiments. HEK-293T cells were loaded via pipette with 2 mm DA and 90 mm Na+, and membrane potential was stepped in 20 mV intervals from −100 to +100 mV in hDAT (squares; n = 3) and hDAT A559V (circles; n = 5) cells. Right, Representative currents evoked from hDAT and hDAT A559V cells. B, hDAT A559V cells produce elevated voltage-dependent, DA amperometric currents. HEK-293T cells were loaded, and efflux of DA was monitored by amperometry after voltage steps as described in A. Recordings were obtained from hDAT (black squares; n = 3) and hDAT A559V (red circles; n = 5) cells with data plotted as mean amperometric current ± SEM. Right, Representative amperometric traces from hDAT and hDAT A559V cells. Both charge and amperometric current traces exhibit significant differences comparing hDAT to hDAT A559V (p < 0.05, 2-way ANOVA).
Consistent with measurements of increased outward charge flux for hDAT A559V, we found that this mutant exhibited exaggerated amperometric currents at depolarized potentials (Fig. 2 B) (p < 0.01, two-way ANOVA). These currents reflect the outward movement of DA, because they are not evident without DA preloading, nor in mock-transfected cells, and they can be blocked by DAT antagonists. In contrast to the voltage-sensitive efflux phenotype of hDAT A559V, hDAT R237Q-mediated currents were not significantly impacted by depolarization (data not shown).
To gain insight into a possible mechanism for elevated DA efflux in hDAT A559V, we next examined the Na+ dependence of DA efflux. DA efflux currents were measured at +40 mV using concentrations of 1, 3, 5, 30, or 60 mm intracellular Na+. Strikingly, hDAT A559V exhibited significantly increased DA efflux compared with hDAT at intracellular Na+ concentrations as low as 3 mm (n = 3; p < 0.05, two-way ANOVA, Bonferroni posttest) (Fig. 3 A,B). Saturation analysis from multiple experiments reveal the change in Na+ sensitivity in hDAT A559V to derive from a decrease in the Na+ K m (hDAT, 11.2 ± 5.1 mm; hDAT A559V, 2.6 ± 0.7 mm) accompanied by an increase in V max (hDAT, 0.05 ± 0.01 pA; hDAT A559V, 0.16 ± 0.01 pA) (Fig. 3 C). Considering the V max/K m ratio from these studies as a measure of internal Na+ coupling efficiency for DA efflux, hDAT A559V exhibits a >12-fold increase in Na+ coupling efficiency for outward DA movement (hDAT, 0.005; A559V, 0.061). The finding that hDAT A559V is able to efflux at low intracellular Na+ concentrations is consistent with our evidence of elevated spontaneous DA efflux at the intracellular Na+ concentrations present in unclamped cells. In fact, it is normally difficult to observe appreciable DA efflux for hDAT at potentials below +60 mV when intracellular Na+ is not raised to ≥30 mm. Analysis of fits of amperometry I–V plots at 5 mm Na+ I to estimate membrane potential at which DA efflux is first evident yields −37.4 mV (r 2 = 0.98) for hDAT versus −51.4 mV (r 2 = 0.98) for hDAT A559V. Together, these data indicate that hDAT A559V rests in a more DA efflux-competent state than hDAT, a state that requires only modest changes in intracellular Na+ or V m to trigger reversal.
Figure 3.
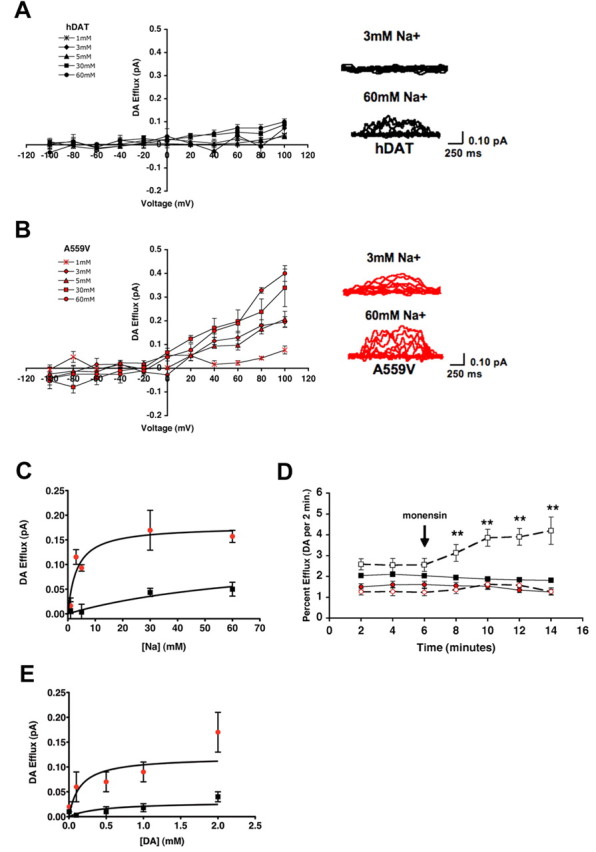
Enhanced intracellular Na+ sensitivity supports increased DA efflux by hDAT A559V. A, B, hDAT (A) or hDAT A559V (B) cells (n = 3, except for 30 mm, when hDAT n = 5 and hDAT A559V n = 4) were loaded with 2 mm DA and varying concentrations of Na+ (1, 3, 5, 30, and 60 mm), and the voltage dependence of DA efflux was measured by amperometry as in Figure 2 B. Representative raw traces are shown beside each plot. C, hDAT A559V exhibits a reduced K m and increased V max for Na+-dependent DA efflux. Hyperbolic fit of Na+ dependence of DA efflux in hDAT (black squares) and hDAT A559V (red circles) cells at +40 mV. D, Monensin triggers DA efflux in hDAT (black squares) but not hDAT A559V (red circles) cells. Superfusion assays examining monensin-triggered (10 μm; open symbols) DA efflux were performed as described in Materials and Methods. **p < 0.01, one-way ANOVA, Bonferroni posttest. E, hDAT A559V DA efflux exhibits an altered V max but equivalent K m for DA. Shown is a hyperbolic fit of DA dependence of DA efflux in hDAT (black squares) and hDAT A559V (red circles) cells at +40 mV.
We next sought a measure of altered intracellular Na+ sensitivity in hDAT A559V in nonclamped cells. The ionophore monensin confers electroneutral Na+/H+ exchange and can be used to elevate intracellular Na+ in cells without altering membrane potential; applications of 5–20 μm monensin have been found to raise intracellular Na+ to 30–50 mm in HEK-293 cells (Chen and Reith, 2004). Therefore, we monitored efflux of preloaded [3H]DA under superfusion before and after stimulation of cells with 10 μm monensin (Fig. 3 D). As expected, treatment of hDAT-transfected cells with monensin rapidly (2 min) stimulated [3H]DA efflux (Fig. 3 D). Under the same conditions, hDAT A559V-transfected cells exhibited a slightly reduced basal level of DA efflux in control medium at the onset of fraction collection, because of more pronounced prior efflux of [3H]DA (data not shown). Strikingly, hDAT A559V cells displayed essentially no response to monensin treatments. Although monensin, in addition to enhancing intracellular Na+ levels, also impacts the pH of trafficking organelles, a trafficking impact would presumably diminish (not trigger) hDAT-mediated DA efflux and would also not be expected to distinguish between hDAT and hDAT A559V. Rather, the loss of monensin action at hDAT A559V is expected if the increased affinity for intracellular Na+ places the mutant already at saturation for DA efflux.
Another mechanism that could lead to enhanced DA efflux in hDAT A559V is elevated affinity of the transporter for intracellular DA. To assess this possibility, we clamped intracellular Na+ at 30 mm and monitored the extent of DA efflux by amperometry as a function of DA concentration when elicited by a voltage step to +40 mV. In cells that were loaded with varying concentrations of DA (3, 100, 500, 1000, and 2000 μm), we obtained saturable efflux in both hDAT and hDAT A559V, with a clear separation between the two evident at 100 μm DA. Our saturation analyses indicate that the elevated efflux of hDAT A559V derives predominantly from an increased DA efflux V max (hDAT, 0.04 ± 0.01 pA; hDAT A559V, 0.17 ± 0.04 pA), because no difference was detected in DA K m (hDAT, 0.36 ± 0.31 mm; hDAT A559V, 0.20 ± 0.19 mm).
Anomalous action of amphetamine to limit DA efflux in hDAT A559V cells
In a review of our hDAT A559V proband's medication history, we were surprised to see that both were successfully treated with the AMPH formulation Adderall (Mazei-Robison et al., 2005). AMPH analogs normally enhance DA efflux (Sulzer et al., 2005) and thus would be expected to exacerbate symptoms. We therefore investigated the effects of AMPH on DA efflux in hDAT and hDAT A559V using amperometry on unclamped cells. In these studies, hDAT cells treated with 10 μm AMPH displayed the expected elevation in hDAT-mediated efflux (Fig. 4 A). In contrast, AMPH treatment of hDAT A559V cells reduced DA efflux currents. Average results using either cocaine or MPH to define DAT-specific DA efflux revealed an absolute magnitude of efflux (as revealed by AMPH block) in hDAT A559V equal to or greater than that triggered by a maximal concentration of AMPH in hDAT (Fig. 4 B). In other words, under the same conditions hDAT A559V spontaneously effluxes DA at a rate equal to or exceeding that achieved with AMPH on hDAT. Finally, AMPH does not appear to block hDAT A559V-supported DA efflux via a nonspecific mechanism, as MPH and cocaine do not further reduce DA efflux currents in the presence of AMPH (Fig. 4 C,D).
Figure 4.
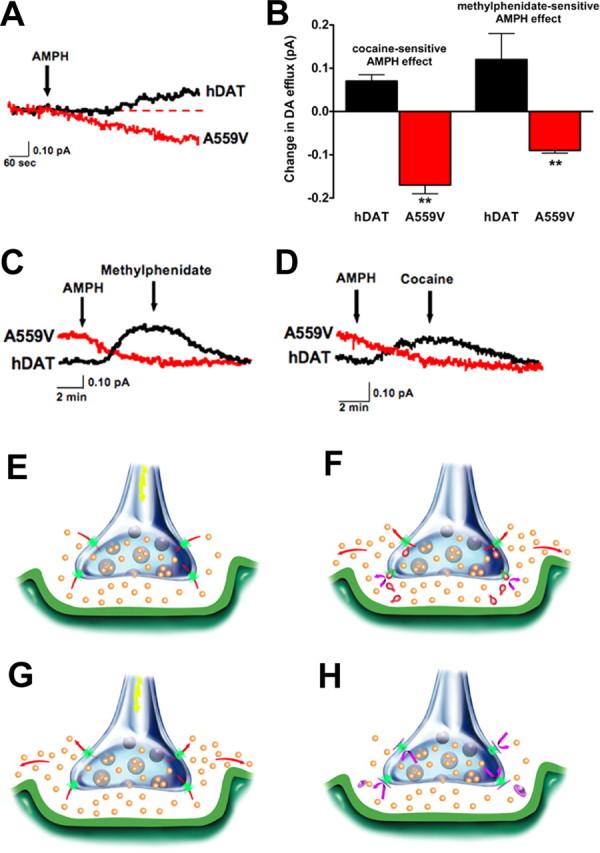
Anomalous AMPH block of DA efflux from hDAT A559V cells. A, AMPH suppresses outward DA efflux as monitored by amperometry. Traces reflect exemplary amperometric recordings of single unclamped hDAT (black) or hDAT A559V (red) cells, loaded with 1 μm DA and stimulated by 10 μm AMPH. B, Bar graph reflects summary data of DA efflux from hDAT or hDAT A559V cells, mean amperometric current ± SEM (cocaine-sensitive: hDAT, n = 10; hDAT A559V, n = 8; methylphenidate-sensitive: hDAT, n = 3; hDAT A559V, n = 4; **p < 0.01, unpaired t tests). C, D, Representative traces illustrating AMPH evoked DA release in hDAT cells (black) is blocked by 10 μm MPH (C) or 10 μm cocaine (D), whereas 10 μm AMPH blocks basal DA efflux in hDAT A559V cells (red) with no further reductions evident with addition of MPH or cocaine. E–H, Illustration of disrupted synaptic transmission induced by transporter-mediated neurotransmitter efflux. E, Under normal circumstances, presynaptic hDAT (green) inactivates and recycles DA (orange) released via vesicular fusion. F, AMPH (red) acts as both a DAT substrate and a reuptake blocker, eliciting reverse transport and blocking normal reuptake, thereby increasing DA levels in and beyond the synaptic cleft. G, With hDAT A559V, DAT-mediated DA efflux becomes a second, unexpected and temporally distinct source of DA release, although normal DA reuptake can still occur. H, Agents such as methylphenidate and cocaine (magenta) that block DAT-mediated DA efflux restrict DA signaling to DA released via vesicular fusion. In hDAT A559V and possibly other enhanced efflux states triggered by regulatory genes, amphetamine blocks DA efflux in a manner similar to methylphenidate acting on hDAT (data not shown).
Discussion
ADHD affects 3–5% of the United States school-age population, making it one of the most common diseases in children (American Psychiatric Association, 1994). Although ADHD is a highly heritable disorder (Thapar et al., 1999), the etiology of the disorder is unknown. Prominent theories posit an alteration of brain DA neurotransmission based, in part, on the importance of DA signaling in circuits involved in motor function, reward, and attention (Carlsson, 1987). Our studies define a novel property of an ADHD-associated mutation, enhancement of nonvesicular, DAT-dependent DA release. DA efflux is now known to be a regulated facet of DAT behavior, triggered not only by alterations in membrane potential and intracellular Na+, but also via the activation of an intracellular signal transduction network supported by PKC (Johnson et al., 2005) and CaMKII (Fog et al., 2006). Our findings, although derived from a single pedigree and currently limited to in vitro studies, are important because they raise the possibility that anomalous DAT-mediated DA efflux could impact risk for ADHD. Other disorders that benefit from blockade of DA receptors, such as schizophrenia, could also be supported by inappropriate, DAT-mediated DA release.
Exactly how the hDAT A559V substitution perturbs transporter structure to enhance intracellular Na+ potency and trigger anomalous DA efflux is at present only partially understood. Importantly, we have recently gathered evidence that hDAT A559V supports anomalous DA efflux from transfected DA neurons, cultured from DAT knock-out mice (E. Bowton, unpublished findings), suggesting that these findings have relevance for native DA systems. Resting membrane potentials of hDAT and hDAT A559V cells do not differ [hDAT (n = 18), −37.2 ± 2.3 mV; hDAT A559V (n = 18), −33.1 ± 1.9 mV], and elevated DA efflux is evident under voltage clamp, at defined Na+ concentrations; thus, DAT-mediated Na+ leak would not appear to explain enhanced DA efflux. Based on the LeuTAa bacterial crystal structure (Yamashita et al., 2005), A559 is predicted to lie at the extracellular end of TM12. Although recent studies have documented the importance of the cytosolic N-tail (Khoshbouei et al., 2004) and C-tail (Fog et al., 2006) for AMPH-induced DA efflux, little work has examined the role of TM regions in this process. Interestingly, a study examining engineered proline-to-alanine substitutions in TM12 of the rat DAT documented an increased V max for DA efflux (Itokawa et al., 2002). More relevant, perhaps, is Ser528Ala (S528A), an engineered mutation in TM11 that was found to increase the outward translocation of substrates without affecting inward transport (Chen and Justice, 2000). Both TMs 11 and 12 have been suggested to contribute to the dimerization interface of LeuTAa, and properly organized dimer interactions appear critical for serotonin efflux (Seidel et al., 2005). The possibility that S528A and A559V may disrupt interactions between hDAT subunits that normally dictate substrate influx/efflux deserves further study.
A striking finding in our current report is the anomalous action of AMPH to block DA efflux in hDAT A559V, rather than enhance it, as it does in hDAT. It seems unlikely that hDAT A559V does not transport AMPH, because AMPH and DA are structurally very similar, and TM12 is not believed to play a role in substrate transport. We prefer, and are currently investigating, a model whereby AMPH-induced changes in intracellular signaling pathways and hDAT phosphorylation (Fog et al., 2006) are constitutively promoted in hDAT A559V, and AMPH application restores an efflux-incompetent conformation. Consistent with this idea, we have preliminary evidence of elevated basal hDAT A559V phosphorylation that can be attenuated by AMPH (D. Sakrikar, E. Bowton, A. Galli, and R. D. Blakely, unpublished findings).
The hDAT A559V variant was first reported in a single female in a cohort with bipolar disorder (n = 45), where the variant was reported to be absent from controls (n = 46) (Grünhage et al., 2000). The variant could not be further tracked with aspects of bipolar disorder or comorbid syndromes, however, because no other living member of the family carried the allele. ADHD is three times more common in relatives of bipolar subjects and exhibits an earlier age of onset. We identified the hDAT A559V variant in two male siblings in a screen of 70 children with ADHD (Mazei-Robison et al., 2005), to our knowledge making hDAT A559V the only nonsynonymous hDAT mutation to be identified in two unrelated families. As with the hDAT A559V bipolar subject, our hDAT A559V ADHD probands are heterozygous carriers. The pedigree comprising our two hDAT A559V probands lacks other known male carriers to permit assessment of gender-matched phenotypic correlations within the family. The mother who transmitted the allele to our male ADHD probands has no remarkable phenotype. However, the maternal grandmother is an hDAT A559V carrier and scored above average on the hyperactivity/restlessness and impulsivity/emotional liability items of the Conner's Adult ADHD rating scale (CAARS-S:L) and, more remarkably, was above the 90th percentile in DSM-IV hyperactive-impulsive symptoms (Mazei-Robison et al., 2005). Expanded single-nucleotide polymorphism discovery efforts in SLC6A3 by our lab, as well as by colleagues in the field, represent surveys of >500 ADHD probands, as well as 275 subjects with other diagnoses, and 253 controls (all predominantly Caucasians like our A559V probands). Thus, considering >2000 chromosomes, only three occurrences of hDAT A559V have been reported (<0.2%), and thus clearly this variant is not a common polymorphism, nor does it comprise genetic risk for ADHD at a population level. The identification of rare, functional variants has proven critical to the elucidation of molecular mechanisms underlying complex diseases including Parkinson's disease and Alzheimer's disease (St George-Hyslop, 2000; Hardy et al., 2003).
An alanine at position 559 in hDAT is observed in all vertebrate DATs (Mazei-Robison and Blakely, 2005), suggesting that even conservative changes at this position could have untoward consequences. Interestingly, research has suggested that rare, missense mutations may cumulatively predominate in diseased individuals over healthy controls (Kryukov et al., 2007). Because DA efflux is under the control of other signaling proteins (Khoshbouei et al., 2004), it seems very likely that mutation of hDAT is but one way for DAT to be placed, inappropriately, in an efflux-competent state. Specifically, mutations in DAT regulatory proteins may represent a reservoir of other risk determinants for ADHD.
Illustrations that describe the potential physiological significance of our findings are presented in Figure 4 E–H. Normally, DA is released via fusion of storage vesicles, triggered by afferent electrical stimulation (Fig. 4 E). AMPH can also trigger elevation of DA via competitive block of DA reuptake and through stimulation of DA efflux (Fig. 4 F). A nonvesicular release of DA through hDAT A559V (Fig. 4 G), evoked in parallel with DA vesicular release, would be expected to mimic the actions of AMPH and lead to a loss in fidelity of DA signaling by altering the relationship between afferent input and changes in extracellular DA levels as well by triggering greater DA synaptic spillover. These changes would also likely dampen the consequences of transitions between tonic and phasic DA neuronal firing (Grace, 2000). Although as yet we are unable to assess the function of hDAT A559V in our probands in vivo, AMPH and methylphenidate are both predicted to act to diminish anomalous DA efflux (Fig. 4 H), restoring the normal relationship between DA neuron excitation and extracellular DA levels. As such, the therapeutic utility of transporter antagonists, currently attributed to blockade of neurotransmitter reuptake, may in some cases arise from cessation of transporter-mediated neurotransmitter efflux. Our findings argue for further inspection of anomalous transporter-mediated neurotransmitter efflux as an unappreciated source of risk for brain disorders.
Footnotes
This work was supported by National Institutes of Health Grants MH067472 (M.S.M.-R.), MH064913 (E.B.), HL56693 (R.D.B.), and DA13975 and DA014684 (A.G.). M.H., M.S., M.F., and H.H.S. were supported by Austrian Science Foundation Grant P18706. We thank Tammy Jessen, Jane Wright, Qiao Han, and Angela Steele for general laboratory maintenance and technical assistance and Louis J. DeFelice for critical review of this manuscript.
References
- American Psychiatric Association. Ed 4. Washington, DC: American Psychiatric; 1994. Diagnostic and statistical manual of mental disorders. [Google Scholar]
- Apparsundaram S, Galli A, DeFelice LJ, Hartzell HC, Blakely RD. Acute regulation of norepinephrine transport: I. PKC-linked muscarinic receptors influence transport capacity and transporter density in SK-N-SH cells. J Pharmacol Exp Ther. 1998;287:733–743. [PubMed] [Google Scholar]
- Arnsten AF. Catecholamine and second messenger influences on prefrontal cortical networks of “representational knowledge”: a rational bridge between genetics and the symptoms of mental illness. Cereb Cortex. 2007;17(Suppl 1):i6–i15. doi: 10.1093/cercor/bhm033. [DOI] [PubMed] [Google Scholar]
- Cargill M, Altshuler D, Ireland J, Sklar P, Ardlie K, Patil N, Shaw N, Lane CR, Lim EP, Kalyanaraman N, Nemesh J, Ziaugra L, Friedland L, Rolfe A, Warrington J, Lipshutz R, Daley GQ, Lander ES. Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat Genet. 1999;22:231–238. doi: 10.1038/10290. [DOI] [PubMed] [Google Scholar]
- Carlsson A. Perspectives on the discovery of central monoaminergic neurotransmission. Annu Rev Neurosci. 1987;10:19–40. doi: 10.1146/annurev.ne.10.030187.000315. [DOI] [PubMed] [Google Scholar]
- Chen N, Justice JB. Differential effect of structural modification of human dopamine transporter on the inward and outward transport of dopamine. Brain Res Mol Brain Res. 2000;75:208–215. doi: 10.1016/s0169-328x(99)00288-0. [DOI] [PubMed] [Google Scholar]
- Chen N, Reith ME. Interaction between dopamine and its transporter: role of intracellular sodium ions and membrane potential. J Neurochem. 2004;89:750–765. doi: 10.1111/j.1471-4159.2004.02409.x. [DOI] [PubMed] [Google Scholar]
- Falkenburger BH, Barstow KL, Mintz IM. Dendrodendritic inhibition through reversal of dopamine transport. Science. 2001;293:2465–2470. doi: 10.1126/science.1060645. [DOI] [PubMed] [Google Scholar]
- Fog JU, Khoshbouei H, Holy M, Owens WA, Vaegter CB, Sen N, Nikandrova Y, Bowton E, McMahon DG, Colbran RJ, Daws LC, Sitte HH, Javitch JA, Galli A, Gether U. Calmodulin kinase II interacts with the dopamine transporter C terminus to regulate amphetamine-induced reverse transport. Neuron. 2006;51:417–429. doi: 10.1016/j.neuron.2006.06.028. [DOI] [PubMed] [Google Scholar]
- Giros B, Jaber M, Jones SR, Wightman RM, Caron MG. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature. 1996;379:606–612. doi: 10.1038/379606a0. [DOI] [PubMed] [Google Scholar]
- Goldberg NR, Beuming T, Soyer OS, Goldstein RA, Weinstein H, Javitch JA. Probing conformational changes in neurotransmitter transporters: a structural context. Eur J Pharmacol. 2003;479:3–12. doi: 10.1016/j.ejphar.2003.08.052. [DOI] [PubMed] [Google Scholar]
- Grace AA. The tonic/phasic model of dopamine system regulation and its implications for understanding alcohol and psychostimulant craving. Addiction. 2000;95(Suppl 2):S119–S128. doi: 10.1080/09652140050111690. [DOI] [PubMed] [Google Scholar]
- Grünhage F, Schulze TG, Müller DJ, Lanczik M, Franzek E, Albus M, Borrmann-Hassenbach M, Knapp M, Cichon S, Maier W, Rietschel M, Propping P, Nöthen MM. Systematic screening for DNA sequence variation in the coding region of the human dopamine transporter gene (DAT1) Mol Psychiatry. 2000;5:275–282. doi: 10.1038/sj.mp.4000711. [DOI] [PubMed] [Google Scholar]
- Hardy J, Cookson MR, Singleton A. Genes and parkinsonism. Lancet Neurol. 2003;2:221–228. doi: 10.1016/s1474-4422(03)00350-8. [DOI] [PubMed] [Google Scholar]
- Itokawa M, Lin Z, Uhl GR. Dopamine efflux via wild-type and mutant dopamine transporters: alanine substitution for proline-572 enhances efflux and reduces dependence on extracellular dopamine, sodium and chloride concentrations. Brain Res Mol Brain Res. 2002;108:71–80. doi: 10.1016/s0169-328x(02)00515-6. [DOI] [PubMed] [Google Scholar]
- Johnson LA, Guptaroy B, Lund D, Shamban S, Gnegy ME. Regulation of amphetamine-stimulated dopamine efflux by protein kinase C beta. J Biol Chem. 2005;280:10914–10919. doi: 10.1074/jbc.M413887200. [DOI] [PubMed] [Google Scholar]
- Khoshbouei H, Wang H, Lechleiter JD, Javitch JA, Galli A. Amphetamine-induced dopamine efflux. A voltage-sensitive and intracellular Na+-dependent mechanism. J Biol Chem. 2003;278:12070–12077. doi: 10.1074/jbc.M212815200. [DOI] [PubMed] [Google Scholar]
- Khoshbouei H, Sen N, Guptaroy B, Johnson L, Lund D, Gnegy ME, Galli A, Javitch JA. N-terminal phosphorylation of the dopamine transporter is required for amphetamine-induced efflux. PLoS Biol. 2004;2:E78. doi: 10.1371/journal.pbio.0020078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryukov GV, Pennacchio LA, Sunyaev SR. Most rare missense alleles are deleterious in humans: implications for complex disease and association studies. Am J Hum Genet. 2007;80:727–739. doi: 10.1086/513473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazei-Robison MS, Blakely RD. Expression studies of naturally occurring human dopamine transporter variants identifies a novel state of transporter inactivation associated with Val382Ala. Neuropharmacology. 2005;49:737–749. doi: 10.1016/j.neuropharm.2005.08.012. [DOI] [PubMed] [Google Scholar]
- Mazei-Robison MS, Blakely RD. ADHD and the dopamine transporter: are there reasons to pay attention? Handb Exp Pharmacol. 2006;175:373–415. doi: 10.1007/3-540-29784-7_17. [DOI] [PubMed] [Google Scholar]
- Mazei-Robison MS, Couch RS, Shelton RC, Stein MA, Blakely RD. Sequence variation in the human dopamine transporter gene in children with attention deficit hyperactivity disorder. Neuropharmacology. 2005;49:724–736. doi: 10.1016/j.neuropharm.2005.08.003. [DOI] [PubMed] [Google Scholar]
- Seidel S, Singer EA, Just H, Farhan H, Scholze P, Kudlacek O, Holy M, Koppatz K, Krivanek P, Freissmuth M, Sitte HH. Amphetamines take two to tango: an oligomer-based counter-transport model of neurotransmitter transport explores the amphetamine action. Mol Pharmacol. 2005;67:140–151. doi: 10.1124/mol.67.1.. [DOI] [PubMed] [Google Scholar]
- St George-Hyslop PH. Molecular genetics of Alzheimer's disease. Biol Psychiatry. 2000;47:183–199. doi: 10.1016/s0006-3223(99)00301-7. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Sonders MS, Poulsen NW, Galli A. Mechanisms of neurotransmitter release by amphetamines: a review. Prog Neurobiol. 2005;75:406–433. doi: 10.1016/j.pneurobio.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Thapar A, Holmes J, Poulton K, Harrington R. Genetic basis of attention deficit and hyperactivity. Br J Psychiatry. 1999;174:105–111. doi: 10.1192/bjp.174.2.105. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang G, Fowler JS, Logan J, Gerasimov M, Maynard L, Ding Y, Gatley SJ, Gifford A, Franceschi D. Therapeutic doses of oral methylphenidate significantly increase extracellular dopamine in the human brain. J Neurosci. 2001;21(RC121):1–5. doi: 10.1523/JNEUROSCI.21-02-j0001.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willams J, Dayan P. Dopamine, learning, and impulsivity: a biological account of attention-deficit/hyperactivity disorder. J Child Adolesc Psychopharmacol. 2005;15:160–179. doi: 10.1089/cap.2005.15.160. [DOI] [PubMed] [Google Scholar]
- Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporters. Nature. 2005;437:215–223. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]