

Abstract
Pancreatic endocrine tumors (PETs) can occur in as part of four inherited disorders including: Multiple Endocrine Neoplasia type 1 (MEN1), von Hippel-Lindau disease (VHL), neurofibromatosis 1(NF-1) [von Recklinghausen’s disease] and the tuberous sclerosis complex (TSC). The relative frequency with which patients with these disorders develop PETs is MEN1>VHL>NF-1>TSC. Over the last few years there have been major advances in the understanding of the genetics and molecular pathogenesis of these disorders as well in the localization, medical and surgical treatment of the PETs in these patients. The study of the PETs in these disorders has not only provided insights into the possible pathogenesis of sporadic PETs, but have also presented a number of unique management and treatment issues, some of which are applicable to patients with sporadic PETs. Therefore the study of PETs in these uncommon disorders has provided valuable insights that in many cases are applicable to the general group of patients with sporadic PETs. In this article these areas are briefly reviewed as well as the current state of knowledge of the PETs in these disorders and the controversies that exist in their management are briefly summarized and discussed.
Keywords: pancreatic endocrine tumors, neuroendocrine tumors, multiple endocrine neoplasia type 1, von Hippel-Lindau disease, neurofibromatosis 1, tuberous sclerosis, gastrinomas, insulinomas, Zollinger-Ellison syndrome
I. Introduction
Pancreatic endocrine tumors (PETs) have long fascinated clinicians because some can release biologically active hormones that cause distinct syndromes that provide important insights into the biological effects of these hormones in man 1–3. PETs can be divided into two general groups; functional PETs associated with symptoms due to ectopic secretion of hormones and nonfunctional PETs (NF-PET), which either don’t secrete or the products secreted do not cause a clinical syndrome (i.e. pancreatic polypeptide [PPoma], chromogranin A, ghrelin, neurotensin, subunits of chorionic gonadotropin, neuron- specific enolase) 1–4. Functional PETs include gastrinomas which ectopically releasing gastrin causing the Zollinger-Ellison syndrome (ZES) characterized by refractory peptic ulcer disease due to the gastric hypersecretion; insulinomas secreting insulin causing hypoglycemia; vipomas (Verner-Morrison syndrome, WHA syndrome, pancreatic cholera) secreting vasoactive intestinal peptide causing severe secretory diarrhea; glucagonomas secreting glucagon causing hyperglycemia, diabetes, and a characteristic skin rash, necrolytic migratory erythema; somatostatinomas secreting somatostatin causing diabetes, steatorrhea and gallstones and GRFomas secreting growth hormone-releasing factor (GRF) causing acromegaly 1,2,5,6. Other less common functional PETs reported to cause specific syndromes secrete luteinizing hormone, erythropoietin, renin and PYY have been described 3, but in general too few cases have been described to fully establish a syndrome. Each patient with a PET has potentially two treatment problems: treatment of the hormone excess state if present, and treatment of the PET itself, because except for insulinomas, >50% of the different PETs are malignant 1,3,7. In the last few years there have been marked advances in the treatment of the hormone excess states, the ability to localize these tumors and fully stage them and in treatment for advanced disease 8–10. Another importance advance is increased understanding that a subset of these tumors has an inherited basis and the insights being provided by study of this subset (Table 1)11,12.
Table 1.
Inherited PET syndromes
| Syndrome | Frequency | Location/type genetic abnormality | Altered protein function(s) | Frequency PETs | Type PET |
|---|---|---|---|---|---|
| Multiple Endocrine Neoplasia type 1 (MEN1)(Wermer’s syndrome) | Prevalence-1–10 per 100,000 | 11q13(encodes 610 amino acid protein, menin) | Nuclear location; exact function unclear- interacts JunD, NFκB, SMAD signaling pathways. Effects cell cycle, growth, genomic stability, and apoptosis. | 80–100% (microscopic)20–80% (clinical) | Nonfunctional (NF)(microscopic)>functional (20–80%) |
| Von Hippel-Lindau disease (VHL) | Prevalence-2–3 per 100,000 | 3p25(encodes 232 amino acid protein, [pVHL]) | Interacts elongins which act as transcriptional regulators that degrades HIF, regulates cell cycle, VEGF | 10–17% | >98% NF |
| Von Recklinghausen’s Disease (neurofibromatosis 1, NF-1) | Prevalence-1 per 4–5,000 | 17q11.2 (encodes2485 amino acid protein neurofibromin) | Ras GTPase-activating activity, bind microtubules, modulates adenylate cyclase, mTor-regulates growth, cell cytoskeleton | Uncommon (0–10%) | Duodenal somatostatinomas, rare PETs |
| Tuberous sclerosis (Bourneville’s disease) | Prevalence-1 per 10,000 | 9q34 (TSC1) (encodes 1164 amino acid protein, hamartin) 16p13(TSC2) (encodes 1807 amino acid protein, tuberin) | Interact PI3K signaling pathway regulating GTPase, mTor which plays a key role in growth, energy regulation, response to hypoxia, nutrients | Uncommon | Rarely develop functional, NF PETs |
Abbreviations. PET-pancreatic endocrine tumor; NF- nonfunctional PET; TSC-tuberous sclerosis complex, mTor-mammalian target of rapamycin; HIF, hypoxia-inducible factor; VEGA-vascular endothelial growth factor; PI3K-phosphoinositide 3-kinase;
There are four well-established diseases associated with inherited pancreatic endocrine tumors (PETs) (Table 1). These include Multiple Endocrine Neoplasia type 1 (MEN1)(Wermer’s syndrome), von Hippel-Lindau disease (VHL), von Recklinghausen’s disease (neurofibromatosis 1)(NF-1) and tuberous sclerosis. Each of these disorders shows autosomal dominant inheritance and the causative genes (MEN1, VHL, NF1, TSC1/2) (Table 1) all function as tumor suppressor genes 11,13–18. While there are occasional reports of other familial occurrences of PETs (insulinomas) 19 or gastrointestinal carcinoid tumors 20, which share many clinical and pathologic similarities to PETs 21, these have not been investigated in detail genetically and it is unclear whether they have a distinct etiology not related to the above syndromes.
Within the last twenty years there have been considerable advances in both the clinical understanding of these diseases as well as their molecular basis and treatment. The genetic defect(s) causing each of these inherited PET syndromes has been elucidated (Table 1) and considerable advances have been made in understanding the cellular roles of the various altered proteins (Table 1) and how alterations might lead to the observed clinical abnormalities seen. These genetic changes also provide important insights into the role of some of these genes in the pathogenesis of sporadic PETs, a group of tumors for which the understanding of their pathogenesis has remained relatively unclear, because it does not depend on the common oncogenes and tumor suppressor genes frequently altered in the common gastrointestinal adenocarcinomas 11,12. In the last few years there have been rapid advances in early diagnosis of these diseases and the associated PETs using genetic studies as well as improved imaging studies. These have led to a number of unique controversies over the treatment of PETs, especially when small, asymptomatic PETs are found in patients. In this article the information available on PETs in these inherited disorders will be briefly summarized, as well as the advances and controversies in diagnosis and treatment of PETs in these syndromes. Because PETs are much more a prominent part of the MEN1 syndrome than the other three syndromes (Table 1) and because the natural history of the PET in MEN1 patients is becoming the leading determinant of long term survival 22–25, the PETs in this disorder will be discussed in more detail.
II. Multiple Endocrine Neoplasia type 1 (MEN1)
II.A. General
MEN1 is characterized classically by tumors/hyperplasia of the parathyroid, enteropancreatic tissues and pituitary 13,14. Increasingly, tumors are being described in a number of different tissues including the adrenal, thyroid, CNS, skin (angiofibromas, collagenomas), smooth muscle as well as foregut carcinoids (thymus, gastric, lung)(Table 2, Fig. 1).
Table 2.
Frequency of general manifestations and of different PETs in MEN1
|
Shown are both the frequency reported in various series and when known the percentage that cause symptoms.
Abbreviations: CNS-central nervous system; VIPoma-vasoactive intestinal peptide secreting tumor; GRFoma-Growth hormone releasing factor secreting tumor;
Figure 1.

Location and frequency of tumors/hyperplasia in patients with MEN1. Location and frequency are graphed from the data for the 130 MEN1 patients reported in 14. Percentage refers to percent of the total 130 patients with the tumor in the indicated location.
II.B. Genetics/molecular pathogenesis
In 1988 26 the Men1 gene was mapped to chromosome region 11q13 using linkage analysis and in 1997 was cloned 27. It spanned 10 exons (9 coding) and encoding for a 610 amino acid nuclear protein, menin, which was widely expressed in endocrine and nonendocrine tissues 14,27 (Fig. 2). The intracellular functions of menin have not been fully elucidated, however an increasing amount is known about the numbers of proteins it interacts with 28,28–33 (Fig. 2). Menin interacts with a large number of proteins, which have important effects on transcriptional regulation, genomic stability, cell division and cell cycle control 28,28–33 (Fig. 2).
Figure 2.

Representation of the genomic organization of the MEN 1 gene, its encoded protein, menin, and the menin regions that interact with different proteins grouped according to their main functional activity. The 10-exon gene has a 1.8-kb coding region (shaded) from 9 exons, which encode for the 610 amino acid protein, menin. Menin has 3 nuclear recognition signals (NLS) and five putative guanosine triphosphatase (GTPase) sites indicated by the bars. Regions which have been implicated in binding to different interacting proteins are shown and they are divided by their main intracellular function. Abbreviations are explained in text and individual menin interactions discussed. Figure drawn form data in 28–33 and references in text.
In transcriptional regulation, menin interacts with the activating protein-1 transcription factor JunD and NF-κB resulting in inhibition of transcription caused by each of these proteins 28,30–32. Menin’s interaction with SMAD proteins (SMAD 3 and SMAD1/5 complex) results in enhanced transcriptional activation and TGFβ signaling as well as interaction with the bone morphogenetic protein(BMP)-2 signaling pathway and it also interacts with Runx2, which is a target of TGFβ and BMP as well as with mouse placental embryonic expression gene protein(Pem)28,32,34,35. Menin’s interacts with the homeobox-containing protein Pem can affect JNK and other MAP kinases signaling cascades 32,36. The forkhead transcription factor CHES1 which forms a transcriptional repressor complex that includes mSin3a, histone deacetetylase (HDAC) and HDAC-2 interacts with menin in a S-phase checkpoint pathway related to the cellular DNA damage response seem after ionizing radiation 37. Menin has been shown to bind in a histone methyltransferase complex containing MLL2, which is homologous to the mixed lineage leukemia (MLL) family of proteins that alter transcriptional activity 28,31,38–40. Menin in cooperative interaction with MLL proteins regulates cyclin-dependent kinase inhibitors, p27 and p18, which in turn regulate the clustered homeobox genes (Hox) which have important effects on proliferation 28,38–40. Menin has been shown to interact directly with the estrogen nuclear receptor (ERalpha) and to acts as a co-activator for ER alpha mediated transcription; therefore it acts as a critical link between recruitment of histone methyltransferase complexes and nuclear receptor –mediated transcription 41 (Fig. 2).
Menin also has been shown to bind directly with double-stranded DNA, which is mediated by the positively charged residues in the NLSs in the COOH terminus of menin (Fig. 2) 42. The NLSs are also important for interaction of menin with the insulin-like growth factor-2 (IGFBP-2) promoter resulting in repression of the expression of IGFBP-2 and in menin-mediated induction of caspases 8 expression, suggesting menin may act as a scaffold protein in coordinating activation and repression of gene transcription 43,44.
Menin associates with chromatin and nuclear matrix and from studies of menin in irradiated cells it has been proposed that menin might be important for repairing DNA damage 28. Menin interacts with a 32-kD subunit of replication protein A (RPA2) which is required for DNA replication, repair and recombination 45 and the FANCD2 protein which is also involved in DNA repair and whose mutations result in the inherited cancer-prone syndrome, Fanconi’s anemia46, suggesting that menin plays an important role in genomic stability (Fig. 2) 45–47. Menin also plays a role in cell division through its interaction with the non-muscle myosin II-A heavy chain protein (NMHC II-A) that is involved in cytokinesis and cell shape changes during cell division 48 and by interacting with the type III intermediate filament proteins, glial fibrillary acidic protein (GFAP) and vimentin 49. The latter interaction demonstrates that the intermediate filament network may serve as a cytoplasmic sequestering network for menin at the S and early G2 phase of the cell cycle 49. Menin also plays a role in cell cycle control through its interaction with the tumor metastases suppressor nm23/nucleoside diphosphate (NDP) kinase which is a multifunctional kinase which has been implicated in cell growth, differentiation, development and suppression of tumor metastases 50, as well as its interaction with the S-phase kinase ASK (Fig. 2) 51. ASK, a component of the Cdc/ASK kinase complex, is essential for cell proliferation, it is repressed by intact menin, but not by disease causing menin mutants, demonstrating a functional link between menin and ASK in the regulation of cell proliferation 51. Studies also suggest functions of menin related to telomeres with menin localizing in telomeres during the prophase 52 and menin inhibiting telomere expression by functioning as a direct repressor of the hTERT, an essential telomeric protein component 53.
Recent data suggest that menin and its interaction with the above proteins play an important role in physiological regulation of growth, control of the cell cycle, genomic stability and are potentially important in bone development and multipotent mesenchymal stem cell differentiation 28,32. Specifically, in the case of the pancreatic islets which may give rise to some PETs in MEN1 54, recent studies 47,55,56 show the loss of menin increases β cell proliferation by disrupting menin’s inhibitory effect on S-phase entry 47 and by disrupting menin-dependent histone methylation that maintains p27 and p18 inhibition of cyclin-dependent kinases 55.
Targeted disruption of the Men1 gene 57–59 resulted in mice with a similar phenotype to the disease that develops in humans with tumors of the pancreas, parathyroid, pituitary, thyroid and adrenal. Conditioned knockout of the Men1 gene 60,61 in pancreatic islets of the mouse, developed β cell hyperplasia and insulinomas in some animals by nine months. This late development of tumors even though both copies of the Men1 gene were removed, suggested that additional somatic events are required for adenoma formation in B cells.
Mutations in the Men1 gene are found in 78–93% of MEN1 families with 1336 different mutations reported which occur throughout the 9 exon-coding region 14,29,62. Overall 23% of the mutations are nonsense mutations, 9% slice-site mutations, 41% frameshift mutations or insertions, 6% in –frame deletions/insertions, 20% missense mutations and 1% whole or partial gene deletions 29. More than 75% of the mutations lead to truncated forms of menin 29. In general there is no genotype phenotype correlation 29. One study reported no association of the MEN1 mutation type with tumor behavior 63, however another study by Bartsch et al 64 reported that truncating mutations in the amino or COOH terminal regions of menin (exons 2,9,10) were associated with a significantly higher rate of malignant PETs (55 vs. 10%, p<0.05) and tended to have a shorter disease-free interval (26 vs. 92 mos, p=0.11).
PETs from patients with sporadic PETs (not due to MEN1) have loss of heterozygosity (LOH) at the MEN1 locus (11q13) in 5–93% and have a mutation in the Men1 gene in 27–39% 13,29,65,66. The presence of an MEN1 mutation in sporadic gastrinomas was not associated with more severe disease 66. These results support the conclusion that alterations in the Men1 gene may be important in the pathogenesis of a subset of sporadic PETs 11,29,65,66.
Global gene expression has been studied in a small number of MEN1 PETs (N=8) and compared to normal islets 67. Four-five genes were over-expressed and 148 genes under-expressed compared to normal islets including 19 apoptosis-related genes [specially, IER3, DDT, PHLDS2, IAPP], which were under-expressed 67.
II.C. MEN1-clinical aspects (Figure 1 and Table 2, top)
The classical manifestations of MEN1 are parathyroid hyperplasia leading to hyperparathyroidism (HPT) (95–100), PETs (symptomatic in 20–70%) and anterior pituitary tumors (54–65%) 13,14,68(Table 2, Fig. 1). Adrenal tumors, thyroid adenomas, carcinoid tumors (thymic, gastric, pulmonary), skin tumors, CNS tumor and smooth muscle tumors also occur in a significant number of MEN1 patients (Table 2, Fig. 1). Classically, almost all patients are reported to initially present with HPT (Fig. 3 panels A, B), however in numerous recent series up to 40% of patients present with a functional PET (usually an insulinoma or gastrinoma) and the patients were frequently initially thought to have sporadic PETs 13,68–72. In some of these cases the HPT is almost certainly missed. This occurs because, in most MEN1 patients, the HPT initially is mild and easily missed if careful assessments of parathyroid function are not performed with serum ionized calcium studies and assessment of intact plasma PTH levels using a sensitive IRMA assay 13,68,73. This point is well shown in a recent study 68 of ZES/MEN1 patients (107 NIH, 1009 literature patients) in which in 44% and 47% of the MEN1 patients, the ZES was clinically evident before the diagnosis of HPT and in 20–25% it was diagnosed 5 years earlier. This study showed if careful assessment of parathyroid function was carried out in these patients only 8% of patients found to have MEN1 who presented with ZES did not have HPT identified at the initial evaluations 68. The mean age of presentation in MEN1 patients not detected by genetic screening or family studies is 29–34 years old, the mean age of diagnosis is a decade later and the mean age of diagnosis for patients detected by biochemical screening in families is 25–29 years 68,74,75
Figure 3.
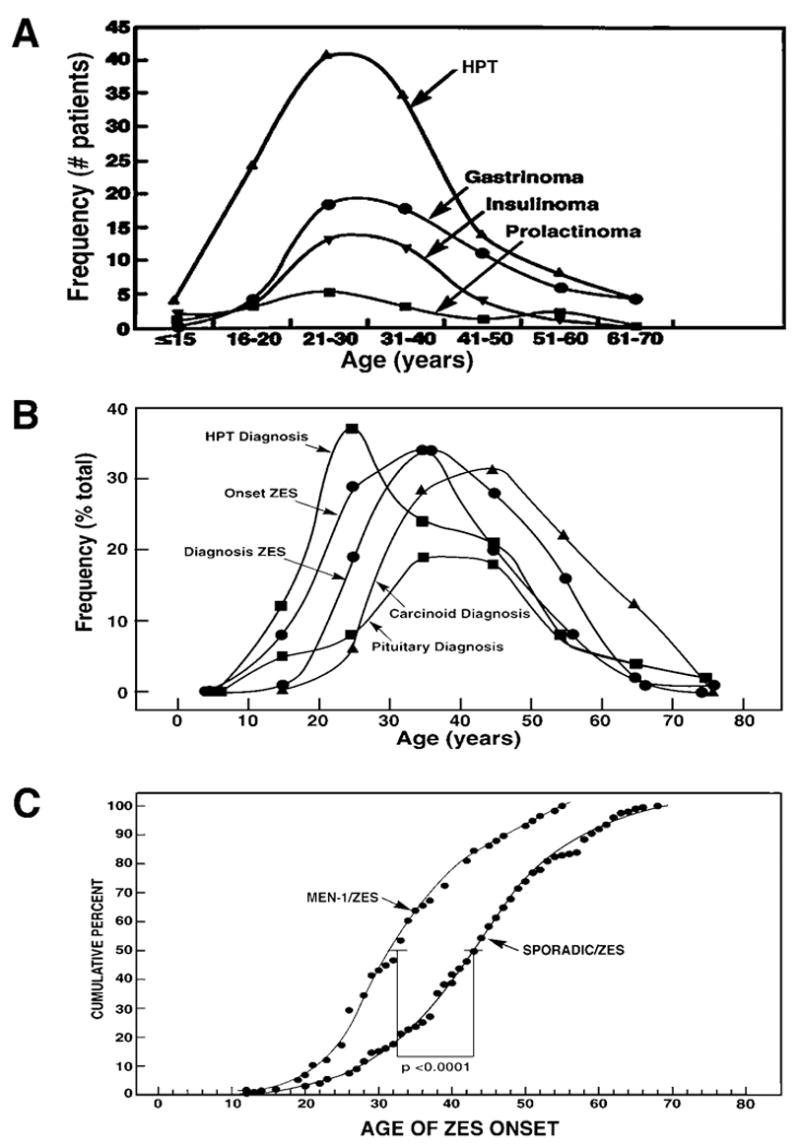
Age of diagnosis or onset of various PETs and other manifestations of MEN1 in all MEN1 patients (A, top) or MEN1/ZES patients (B, Middle) and comparison of age onset of ZES in MEN1 patients and patients with sporadic ZES. Panel A is drawn form data in 14, panel B from 68 and panel C from 85.
II.D. PET spectrum in MEN1 (Table 2, bottom)
II.D.1. PETs in MEN1: General
The most common PETs in MEN1 are nonfunctional tumors (80–100%), gastrinomas (mean-54% [range-20–61] and insulinomas (mean 18%[range-7–31%]) (Table 2). MEN1 is found in 10–54% (mean-25%) of all patients with gastrinomas 68,76 and 4% of insulinomas 77. Other functional PETs also occur but are present in <5% (Table 2) 78. The PETs characteristically are diagnosed between ages 30–50 68,78–83 (Fig. 3A,B) although a wide range exists varying from 5–15 years for the youngest with the different PETs to 68 to 80 years for the oldest 68,78,81,84. Both insulinomas and gastrinomas occur earlier in MEN1 patients than in patients with sporadic PETs and in a detailed study 85 in gastrinomas the difference was one decade (43.5 vs. 33.2 years) (Fig. 3C).
II.D.2. PETs in MEN1: pathology/origin of PETs
Except for gastrinomas and a rare somatostatinoma, all the other classes of PETs in MEN1 (Table 2) occur exclusively within the pancreas. In patients with ZES/MEN1 duodenal gastrinomas are present in 80–100% (Fig. 4A) and pancreatic gastrinomas in 0–15% in different series 79,86–89. The most characteristic MEN1 pancreatic lesion is the presence of diffuse microadenomatosis (lesions <0.5 mm) (Fig. 5D) which are present in almost every case in various series (Fig. 5D) 89–91. Macroadenomas frequently also occur (Fig. 5,A–D). The microadenomas primarily display a trabecular or mixed trabecular growth pattern surrounded by or interspersed with dense connective tissue 90,91. Most of the micro- and macroadenomas produce multiple hormones/peptides on immunohistochemical staining with 100% positive for generic neuroendocrine markers (neurons-specific enolase, chromogranin A, synaptophysin) followed by glucagon producing tumors (37–52%), insulin (15–27%), pancreatic polypeptide (17–20%), somatostatin-producing (5%), gastrin (1%) and VIP-producing (1%) 90–92. Studies of the duodenal gastrinomas in patients with ZES demonstrate they are usually small (<1 cm in diameter); they are multicentric (Fig. 4A–C), show trabecular and pseudoglandular histological patterns and stain positive for gastrin and occasionally for somatostatin 86,93,94. Lymph node metastases occur in 45–85%% of gastrinomas despite their small size, although liver metastases are uncommon from the duodenal gastrinomas 86,94–96.
Figure 4.
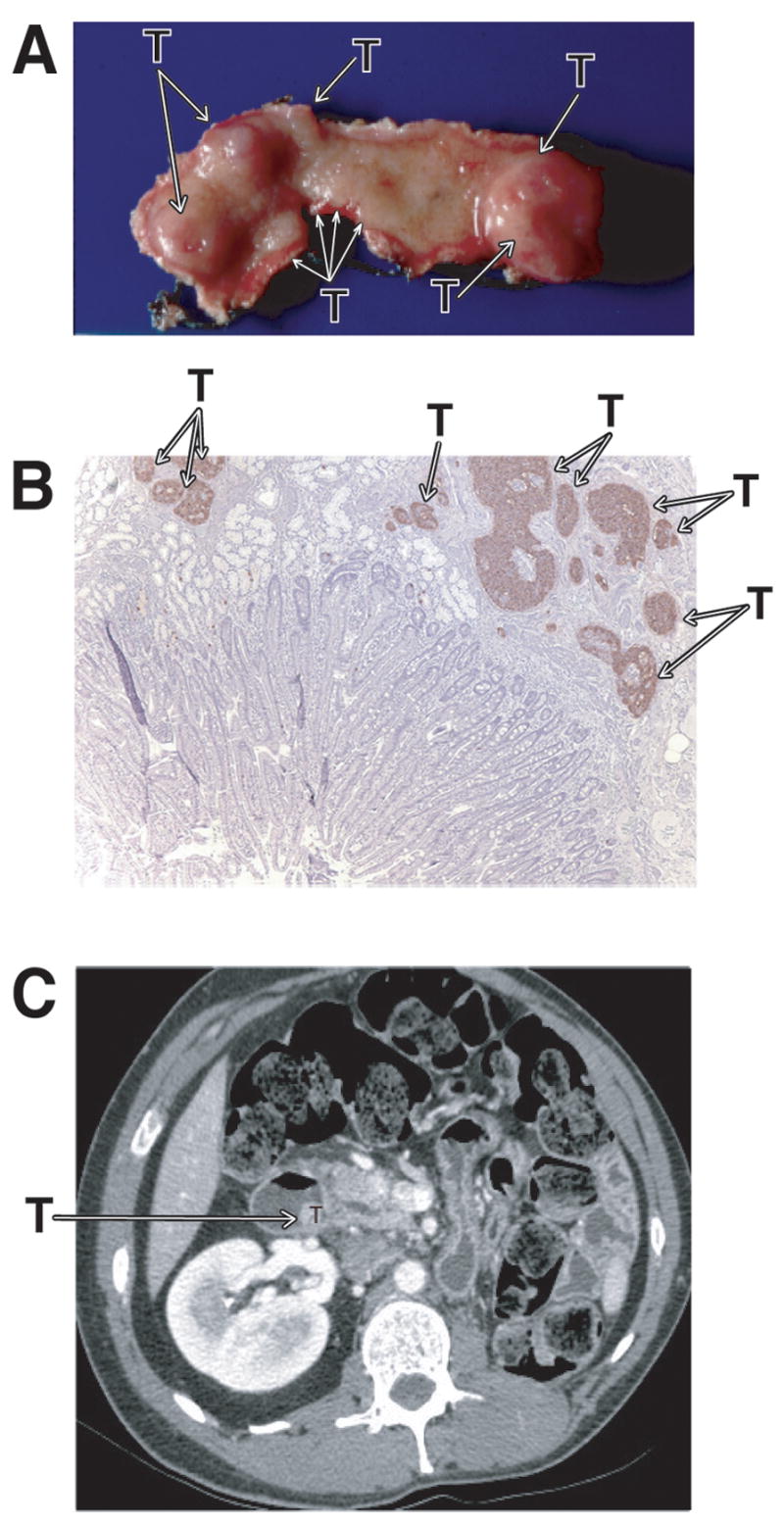
Characteristic multiplicity of duodenal gastrinomas PETs in MEN1/ZES patients and its appearance on CT scan. Panels A (gross specimen) and B (IHC-gastrin stain) show multiple small and larger duodenal gastrinomas (T=tumors) which limits the ability to cure patients MEN1/ZES patients with tumor enucleation 95 (see Figure 2B). Panel 1C shows the appearance of a duodenal gastrinoma which is projecting into the lumen but arises submucosally 93,98,201.
Figure 5.

Characteristic multiplicity of pancreatic PETs in MEN1 patients and the ability to image them. Panel 1A show an anterior somatostatin receptor scintigraphy (SRS) SPECT image demonstrating 4 tumors in this patient with MEN1/ZES. The tumor in the lower right was not in the pancreas but a gastric carcinoid, demonstrating the importance of not assuming all positive abdominal lesions are PETs in MEN1 patients. Panels 1B (SRS), 1C (CT) and 1D (histology) from a different patient show the multiple, intrapancreatic PETs (T=tumor) that are frequently seen in MEN1 patients.
There is no agreement on which cell gives rise to PETs in MEN1. On study 97 concluded that the pancreatic PETs arise from acinar/ductal structures, not from pancreatic islet tissue. Another study identified monohormonal endocrine cell clusters in MEN1 pancreas 54 and found that all pancreatic microadenomas and 95% of the hormonal cell clusters had loss of one Men1 allele and concluded that the microadenomas originated from islets. In a detailed study 98 of duodenal mucosa in patients with MEN1/ZES, all patients found to have proliferative, hyperplasic lesions consisting of gastrin cells in the nontumoral duodenal mucosa, similar to the gastric ECL cells seen in the gastric mucosa in patients with hypergastrinemia, as well as many having small microadenomas. Approximately 50% of the MEN1 duodenal microadenomas showed allelic deletion of the Men1 gene, whereas the endocrine cell precursor lesions retained heterozygosity 99. These lesions were not seen in MEN1 patients without ZES or sporadic ZES patients, which led the authors to propose that these proliferative lesions likely preceded the development of the duodenal gastrinomas in MEN1 patients but not patients with sporadic duodenal gastrinomas 98.
The mean size of PETs in various studies was 2.4–4 cm 23,78,81,83,100,101. However, the mean size varies markedly in the different PETs in MEN1 patients with gastrinomas averaging 0.7–1.2 cm in diameter (usually duodenal)78,98,101, insulinomas 0.2–2 cm in diameter 78,102, whereas with NF-PETs it was 3.1 cm 81,101, glucagonomas 3.25 cm 78, Vipomas 4 cm in diameter 78 and with somatostinomas 7 cm 78. In most studies multiple pancreatic PETs are found at surgery (mean 2.9–4.8, range 1–10) 23,81,101.
II.D.3. PETs in MEN1: Natural history/prognostic factors
The age-specific clinical penetrance or recognition of any manifestation of MEN1 (positive clinically or biochemically), in patients with known MEN1, for patients <20 years old is 10% and 52%, respectively; for patients < 30 years old it is 25% and 87%, respectively and by age 50 years old, 85–80% of patients with MEN1 had clinical symptoms 103. In one study the age specific penetrance of PETs in MEN1 patients was 15% at age 30, 49% at age 50 and 68% at age 70 104 and in the large GTE study (134 patients) 81 it was 9% at 20 years, 53% at 50 years and 84% at 80 years. The type of Men1 gene mutation did not affect the time course of penetrance of PETs 84. The survival is reported to differ significantly (p=0.007) in one study 78 between MEN1 patients with different PET subtypes. Specifically, the relative 10 year survival rates were gastrinomas, insulinomas (82–91%)>NF-PETs (62%)>glucagonomas, VIPomas, somatostatinomas (54%) 78 (Fig. 6, panel 1A). This result is also mirrored in the summary data of survival for any PET or different PETs from a number of studies in the literature (Table 4, Top). In patients with nonfunctional PETs, treatment options for the PET itself have to be considered because these are malignant in a percentage of patients (13%) (Table 3, bottom). Patients with functional PETs require two aspects of treatment: the hormone excess state must be treated and because a proportion of these tumors are also malignant (11–50% with the different PETs) (Table 3, bottom), in addition, treatment needs to be considered for the PET itself 1–3,23,105.
Figure 6.

Effect of various factors on survival in patients with PETs with MEN1 (1A, 1B, 2B, 3A, 3B) or without MEN1 (2A, 2B). Panels 1A, 3A show the effect of type of PET or lack of size of NF-PET on survival in MEN1 patients and are drawn from data in 81,128. Panels 1B, shows survival in patients with MEN1/ZES with (15%) or without (85%) aggressive tumors and is drawn from data in 105. Panel 2A shows the effect presence or extent of liver metastases in patients with sporadic ZES and is drawn form data in 96,215. Panel 2B compares the disease-free survival rate in patients with ZES with sporadic ZES or MEN1/ZES and is drawn from 95. Panel 3B shows the difference in survival for MEN1/ZES patients with no surgery with tumors <2 cm, (Group 1); group 2 with surgery with no liver metastases with a single (group 1A) or >2 PETs (2B) prior to surgery and group 3 with diffuse liver metastases and no surgery (drawn from data in 23.
Table 4.
Survival and cause of death in patients with MEN1 with PETs
| A. Survival in MEN1 patients with different PETs
| |||||||
|---|---|---|---|---|---|---|---|
| Survival (%)
|
|||||||
| PET
|
Years Follow-up
|
References
|
|||||
| 5
|
10
|
20
|
|||||
| Any PET | 82 ± 6 | 75 ± 5 | 52 ± 13 | 100,100,104,110,125,360 | |||
| NF-PET | 74 ± 16 | 49 ± 14 | ND | 78,81,100,125 | |||
| Gastrinoma | 89 ± 8 | 83 ± 9 | 67 ± 18 | 78,80,81,105,126,218,265,361 | |||
| Insulinoma | 95± 8 | 94 ± 1 | 92 ± 3 | 78,82,101,244 | |||
| Glucagonoma, | 78 | ||||||
| VIPoma, SSoma | 100 | 54 | 54 | ||||
|
B. Cause of death in MEN1 patients | |||||||
| Died/total
(%) |
MEN1 Related death
(%) |
PET related death
(%) |
ZES related death
(%) |
Malignant PET related death
(%) |
Mean age death
(%) |
Year of study | ref |
|
|
|
|
|||||
| 8/42 (19) | 100 | 100 | 88 | 12 | - | 1964 | 106 |
| 12/36 (33) | 100 | 83 | 75 | 16 | - | 1978 | 107 |
| 11/- | 100 | 100 | 91 | 9 | - | 1979 | 108 |
| 15/52 (29) | 100 | 87 | 73 | 14 | 41 | 1989 | 71 |
| 46/- | 43 | 30 | 4 | 26 | 51 | 1993 | 109 |
| 59/- | 46 | 30 | 10 | 20 | 55 | 1998 | 25 |
| 7/34 (20) | 86 | 71 | 0 | 71 | 50 | 1998 | 104 |
| 60/233 (26) | 28 | 19 | 2 | 17 | - | 2000 | 110 |
| 8/57 (14) (a) | 38 | 38 | 0 | 38 | - | 2002 | 105 |
| 30/84 (34) | 57 | 33 | 13 | 20 | 51 | 2003 | 111 |
| 16/55 (29) (b) | 81 | 62 | - | 62 | - | 2006 | 100 |
| 16/82 (20) | 68 | 82 | 27 | 73 | 60 | 2007 | 112 |
Abbreviations: SSoma-somatostatinoma, NF-PET-nonfunctional PET
all patients in this series had gastrinomas;
All patients in this series had a PET
Table 3.
Frequency of liver metastases and tumor extent at diagnosis in MEN1 patients
| I. Conventional imaging results (CT, MRI, Ultrasound) and/or laparotomy(1) | |||||
|---|---|---|---|---|---|
| No. MEN1 patients
|
% PET imaging
|
Imaging used
|
% with liver Metastases
|
Mean Age (yrs)
|
Year, Reference
|
| 41 | 39% | CT, US | 10% | 1996, 270 | |
| 69 | 40% | CT, US, Lap | 20% | 46 | 1998, 130 |
| 20 | 70% | CT, US, MRI | 20 | 29 | 1998, 75 |
| 8 | 38% | CT, MRI | 15% | 1998, 104 | |
| 130 | 66% | CT, US, MRI | 1998, 14 | ||
| 36 | 55% | CT, US, MRI | 51 | 1999, 233 | |
| 580 | 53% | CT, US, MRI | 8.2% | 40 | 2004 78 |
| 118 | 56% | CT, US, MRI | 5.1% | 37 | 2004 100 |
|
II. Metastatic PET to liver at Diagnosis in MEN1 compared to patients without MEN1 (sporadic)(1) | |||||
|
% with Liver Mets
|
|||||
| PET | Sporadic(1) | MEN1 | MEN1 reference | ||
|
|
|
|
|||
| A. Functional | |||||
| Insulinoma | 5% [5–15] | 0%–20% | 24,64,82,197,244,245 | ||
| Gastrinoma | 34% [13–53] | 11% | 23,24,68,79,80,88,218,358 | ||
| Vipoma | 60% [40–70] | 0 (0/2)–33%(1/3) | 24,78 | ||
| Glucagonoma | 60% [50–80] | 40% (2/5) | 78 | ||
| Somastatinoma | >70% | 50% (1/2) | 233 | ||
| GRFoma | >60% | unknown | |||
| B. Nonfunctional | 100 | ||||
| (NF-PET, PPoma, NToma, ghrelin-secreting, etc). | 60% [9–91] | 19% | 24,63,81,100 | ||
Series reporting percentage of total MEN1 patients with PET by imaging study with percentage with liver metastases and age of patient at time of identification of PET.
Abbreviations: NF-PET-nonfunctional PET; NToma-neurotensin-secreting PET; PPoma-pancreatic polypeptide-secreting PET)
In the past the main cause of death was the hormone excess states including renal failure due to uncontrolled hyperparathyroidism and the inability to control the PET hormone excess-state in patients with functional PETs 72,106–110 (Table 4, bottom). This has largely changed over the last 20 years with effects of uncontrolled hyperparathyroidism, advanced pituitary disease or failure to adequately treat the PET hormone excess state becoming an uncommon cause of death 25,100,104,105,110–112. This change has occurred because of increased use of careful serial assessments of parathyroid function, the use of appropriate surgical parathyroid resections (i.e. 3.5–4 gland resections), recognition with effective medical and surgical treatments of the pituitary disease, the increased diagnosis and effective surgical treatment of insulinomas and the development of effective medical treatments for the hormone excess states 88,113–115. The latter includes particularly the development of effective drugs for treatment of the gastric acid hypersecretion seen in ZES and the development of long-acting somatostatin analogues (lanreotide, octreotide), which control the symptoms of most of the other functional PETs 3,116. This was a particularly important advance in the case of ZES, which used to be the leading cause of MEN1 deaths (Table 4, bottom), because of its occurrence in more than one-half of MEN1 patients in most series (Table 2) and because of its high morbidity and mortality from untreated severe gastric acid hypersecretion 76,117–119 that occurs in most ZES patients 68,85,120. The result of these changes is that increasingly the natural history of the PET is becoming the main determining factor for survival (Table 4, bottom) 23,105,109,110. A second important determinant of survival in subset of patients (3–8%) recently recognized is the development of thymic carcinoid tumors, especially in males (i.e. 89–100% MEN1/thymic carcinoids in males), which occur later in the disease course usually after the onset of PETs or hyperparathyroidism (mean age diagnosis-50 years)(Fig. 1B) and which are particularly aggressive 68,109,121–123.
The current natural history, with more effective treatments as outline above, of MEN1 patients with or without PETs (except for those with MEN1/ZES, discussed in latter section) is not well defined by prospective studies. Patients with MEN1 are reported to have an increased incidence of premature death (mean age –46–54 years) with 28–100% of the patients dying from a MEN1-related cause and with PETs accounting for 19–100% of the MEN1 related deaths (Table 4, bottom) 104,109,110. The overall 10 and 20 year survival from diagnosis for all MEN1 patients is 85 % and 64%% 100,110 and patients with PETs have a decreased survival (Fig. 6, panel 1A) 72,81,109,110. The mean age at death in patients with NF-PETs is 43 81 and with MEN1 with or without a PET is 47 years old 25. The 5 and 10 year survival for MEN1 patients with any PET from a number of series is 82% [range 79–100%] and 75%, respectively (Table 4, top)(Fig. 6, panel 1A)81,100,104,110,124,125. In most 96,124,126 but not all 127 studies the survival rate of MEN1 patients with PETs is better than that of patients with sporadic PETs. At present the basis for this difference is not clear.
The extent of PET in MEN1 patients without gastrinomas in the literature is generally poorly studied. This has occurred because it has rarely been examined prospectively, all patients do not routinely undergo abdominal surgical exploration, and/or sensitive imaging modalities such positron emission tomography or somatostatin receptor scintigraphy (SRS) with SPECT imaging and CT/MRI fusing imaging, which might allow localization of small primaries and metastases, is not routinely available or used. In 8 different studies of MEN1 patients (Table 3, top) 52 ± 12%[range-38–70%] had a PET identified by conventional imaging studies (CT, MRI, Ultrasound) and 12 ± 6% [range 5.1–20%] had liver metastases detected. When the frequency of metastatic disease to the liver is compared in MEN1 patients with various functional or nonfunctional PETs, to the frequency seen in patients with sporadic PETs, in general it is reduced by more than one-half (Table 3, bottom panel). Specifically, the frequency of liver metastases in MEN1 patients with gastrinomas compared to patients with sporadic gastrinomas is reduced by 62%, with VIPomas or glucagonomas by almost 50%, with somatostatinomas by 30% and with nonfunctional PETs by 80% (Table 3, bottom panel). In contrast, the frequency of liver metastases in patients with insulinomas with MEN1 is not lower than that seen in series of patients with sporadic insulinomas, and may even be higher (i.e. 14% vs. 5%, Table 3, lower panel). At exploratory laparotomy in MEN1 patients with PETs without ZES the frequency of lymph node metastases is 16–33% 81,128,129.
Clinical prognostic factors for the growth of the PET/survival or development of liver metastases in MEN1 include (Table 5, top; Fig. 6, panel 3B): age>35 yrs at PET diagnosis 100,130; history of a first degree relative with a PET malignancy 130; presence of a nonfunctional PET or gastrinoma (Fig. 6, panel 1A) 81,100,130; presence of a large PET (i.e. > 3 cm) in some studies 78,81, but not others 24 (Fig. 6, panel 3A); presence of liver metastases 81,100 (Fig. 6, panel 3B); or history of an unsuccessful curative resection 81,100. Some 64,100 but not other 129 molecular studies have reported mutations of exon 2, 9 or 10 (truncating/frameshift) 64 or exon 2 100 are associated with a more aggressive tumors and decreased survival. These results in MEN1 patients generally show similarities to the results of prognostic studies in sporadic PETs (Fig. 6, panel 2A), which have been much more extensively studied 7,12,124,131–135. In sporadic PETs with multivariate analysis the presence of liver metastases (initially, their development, extent) has been the most important predictor of survival (p<0.00001) and to a lesser extent, poor differentiation and incomplete resection. In general, the presence of lymph node metastases has had minimal, if any, effect on survival in most studies 7,12,124,131–137, but not all 124,132,135. In most studies the presence of liver metastases is associated with larger primary tumor size (>2–3 cm) 7,12,124,131–134,136, but not all 124. Other important predictors in a number of studies are the presence of distant metastases, incomplete tumor resections, or the presence of a nonfunctional PET rather than a functional PET 7,125,131,132,132,134,138 (Table 5, top). In contrast to MEN1 PETs, sporadic PETs have been well-studied for histological predictors of aggressive behavior (i.e. high proliferative index [Ki67, PCNA] or mitotic index, vascular/neural/capsular invasion, nuclear pleomorphism) 7,131–135,139 and numerous molecular studies have identified predictive factors (p53 over-expression, LOH or chromosomal losses at certain loci [1,3p, 3q, 6q] or chromosomal gains [17p, 17q, 20q], presence of aneuploidy, and altered expression of a number of genes detected by gene array expression studies 11,12,62,131,135,140–143. In one study comparing sporadic PETs to normal islets, 667 genes were up-regulated [particularly BIN1, SERPINA10, LCK, BST2] and 223 genes down-regulated 143. Comparison of metastatic to primary tumors showed no gene expression differences in one study 143, whereas in a second study 144 65 genes were found overexpressed in metastatic PETs compared to well-differentiated non-metastatic PETs and 57 genes under-expressed. A number of different classes of genes were altered including growth factor-related genes (IGFBP1, IGFBP3, MET), developmental factors (TBX3, MEIS2), cytoskeletal factors (B1 tubulin, ACTN2), intracellular signaling and methytransferases (DYRK1A, PK1B, AK2, MGMT, GAMT) and DNA repair/regulatory molecules (CHEK1, ZNF198) 144.
Table 5.
Prognostic factors/characteristics of aggressive PETs and Gastrinomas in MEN1
| I. All PETs in MEN1 patients |
| A. Clinical/histologic Tumoral Features |
| A.1. Univariate analysis |
| Presence of a 1st degree relative with PET malignancy (odds ratio=3.7, p<0.05) 130 |
| Advanced age (p<0.05) 100,130 |
| Presence of a NF-PET (p<0.01) 81,100 |
| Presence of a gastrinoma (p<0.01) 81,130 |
| Primary tumor size >3 cm (p<0.01)78,81,134,218 |
| Presence of liver metastases (p<0.01) 81,134 |
| Noncurative resections (p<0.01) 81 |
| Presence of a VIPoma, SSoma, glucagonoma (p=0.01) 78 |
| Poor tumor differentiation (p=0.019) 134 |
| A.2. Multivariate analysis |
| Liver metastases (p=0.002)100,134 |
| Advanced age (0.0003) 100 |
| Presence of a NF-PET (p=0.037) 100 |
| Successful surgical resection (p=0.0043) 100 |
| B. Molecular Biologic Features |
| Presence of MEN1 gene truncations/frameshift mutation in exon 2,9,1064 or exon 2 mutation 100 |
| II. Gastrinomas in MEN1 patientsa |
| A. Clinical Tumoral Features |
| A.1. Univariate analysis |
| Presence of a large gastrinoma (>3 cm) (p<0.005) 218 |
| Male gender (p=0.022) 218 |
| Young age (<30 yrs old) of onset or diagnosis of ZES (p=0.03) 105 |
| Diagnosis 20 yrs prior to report (p=0.008) 218 |
| Development/presence of liver metastases (p=0.0012 23,105,218 |
| A.2. Multivariate analysis |
| Presence of a large primary gastrinoma (>3 cm) (p<0.0001) 105,218 |
| Development/presence of liver metastases (p=0.0012 23,105,218 |
II.D.4. PETs in MEN1: Imaging
Numerous studies in both MEN1 patients and patients with sporadic PETs demonstrate that the relative order of sensitivity of conventional imaging studies for localizing a PET is angiography>CT, MRI> transabdominal ultrasound (US)23,145–147. These studies show sensitivity is in large part dependent on primary tumor size, with detection rates of <20–30% of PETs<1 cm in diameter, 30–50% 1–3 cm and >70% for >3 cm 146,147. Therefore conventional imaging studies will miss most small PETs (<1 cm) and will not detect most duodenal gastrinomas 23,95,148–153 which are very frequently <0.5cm (Fig. 4A,B) 94,95,150. Greater than 80% of PETs both in patients with or without MEN1 over-express somatostatin receptors (sst 2,3,5) that bind the synthetic somatostatin analogues, lanreotide or octreotide with high affinity and this can be used to image these tumors (using 111Indium –labeled analogues) (Fig. 5A,B), to deliver somatostatin receptor-mediated radiotherapy (using 111Indium, 90Yttrium, 177Lutetium-labeled analogues) and to treat the hormone excess state 146,154–160. Somatostatin receptor scintigraphy (SRS) has greater sensitivity than conventional imaging studies in most reports 23,146,148,149,155,156,159, however, its sensitivity is also size dependent and it will miss 50% of small PETs/duodenal gastrinomas < 1 cm in diameter 149. SRS is also the most sensitive modality for detection of liver metastases 23,156,161–163, the detection of distant metastases (bone, distant lymph nodes, soft tissues) 164,165 and has the advantage of allowing whole body imaging at one time. SRS is equally sensitive in all PETs except for insulinomas, which because of the low density of somatostatin receptors that bind 111In penetreotide with high affinity, SRS is generally positive in <50% of the cases 155,157,166,167. The presence of MEN1 can introduce an added element of confusion in the interpretation of the SRS for PET localization in these patients. Increased densities of somatostatin receptors not only occur on PETs, but they can also occur on other MEN1 related neuroendocrine tumors (carcinoids [gastric, thymic, bronchial] (Fig. 5A), CNS tumors, thyroid tumors)122,155,157,168–171, as well as other neoplasms (breast, prostate, lymphomas) and in other diseases (granulomatous disease, inflammatory disorders, thyroid disease) 155,157,168. These can lead to false positive SRS’s for PETs, as well as various procedural aspects (isotope spill, retained isotope in gallbladder or intestine) or overexpression by normal tissues such as lymphocytes in injury (wound infection, abscess) 146,168. In one prospective study 168 of patients with PETs primarily without MEN1, 12% of all SRSs were false positive, however with proper clinical correlation this was reduced to 3%. Therefore in MEN1 patients it is important to consider that the uptake in the upper abdomen or at a distant site suggesting distant metastases in a patient with a PET could be another MEN1 related tumor (especially gastric carcinoid) (Fig. 5A), or another false positive cause. An example of such a patient is shown in Figure 4 (panel A) who had MEN1/ZES with SRS showing possible tumor localization in four areas in the upper abdomen of which 3 were due to pancreatic PETs, but the fourth was due to gastric carcinoid.
Increasingly endoscopic ultrasound (EUS) is being used to evaluate the number, size, location and presence of PETs in patients with MEN1 160,172–174. EUS shows excellent sensitivity for detection of intra-pancreatic tumors (82–100%), however it is less useful for duodenal gastrinomas. which it frequently misses (>50% in most studies) 131,152,172,173,175–177. In numerous studies EUS has greater sensitivity than SRS or conventional imaging studies for localizing primary PETs in patients with MEN1 160,173,174. EUS is able to detect smaller PETs than seen on SRS or conventional imaging studies in MEN1 patients, with a mean size of 0.6–1.1 cm [range 0.4–4 cm] in different studies 172,173,176, however in one study it missed a number of small PETs with a mean diameter of 3.2 mm (rang 2–6 mm) 173. Furthermore, EUS was able to detect multiple PETs (mean 3 [range 1–9]) in 55–100% of MEN1 patients even when asymptomatic 160,172,176,176,178,179. In various studies EUS detected PETs in 54–85% of MEN1 patients with non-functional PETs 172,176. Recent studies show EUS also has excellent specificity and reproducibility with a mean coefficient of variation with serial measurements of 5.5 ± 4.6% for PETs <5 mm in diameter and 4.4 ± 2.6% for PETs > 5 mm in diameter 160,179. This latter point is particularly important in MEN1 patients, because it has been proposed that serial EUS studies can be used to assess the growth of small PETs and determine when intervention should be considered 160,172,174,176,179. This latter point will be discussed in more detail in the later sections [Sections II.E.3.b and II.F.2.c] dealing with the surgical treatment of small PETs in MEN1 patients.
Recently two other imaging approaches are being increasingly used which will likely play an important role in the future management of PETs in patients with MEN1: the use of positron emission tomographic scanning 180–183 and the use of image fusion or hybrid techniques that allow the SRS image to be fused with a CT or MRI image 184,185. One of the main limitations of SRS is that only the anatomical area of the lesion is defined (i.e. pancreatic head area) and it does not allow clear definition of the exact anatomical location of the lesion seen (i.e. gastrinoma in pancreas or duodenal wall). The recent introduction of dual gamma camera/CT imaging systems allows the fusion of the CT result with the SRS result (SPECT/CT hybrid systems) allowing 3-dimensional localization that can define the exact location of the lesion seen 184,185. Preliminary results suggest this methodology many alter the clinical approach 184,185 and affect the management of the PET in up to 64% of patients 184. Positron emission tomographic scanning with 18F-labelled deoxyglucose (FDG), which is widely used in various common tumors, does not work well for PET imaging with the exception of tumors with high proliferative activity and low differentiation, which are the minority 186,187. Recent studies using 11C-5-HTP, 11C-labeled L-DOPA, 18F-labeled L-DOPA or 68Ga-DOTA-Tyr3-octreotide demonstrate increased sensitivity over SRS or conventional imaging studies for detecting both the primary PET site and the presence of liver metastases or distant metastases 180,181,183. In the future both the SRS/CT hybrid scanning and positron emission tomography using these radiolabeled analogues, and particularly 68Ga labelled somatostatin analogues which can be prepared using a reactor instead of requiring a cyclotron, will likely play an important role in the management of these patients.
II.E. Specific PET syndromes in MEN1
II.E.1. General: Specific PET syndromes
Three PET syndromes (gastrinomas, NF-PETs, insulinomas) present a number of special problems or issues either because of their frequency, insights provided or unique management issues and will be considered separately in the next section. The medical and surgical treatment of all MEN1-related PETs as well as controversies in these treatments will be considered in separate latter sections.
II.E.2. Gastrinomas
II.E.2.a Gastrinomas in MEN1: General
Gastrinomas are PETs, which ectopically secrete gastrin-causing hypergastrinemia, which results in the Zollinger-Ellison syndrome (ZES), which is due to the gastric acid hypersecretion resulting in refractory peptic ulcer disease 68,79,85,113,120. In addition, the hypergastrinemia has trophic effects on the gastric mucosa resulting in increased parietal cell mass and proliferation of gastric enterochromaffin-like cells (ECL cells), which can result in gastric carcinoid tumors, a proportion (20–30%) of which are malignant 170,188–192. Gastrinomas are the most common functional PET occurring in MEN1 patients and occur 3 times as frequent as insulinomas (54% vs. 18%) (Table 2) which is the reverse of their relative frequency in sporadic cases, where insulinomas are generally reported more frequently than gastrinomas 3,113. One peculiarity of the gastrinoma in MEN1 patients compared to sporadic cases, is the presence of the hypercalcemia due to the concomitant hyperparathyroidism that is frequently present in the MEN1 patients (Fig. 1, 2; Table 2), can influence the secretory behavior of the gastrinoma 88,115,193. After a successful parathyroidectomy in MEN1 patients with ZES, fasting gastrin levels and gastric acid secretory rates frequently fall, sometimes to normal ranges 88,115,193, and the secretin provocative test used to diagnose ZES 194 can become negative 193; making it difficult to diagnose the gastrinoma post parathyroidectomy in some patients. Whether correction of the hyperparathyroidism affects the gastrinoma growth in MEN1 patients is unknown.
II.E.2.b. Gastrinomas in MEN1: Pathology
MEN1 is the cause of ZES in 20–25% of all ZES patients in most series [range 10–48%]85,87,113 and gastrinomas are the most common, functional tumor in MEN1 patients, occurring in slightly more than one-half of the patients (Table 2). The gastrinomas are primarily in the duodenum (65–100% in most series), are almost invariably multiple, are usually small (usually < 1 cm), and are most common in the first and second parts of the duodenum 23,86,95,105,195–199 (Fig. 4A–C). Recent studies in patients with sporadic gastrinomas demonstrate these tumors show a clear proximal distal gradient of occurrence in the duodenum with a frequency of: 1st part of the duodenum (D1) (56% of all sporadic duodenal gastrinomas)>D2 (31%) >D3>(7%), (D4 (6%)150,200–203. Recent data suggest a similar proximal distal localization gradient occurs for duodenal gastrinomas in MEN1 patients 199. The duodenal gastrinomas are primarily submucosal in location, 34–85% are associated with lymph node metastases, whereas liver metastases are uncommon and only occur in 2–14 % of all patients (Table 2, bottom) at some time in the disease course and in 7% at diagnosis 23,80,86,95,105,195,198,203,204.
II.E.2.c. Gastrinomas in MEN1: Clinical
The mean age of onset of ZES in MEN1 patients is 33–39 years 80,85, and in one study was 10 years earlier than seen in patients with sporadic ZES (i.e., 33.2 vs.43.5 yrs)85 (Fig. 3B, C). The presenting clinical symptoms of ZES in MEN1 patients do not differ generally from those in patients with sporadic ZES with pain, diarrhea (66–76%)>heartburn> nausea/vomiting> weight loss 85. Similar to sporadic ZES, the vast majority have prominent gastric folds on endoscopy (95%), peptic ulcer disease (PUD) occurs in the majority (62%), only a minority develop PUD complications [perforation, bleeding (7–12%)], however they differ from sporadic ZES in manifestations of hyperparathyroidism (nephrolithiases-47% vs. 4%), or history of endocrinopathies (81% vs. 14%) 85. The delay in diagnosis of ZES in MEN1 patients is not different from that for sporadic disease (5.5 years) 85,205. Recently 206 MEN1/ZES patients were shown to have a 3-fold higher risk of severe esophageal reflux disease (GERD), 5-fold higher for development of the pre-malignant condition, Barrett’s esophagus, and 8-fold higher for developing esophageal dysplasia. A detailed analysis 206 suggested the earlier onset of ZES in MEN1 patients and inadequate as well as delayed treatment of the GERD, which can be more difficult to treat in MEN1/ZES patients than sporadic cases 113,207–209, were important contributing factors.
II.E.2.d. Gastrinomas in MEN1: Diagnosis of ZES
The diagnosis of ZES in MEN1 patients is similar to that in sporadic disease 210 in that it requires the demonstration of hypergastrinemia in the presence of hyperchlorhydria combined with other clinical or laboratory findings 5,87,210. Similar to sporadic ZES, 97–99% of MEN1/ZES patients have elevated fasting serum gastrin (FSG) levels with a median increase of 6-fold 205, so this is usually the initial determination 87,205,210. An exception to this is post successful parathyroidectomy, in MEN1/ZES patients, the FSG may return to normal, as well as the basal acid output and the secretin test becomes negative 88,88,115,193 masking the diagnosis of gastrinoma. No level of FSG alone is diagnostic of ZES because achlorhydria, presence of antral G cell hyperplasia, other diseases or the presence of H. pylori can all increase FSG 5,113,210–213. Similar to sporadic ZES, MEN1/ZES patients have marked basal acid hypersecretion 120 (mean increase -4 fold, 37.5 mEq/hr), however acid secretion rates are rarely measured, so this assessment is uncommonly used in the diagnosis. Recent studies suggest the following criteria for diagnosis of ZES in MEN1 patients; if the FSG≥10fold increased (40% of ZES) and the gastric pH is ≤2 (occurs 100% ZES patients), the diagnosis is established (after excluding retained antrum syndrome by history) 5,87,120,194,205,210,214; if the FSG is ≤10 fold increased and gastric pH≤2, then a secretin test (≥120 pg/ml increase is positive 194) and basal acid output (positive >15 mEq/hr) should be done 5,87,120,194,205,210. Using the above criteria for the secretin provocative test 94% of MEN patients will have a positive test with 100% specificity 194. Besides confusion post parathyroidectomy, as discussed above, the use of potent acid suppressant drugs such as proton pump inhibitors (omeprazole, lansoprazole, pantoprazole, rabeprazole, esomeprazole) (PPIs) can confound the diagnosis, because they can cause hypergastrinemia secondary to achlorhydria/hypochlorhydria, and thus they need to be carefully stopped for at least a week to allow assessment 5,210,212.
II.E.2.e. Gastrinomas in MEN1: natural history and prognostic factors (Table 5)
The natural history/prognostic factors for gastrinomas are considered separately from the other PETs, for a number of reasons. First, they are the most frequent malignant tumor causing clinical problems in MEN1 patients (Table 2). Second, they arise from the duodenum not the pancreas as the other PETs. Third, there have been extensive studies (clinical, laboratory, molecular) of the natural history/prognosis in sporadic ZES 7,12,96,131,215–217, which is also caused by primarily duodenal gastrinomas (>60% recent studies) 86,87,93,95,200, that can be used for comparison. Overall survival in MEN1/ZES in is 90–96% at 5 yrs, 75–95% at 10 years and 58–90% at 20 years. 80,96,105,126,218. In the NIH series where no patient died of acid-related conditions the survival in MEN1/ZES patients was 5 years-96%, 10 years 96% and 20 years (90%) 23,96,105.
Prognostic factors for MEN1/ZES include the presence of liver metastases (p=0.0012) and on univariate analysis the presence of male gender, the development of liver metastases (p=0.012), young age of onset or diagnosis (i.e. <30 years) (p=0.03) or the presence of a large primary tumor (>3 cm) (Table 5, bottom). These results are similar to studies with sporadic ZES where the presence, development or extent of liver metastases (Fig. 6, panel 2A) are the most important predictors of survival 96,105,113,157,215,219. In addition numerous predictors of aggressive tumor growth and/or the development liver metastases or survival have been described for sporadic gastrinomas including clinical factors [female gender, short disease duration at diagnosis of ZES, development of bone metastases and/or ectopic Cushing’s syndrome, control of acid hypersecretion]; laboratory [marked increase gastrin]; histopathological [low % nontetraploid aneuploid or high stem-line aneuploidy, nonduodenal location, tumor size>3 cm, rapid tumor growth], and molecular [high HER2/neu expression, high 1q LOH, high EGF receptor expression] 96,105,131,164,215,216,220–229. In three studies the presence of lymph node metastases had no effect on survival 96,229,230, in a second study 215 their presence had a weak effect on survival (p=0.03) and in a third study the 20 year survival was 83% in patients with lymph node metastases only 231. A particular important predictor for development of liver metastases in patients with sporadic ZES is the location of the primary gastrinoma 96,204,215,229. Duodenal and pancreatic gastrinomas showed equal rates of metastases to the lymph nodes (47–48%) 96, however, liver metastases occurred much more frequently in pancreatic gastrinomas than duodenal gastrinomas (47% vs. 5%, p<0.00001, 96. In general pancreatic gastrinomas are large (95% > 3 cm) 96 while duodenal gastrinomas are small (80%<1cm) 96 and it was unclear 96 whether the gastrinoma size or location was the critical determinant of the high metastatic rate to the liver. It is also unclear whether gastrinomas in the duodenum and pancreas in MEN1/ZES patients behave in a similar fashion to the sporadic cases, because there are too few pancreatic gastrinomas in MEN1/ZES described to determine the behavior pattern.
In some studies ZES in MEN1 patients was associated with a significantly lower metastatic rate to the liver compared to the sporadic form of ZES (6% vs. 22%) 96, whereas in others the liver metastatic rate did not differ 230. However during follow-up over a 9 year period both groups of ZES had the same rate of development of metastatic liver disease ((9%-MEN1/ZES and 5% for sporadic ZES) 96.
Prospective studies of both sporadic ZES and MEN1/ZES (Fig. 6, panel 1B) provide evidence for two forms of the disease: one characterized by aggressive tumor growth and the other by either no tumor growth or nonaggressive growth 96,105,215,216,232. With MEN1/ZES 14% of patients have tumors demonstrating aggressive growth) Fig. 6, panel 2A) 105, whereas with sporadic ZES in 24% aggressive growth is seen 96,215.
II.E.3. Nonfunctional PETs (NF-PETs) in MEN1
II.E.3.a NF-PETs in MEN1: General
NF-PETs are PETs, which cause no hormonal symptoms. Immunocytochemically almost all synthesize multiple peptides and many secrete pancreatic polypeptide (50–70%) (PPomas), chromogranin A (44%) 233,234 and other hormones which don’t cause symptoms 235–238. NF-PETs occur histologically in 80–100% of MEN1 pancreases as multiple, microadenomas (<0.5 cm) 89–91 with 55–82% of patients also having macroadenomas (>1 cm) 90,91. These pancreatic microadenomas on immunocytochemistry (IHC) show multiple hormone production in 38–100% with neuron-specific enolase IHC seen in 100%, PP (20–75%), glucagon (24–52%), insulin (15–42%), somatostatin (3–58%), gastrin (4–33%) and <1–8% (VIP, neurotensin) 89–92. As is apparent the term NF-PET is a misnomer in that even though these PETs usually produce by IHC multiple peptides, because no clinical symptoms of hormone excess occur, they are clinically NF-PETs. An additional unusual feature is that even though these adenomas are found histologically in almost every MEN1 patient, in series reporting the presence of NF-PETs detected by conventional imaging studies they are present in only 1.1%74, 4% 14 (Fig. 1) and 18% 81 of all the MEN1 patients. EUS has increased sensitivity and is able to detect multiple PETs (mean 3 [range 1–9]) in 55–100% of MEN1 patients even when asymptomatic160,172,176,176,178,179.
II.E.3.b NF-PETs in MEN1: clinical/pathology
In the GTE series of 579 MEN1 patients, NF-PETs were the most frequent PET, occurring in 3% of patient’s age 20 yrs, 34% aged 50 yrs and 53% of patients aged 80 yrs 81. NF-PETs are diagnosed at an average age of 36.2 years with and average time from MEN1 diagnosis to NF-PET diagnosis of 5 years [range 0–33] 81. Most were primarily discovered by imaging studies with symptoms occurring in only 0–13% (Table 2) 81. In various series only 9–19% of the NF-PETs were associated with liver metastases (Table 3, bottom), 14–33% with lymph node metastases 63,81 and in the GTE series of 108 NF-PETs/MEN1, there was a correlation of primary size (p<0.01) with the frequency of liver metastases. Specifically, only 4% of patients with NF-PETs ≤1 cm in diameter had liver metastases, 10% with 1.1–2 cm, 18% with 2.1–3 cm and 43% with NF-PETs> 3cm 81. Patients with NF-PETs with MEN1 had a similar survival curve to patients with gastrinomas with MEN1 and each had a significantly shorter life expectancy than MEN1 patients with no PET (Fig. 6, panel 1A) 81. However, MEN1 patients with NF-PETs ≤2 cm did not have a decreased survival (Fig. 6, panel 3A) 128 so the decrease in overall survival of NF-PET patients was due to decreased survival occurring in the patients with larger NF-PETs 81,128. Biochemical screening by some using a standardized meal test 236,239, but not others 235,240, as well as the routine use of EUS 160,172,174,175,178, has allowed the detection of NF-PETs in patients with negative conventional imaging modalities and at an earlier age, often in the third decade.
Recently, using serial imaging studies (especially EUS), the natural history of NF-PETs in MEN1 patients has been examined in a number of studies 174,176,241. In various studies 128,241 8% of the NF-PETs and in 7% 241 and 38% 176 of the patients with a NF-PET, there was an increased in tumor size over a 4–7 year follow-up. In one study 241 none of the PETs ≤2cm in size increased in size over a 6 year follow-up, whereas in a second study 174 of 20 MEN1 patients with NF-PETs<1.5 cm followed over a 20 month period, the largest tumor increased 1.3%/mo with an annual tumor incidence rate of 0.62 new tumor/patient/year. Over a 4–7 year period 38–100% of the patients developed new NF-PETs and in some patients the growth was rapid 174,176,241. The conclusion of a number of these studies is that small NF-PETs appear to generally show indolent growth, however, in some patients faster growing tumors occur 174,241, and thus regular follow-up EUS’s were recommended with the frequency depending of the NF-PET’s size. The results above demonstrate that although 55–82% of MEN1 patients have macroadenomas histologically, in most series they are not detected by routine conventional imaging studies, they are rarely symptomatic, however with EUS they can be detected in most patients. At present the treatment of NF-PETs, especially if small (≤ 2cm in diameter) is controversial 63,64,64,81,100,128,174,176,198,239,241,242 and will be discussed in the general surgery section below.
II.E.4. Insulinomas in MEN1
II.E.4.a Insulinomas in MEN1: General/Clinical/pathology
Insulinomas are PETs secreting insulin, which results in hypoglycemic symptoms 6,243. Insulinomas occur in slightly less that one-fifth of MEN1 patients (18%, Table 2, bottom) and MEN1 is an uncommon cause of insulinoma in most large series of insulinomas patients (sporadic and MEN1) accounting for only 4% 77. Insulinomas are diagnosed at a mean age of 36.4 yrs in MEN1 patients in one study 82, at 24 yrs old in a second study 244 and at 34 yrs in a review of 53 cases in the literature 245. In 14–29.5% of patients the insulinoma was the initial revelation of MEN1 244,245 and the insulinoma was associated with another functional tumor in 34% of patients (ZES>glucagonoma≫VIPoma) in one study 82, in 10% in a second study (ZES, glucagonoma-1 case each) 244 and in 42% in a third study (all ZES) 245.
Insulinomas in MEN1 patients, similar to patients with sporadic insulinomas (Table 3, bottom) have a low rate of malignancy, with 14% of patients showing liver metastases in the large GTE study of 44 MEN1 insulinoma patients 82, 5% (1/19 patients) in a review of the Mayo clinic cases 244, 0% (0/7 and 0/10, patients) in three studies 24,64,197 and in 20% of 43 cases in the literature 245. In two series 24,244 10% of the insulinoma patients had lymph node metastases, whereas in another study (0%) (0/6 patients 64 had lymph node metastases. In MEN1, in contrast to sporadic cases 6,77,243, in most studies the insulinomas are reported to be multiple (76–89%) 244,245 and they are usually small (mean size-0.6 cm [range 0.2–1.6 cm]) 244. The insulinomas are relatively evenly distributed throughout the pancreas similar to their localization in sporadic disease 6,77,243–245.
The importance of tumor localization, medical and surgical treatment will be consider in the following sections dealing with treatment of PETs in MEN1.
II.F. Treatment of PETs in MEN1
II.F.1. Treatment of PETs: General
In functional PETS the hormone excess-state needs to be diagnosed and controlled acutely and long-term until possible tumor resection. In both functional and NF-PETs treatment directed at the PET per se needs to be considered, because a percentage of these tumors are malignant (Table 3, bottom) and they are an increasingly prominent cause of death in these patients (Table 4, bottom; Fig. 6, panels 1A, 1B, 3B). Furthermore, in the case of some functional PETs (insulinomas, VIPomas, glucagonomas, somatostatinoma), surgical resection has a high probability of cure 64,78,82,102,197,244–248.
PETs in MEN1 present special diagnostic and treatment problems because of their invariable multiplicity. This leads to the need to determine which of the multiple PETs that might be present is the functional PET(s) responsible for the functional syndrome so it can be removed at surgery. Furthermore, the multiplicity of the PETs in the pancreas means that cure of all NF-PETs is not possible without total pancreatectomy. Furthermore, the invariable multiplicity of duodenal gastrinomas is one of the main contributors to the inability to cure patients with MEN1/ZES patients long-term (Fig. 6, panel 2A) 23,64,94,95,131,197,199,249 without aggressive resections such as a Whipple procedure (Table 6).
Table 6.
Surgical Cure Rate in MEN1 and ZES with and without pancreaticoduodenectomy, and MEN1 and insulinoma.
| MEN1/ZES - NON-PANCREATICODUODENECTOMY
| ||
|---|---|---|
| Author (year)
|
# patients
|
Long-term # disease –free (%)
|
| van Heerden (1986) 275 | 25 | 0 (0)) |
| Samaan (1989) 75 | 5 | 0 (0)} |
| Pipeleers-Marichal (1990) 86 | 4 | 2 (50) |
| Cherner (1992) 362 | 1 | 0 (0) |
| Farley (1992) 127 | 15 | 0 (0) |
| Grama (1992) 197 | 12 | 0 (0) |
| Melvin (1993) 126 | 19 | 1 (5) |
| Mignon (1995) 361 | 36 | 1 (3) |
| MacFarlane (1995) 94 | 10 | 0 (0) |
| Thompson (1997, 1998) 248,271 | 27 | 9 (33) |
| Kisker (1998) 363 | 2 | 0 (0) |
| Jordan (1999) 364 | 3 | 0 (0) |
| Norton (1999) 95 | 28 | 2 (6) |
| Bartsch (2000) 64 | 7 | 3 (42) |
| Kato (2000) 365 | 2 | 2 (100) |
| Norton (2001) 23 | 48 | 0 (0) |
| Thodiyl (2001) 366 | 1 | 0 (0) |
| Gauger and Thompson (2001) 367 | 37 | 13 (33) |
|
| ||
|
MEN1/ZES - PANCREATICODUODENECTOMY
| ||
| Waddell (1968) 368 | 1 | 1 (100) |
| Lind (1989) 369 | 1 | 1 (100) |
| Pipeleers-Marichal (1990) 86 | 2 | 2 (100) |
| Delcore (1992) 370 | 2 | 2 (100) |
| Stadil (1995) 283 | 3 | 3 (100) |
| Schroder (1996) 371 | 1 | 1 (100) |
| Phan (1997) 284 | 6 | 6 (100) |
| Jordan (1999) 363 | 1 | 0 (0) |
| Bartsch (2000) 64 | 3 | 3 (100) |
| Kato (2000) 365 | 2 | 2 (100) |
| Norton (2001) 23 | 2 | 0 (0) |
| Sarmiento (2002) 372 | 1 | 1 (100) |
| Tonelli (2006) 101 | 13 | 10 (77) |
| Bartsch (2005) 373 | 4 | 4 (100) |
|
| ||
|
MEN1/INSULINOMA
| ||
| Rasbach (1985) 102 | 12 | 10 (83) |
| Sheppard (1989) 196 | 3 | 3 (100) |
| Jadoul (1990) 247 | 2 | 2 (100) |
| Demeure (1993) 245 | 6 | 5 (83) |
| O’Riordain (1994) 244 | 19 | 17 (89) |
| Thompson (1998) 248 | 6 | 6 (100) |
| Cougard (2000) 82 | 44 | 27 (61) |
| Bartsch (2005) 101 | 4 | 4 (100) |
| Fernandez-Cruz (2005)373 | 2 | 2 (100) |
| Tonelli (2006) 198 | 3 | 3 (100) |
| Norton (2006) 246 | 7 | 6 (86) |
II.F.2. Treatment of PETs hormone-excess state: Medical
II.F.2.a Gastrinomas
In ZES/MEN1 patients in older studies (Table 4, bottom), the complications of the severe peptic ulcer disease due to the unchecked gastric acid hypersecretion, were the main cause of death and total gastrectomy was the only proven effective treatment 117–119. With the development of potent acid suppressant drugs such as the histamine H2 receptor antagonists in the 1970 (cimetidine, ranitidine and later others) and more recently inhibitors of the H+K+ ATPase (or proton pump inhibitors [PPIs])(omeprazole, lansoprazole, esomeprazole, pantoprazole, rabeprazole), the acid secretion can now be controlled in almost every patient by once or twice a day dosing 5,116,207,209,250–253. Acute control of the gastric hypersecretion is now simplified with the availability of parental PPIs which can be given every 4–8 hours and which have largely replaced the parenteral histamine H2 receptor antagonists, which were effective, but high doses administered by continuous infusion were required 254–259. Oral PPIs are safe and effective long-term in MEN1/ZES patients and tachyphylaxis does not develop, however the hypercalcemia due to the hyperparathyroidism makes the patients less sensitive to antisecretory drugs 88,115,193, with the result higher doses with more frequent PPI dosing is required, both initially and long -term in MEN1/ZES patients than in patients with sporadic ZES 207,209,250.
II.F.2.a Other PETs-treatment of hormone excess-state
Insulinomas in MEN1 patients, similar to sporadic disease, are almost invariably treated surgically, except for the small percentage with metastatic disease to the liver (Table 3, bottom) 6,243,260. Initially, to control the hypoglycemia prior to surgery, diazoxide combined with frequent feedings is generally used (50–60% respond)6,243,260 and some use long-acting somatostatin analogues such as octreotide or lanreotide 6,243,260. The somatostatin analogues need to be used with care, because in some cases they may make the hypoglycemia worse by suppressing counter-regulatory hormones (glucagon, etc) and also some insulinomas have very low densities of sst2, 3,5 somatostatin receptors that bind octreotide/lanreotide with high affinity and thus these agents are effective in only 40–50% of patients6,155,157,166,167,243,260–262.
With all other functional PETs in MEN1 (VIPomas, glucagonomas, GRFomas, somatostatinomas), the drugs of choice for control of the hormone excess-state prior to surgery or in patients who are not cured surgically are long acting somatostatin analogues (octreotide/lanreotide) 1,263,264. For long-term treatment, depot forms of these two drugs are now increasingly used which require once a month dosing (octreotide-LAR, lanreotide autogel)1,263,264.
II.F.2. Treatment of PETs: Surgical
II.F.2.a General: controversies
Whereas there is no controversy in the need for surgical treatment of a PET causing direct tumor -related symptoms or for a number of functional PETs in MEN1 (insulinoma, glucagonoma, vipoma, somatostatinoma, GRFoma) which can frequently be cured 64,78,82,102,197,244–248, the role of surgery in patients with some familial MEN1 pancreatic and duodenal neuroendocrine tumor (NET) syndromes, especially with small NF-PETs (<2 cm) or gastrinomas is controversial63,64,64,81,83,95,100,128,131,174,176,198,239,241,242,246,265,266. The controversy not only involves the timing of surgery, if any, but also the type of operation that should be routinely performed. This controversy has resulted in some groups recommending surgery not be routinely done in patients with MEN1/ZES or NF-PETs without direct tumor symptoms (pain, etc) 267,268, others recommend surgery only be performed if PETs are found of a certain size on preoperative imaging studies (>2 or >3 cm)23,94,95,128,218, whereas still others recommend that all such MEN1 patients with a PET undergo surgical exploration 269,270. The controversy on the type of operation involves how aggressive surgery should be for gastrinomas in the pancreatic head area and also whether routine distal pancreatectomy should be performed at the time of surgery in any MEN1 patient, to reduce the amount of pancreatic tissue that could potentially develop new PETs in the future 23,64,95,271,272. These controversies have occurred because the hormone-excess state in MEN1/ZES/NF-PET patients can be either controlled or requires no treatment; the tumors are invariably multiple and frequently small, making cure unlikely without aggressive surgery; most patients with small gastrinomas/NF-PETs have a 15 year life-expectancy > 90% (Fig. 1B, 3A, 3B)23,83,96,105,128 and because the natural history of these PETs is to a large extent unknown, as well as their importance as a cause of death in MEN1 patients. Recent studies show that malignant neuroendocrine tumors (NETs) are becoming the main determinant of long-term survival in MEN1 patients and a prominent role is played by malignant PETs (Table 4, bottom). Because numerous studies show a subset of these patients develop aggressive PETs or PETs with rapid growth 23,25,105,109,128,130,273,274 and because of the increasing importance of PETs as a determinant of survival (Table 4), almost all investigators agree surgical treatment is indicated for some patients with MEN1/ZES or NF-PETs, but there is no agreement on which group of these patients.
The role of surgery is becoming more clearly defined, in some cases, as we have better understanding of surgical outcomes and the natural history of neuroendocrine tumors in the familial setting. For example, in patients with ZES/MEN1 controversies about the role of surgery has clearly changed with the development of new drugs to treat gastric acid hypersecretion. Initially surgery for ZES in MEN-1 patients was designed to control the severe peptic ulcer disease and total gastrectomy was required, as lesser procedures resulted in recurrent symptoms 76,117,119. Initially, the gastrinomas were seldom found at surgery because surgeons focused primarily on the pancreas 113,118,196,275,276. Now it is clear that the most MEN1 gastrinomas (>80%) are in the duodenum and not the pancreas 86,93,94,248. This inability to find tumor in the early operations, further supported the use of total gastrectomy. However, since PPIs have been developed to effectively control the acid hypersecretion without symptoms or complications in all patients with ZES, surgery can now focus on removal of the gastrinoma for potential cure of ZES and control of the tumoral process. This is still controversial, because recent studies show that MEN1 ZES patients are seldom cured by simple resection of duodenal tumor (Table 6, top panel).
However, the role of surgery is becoming less well defined in other cases, such as with NF-PETs in MEN1 patients or the role of when patients should be re-operated. With the increased routine use of sensitive imaging modalities in MEN1 patients, especially with endoscopic ultrasound, the presence and location of NF-PETs is being better defined and the detection rate is approaching the 80% penetrance seen in some autopsy studies 160,172,176,176,178,179. This has raised a new set of controversies about whether these small asymptomatic PETs should be routinely removed or how they should be managed 128,160,172,176,176,178,179,273. With an increasing numbers of MEN1 patients with PETs undergoing initial surgery that does not result in cure (i.e. especially patients with gastrinomas, NF-PETs), there is an increasing question of when re-operation for a PET is indicated 277–279. At present there are no studies specifically on MEN1 patients with PETs that provide enough information on this point to address this question, whose importance will increase in the future as these patients continue to live longer.. A similar question exists in patients with sporadic PETs (especially gastrinomas where long-term up >50% are not cured) 277–279, however the limited information is unlikely to be applicable to MEN1 patients with PETs because of the difference in multiplicity of tumors, different PET natural history and differ surgical cure rates with PETs (especially gastrinomas/NF-PETs). Because the controversies and role of surgery differ in the different PETs in MEN1 patients, they will be considered briefly, separately below.
II.F.2.b Surgery for localized gastrinomas
Since 1990 it is appreciated that the majority (>80%) of gastrinomas in patients with MEN-1/ZES arise in the duodenum 86,87,276. Prior to 1990 this lack of knowledge of the location of the primary gastrinoma very likely contributed to the low cure rate in MEN1/ZES patients (Table 6, top) and the controversy at that time about the value of routine surgical exploration in MEN1/ZES. With this increased appreciation of the duodenal location of the gastrinomas and the routine use of duodenotomy at the time of surgery, which increases the number of duodenal tumors found and cure-rate in sporadic ZES patients 95,150,150,153,276,280–282, it was hoped that a similar increase in cure rate would be seen in MEN1/ZES patients. However, this was not the case 23,94,95, because even with a duodenotomy the 5 year cure rate in MEN1/ZES patients is very low or zero, without aggressive resections such as pancreaticoduodenectomy (Fig. 3B). This likely occurs because duodenal gastrinomas in MEN1/ZES patients, in contrast to sporadic ZES, are invariably multiple, frequently very small and thus difficult to find even with a duodenotomy (Fig. 4A, B), and are associated with lymph node metastases in 50–86% of cases 23,86,93–96,150,201. Since gastrinomas in MEN-1 are usually in the gastrinoma triangle 268 that includes the head of the pancreas and the duodenum, some advocate the use of more radical procedures like Whipple pancreaticoduodenectomy for improved cure-rate 150,198,269,283,284.
Pancreaticoduodenectomy has been performed in a limited of MEN1/ZES patients and generally results in a high (77–100%) cure rate (Table 6). However, most experts do not recommend Whipple pancreaticoduodenectomy for the surgical management of gastrinoma 22,23,83,129,150,248. This recommendation is made both because of the increased understanding of the natural history of MEN1/ZES patients with PET <2 cm on imaging and because of the possible side-effects of Whipple resections. A recent study 23 reports the disease-related survival in patients with MEN1/ZES with PETs<2 cm on imaging and no surgery is 100% at 15 years 23. Numerous studies show that lymph node metastases may develop in a proportions of these patients 23,86,94 however in a most studies, they alone are not associated with decreased survival 96,113,218,231,232, only the development of liver metastases 7,113,215,218. A large prospective study showed duodenal gastrinomas metastasize to the liver with a 10-fold lower frequency than pancreatic PETs and liver metastases are uncommon in patients with duodenal gastrinomas (1–5%) 88,96. A Whipple procedure is clearly associated with a higher complication and death rate than simple excision or enucleation of pancreatic and duodenal tumors. Whipple resections may have long-term complications like weight loss, diabetes and malabsorption and the frequency of these or other late complications is largely unknown. Furthermore, Whipple resection makes subsequent re-operation much more difficult. Finally, liver metastases occurring after Whipple procedure cannot be treated with chemo-embolization, because of an increased risk of ascending infection. Nevertheless, because of the dramatic increase in cure rate and the ability to totally remove the entire gastrinoma triangle, Whipple pancreaticoduodenectomy is clearly indicated in some patients with gastrinoma. It is recommended to be considered in MEN-1 ZES patients with good health and a large pancreatic head or duodenal tumor that is not able to be simply excised; multiple localized lymph nodes; or if the patient is not cured after removal of a duodenal or pancreatic head gastrinoma and develops recurrent tumor that is increasing in size and localized to the gastrinoma triangle.
At present the operative approach that is most widely used in patients with MEN1/ZES includes the use of intra-operative ultrasound, enucleation of tumors in the pancreatic head, duodenotomy and excision of duodenal wall tumors, systematic removal of lymph nodes around the pancreas, along the celiac trunk and hepatic ligament with or without routine distal pancreatectomy-splenectomy 23,64,95,271,272. The role of the routine distal pancreatectomy is controversial and will be discussed in the surgical treatment of NF-PETs below. At present most recommend that 23,94,95,218,285 an operation be performed only in MEN1/ZES patients with imaged tumor>2cm, because of the low possibility of cure (Table 6), their excellent long term survival and quality of life with smaller tumors 23, the morbidity of more advanced surgical procedures and the lack of evidence that routine exploration in this group of patients extends life. Others 248,286 recommend such a procedure be performed in all MEN1/ZES patients. At present without long-term prospective studies and a clear definition of the importance of duodenal gastrinomas in altering survival these different approaches will not be easily resolved.
II.F.2.c Surgery for NF-PETs in MEN1
Similar to gastrinomas in MEN1/ZES patients there is no general agreement on the approach to small NF-PETs (<2 cm) in MEN1 patients 63,128,160,172,176,176,178,179,241,266,270. All agree that NF-PETs >2–3 cm should be routinely removed from MEN1 patients, even if asymptomatic, because numerous studies demonstrate that pancreatic PETs of this size are associated with an increased incidence of liver metastases/decreased survival 7,24,96,215,218,241. Furthermore, studies show that the decreased survival of MEN1 patients with NF-PETs (Fig. 6, panel 1A)273 is not due to the PETs<2 cm (Fig. 6, panel 3A) 128, but instead to the inclusion of patients with NF-PETs >2 cm in diameter. It is not proven in MEN1 patients that the removal of larger PETs (>2 cm) will prevent the subsequent development of liver metastases and increase disease-related survival, however recent studies in patients with sporadic gastrinoma show the routine surgical removal of the primary PET decreases the subsequent rate of developing liver metastases and increases survival 225,226. Controversy exists for the small NF-PETs (<2 cm) in asymptomatic MEN1 patients, because a recent GTE study 128, demonstrated that, similar to the finding with small gastrinomas in MEN1 patients 23, there was no decrease in survival in patients with small NF-PETs (<2 cm) with MEN1 (Fig. 6, panel 3A) 128. Furthermore, a second study 241demonstrated no growth of NF-PETs<2cm in MEN1 patients and no patient developed liver metastases on follow-up. Recently it has been proposed that serial EUS studies could be used to assess the growth of small NF-PETs and determine when intervention should be considered 160,172,174,176,179. Recent studies show EUS also has excellent specificity and reproducibility for assessing the size of pancreatic PETs with a mean coefficient of variation with serial measurements of 5.5 ± 4.6% for PETs <5 mm in diameter and 4.4 ± 2.6% for PETs > 5 mm in diameter 160,179. Such serial studies may be helpful in identifying the subset of small NF-PETs that demonstrate rapid growth 241, however at present no study has established criteria that have been evaluated serially in a large number of patients.
There is also controversy whether the standard operation in MEN1 patients with NF-PETs or for any PET, should include a routine distal pancreatectomy with some advocating this procedure 22,64,248,271,272 and others not 23,94,287. The National Comprehensive Cancer network guidelines for surgery for MEN1 patients with PETs recently recommend the use of routine distal pancreatectomy 288. Although it seems logical that this approach may reduce the subsequent development of PETs that could be clinically significant, it has not been shown to extend life or decreases the liver metastases rate that is the primary determinant of long-term survival. In one study 23 in which distal pancreatectomy was not routinely performed, the 15 year survival rate was 92%. It was recommended that distal pancreatectomy not be performed, because of the excellent survival without it, its possibility of leading to the development of glucose intolerance, increasing the complication rate and reducing the possibility of a subsequent pancreaticoduodectomy, if it is needed 23. The possibility of such routine resections leading to glucose intolerance or diabetes is receiving increased attention, because a recent study shows MEN1 patients have an increased occurrence of glucose intolerance and diabetes 289. Furthermore, whereas it is reported that such surgery rarely results in the development of diabetes by some 22, a number of studies indicate that impairment of glucose intolerance after such a procedure is relatively common and therefore raise questions about its routine use 100,241,289.
II.F.2.d Surgery for Insulinomas in MEN1
The surgical cure rate in patients with sporadic insulinoma is nearly 100% as they are usually benign and simply enucleated or resected 6,264,290. With MEN1, most patients (61–100%) are still cured by removal of a dominant pancreatic PET (Table 6), but some will develop recurrent hypoglycemia or show persistent hypoglycemia suggesting the presence of other or multiple pancreatic insulinomas (Table 6) 82,102,244–246. The role of surgery is more critical in these patients than those with gastrinomas, as the medical management of the hypoglycemia is less efficacious than the acid management of gastrinoma and the possibility of long-term cure is much higher with simple PET enucleation (Table 6). Although a number of studies 244,245 report multiple PETs removed at surgery in MEN1 patients with insulinomas and the implication is that most of these patients have multiple insulinomas, this is not clearly established. Insulinoma patients typically have a dominant NET that secretes excessive insulin and causes the neuroglycopenic symptoms and the resected pancreas contains other smaller PETs. However, the other NETs may not be secreting insulin. Immunohistochemistry for insulin is seldom done in prior series so it is impossible to determine if these other tumors are insulinomas.
II.F.2.d Surgery for other PETs in MEN1
Even though Vipomas, somatostatinomas glucagonomas and GRFomas are uncommon in MEN1 patients (Table 2) they can cause severe symptoms, which may or may not be well controlled with long acting somatostatin analogues. These patients may be cured 70,78,101,196 and therefore if the PET or its metastases are resectable, these patients should have a surgical exploration and tumor resection whenever possible.
II.F.3. Treatment of advanced disease in patients with PETs/MEN1
Metastatic disease to the liver or distant sites occurs in a proportion of MEN1 patients with different PETs, although it is at a lower rate in MEN1 PETs than seen in sporadic PETs. (Table 3). In many patients the metastatic disease shows either no growth or indolent growth and no immediate anti-tumor treatment, except to control the hormone excess-state, is needed 105,216,218. However, in a subset of patients (Fig 5, panel 1B) aggressive tumor growth occurs 100,105,197,218. Both in these patients, those with tumor symptoms due to the metastatic disease or those with the hormone-excess state poorly controlled, anti-tumor treatment is indicated. In most patients the liver metastases are diffuse and surgical resection is only possible in <15% 105,131,291,292. Numerous treatments are reported to be effective including cytoreductive surgery, chemotherapy [primarily streptozotocin/doxorubicin], biotherapy with somatostatin analogues or interferon, chemoembolization of the liver, treatment with 111Indium-, 90Yttrium-, 177 Lutetium-labeled somatostatin analogues, and various new agents (temozolamide, thalidomide, VEGFR inhibitors, tyrosine kinase inhibitors, mTor inhibitors). These treatments will not be further discussed here, because they are similar to those used in malignant sporadic PETs and have been covered in a number of recent reviews 8,18,154,291–295.
IV. Von Hippel-Lindau Disease (VHL)
IV.A. General
VHL is an autosomal dominant neoplastic disorder due to germline mutations in the VHL gene on chromosome 3p25 (Table 1) 15,296. Germline mutations in this gene lead to benign and malignant tumors in a number of tissues as well as cyst development in a number of organs 15,296. Characteristically, VHL patients develop hemangioblastomas of the retina (25–60%) and craniospinal region [cerebellum (44–72%), brainstem (10–25%), spinal cord (13–50%)]; endolymphatic sac tumors (10%); renal cell carcinomas or cysts (25–60%); pheochromocytomas (10–20%); and epididymal cytadenomas (25–60%) 15,296. Pancreatic tumors or cysts develop in 35–77% of patients in most series with the majority being cysts, with the pancreatic tumors including cytadenomas (12 %), hemangioblastomas (<1%), adenocarcinomas (<1%) and PETs (Table 1)15,296–305,305–310
IV.B. Genetics/molecular pathogenesis
The VHL gene encodes for a 232 amino acid protein, pVHL which forms a complex with various proteins including elongin B and C and cullin 2 which determines ubiquitin-dependent proteolysis of large cellular proteins (Table 1) 15,296. In normal oxygen levels, this complex targets and degrades hypoxia-inducible factors 1 and 2 (HIF1 and 2). Altered pVHL, as occurs in VHL, can lead to increased HIF effects because of failure to degrade it, which include increased glucose uptake, increased expression of angiogenic, growth and mitogenic factors including VEGF, PDGFβ, TGF α and erythropoietin 15,296. The exact mechanism by which this results in the formation of PETs is not clear.
Although PETs develop in VHL patients there is no evidence of mutations in the VHL gene in a subset of sporadic PETs 12,142. In one study of sporadic PETs 142, 33% had 3p LOH which is the region of the VHL gene, but no mutations were found in any sporadic PET in the VHL gene, leading to the proposal that there may be another tumor suppressor gene adjacent to VHL gene that could play a role in the PET formation in this subset of patients. This latter proposal was supported by a study of a VHL kindred 311 with prominent PET formation, which showed 3p LOH proximal to the VHL locus, also suggesting this area might contain an unknown tumor suppressor gene, besides VHL, which is critical for the PET formation in VHL patients.
IV.B. PETs in VHL
IV.B.1. PETs in VHL.General/pathology
PETs develop in 10–17% of VHL patients (Table 1) and they are almost invariably nonfunctional and usually asymptomatic, although they may occasional cause pancreatitis or a mass causing pain 15,296–309,312. In 7.6% of patients only the pancreas is affected 297. In almost every series 100% of the PETs are NF-PETs 297,300,306,312. The mean age of diagnosis of the PET is 29–38 years and the majority of patients, in contrast to those with MEN1, have a single PET (67–70%) 299–301,306,312. Malignant PETs occur in 8%–50 % with metastases to the liver in 9–37% 297,299–302,306,312. The mean size of the PETs is 2.6–4.3 cm (range-1–10 cm) 301,306 and in various studies 52–70%, 28–33% and 7–21% are located in the pancreatic head, body and tail, respectively 297,298,301,306. IHC demonstrated chromogranin A in 78–100%, synaptophysin (100%), neuron specific enolase (100%), with 35% demonstrating focal staining for pancreatic polypeptide, insulin, somatostatin, and/or glucagon and 0% for gastrin 297,306,307.
IV.B.2 PETs in VHL. Natural history and prognosis
In one study of 30 patients with VHL with PETs, 7%(2/30) died of metastatic disease due to the PET during follow-up 306 and in a second study 312 2% (2/108) died of metastatic disease due to the PET. In both studies 306,312 the size of the primary PET correlated with the presence of hepatic metastases (mean- 5 cm-liver metastases vs. 2 cm, no metastases, p=0.0013). No patient in these two studies 298,306 with a primary <3 cm had hepatic metastases and in a third study 312 metastases were much more frequent in patients with primary tumors >3 cm (69% vs. 19%, P<0.005). PETs <3 cm demonstrated a longer mean doubling time (mean 2.5 vs. 1 yr) than did larger PETs (≥3cm) 298 and the mean doubling time of the primary tumor was 8-times faster in patients with metastatic disease than those without metastatic disease (334 days vs. 2628 days, p<0.001)312. The mean age of death of VHL patients from any cause is 40–48 years 313–315 and the PET is, in general, an uncommon cause of death in VHL patients, with most dying of metastatic renal cell cancer or complications of the cerebellar hemangioblastomas 18,313–315.
IV.B.3 PETs in VHL. Treatment of the PETS
Similar to NF-PETs, the indications for treatment of PETs in VHL depends on two factors: whether the PET is symptomatic and the perceived risk of malignancy. Functional PETs have only very rarely been reported in VHL 300,301,315 and thus 100% are NF-PETs clinically. Nevertheless, they can on occasion (<5% in most series) cause symptoms because of expansion or local invasion (pancreatitis, pain, etc) 297,300,309 and this would be an indication for surgical treatment. Because most PETs in VHL remain asymptomatic, show indolent growth, and no liver metastases are reported in patients with PETs <3 cm, three series recommend that only PETs≥ 3 cm should be routinely resected 297,306,312. A recent study 312 of 108 patients with VHL and PETs reported all patients with metastatic disease had at least two of three important factors associated with malignancy: a primary tumor>3 cm, mutation in exon 3 of the VHL gene or a primary tumor doubling time >500 days. This study 312 recommended that VHL patients without 2 of these three PET prognostic factors could be followed. One study 306 recommended that VHL patients with PETs < 1cm be followed at 12-month intervals with serial CT scans and patients with PETs 1–3 cm should be monitored and resection is recommended if growth to >2 cm. With the accuracy of EUS and its increased availability, the growth of these PETs could be much more carefully assessed and it is likely in the future, similar to the case with NF-PETs in MEN1 patients discussed above, that criteria could be developed to better select these patients for surgery.
V. Von Recklinghausen’s syndrome [neurofibromatosis 1 (NF-1)]
V.A. General
NF-1 is a relatively common disorder occurring in 1-4,000-5000 live births and is due to alterations in the NF-1 gene on chromosome 17q11.2, which encodes for a 2485 amino acid protein, neurofibromin (Table 1) 16,316,317. The principal clinical manifestations are: cafe au lait macules (>99%), neurofibromas (cutaneous>99%, deep-seated-44%), skin-fold freckling (85%), iris Lisch nodules (iris hamartomas seen on slit lamp examination) (>95%), optic pathway gliomas (15%) and bony dysplasia (sphenoid wing and bowing of long bones of the bones) 16,316. CNS abnormalities are common with learning problems (30–60%), attention deficit hyperactivity disorder (38%) and epilepsy (6–7%) 16,316,317. Renal artery stenosis (2%), pheochromocytomas (2%) and an increase risk of hypertension are important clinically in some patients 16,316,317.
V.B. Genetics/molecular pathogenesis
Neurofibromin, acts as a tumor suppressor, and is especially widely expressed in the nervous system. Neurofibromin affects cell proliferation/growth and signaling by regulating the activation of p21 ras by its Ras GTPase protein-activating activity, binding microtubules, modulating adenylate cyclase activity, interacting with the cellular cytoskeleton and by interacting through a signaling pathway with tuberin, the TSC2 gene product, it regulates mTor, which is a serine-threonine kinase that regulates cell growth and proliferation (Table 1) 316–318.
V.C. PETs in NF-1
V.C.1 PETs in NF-1: Clinical, pathology
PETs, almost exclusively duodenal somatostatinomas, occur in NF-1 patients, but are relatively uncommon (0–10%) (Table 1) 87,319–323. However, NF-1 accounted for 48% of the total duodenal somatostatinomas reported in the literature in one review 322 and 23% 324 of all ampullary carcinoid tumors. In NF-1 rare patients are reported with pancreatic somatostatinomas (16 times less common than duodenal somatostinomas) 322, ZES 325,326, an insulinoma 327–329 or a nonfunctioning pancreatic PET 330 and increasing numbers are reported with GI stromal tumors 331,332 which have been suggested to be the most common NF-1 associated GI tumor.
Characteristically these duodenal somatostatinomas occur in the periampullary region 87,319–323 and have a mean size of 2.8 cm (range 1–5 cm) 322. In one series of 71 cases of ampullary PETs reviewed in the literature 333 18% were caused by NF-1. These ampullary somatostinomas are almost always hormonally silent and do not cause a functional somatostatinoma syndrome 87,333,334. In contrast to other duodenal neuroendocrine tumors, these periampullary tumors more frequently present with jaundice, cause biliary dilatation, pancreatitis and can cause pain, nausea, bleeding or vomiting 87,332,335,336. Duodenal somatostatinomas my be suspected histologically because they may show a glandular pattern with psammoma bodies (63%) 87,322,334,337,338. Metastases to liver and/or lymph nodes occur in 30% 322. Duodenal somatostatinomas in patients with NF-1 are similar to sporadic duodenal somatostatinomas in most respects [size, psammoma body presence, metastatic rate], however they are less likely to produce other hormones on IHC examination (18% vs. 43%) 322. In contrast, both NF-1 and sporadic duodenal somatostatinomas differ from sporadic pancreatic somatostatinomas in a number of respects 322 including: frequency of producing the clinical somatostatinoma syndrome (1–2% vs. 66%), tumor size (2.8 vs. 5.9 cm), psammoma body production (61% vs. 0%) and presence of metastases (30% vs. 71%) 322.
V.C.2 PETs in NF-1: Treatment
NF1 patients have a 10–15 year decrease in life-span (median age of death-59 years) 339, with the most common cause of death being malignancy 339–341. In one series 1 of 11 deaths (9%) was due to a PET 341, whereas in a large population study 339 malignant neoplasms of the connective tissue or other soft tissues were an increased cause of death by 34 times compared to a non-NF-1 patients and CNS tumor 5.5 times increased, but GI/pancreatic tumors were not an increased cause of death 339. Nevertheless, because duodenal somatostatinomas are malignant in 30%, because of their periampullary location they can grow and cause symptoms, and because tumor size is not a good predictor of tumor behavior with periampullary tumors 333,342, many experts recommend either a Whipple resection or pylorus-sparing pancreatico-duodenectomy be performed, particularly for PETs>2cm 324,343. However, satisfactory local resections 324,343,344 (especially with PETs <2 cm) inducing endoscopic resection 344, have been reported. Therefore, if after careful EUS examination, only local disease is seen which is thought locally resectable, then a more conservative approach could be considered.
VI. Tuberous sclerosis [Bourneville’s disease)
VI.A. General/genetics/molecular pathogenesis
Tuberous sclerosis is an autosomal dominant neuro-cutaneous multi-system disorder, which is due to mutations in one of two genes, the tuberous sclerosis complex 1 gene (TSC1)(encoding for hamartin) or TSC2 (encoding for tuberin) (Table 1) 17,345,346. TSC is characterized by the development of hamartomas in almost every organ and patients develop disabling neurological features (epilepsy, mental retardation, autism), dermatological features (facial angiofibromas, hypomelanotic macules, shagreen patch, ungula fibromas); and tumor-like hamartomatous lesions (cortical tubers, cardiac rhabdomyomas, subependymal nodules, renal angiomyolipomas and lymphangiomyomastosis) 17,345,346.
The products of the TSC1 and TSC2 genes, hamartin and tuberin form a dimer, which is important in the PI3K signaling pathway 17,345,346. They are also important for regulation of the small GTPase, rheb, which is turn is involved in regulating mTOR activity and through this cascade and others can affect protein translation and protein synthesis, growth and proliferation, cellular energy levels and nutrient availability (Table 1) 17,345,346.
VI.B. PETs in TSC
PETs both functional 347–352 and nonfunctional 353–356 are reported in a small percentage of patients with TSC usually occurring in patients with TSC2 354. In TSC patients with functional PETs,, tumors reported include gastrinomas causing ZES 347 and insulinomas 348–352. In patients with a functional tumor, the hormone excess state needs to be controlled medically and/or surgically as discussed above with MEN1. Furthermore, in some TSC patients the PETs are malignant 353,355 so that appropriate surgical treatment of the PETs should be considered.
Acknowledgments
This work was partially supported by intramural funds of NIDDK, NIH and R01 CA 096645-01
References
- 1.Oberg K, Eriksson B. Endocrine tumours of the pancreas. Best Pract Res Clin Gastroenterol. 2005;19:753–81. doi: 10.1016/j.bpg.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 2.Mignon M, Jensen RT, editors. Endocrine Tumors of the Pancreas: Recent Advances in Research and Management. Vol. 23. Basel, Switzerland: S. Karger; 1995. [Google Scholar]
- 3.Jensen RT, Norton JA. Endocrine tumors of the panceas and gastrointestinal tract. In: Feldman M, Friedman LS, Brandt LJ, editors. Sleisinger and Fordtrans’s Gastrointestinal and Liver Disease. 8. Philadelphia: Saunders; 2006. pp. 625–66. [Google Scholar]
- 4.Falconi M, Plockinger U, Kwekkeboom DJ, et al. Well-differentiated pancreatic nonfunctioning tumors/carcinoma. Neuroendocrinology. 2006;84:196–211. doi: 10.1159/000098012. [DOI] [PubMed] [Google Scholar]
- 5.Jensen RT, Niederle B, Mitry E, et al. Gastrinoma (duodenal and pancreatic) Neuroendocrinology. 2006;84:173–82. doi: 10.1159/000098009. [DOI] [PubMed] [Google Scholar]
- 6.Grant CS. Insulinoma. Best Pract Res Clin Gastroenterol. 2005;19:783–98. doi: 10.1016/j.bpg.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 7.Jensen RT. Natural history of digestive endocrine tumors. In: Mignon M, Colombel JF, editors. Recent advances in pathophysiology and management of inflammatory bowel diseases and digestive endocrine tumors. Paris, France: John Libbey Eurotext Publishing Co.; 1999. pp. 192–219. [Google Scholar]
- 8.Arnold R, editor. Endocrine tumors of the Gastrointestinal Tract: Part 1. 4. Vol. 19. 2005. [Google Scholar]
- 9.Arnold R, editor. Endocrine Tumors of the Gastrointestinal Tract: Part 11. 19. Vol. 5. 2005. [Google Scholar]
- 10.Jensen RT, Doherty GM. Carcinoid tumors and the carcinoid syndrome. In: DeVita VT Jr, Hellman S, Rosenberg SA, editors. Cancer: Principles and practice of Oncology. 7. Philadelphia: Lippincott Williams and Wilkins; 2005. pp. 1559–74. [Google Scholar]
- 11.Duerr EM, Chung DC. Molecular genetics of neuroendocrine tumors. Best Pract Res Clin Endocrinol Metab. 2007;21:1–14. doi: 10.1016/j.beem.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 12.Corleto VD, Delle Fave G, Jensen RT. Molecular insights into gastrointestinal neuroendocrine tumors: importance and recent advances. Dig Liver Dis. 2002;34:668–80. doi: 10.1016/s1590-8658(02)80212-2. [DOI] [PubMed] [Google Scholar]
- 13.Thakker RV. Multiple endocrine neoplasia type 1. Endocrinol Metab Clin North Am. 2000;29:541–67. doi: 10.1016/s0889-8529(05)70150-x. [DOI] [PubMed] [Google Scholar]
- 14.Marx S, Spiegel AM, Skarulis MC, et al. Multiple endocrine neoplasia type 1: clinical and genetic topics. Ann Intern Med. 1998;129:484–94. doi: 10.7326/0003-4819-129-6-199809150-00011. [DOI] [PubMed] [Google Scholar]
- 15.Lonser RR, Glenn GM, Walther M, et al. von Hippel-Lindau disease. Lancet. 2003;361:2059–67. doi: 10.1016/S0140-6736(03)13643-4. [DOI] [PubMed] [Google Scholar]
- 16.Ferner RE. Neurofibromatosis 1. Eur J Hum Genet. 2007;15:131–8. doi: 10.1038/sj.ejhg.5201676. [DOI] [PubMed] [Google Scholar]
- 17.Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006;355:1345–56. doi: 10.1056/NEJMra055323. [DOI] [PubMed] [Google Scholar]
- 18.Alexakis N, Connor S, Ghaneh P, et al. Hereditary pancreatic endocrine tumours. Pancreatology. 2004;4:417–35. doi: 10.1159/000079616. [DOI] [PubMed] [Google Scholar]
- 19.Maioli M, Ciccarese M, Pacifico A, et al. Familial insulinoma: description of two cases. Acta Diabetol. 1992;29:38–40. doi: 10.1007/BF00572828. [DOI] [PubMed] [Google Scholar]
- 20.Hemminki K, Li X. Familial carcinoid tumors and subsequent cancers: a nation-wide epidemiologic study from Sweden. Int J Cancer. 2001;94:444–8. doi: 10.1002/ijc.1473. [DOI] [PubMed] [Google Scholar]
- 21.Rindi G, Kloppel G. Endocrine tumors of the gut and pancreas tumor biology and classification. Neuroendocrinology. 2004;80:12–5. doi: 10.1159/000080733. [DOI] [PubMed] [Google Scholar]
- 22.Akerstrom G, Hessman O, Hellman P, et al. Pancreatic tumours as part of the MEN-1 syndrome. Best Pract Res Clin Gastroenterol. 2005;19:819–30. doi: 10.1016/j.bpg.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 23.Norton JA, Alexander HR, Fraker DL, et al. Comparison of surgical results in patients with advanced and limited disease with multiple endocrine neoplasia type 1 and Zollinger-Ellison syndrome. Ann Surg. 2001;234:495–506. doi: 10.1097/00000658-200110000-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lowney JK, Frisella MM, Lairmore TC, et al. Pancreatic islet cell tumor metastasis in multiple endocrine neoplasia type 1: correlation with primary tumor size. Surgery. 1999;125:1043–9. doi: 10.1067/msy.1998.92561. [DOI] [PubMed] [Google Scholar]
- 25.Doherty GM, Olson JA, Frisella MM, et al. Lethality of multiple endocrine neoplasia Type I. World J Surg. 1998;22:581–7. doi: 10.1007/s002689900438. [DOI] [PubMed] [Google Scholar]
- 26.Larsson C, Skogseid B, Oberg K, et al. Multiple endocrine neoplasia type 1 gene maps to chromosome 11 and is lost in insulinoma. Nature. 1988;332:85–7. doi: 10.1038/332085a0. [DOI] [PubMed] [Google Scholar]
- 27.Chandrasekharappa SC, Guru SC, Manickam P, et al. Positional cloning of the gene for multiple endocrine neoplasia-Type 1. Science. 1997;276:404–7. doi: 10.1126/science.276.5311.404. [DOI] [PubMed] [Google Scholar]
- 28.Balogh K, Racz K, Patocs A, et al. Menin and its interacting proteins: elucidation of menin function. Trends Endocrinol Metab. 2006;17:357–64. doi: 10.1016/j.tem.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 29.Lemos MC, Thakker RV. Multiple endocrine neoplasia type 1 (MEN1): analysis of 1336 mutations reported in the first decade following identification of the gene. Hum Mutat. 2007 doi: 10.1002/humu.20605. [DOI] [PubMed] [Google Scholar]
- 30.Agarwal SK, Kennedy PA, Scacheri PC, et al. Menin molecular interactions: insights into normal functions and tumorigenesis. Horm Metab Res. 2005;37:369–74. doi: 10.1055/s-2005-870139. [DOI] [PubMed] [Google Scholar]
- 31.Yang Y, Hua X. In search of tumor suppressing functions of menin. Mol Cell Endocrinol. 2007;265–266:34–41. doi: 10.1016/j.mce.2006.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Busygina V, Bale AE. Multiple Endocrine Neoplasia Type 1 (MEN1) as a Cancer Predisposition Syndrome: Clues into the Mechanisms of MEN1-related Carcinogenesis. Yale J Biol Med. 2006;79:105–14. [PMC free article] [PubMed] [Google Scholar]
- 33.Dreijerink KM, Hoppener JW, Timmers HM, et al. Mechanisms of disease: multiple endocrine neoplasia type 1-relation to chromatin modifications and transcription regulation. Nat Clin Pract Endocrinol Metab. 2006;2:562–70. doi: 10.1038/ncpendmet0292. [DOI] [PubMed] [Google Scholar]
- 34.Hendy GN, Kaji H, Sowa H, et al. Menin and TGF-beta superfamily member signaling via the Smad pathway in pituitary, parathyroid and osteoblast. Horm Metab Res. 2005;37:375–9. doi: 10.1055/s-2005-870152. [DOI] [PubMed] [Google Scholar]
- 35.Sowa H, Kaji H, Hendy GN, et al. Menin is required for bone morphogenetic protein 2- and transforming growth factor beta-regulated osteoblastic differentiation through interaction with Smads and Runx2. J Biol Chem. 2004;279:40267–75. doi: 10.1074/jbc.M401312200. [DOI] [PubMed] [Google Scholar]
- 36.Lemmens IH, Forsberg L, Pannett AA, et al. Menin interacts directly with the homeobox-containing protein Pem. Biochem Biophy Res Commun. 2001;286:426–31. doi: 10.1006/bbrc.2001.5405. [DOI] [PubMed] [Google Scholar]
- 37.Busygina V, Kottemann MC, Scott KL, et al. Multiple endocrine neoplasia type 1 interacts with forkhead transcription factor CHES1 in DNA damage response. Cancer Res. 2006;66:8397–403. doi: 10.1158/0008-5472.CAN-06-0061. [DOI] [PubMed] [Google Scholar]
- 38.Milne TA, Hughes CM, Lloyd R, et al. Menin and MLL cooperatively regulate expression of cyclin-dependent kinase inhibitors. Proc Natl Acad Sci U S A. 2005;102:749–54. doi: 10.1073/pnas.0408836102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yokoyama A, Somervaille TC, Smith KS, et al. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell. 2005;123:207–18. doi: 10.1016/j.cell.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 40.Hughes CM, Rozenblatt-Rosen O, Milne TA, et al. Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol Cell. 2004;13:587–97. doi: 10.1016/s1097-2765(04)00081-4. [DOI] [PubMed] [Google Scholar]
- 41.Dreijerink KM, Mulder KW, Winkler GS, et al. Menin links estrogen receptor activation to histone H3K4 trimethylation. Cancer Res. 2006;66:4929–35. doi: 10.1158/0008-5472.CAN-05-4461. [DOI] [PubMed] [Google Scholar]
- 42.La P, Silva AC, Hou Z, et al. Direct binding of DNA by tumor suppressor menin. J Biol Chem. 2004;279:49045–54. doi: 10.1074/jbc.M409358200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.La P, Schnepp RW, Petersen D, et al. Tumor suppressor menin regulates expression of insulin-like growth factor binding protein 2. Endocrinology. 2004;145:3443–50. doi: 10.1210/en.2004-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.La P, Desmond A, Hou Z, et al. Tumor suppressor menin: the essential role of nuclear localization signal domains in coordinating gene expression. Oncogene. 2006;25:3537–46. doi: 10.1038/sj.onc.1209400. [DOI] [PubMed] [Google Scholar]
- 45.Sukhodolets KE, Hickman AB, Agarwal SK, et al. The 32-kilodalton subunit of replication protein A interacts with menin, the product of the MEN1 tumor suppressor gene. Mol Cell Biol. 2003;23:493–509. doi: 10.1128/MCB.23.2.493-509.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jin S, Mao H, Schnepp RW, et al. Menin associates with FANCD2, a protein involved in repair of DNA damage. Cancer Res. 2003;63:4204–10. [PubMed] [Google Scholar]
- 47.Schnepp RW, Chen YX, Wang H, et al. Mutation of tumor suppressor gene Men1 acutely enhances proliferation of pancreatic islet cells. Cancer Res. 2006;66:5707–15. doi: 10.1158/0008-5472.CAN-05-4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Obungu VH, Lee BA, Agarwal SK, et al. Menin, a tumor suppressor, associates with nonmuscle myosin II-A heavy chain. Oncogene. 2003;22:6347–58. doi: 10.1038/sj.onc.1206658. [DOI] [PubMed] [Google Scholar]
- 49.Lopez-Egido J, Cunningham J, Berg M, et al. Menin’s interaction with glial fibrillary acidic protein and vimentin suggests a role for the intermediate filament network in regulating menin activity. Exp Cell Res. 2002;278:175–83. doi: 10.1006/excr.2002.5575. [DOI] [PubMed] [Google Scholar]
- 50.Ohkura N, Kishi M, Tsukada T, et al. Menin, a gene product responsible for multiple endocrine neoplasia type 1, interacts with the putative tumor metastasis suppressor nm23. Biochem Biophy Res Commun. 2001;282:1206–10. doi: 10.1006/bbrc.2001.4723. [DOI] [PubMed] [Google Scholar]
- 51.Schnepp RW, Hou Z, Wang H, et al. Functional interaction between tumor suppressor menin and activator of S-phase kinase. Cancer Res. 2004;64:6791–6. doi: 10.1158/0008-5472.CAN-04-0724. [DOI] [PubMed] [Google Scholar]
- 52.Suphapeetiporn K, Greally JM, Walpita D, et al. MEN1 tumor-suppressor protein localizes to telomeres during meiosis. Genes Chromosomes Cancer. 2002;35:81–5. doi: 10.1002/gcc.10113. [DOI] [PubMed] [Google Scholar]
- 53.Lin SY, Elledge SJ. Multiple tumor suppressor pathways negatively regulate telomerase. Cell. 2003;113:881–9. doi: 10.1016/s0092-8674(03)00430-6. [DOI] [PubMed] [Google Scholar]
- 54.Perren A, Anlauf M, Henopp T, et al. Multiple endocrine neoplasia type 1 (MEN1): loss of one MEN1 allele in tumors and monohormonal endocrine cell clusters but not in islet hyperplasia of the pancreas. J Clin Endocrinol Metab. 2007;92:1118–28. doi: 10.1210/jc.2006-1944. [DOI] [PubMed] [Google Scholar]
- 55.Karnik SK, Hughes CM, Gu X, et al. Menin regulates pancreatic islet growth by promoting histone methylation and expression of genes encoding p27Kip1 and p18INK4c. Proc Natl Acad Sci U S A. 2005;102:14659–64. doi: 10.1073/pnas.0503484102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Karnik SK, Chen H, McLean GW, et al. Menin controls growth of pancreatic beta-cells in pregnant mice and promotes gestational diabetes mellitus. Science. 2007;318:806–9. doi: 10.1126/science.1146812. [DOI] [PubMed] [Google Scholar]
- 57.Crabtree JS, Scacheri PC, Ward JM, et al. A mouse model of multiple endocrine neoplasia, type 1, develops multiple endocrine tumors. Proc Natl Acad Sci U S A. 2001;98:1118–23. doi: 10.1073/pnas.98.3.1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bertolino P, Tong WM, Galendo D, et al. Heterozygous Men1 mutant mice develop a range of endocrine tumors mimicking multiple endocrine neoplasia type 1. Molecular endocrinology (Baltimore, Md. 2003;17:1880–92. doi: 10.1210/me.2003-0154. [DOI] [PubMed] [Google Scholar]
- 59.Loffler KA, Biondi CA, Gartside M, et al. Broad tumor spectrum in a mouse model of multiple endocrine neoplasia type 1. Int J Cancer. 2007;120:259–67. doi: 10.1002/ijc.22288. [DOI] [PubMed] [Google Scholar]
- 60.Crabtree JS, Scacheri PC, Ward JM, et al. Of mice and MEN1: Insulinomas in a conditional mouse knockout. Molecular and cellular biology. 2003;23:6075–85. doi: 10.1128/MCB.23.17.6075-6085.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Biondi CA, Gartside MG, Waring P, et al. Conditional inactivation of the MEN1 gene leads to pancreatic and pituitary tumorigenesis but does not affect normal development of these tissues. Molecular and cellular biology. 2004;24:3125–31. doi: 10.1128/MCB.24.8.3125-3131.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Giraud S, Zhang CX, Serova-Sinilnikova O, et al. Germ-line mutation analysis in patients with multiple endocrine neoplasia type 1 and related disorders. Am J Hum Genet. 1998;63:455–67. doi: 10.1086/301953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lairmore TC, Wells SA., Jr . Cancer of the endocrine system. In: DeVita VT Jr, Hellman S, Rosenberg SA, editors. Cancer: Principles & Practice of Oncology. 5. Philadelphia: Lippincott-Raven Publishers; 1997. pp. 1617–28. [Google Scholar]
- 64.Bartsch DK, Langer P, Wild A, et al. Pancreaticoduodenal endocrine tumors in multiple endocrine neoplasia type 1: Surgery or surveillance? Surgery. 2000;128:958–66. doi: 10.1067/msy.2000.109727. [DOI] [PubMed] [Google Scholar]
- 65.Debelenko LV, Zhuang ZP, Emmert-Buck MR, et al. Allelic deletions on chromosome 11q13 in Multiple Endocrine Neoplasia Type-I-associated sporadic gastrinomas and pancreatic endocrine tumors. Cancer Res. 1997;57:2238–43. [PubMed] [Google Scholar]
- 66.Goebel SU, Heppner C, Burns AD, et al. Geneotype/phenotype correlations of MEN1 gene mutations in sporadic gastrinoma. J Clin Endocrinol Metab. 2000;85:116–23. doi: 10.1210/jcem.85.1.6260. [DOI] [PubMed] [Google Scholar]
- 67.Dilley WG, Kalyanaraman S, Verma S, et al. Global gene expression in neuroendocrine tumors from patients with the MEN1 syndrome. Mol Cancer. 2005;4:9. doi: 10.1186/1476-4598-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gibril F, Schumann M, Pace A, et al. Multiple endocrine neoplasia type 1 and Zollinger-Ellison syndrome. A prospective study of 107 cases and comparison with 1009 patients from the literature. Medicine (Baltimore) 2004;83:43–83. doi: 10.1097/01.md.0000112297.72510.32. [DOI] [PubMed] [Google Scholar]
- 69.Benya RV, Metz DC, Venzon DJ, et al. Zollinger-Ellison syndrome can be the initial endocrine manifestation in patients with multiple endocrine neoplasia-type 1. Am J Med. 1994;97:436–44. doi: 10.1016/0002-9343(94)90323-9. [DOI] [PubMed] [Google Scholar]
- 70.Skogseid B, Ericksson B, Lundqvist G, et al. Multiple endocrine neoplasia type I: A 10-year prospective screening study in four kindreds. J Clin Endocrinol Metab. 1991;73(2):281–7. doi: 10.1210/jcem-73-2-281. [DOI] [PubMed] [Google Scholar]
- 71.Vasen HF, Lamers CB, Lips CJ. Screening for the multiple endocrine neoplasia syndrome type I. A study of 11 kindreds in the Netherlands. Arch Intern Med. 1989;149:2717–22. [PubMed] [Google Scholar]
- 72.Shepherd JJ. The natural history of multiple endocrine neoplasia type I. Highly uncommon or highly unrecognized. Arch Surg. 1991;126:935–52. doi: 10.1001/archsurg.1991.01410320017001. [DOI] [PubMed] [Google Scholar]
- 73.Cadiot G, Houillier P, Allouch A, et al. Oral calcium tolerance test in the early diagnosis of primary hyperparathyroidism and multiple endocrine neoplasia type 1 in patients with the Zollinger-Ellison syndrome. Groupe de Recherche et d’Etude du Syndrome de Zollinger-Ellison. Gut. 1996;39:273–8. doi: 10.1136/gut.39.2.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Trump D, Farren B, Wooding C, et al. Clinical studies of multiple endocrine neoplasia type 1 (MEN1) QJM. 1996;89:957–8. doi: 10.1093/qjmed/89.9.653. [DOI] [PubMed] [Google Scholar]
- 75.Samaan NA, Ouais S, Ordonez NG, et al. Multiple endocrine syndrome type I. Cancer. 1989;64:741–52. doi: 10.1002/1097-0142(19890801)64:3<741::aid-cncr2820640329>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 76.Jensen RT, Doppman JL, Gardner JD. Gastrinoma. In: Go VLW, Brooks FA, DiMagno EP, Gardner JD, Lebenthal E, Scheele GA, editors. The Exocrine Pancreas: Biology, Pathobiology and Disease. 1. New York: Raven Press; 1986. pp. 727–44. [Google Scholar]
- 77.Stefanini P, Carboni M, Patrassi N, et al. Beta-islet cell tumors of the pancreas: results of a study on 1,067 cases. Surgery. 1974;75:597–609. [PubMed] [Google Scholar]
- 78.Levy-Bohbot N, Merle C, Goudet P, et al. Prevalence, characteristics and prognosis of MEN 1-associated glucagonomas, VIPomas, and somatostatinomas: study from the GTE (Groupe des Tumeurs Endocrines) registry. Gastroenterol Clin Biol. 2004;28:1075–81. doi: 10.1016/s0399-8320(04)95184-6. [DOI] [PubMed] [Google Scholar]
- 79.Ruszniewski P, Podevin P, Cadiot G, et al. Clinical, anatomical, and evolutive features of patients with the Zollinger-Ellison syndrome combined with type I multiple endocrine neoplasia. Pancreas. 1993;8:295–304. doi: 10.1097/00006676-199305000-00003. [DOI] [PubMed] [Google Scholar]
- 80.Goudet P, Peschaud F, Mignon M, et al. Les gastrinomes dans les neoplasies endocriniennes multiples de type 1. Une etude de cohorte de 127 cas du groupe des tumeurs endocrines (GTE) Ann Chir. 2004;129:149–55. doi: 10.1016/j.anchir.2003.11.013. [DOI] [PubMed] [Google Scholar]
- 81.Triponez F, Dosseh D, Goudet P, et al. Epidemiology Data on 108 MEN 1 Patients From the GTE With Isolated Nonfunctioning Tumors of the Pancreas. Ann Surg. 2006;243:265–72. doi: 10.1097/01.sla.0000197715.96762.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cougard P, Goudet P, Peix JL, et al. [Insulinomas in multiple endocrine neoplasia type 1. Report of a series of 44 cases by the multiple endocrine neoplasia study group] Ann Chir. 2000;125:118–23. doi: 10.1016/s0001-4001(00)00112-4. [DOI] [PubMed] [Google Scholar]
- 83.You YN, Thompson GB, Young WF, Jr, et al. Pancreatoduodenal surgery in patients with multiple endocrine neoplasia type 1: Operative outcomes, long-term function, and quality of life. Surgery. 2007;142:829–36. doi: 10.1016/j.surg.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 84.Machens A, Schaaf L, Karges W, et al. Age-related penetrance of endocrine tumours in multiple endocrine neoplasia type 1 (MEN1): a multicentre study of 258 gene carriers. Clin Endocrinol (Oxf) 2007;67:613–22. doi: 10.1111/j.1365-2265.2007.02934.x. [DOI] [PubMed] [Google Scholar]
- 85.Roy P, Venzon DJ, Shojamanesh H, et al. Zollinger-Ellison syndrome: clinical presentation in 261 patients. Medicine (Baltimore) 2000;79:379–411. doi: 10.1097/00005792-200011000-00004. [DOI] [PubMed] [Google Scholar]
- 86.Pipeleers-Marichal M, Somers G, Willems G, et al. Gastrinomas in the duodenums of patients with multiple endocrine neoplasia type 1 and the Zollinger-Ellison syndrome. N Engl J Med. 1990;322:723–7. doi: 10.1056/NEJM199003153221103. [DOI] [PubMed] [Google Scholar]
- 87.Hoffmann KM, Furukawa M, Jensen RT. Duodenal neuroendocrine tumors: Classification, functional syndromes, diagnosis and medical treatment. Best Pract Res Clin Gastroenterol. 2005;19:675–97. doi: 10.1016/j.bpg.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 88.Jensen RT. Management of the Zollinger-Ellison syndrome in patients with multiple endocrine neoplasia type 1. J Intern Med. 1998;243:477–88. doi: 10.1046/j.1365-2796.1998.00281.x. [DOI] [PubMed] [Google Scholar]
- 89.Thompson NW, Lloyd RV, Nishiyama RH, et al. MEN I pancreas: a histological and immunohistochemical study. World J Surg. 1984;8:561–74. doi: 10.1007/BF01654938. [DOI] [PubMed] [Google Scholar]
- 90.Kloppel G, Willemar S, Stamm B, et al. Pancreatic lesions and hormonal profile of pancreatic tumors in multiple endocrine neoplasia type I. An immunocytochemical study of nine patients. Cancer. 1986;57:1824–32. doi: 10.1002/1097-0142(19860501)57:9<1824::aid-cncr2820570920>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 91.Anlauf M, Schlenger R, Perren A, et al. Microadenomatosis of the endocrine pancreas in patients with and without the multiple endocrine neoplasia type 1 syndrome. Am J Surg Pathol. 2006;30:560–74. doi: 10.1097/01.pas.0000194044.01104.25. [DOI] [PubMed] [Google Scholar]
- 92.LeBodic MF, Heymann MF, Lecomte M, et al. Immunohistochemical study of 100 pancreatic tumors in 28 patients with Multiple Endocrine Neoplasia, Type 1. Am J Surg Pathol. 1996;20:1378–84. doi: 10.1097/00000478-199611000-00009. [DOI] [PubMed] [Google Scholar]
- 93.Anlauf M, Garbrecht N, Henopp T, et al. Sporadic versus hereditary gastrinomas of the duodenum and pancreas: distinct clinico-pathological and epidemiological features. World J Gastroenterol. 2006;12:5440–6. doi: 10.3748/wjg.v12.i34.5440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.MacFarlane MP, Fraker DL, Alexander HR, et al. A prospective study of surgical resection of duodenal and pancreatic gastrinomas in multiple endocrine neoplasia-Type 1. Surgery. 1995;118:973–80. doi: 10.1016/s0039-6060(05)80102-3. [DOI] [PubMed] [Google Scholar]
- 95.Norton JA, Fraker DL, Alexander HR, et al. Surgery to cure the Zollinger-Ellison syndrome. N Engl J Med. 1999;341:635–44. doi: 10.1056/NEJM199908263410902. [DOI] [PubMed] [Google Scholar]
- 96.Weber HC, Venzon DJ, Lin JT, et al. Determinants of metastatic rate and survival in patients with Zollinger-Ellison syndrome: a prospective long-term study. Gastroenterology. 1995;108:1637–49. doi: 10.1016/0016-5085(95)90124-8. [DOI] [PubMed] [Google Scholar]
- 97.Vortmeyer AO, Huang S, Lubensky I, et al. Non-islet origin of pancreatic islet cell tumors. J Clin Endocrinol Metab. 2004;89:1934–8. doi: 10.1210/jc.2003-031575. [DOI] [PubMed] [Google Scholar]
- 98.Anlauf M, Perren A, Meyer CL, et al. Precursor lesions in patients with multiple endocrine neoplasia type 1-associated duodenal gastrinomas. Gastroenterology. 2005;128:1187–98. doi: 10.1053/j.gastro.2005.01.058. [DOI] [PubMed] [Google Scholar]
- 99.Anlauf M, Perren A, Henopp T, et al. Allelic deletion of the MEN1 gene in duodenal gastrin and somatostatin cell neoplasms and their precursor lesions. Gut. 2007;56:637–44. doi: 10.1136/gut.2006.108910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kouvaraki MA, Shapiro SE, Cote GJ, et al. Management of pancreatic endocrine tumors in multiple endocrine neoplasia type 1. World J Surg. 2006;30:643–53. doi: 10.1007/s00268-006-0360-y. [DOI] [PubMed] [Google Scholar]
- 101.Bartsch DK, Fendrich V, Langer P, et al. Outcome of duodenopancreatic resections in patients with multiple endocrine neoplasia type 1. Ann Surg. 2005;242:757–64. doi: 10.1097/01.sla.0000189549.51913.d8. discussion. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rasbach DA, vanHeerden JA, Telander RL, et al. Surgical management of hyperinsulinism in the multiple endocrine neoplasia, type 1 syndrome. Arch Surg. 1985;120:584–9. doi: 10.1001/archsurg.1985.01390290062010. [DOI] [PubMed] [Google Scholar]
- 103.Bassett JH, Forbes SA, Pannett AA, et al. Characterization of mutations in patients with multiple endocrine neoplasia type 1. Am J Hum Genet. 1998;62:232–44. doi: 10.1086/301729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Carty SE, Helm AK, Amico JA, et al. The variable penetrance and spectrum of manifestations of multiple endocrine neoplasia type 1. Surgery. 1998;124:1106–14. doi: 10.1067/msy.1998.93107. [DOI] [PubMed] [Google Scholar]
- 105.Gibril F, Venzon DJ, Ojeaburu JV, et al. Prospective study of the natural history of gastrinoma in patients with MEN1: Definition of an aggressive and a nonaggressive form. J Clin Endocrinol Metab. 2001;86:5282–93. doi: 10.1210/jcem.86.11.8011. [DOI] [PubMed] [Google Scholar]
- 106.Ballard HS, Frame B, Harstock RT. Familial endocrine adenoma-peptic ulcer disease. Medicine (Baltimore) 1964;43:481–515. [PubMed] [Google Scholar]
- 107.Lamers CB. Familial multiple endocrine neoplasia type I (Wermer’s syndrome) Neth J Med. 1978;21:270–4. [PubMed] [Google Scholar]
- 108.Majewski JT, Wilson SD. The MEN-I syndrome: an all or none phenomenon? Surgery. 1979;86:475–84. [PubMed] [Google Scholar]
- 109.Wilkinson S, Teh BT, Davey KR, et al. Cause of death in multiple endocrine neoplasia type 1. Arch Surg. 1993;128:683–90. doi: 10.1001/archsurg.1993.01420180085016. [DOI] [PubMed] [Google Scholar]
- 110.Dean PG, vanHeerden JA, Farley DR, et al. Are patients with multiple endocrine neoplasia type I prone to premature death? World J Surg. 2000;24:1437–41. doi: 10.1007/s002680010237. [DOI] [PubMed] [Google Scholar]
- 111.Geerdink EAM, van der Luijt RB, Lips CJ. Do patients with multiple endocrine neoplasia syndrome type 1 benefit from periodical screening? Eur J Endocrinol. 2003;149:577–82. doi: 10.1530/eje.0.1490577. [DOI] [PubMed] [Google Scholar]
- 112.Vierimaa O, Ebeling TM, Kytola S, et al. Multiple endocrine neoplasia type 1 in Northern Finland; clinical features and genotype phenotype correlation. Eur J Endocrinol. 2007;157:285–94. doi: 10.1530/EJE-07-0195. [DOI] [PubMed] [Google Scholar]
- 113.Jensen RT, Gardner JD. Gastrinoma. In: Go VLW, DiMagno EP, Gardner JD, Lebenthal E, Reber HA, Scheele GA, editors. The Pancreas: Biology, Pathobiology and Disease. 2. New York: Raven Press Publishing Co.; 1993. pp. 931–78. [Google Scholar]
- 114.Carling T, Udelsman R. Parathyroid surgery in familial hyperparathyroid disorders. J Intern Med. 2005;257:27–37. doi: 10.1111/j.1365-2796.2004.01428.x. [DOI] [PubMed] [Google Scholar]
- 115.Norton JA, Venzon DJ, Berna MJ, et al. Prospective study of surgery for primary hyperaparathyroidism (HPT) in Multiple Endocrine Neoplasia type 1 (MEN1), and Zollinger-Ellison syndrome (ZES): longterm outcome of a more virulent form of HPT. Ann Surgery. 2007 doi: 10.1097/SLA.0b013e31815efda5. Ref Type: In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jensen RT. Use of omeprazole and other proton pump inhibitors in the Zollinger-Ellison syndrome. In: Olbe L, editor. Milestones in Drug Therapy. Basel, Switzerland: Birkhauser Verlag AG Publish. Co.; 1999. pp. 205–21. [Google Scholar]
- 117.Ellison EH, Wilson SD. The Zollinger-Ellison syndrome: Re-appraisal and evaluation of 260 registered cases. Ann Surg. 1964;160:512–30. doi: 10.1097/00000658-196409000-00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Jensen RT, Gardner JD, Raufman JP, et al. Zollinger-Ellison syndrome: current concepts and management. Ann Intern Med. 1983;98:59–75. doi: 10.7326/0003-4819-98-1-59. [DOI] [PubMed] [Google Scholar]
- 119.Zollinger RM, Ellison EH. Primary peptic ulcerations of the jejunum associated with islet cell tumors of the pancreas. Ann Surg. 1955;142:709–28. [PMC free article] [PubMed] [Google Scholar]
- 120.Roy P, Venzon DJ, Feigenbaum KM, et al. Gastric secretion in Zollinger-Ellison syndrome: correlation with clinical expression, tumor extent and role in diagnosis - A prospective NIH study of 235 patients and review of the literature in 984 cases. Medicine(Baltimore) 2001;80:189–222. doi: 10.1097/00005792-200105000-00005. [DOI] [PubMed] [Google Scholar]
- 121.Burgess JR, Giles N, Shepherd JJ. Malignant thymic carcinoid is not prevented by transcervical thymectomy in multiple endocrine neoplasia type 1. Clin Endocrinol (Oxf) 2001;55:689–93. doi: 10.1046/j.1365-2265.2001.01348.x. [DOI] [PubMed] [Google Scholar]
- 122.Gibril F, Chen Y-J, Schrump DS, et al. Prospective study of thymic carcinoids in patients with Multiple Endocrine Neoplasia Type 1. J Clin Endocrinol Metab. 2003;88:1066–81. doi: 10.1210/jc.2002-021314. [DOI] [PubMed] [Google Scholar]
- 123.Teh BT, McArdle J, Chan SP, et al. Clinicopathologic studies of thymic carcinoids in multiple endocrine neoplasia type 1. Medicine(Baltimore) 1997;76:21–9. doi: 10.1097/00005792-199701000-00002. [DOI] [PubMed] [Google Scholar]
- 124.Tomassetti P, Campana D, Piscitelli L, et al. Endocrine pancreatic tumors: factors correlated with survival. Ann Oncol. 2005;16:1806–10. doi: 10.1093/annonc/mdi358. [DOI] [PubMed] [Google Scholar]
- 125.Phan GQ, Yeo CJ, Hruban RH, et al. Surgical experience with pancreatic and peripancreatic neuroendocrine tumors: review of 125 patients. J Gastrointest Surg. 1998;2:472–82. [PubMed] [Google Scholar]
- 126.Melvin WS, Johnson JA, Sparks J, et al. Long-term prognosis of Zollinger-Ellison syndrome in multiple endocrine neoplasia. Surgery. 1993;114:1183–8. [PubMed] [Google Scholar]
- 127.Farley DR, Van Heerden JA, Grant CS, et al. The Zollinger-Ellison syndrome. A collective surgical experience. Ann Surg. 1992;215:561–9. doi: 10.1097/00000658-199206000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Triponez F, Goudet P, Dosseh D, et al. Is surgery beneficial for MEN1 patients with small (< or = 2 cm), nonfunctioning pancreaticoduodenal endocrine tumor? An analysis of 65 patients from the GTE. World J Surg. 2006;30:654–62. doi: 10.1007/s00268-005-0354-9. [DOI] [PubMed] [Google Scholar]
- 129.Lairmore TC, Chen VY, DeBenedetti MK, et al. Duodenopancreatic resections in patients with multiple endocrine neoplasia type 1. Ann Surg. 2000;231:909–18. doi: 10.1097/00000658-200006000-00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Burgess JR, Greenaway TM, Parameswaran V, et al. Enteropancreatic malignancy associated with multiple endocrine neoplasia type 1. Cancer. 1998;83:428–34. doi: 10.1002/(sici)1097-0142(19980801)83:3<428::aid-cncr10>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 131.Norton JA, Jensen RT. Resolved and unresolved controversies in the surgical management of patients with Zollinger-Ellison syndrome. Ann Surg. 2004;240:757–73. doi: 10.1097/01.sla.0000143252.02142.3e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Madeira I, Terris B, Voss M, et al. Prognostic factors in patients with endocrine tumours of the duodenopancreatic area. Gut. 1998;43:422–7. doi: 10.1136/gut.43.3.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.La Rosa S, Sessa F, Capella C, et al. Prognostic criteria in nonfunctioning pancreatic endocrine tumours. Virchows Arch. 1996;429:323–33. doi: 10.1007/BF00198436. [DOI] [PubMed] [Google Scholar]
- 134.Panzuto F, Nasoni S, Falconi M, et al. Prognostic factors and survival in endocrine tumor patients: comparison between gastrointestinal and pancreatic localization. Endocr Relat Cancer. 2005;12:1083–92. doi: 10.1677/erc.1.01017. [DOI] [PubMed] [Google Scholar]
- 135.Rigaud G, Missiaglia E, Moore PS, et al. High resolution allelotype of nonfunctional pancreatic endocrine tumors: identification of two molecular subgroups with clinical implications. Cancer Res. 2001;61:285–92. [PubMed] [Google Scholar]
- 136.Gullo L, Migliori M, Falconi M, et al. Nonfunctioning pancreatic endocrine tumors: a multicenter clinical study. Am J Gastroenterol. 2003;98:2435–9. doi: 10.1111/j.1572-0241.2003.07704.x. [DOI] [PubMed] [Google Scholar]
- 137.Chu QD, Hill HC, Douglass HO, Jr, et al. Predictive factors associated with long-term survival in patients with neuroendocrine tumors of the pancreas. Ann Surg Oncol. 2002;9:855–62. doi: 10.1007/BF02557521. [DOI] [PubMed] [Google Scholar]
- 138.Eriksson B, Oberg K, Skogseid B. Neuroendocrine pancreatic tumors. Clinical findings in a prospective study of 84 patients. Acta Oncol. 1989;28:373–7. doi: 10.3109/02841868909111209. [DOI] [PubMed] [Google Scholar]
- 139.Bordi C, Viale G. Analysis of cell proliferation and tumor antigens of prognostic significance in pancreatic endocrine tumors. In: Mignon M, Jensen RT, editors. Endocrine Tumors of the Pancreas: Recent advances in research and management. Series: Frontiers of Gastrointestinal Research. Vol. 23. Basel, Switzerland: S. Karger; 1995. pp. 45–59. [Google Scholar]
- 140.Stumpf E, Aalto Y, Hoog A, et al. Chromosomal alterations in human pancreatic endocrine tumors. Genes Chromosomes Cancer. 2000;29:83–7. doi: 10.1002/1098-2264(2000)9999:9999<::aid-gcc1011>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 141.Speel EJ, Richter J, Moch H, et al. Genetic differences in endocrine pancreatic tumor subtypes detected by comparative genomic hybridization. Am J Pathol. 1999;155:1787–94. doi: 10.1016/S0002-9440(10)65495-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Chung DC, Smith AP, Louis DN, et al. A novel pancreatic endocrine tumor suppressor gene locus on chromosome 3p with clinical prognostic implications. J Clin Invest. 1997;100:404–10. doi: 10.1172/JCI119547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Capurso G, Lattimore S, Crnogorac-Jurcevic T, et al. Gene expression profiles of progressive pancreatic endocrine tumours and their liver metastases reveal potential novel markers and therapeutic targets. Endocr Relat Cancer. 2006;13:541–58. doi: 10.1677/erc.1.01153. [DOI] [PubMed] [Google Scholar]
- 144.Hansel DE, Rahman A, House M, et al. Met proto-oncogene and insulin-like growth factor binding protein 3 overexpression correlates with metastatic ability in well-differentiated pancreatic endocrine neoplasms. Clin Cancer Res. 2004;10:6152–8. doi: 10.1158/1078-0432.CCR-04-0285. [DOI] [PubMed] [Google Scholar]
- 145.Pisegna JR, Doppman JL, Norton JA, et al. Prospective comparative study of ability of MR imaging and other imaging modalities to localize tumors in patients with Zollinger-Ellison syndrome. Dig Dis Sci. 1993;38:1318–28. doi: 10.1007/BF01296084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Gibril F, Jensen RT. Diagnostic uses of radiolabelled somatostatin-receptor analogues in gastroenteropancreatic endocrine tumors. Dig Liver Dis. 2004;36:S106–S120. doi: 10.1016/j.dld.2003.11.024. [DOI] [PubMed] [Google Scholar]
- 147.Orbuch M, Doppman JL, Strader DB, et al. Imaging for pancreatic endocrine tumor localization: recent advances. In: Mignon M, Jensen RT, editors. Endocrine Tumors of the Pancreas: Recent advances in research and management. Frontiers of Gastrointestinal Research. Vol. 23. Basel, Switzerland: S. Karger; 1995. pp. 268–81. [Google Scholar]
- 148.Skogseid B, Oberg K, Akerstrom G, et al. Limited tumor involvement found at multiple endocrine neoplasia type I pancreatic exploration: can it be predicted by preoperative tumor localization? World J Surg. 1998;22:673–8. doi: 10.1007/s002689900451. [DOI] [PubMed] [Google Scholar]
- 149.Alexander HR, Fraker DL, Norton JA, et al. Prospective study of somatostatin receptor scintigraphy and its effect on operative outcome in patients with Zollinger-Ellison syndrome. Ann Surg. 1998;228:228–38. doi: 10.1097/00000658-199808000-00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Norton JA, Alexander HR, Fraker DL, et al. Does the use of routine duodenotomy (DUODX) affect rate of cure, development of liver metastases or survival in patients with Zollinger-Ellison syndrome (ZES)? Ann Surg. 2004;239:617–26. doi: 10.1097/01.sla.0000124290.05524.5e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Cadiot G, Bonnaud G, Lebtahi R, et al. Usefulness of somatostatin receptor scintigraphy in the management of patients with Zollinger-Ellison syndrome. Gut. 1997;41:107–14. doi: 10.1136/gut.41.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Cadiot G, Lebtahi R, Sarda L, et al. Preoperative detection of duodenal gastrinomas and peripancreatic lymph nodes by somatostatin receptor scintigraphy. Gastroenterology. 1996;111:845–54. doi: 10.1016/s0016-5085(96)70052-5. [DOI] [PubMed] [Google Scholar]
- 153.Norton JA, Doppman JL, Jensen RT. Curative resection in Zollinger-Ellison syndrome: Results of a 10-year prospective study. Ann Surg. 1992;215:8–18. doi: 10.1097/00000658-199201000-00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Teunissen JJ, Kwekkeboom DJ, de Jong M, et al. Peptide receptor radionuclide therapy. Best Pract Res Clin Gastroenterol. 2005;19:595–616. doi: 10.1016/j.bpg.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 155.Krenning EP, Kwekkeboom DJ, Bakker WH, et al. Somatostatin receptor scintigraphy with [111In-DTPA-D-Phe1]- and [123I-Tyr3]-octreotide: the Rotterdam experience with more than 1000 patients. Eur J Nucl Med. 1993;20:716–31. doi: 10.1007/BF00181765. [DOI] [PubMed] [Google Scholar]
- 156.Gibril F, Reynolds JC, Doppman JL, et al. Somatostatin receptor scintigraphy: its sensitivity compared with that of other imaging methods in detecting primary and metastatic gastrinomas: a prospective study. Ann Intern Med. 1996;125:26–34. doi: 10.7326/0003-4819-125-1-199607010-00005. [DOI] [PubMed] [Google Scholar]
- 157.Jensen RT. Peptide therapy. Recent advances in the use of somatostatin and other peptide receptor agonists and antagonists. In: Lewis JH, Dubois A, editors. Current Clinical Topics in Gastrointestinal Pharmacology. Malden, MA: Blackwell Science, Inc.; 1997. pp. 144–223. [Google Scholar]
- 158.Van Essen M, Krenning EP, de Jong M, et al. Peptide Receptor Radionuclide Therapy with radiolabelled somatostatin analogues in patients with somatostatin receptor positive tumours. Acta Oncol. 2007;46:723–34. doi: 10.1080/02841860701441848. [DOI] [PubMed] [Google Scholar]
- 159.Yim JH, Siegel BA, DeBenedetti MK, et al. Prospective study of the utility of somatostatin-receptor scintigraphy in the evaluation of patients with multiple endocrine neoplasia type 1. Surgery. 1998;124:1037–42. doi: 10.1067/msy.1998.92553. [DOI] [PubMed] [Google Scholar]
- 160.Langer P, Kann PH, Fendrich V, et al. Prospective evaluation of imaging procedures for the detection of pancreaticoduodenal endocrine tumors in patients with multiple endocrine neoplasia type 1. World J Surg. 2004;28:1317–22. doi: 10.1007/s00268-004-7642-7. [DOI] [PubMed] [Google Scholar]
- 161.Termanini B, Gibril F, Reynolds JC, et al. Value of somatostatin receptor scintigraphy: A prospective study in gastrinoma of its effect on clinical management. Gastroenterology. 1997;112:335–47. doi: 10.1053/gast.1997.v112.pm9024287. [DOI] [PubMed] [Google Scholar]
- 162.Schillaci O, Spanu A, Scopinaro F, et al. Somatostatin receptor scintigraphy in liver metastasis detection from gastroenteropancreatic neuroendocrine tumors. J Nucl Med. 2003;44:359–68. [PubMed] [Google Scholar]
- 163.Schillaci O, Spanu A, Scopinaro F, et al. Somatostatin receptor scintigraphy with 111In-pentetreotide in nonfunctioning gastroenteropancreatic neuroendocrine tumors. Int J Oncol. 2003;23:1687–95. [PubMed] [Google Scholar]
- 164.Gibril F, Doppman JL, Reynolds JC, et al. Bone metastases in patients with gastrinomas: a prospective study of bone scanning, somatostatin receptor scanning, and MRI in their detection, their frequency, location and effect of their detection on management. J Clin Oncol. 1998;16:1040–53. doi: 10.1200/JCO.1998.16.3.1040. [DOI] [PubMed] [Google Scholar]
- 165.Lebtahi R, Cadiot G, Delahaye N, et al. Detection of bone metastases in patients with endocrine gastroenteropancreatic tumors: bone scintigraphy compared with somatostatin receptor scintigraphy. J Nucl Med. 1999;40:1602–8. [PubMed] [Google Scholar]
- 166.de Sa SV, Correa-Giannella ML, Machado MC, et al. Somatostatin receptor subtype 5 (SSTR5) mRNA expression is related to histopathological features of cell proliferation in insulinomas. Endocr Relat Cancer. 2006;13:69–78. doi: 10.1677/erc.1.00962. [DOI] [PubMed] [Google Scholar]
- 167.Bertherat J, Tenenbaum F, Perlemoine K, et al. Somatostatin receptors 2 and 5 are the major somatostatin receptors in insulinomas: an in vivo and in vitro study. J Clin Endocrinol Metab. 2003;88:5353–60. doi: 10.1210/jc.2002-021895. [DOI] [PubMed] [Google Scholar]
- 168.Gibril F, Reynolds JC, Chen CC, et al. Specificity of somatostatin receptor scintigraphy: a prospective study and the effects of false positive localizations on management in patients with gastrinomas. J Nucl Med. 1999;40:539–53. [PubMed] [Google Scholar]
- 169.Gibril F, Reynolds JC, Roy PK, et al. Ability of somatostatin receptor scintigraphy to identify patients with localized gastric carcinoids: A prospective study. J Nucl Med. 2000;41:1646–56. [PubMed] [Google Scholar]
- 170.Norton JA, Melcher ML, Gibril F, et al. Gastric carcinoid tumors in multiple endocrine neoplasia-1 patients with Zollinger-Ellison syndrome can be symptomatic, demonstrate aggressive growth, and require surgery. Surgery. 2004;136:1267–74. doi: 10.1016/j.surg.2004.06.057. [DOI] [PubMed] [Google Scholar]
- 171.Asgharian B, Chen YJ, Patronas NJ, et al. Meningiomas may be a component tumor of MEN1. Clin Cancer Res. 2004;10:869–80. doi: 10.1158/1078-0432.ccr-0938-3. [DOI] [PubMed] [Google Scholar]
- 172.Wamsteker EJ, Gauger PG, Thompson NW, et al. EUS detection of pancreatic endocrine tumors in asymptomatic patients with type 1 multiple endocrine neoplasia. Gastrointest Endosc. 2003;58:531–5. doi: 10.1067/s0016-5107(03)01965-5. [DOI] [PubMed] [Google Scholar]
- 173.Hellman P, Hennings J, Akerstrom G, et al. Endoscopic ultrasonography for evaluation of pancreatic tumours in multiple endocrine neoplasia type 1. Br J Surg. 2005;92:1508–12. doi: 10.1002/bjs.5149. [DOI] [PubMed] [Google Scholar]
- 174.Kann PH, Balakina E, Ivan D, et al. Natural course of small, asymptomatic neuroendocrine pancreatic tumours in multiple endocrine neoplasia type 1: an endoscopic ultrasound imaging study. Endocr Relat Cancer. 2006;13:1195–202. doi: 10.1677/erc.1.01220. [DOI] [PubMed] [Google Scholar]
- 175.Ruszniewski P, Amouyal P, Amouyal G, et al. Localization of gastrinomas by endoscopic ultrasonography in patients with Zollinger-Ellison syndrome. Surgery. 1995;117:629–35. doi: 10.1016/s0039-6060(95)80005-0. [DOI] [PubMed] [Google Scholar]
- 176.Thomas-Marques L, Murat A, Delemer B, et al. Prospective endoscopic ultrasonographic evaluation of the frequency of nonfunctioning pancreaticoduodenal endocrine tumors in patients with multiple endocrine neoplasia type 1. Am J Gastroenterol. 2006;101:266–73. doi: 10.1111/j.1572-0241.2006.00367.x. [DOI] [PubMed] [Google Scholar]
- 177.McLean AM, Fairclough PD. Endoscopic ultrasound in the localisation of pancreatic islet cell tumours. Best Pract Res Clin Endocrinol Metab. 2005;19:177–93. doi: 10.1016/j.beem.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 178.Gauger PG, Scheiman JM, Wamsteker EJ, et al. Role of endoscopic ultrasonography in screening and treatment of pancreatic endocrine tumours in asymptomatic patients with multiple endocrine neoplasia type 1. Br J Surg. 2003;90:748–54. doi: 10.1002/bjs.4142. [DOI] [PubMed] [Google Scholar]
- 179.Kann PH, Kann B, Fassbender WJ, et al. Small neuroendocrine pancreatic tumors in multiple endocrine neoplasia type 1 (MEN1): least significant change of tumor diameter as determined by endoscopic ultrasound (EUS) imaging. Exp Clin Endocrinol Diabetes. 2006;114:361–5. doi: 10.1055/s-2006-924322. [DOI] [PubMed] [Google Scholar]
- 180.Eriksson B, Orlefors H, Oberg K, et al. Developments in PET for the detection of endocrine tumours. Best Pract Res Clin Endocrinol Metab. 2005;19:311–24. doi: 10.1016/j.beem.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 181.Orlefors H, Sundin A, Garske U, et al. Whole-body (11)C-5-hydroxytryptophan positron emission tomography as a universal imaging technique for neuroendocrine tumors: comparison with somatostatin receptor scintigraphy and computed tomography. J Clin Endocrinol Metab. 2005;90:3392–400. doi: 10.1210/jc.2004-1938. [DOI] [PubMed] [Google Scholar]
- 182.Kauhanen S, Seppanen M, Minn H, et al. Fluorine-18-L-dihydroxyphenylalanine (18F-DOPA) positron emission tomography as a tool to localize an insulinoma or beta-cell hyperplasia in adult patients. J Clin Endocrinol Metab. 2007;92:1237–44. doi: 10.1210/jc.2006-1479. [DOI] [PubMed] [Google Scholar]
- 183.Ambrosini V, Tomassetti P, Rubello D, et al. Role of 18F-dopa PET/CT imaging in the management of patients with 111In-pentetreotide negative GEP tumours. Nucl Med Commun. 2007;28:473–7. doi: 10.1097/MNM.0b013e328182d606. [DOI] [PubMed] [Google Scholar]
- 184.Hillel PG, van Beek EJ, Taylor C, et al. The clinical impact of a combined gamma camera/CT imaging system on somatostatin receptor imaging of neuroendocrine tumours. Clin Radiol. 2006;61:579–87. doi: 10.1016/j.crad.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 185.Krausz Y, Keidar Z, Kogan I, et al. SPECT/CT hybrid imaging with 111In-pentetreotide in assessment of neuroendocrine tumours. Clin Endocrinol (Oxf) 2003;59:565–73. doi: 10.1046/j.1365-2265.2003.01885.x. [DOI] [PubMed] [Google Scholar]
- 186.Adams S, Baum R, Rink T, et al. Limited value of fluorine-18 fluorodeoxyglucose positron emission tomography for the imaging of neuroendocrine tumours. Eur J Nucl Med. 1998;25:79–83. doi: 10.1007/s002590050197. [DOI] [PubMed] [Google Scholar]
- 187.Pasquali C, Rubello D, Sperti C, et al. Neuroendocrine tumor imaging: can 18F-fluorodeoxyglucose positron emission tomography detect tumors with poor prognosis and aggressive behavior? World J Surg. 1998;22:588–92. doi: 10.1007/s002689900439. [DOI] [PubMed] [Google Scholar]
- 188.Lehy T, Cadiot G, Mignon M, et al. Influence of multiple endocrine neoplasia type 1 on gastric endocrine cells in patients with the Zollinger-Ellison syndrome. Gut. 1992;33:1275–9. doi: 10.1136/gut.33.9.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Peghini PL, Annibale B, Azzoni C, et al. Effect of chronic hypergastrinemia on human enterochromaffin-like cells: insights from patients with sporadic gastrinomas. Gastroenterology. 2002;123:68–85. doi: 10.1053/gast.2002.34231. [DOI] [PubMed] [Google Scholar]
- 190.D’Adda T, Corleto V, Pilato FP, et al. Quantitative ultrastructure of endocrine cells of oxyntic mucosa in Zollinger-Ellison syndrome. Correspondence with light microscopic findings. Gastroenterology. 1990;99:17–26. doi: 10.1016/0016-5085(90)91224-t. [DOI] [PubMed] [Google Scholar]
- 191.Rindi G, Bordi C, Rappel S, et al. Gastric carcinoids and neuroendocrine carcinomas: pathogenesis, pathology, and behavior. World J Surg. 1996;20:168–72. doi: 10.1007/s002689900026. [DOI] [PubMed] [Google Scholar]
- 192.Berna MJ, Annibale B, Marignani M, et al. A prospective study of gastric carcinoids and enterochromaffin-like cells changes in Multple Endocrine Neoplaisa Type 1 and Zollinger-Ellison syndrome: Identification of risk factors. J Clin Endocrinol Metab. 2008 doi: 10.1210/jc.2007-2279. Ref Type: In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Norton JA, Cornelius MJ, Doppman JL, et al. Effect of parathyroidectomy in patients with hyperparathyroidism, Zollinger-Ellison syndrome and multiple endocrine neoplasia Type I: A prospective study. Surgery. 1987;102:958–66. [PubMed] [Google Scholar]
- 194.Berna MJ, Hoffmann KM, Long SH, et al. Serum gastrin in Zollinger-Ellison syndrome: II. Prospective study of gastrin provocative testing in 293 patients from the National Institutes of Health and comparison with 537 cases from the literature. evaluation of diagnostic criteria, proposal of new criteria, and correlations with clinical and tumoral features. Medicine (Baltimore) 2006;85:331–64. doi: 10.1097/MD.0b013e31802b518c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Hirschowitz BI. Clinical course of nonsurgically treated Zollinger-Ellison syndrome. In: Mignon M, Jensen RT, editors. Endocrine Tumors of the Pancreas: Recent advances in research and management. Frontiers of Gastrointestinal Research. Vol. 23. Basel, Switzerland: S. Karger; 1995. pp. 360–71. [Google Scholar]
- 196.Sheppard BC, Norton JA, Doppman JL, et al. Management of islet cell tumors in patients with Multiple Endocrine Neoplasia: A prospective study. Surgery. 1989;106:1108–17. [PubMed] [Google Scholar]
- 197.Grama D, Eriksson B, Martensson H, et al. Clinical characteristics, treatment and survival in patients with pancreatic tumors causing hormonal syndromes. World J Surg. 1992;16:632–9. doi: 10.1007/BF02067341. [DOI] [PubMed] [Google Scholar]
- 198.Tonelli F, Fratini G, Nesi G, et al. Pancreatectomy in Multiple Endocrine Neoplasia Type 1-Related Gastrinomas and Pancreatic Endocrine Neoplasias. Ann Surg. 2006;244:61–70. doi: 10.1097/01.sla.0000218073.77254.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Tonelli F, Fratini G, Falchetti A, et al. Surgery for gastroenteropancreatic tumours in multiple endocrine neoplasia type 1: review and personal experience. J Intern Med. 2005;257:38–49. doi: 10.1111/j.1365-2796.2004.01424.x. [DOI] [PubMed] [Google Scholar]
- 200.Gibril F, Jensen RT. Advances in evaluation and management of gastrinoma in patients with Zollinger-Ellison syndrome. Curr Gastroenterol Rep. 2005;7:114–21. doi: 10.1007/s11894-005-0049-2. [DOI] [PubMed] [Google Scholar]
- 201.Thom AK, Norton JA, Axiotis CA, et al. Location, incidence and malignant potential of duodenal gastrinomas. Surgery. 1991;110:1086–93. [PubMed] [Google Scholar]
- 202.Zogakis TG, Gibril F, Libutti SK, et al. Management and outcome of patients with sporadic gastrinomas arising in the duodenum. Ann Surg. 2003;238:42–8. doi: 10.1097/01.SLA.0000074963.87688.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Delcore R, Jr, Cheung LY, Friesen SR. Characteristics of duodenal wall gastrinomas. Am J Surg. 1990;160:621–3. doi: 10.1016/s0002-9610(05)80760-3. [DOI] [PubMed] [Google Scholar]
- 204.Donow C, Pipeleers-Marichal M, Schroder S, et al. Surgical pathology of gastrinoma: site, size, multicentricity, association with multiple endocrine neoplasia type 1, and malignancy. Cancer. 1991;68:1329–34. doi: 10.1002/1097-0142(19910915)68:6<1329::aid-cncr2820680624>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 205.Berna MJ, Hoffmann KM, Serrano J, et al. Serum gastrin in Zollinger-Ellison syndrome: I. Prospective study of fasting serum gastrin in 309 patients from the National Institutes of Health and comparison with 2229 cases from the literature. Medicine (Baltimore) 2006;85:295–330. doi: 10.1097/01.md.0000236956.74128.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Hoffmann KM, Gibril F, Entsuah LK, et al. Patients with multiple endocrine neoplasia type 1 with gastrinomas have an increased risk of severe esophageal disease including stricture and the premalignant condition, Barrett’s esophagus. J Clin Endocrinol Metab. 2006;91:204–12. doi: 10.1210/jc.2005-1349. [DOI] [PubMed] [Google Scholar]
- 207.Metz DC, Pisegna JR, Fishbeyn VA, et al. Currently used doses of omeprazole in Zollinger-Ellison syndrome are too high. Gastroenterology. 1992;103:1498–508. doi: 10.1016/0016-5085(92)91170-9. [DOI] [PubMed] [Google Scholar]
- 208.Miller LS, Vinayek R, Frucht H, et al. Reflux esophagitis in patients with Zollinger-Ellison syndrome. Gastroenterology. 1990;98:341–6. doi: 10.1016/0016-5085(90)90823-j. [DOI] [PubMed] [Google Scholar]
- 209.Metz DC, Jensen RT. Advances in gastric antisecretory therapy in Zollinger-Ellison syndrome. In: Mignon M, Jensen RT, editors. Endocrine Tumors of the Pancreas: Recent advances in research and management. Series: Frontiers of Gastrointestinal Research. Vol. 23. Basel, Switzerland: S. Karger; 1995. pp. 240–57. [Google Scholar]
- 210.Gibril F, Jensen RT. Zollinger-Ellison syndrome revisited: diagnosis, biologic markers, associated inherited disorders, and acid hypersecretion. Curr Gastroenterol Rep. 2004;6:454–63. doi: 10.1007/s11894-004-0067-5. [DOI] [PubMed] [Google Scholar]
- 211.Wolfe MM, Jensen RT. Zollinger-Ellison syndrome: Current concepts in diagnosis and management. N Engl J Med. 1987;317:1200–9. doi: 10.1056/NEJM198711053171907. [DOI] [PubMed] [Google Scholar]
- 212.Corleto VD, Annibale B, Gibril F, et al. Does the widespread use of proton pump inhibitors mask, complicate and/or delay the diagnosis of Zollinger-Ellison syndrome? Aliment Pharmacol Ther. 2001;15:1555–61. doi: 10.1046/j.1365-2036.2001.01085.x. [DOI] [PubMed] [Google Scholar]
- 213.Corleto VD, Minisola S, Moretti A, et al. Prevalence and causes of hypergastrinemia in primary hyperparathyroidism: a prospective study. J Clin Endocrinol Metab. 1999;84:4554–8. doi: 10.1210/jcem.84.12.6193. [DOI] [PubMed] [Google Scholar]
- 214.Gibril F, Lindeman RJ, Abou-Saif A, et al. Retained gastric antrum syndrome. A forgotten, treatable cause of refractory peptic ulcer disease. Dig Dis Sci. 2001;46:610–7. doi: 10.1023/a:1005667719847. [DOI] [PubMed] [Google Scholar]
- 215.Yu F, Venzon DJ, Serrano J, et al. Prospective study of the clinical course, prognostic factors and survival in patients with longstanding Zollinger-Ellison syndrome. J Clin Oncol. 1999;17:615–30. doi: 10.1200/JCO.1999.17.2.615. [DOI] [PubMed] [Google Scholar]
- 216.Sutliff VE, Doppman JL, Gibril F, et al. Growth of newly diagnosed, untreated metastatic gastrinomas and predictors of growth patterns. J Clin Oncol. 1997;15:2420–31. doi: 10.1200/JCO.1997.15.6.2420. [DOI] [PubMed] [Google Scholar]
- 217.Mignon M. Natural history of neuroendocrine enteropancreatic tumors. Digestion. 2000;62:51–8. doi: 10.1159/000051856. [DOI] [PubMed] [Google Scholar]
- 218.Cadiot G, Vuagnat A, Doukhan I, et al. Prognostic factors in patients with Zollinger-Ellison syndrome and multiple endocrine neoplasia type 1. Gastroenterology. 1999;116:286–93. doi: 10.1016/s0016-5085(99)70124-1. [DOI] [PubMed] [Google Scholar]
- 219.Jensen RT. Zollinger-Ellison syndrome. In: Doherty GM, Skogseid B, editors. Surgical Endocrinology: Clinical Syndromes. Philadelphia: Lippincott Williams & Wilkins; 2001. pp. 291–344. [Google Scholar]
- 220.Metz DC, Kuchnio M, Fraker DL, et al. Flow cytometry and Zollinger-Ellison syndrome: relationship to clinical course. Gastroenterology. 1993;105:799–813. doi: 10.1016/0016-5085(93)90898-m. [DOI] [PubMed] [Google Scholar]
- 221.Goebel SU, Iwamoto M, Raffeld M, et al. HER-2/neu expression and gene amplification in gastrinomas: correlations with tumor biology, growth, and aggressiveness. Cancer Res. 2002;62:3702–10. [PubMed] [Google Scholar]
- 222.Chen Y-J, Vortmeyer A, Zhuang ZP, et al. Loss of heterozygosity of chromosome 1q in gastrinomas: occurrence and prognostic significance. Cancer Res. 2003;63:817–23. [PubMed] [Google Scholar]
- 223.Yu F, Jensen RT, Lubensky IA, et al. Survey of genetic alterations in gastrinomas. Cancer Res. 2000;60:5536–42. [PubMed] [Google Scholar]
- 224.Peghini PL, Iwamoto M, Raffeld M, et al. Overexpression of epidermal growth factor and hepatocyte growth factor receptors in a proportion of gastrinomas correlates with aggressive growth and lower curability. Clin Cancer Res. 2002;8:2273–85. [PubMed] [Google Scholar]
- 225.Fraker DL, Norton JA, Alexander HR, et al. Surgery in Zollinger-Ellison syndrome alters the natural history of gastrinoma. Ann Surg. 1994;220:320–30. doi: 10.1097/00000658-199409000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Norton JA, Fraker DL, Alexander HR, et al. Surgery increases survival in patients with gastrinoma. Ann Surg. 2006;244:410–9. doi: 10.1097/01.sla.0000234802.44320.a5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Maton PN, Gardner JD, Jensen RT. Cushing’s syndrome in patients with Zollinger-Ellison syndrome. N Engl J Med. 1986;315:1–5. doi: 10.1056/NEJM198607033150101. [DOI] [PubMed] [Google Scholar]
- 228.Ellison EC, O’Dorisio TM, Woltering EA, et al. Suppression of gastrin and gastric acid secretion in the Zollinger-Ellison syndrome by long-acting somatostatin (SMS 201–995) Scand J Gastroenterol. 1986;21(Suppl 119):206–11. [Google Scholar]
- 229.Ellison EC. Forty-year appraisal of gastrinoma. Ann Surg. 1995;222:511–24. doi: 10.1097/00000658-199522240-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Ellison EC, Sparks J, Verducci JS, et al. 50-year appraisal of gastrinoma: recommendations for staging and treatment. J Am Coll Surg. 2006;202:897–905. doi: 10.1016/j.jamcollsurg.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 231.Delcore R, Jr, Cheung LY, Friesen SR. Outcome of lymph node involvement in patients with Zollinger- Ellison syndrome. Ann Surg. 1988;206:291–8. doi: 10.1097/00000658-198809000-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Stabile BE, Passaro E., Jr Benign and malignant gastrinoma. Am J Surg. 1985;49:144–50. doi: 10.1016/s0002-9610(85)80024-6. [DOI] [PubMed] [Google Scholar]
- 233.Granberg D, Stridsberg M, Seensalu R, et al. Plasma chromogranin A in patients with multiple endocrine neoplasia type 1. J Clin Endocrinol Metab. 1999;84:2712–7. doi: 10.1210/jcem.84.8.5938. [DOI] [PubMed] [Google Scholar]
- 234.Peracchi M, Conte D, Gebbia C, et al. Plasma chromogranin A in patients with sporadic gastro-entero-pancreatic neuroendocrine tumors or multiple endocrine neoplasia type 1. Eur J Endocrinol. 2003;148:39–43. doi: 10.1530/eje.0.1480039. [DOI] [PubMed] [Google Scholar]
- 235.Migliori M, Tomassetti P, Campana D, et al. A meal stimulation test in the diagnosis of pancreatic endocrine tumors in multiple endocrine neoplasia type 1. Endocrine. 2002;17:229–32. doi: 10.1385/ENDO:17:3:229. [DOI] [PubMed] [Google Scholar]
- 236.Skogseid B, Oberg K, Benson L, et al. A standardized meal stimulation test of the endocrine pancreas for early detection of pancreatic endocrine tumors in multiple endocrine neoplasia type 1 syndrome: five years experience. J Clin Endocrinol Metab. 1987;64:1233–40. doi: 10.1210/jcem-64-6-1233. [DOI] [PubMed] [Google Scholar]
- 237.Langstein HN, Norton JA, Chiang V, et al. The utility of circulating levels of human pancreatic polypeptide as a marker for islet cell tumors. Surgery. 1990;108:1109–15. [PubMed] [Google Scholar]
- 238.Friesen SR, Tomita T, Kimmel JR. Pancreatic polypeptide update: its role in detection of the trait for multiple endocrine adenopathy syndrome, type 1 and pancreatic polypeptide-secreting tumors. Surgery. 1983;94:1028–37. [PubMed] [Google Scholar]
- 239.Skogseid B, Rastad J, Akerstrom G. Pancreatic endocrine tumors in multiple endocrine neoplasia type 1. In: Doherty GM, Skogseid B, editors. Surgical Endocrinology. Philadelphia: Lippincott Williams & Wilkins; 2001. pp. 511–24. [Google Scholar]
- 240.Langer P, Wild A, Celik I, et al. Prospective controlled trial of a standardized meal stimulation test in the detection of pancreaticoduodenal endocrine tumours in patients with multiple endocrine neoplasia type 1. Br J Surg. 2001;88:1403–7. doi: 10.1046/j.0007-1323.2001.01874.x. [DOI] [PubMed] [Google Scholar]
- 241.Sakurai A, Katai M, Yamashita K, et al. Long-term follow-up of patients with multiple endocrine neoplasia type 1. Endocr J. 2007;54:295–302. doi: 10.1507/endocrj.k06-147. [DOI] [PubMed] [Google Scholar]
- 242.Brandi ML, Gagel RF, Angeli A, et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab. 2001;86:5658–71. doi: 10.1210/jcem.86.12.8070. [DOI] [PubMed] [Google Scholar]
- 243.Jensen RT. Endocrine tumors of the gastrointestinal tract and pancreas. In: Kasper DL, Braunwald E, Fauci AS, Hauser SL, Longo DL, Jameson JL, editors. Harrison’s Principles of Internal Medicine. 16. New York: McGraw-Hill Medical Publishing Division; 2004. pp. 2220–1. [Google Scholar]
- 244.O’Riordain DS, O’Brien T, Van Heerden JA, et al. Surgical management of insulinoma associated with multiple endocrine neoplasia type 1. World J Surg. 1994;18:488–93. doi: 10.1007/BF00353743. [DOI] [PubMed] [Google Scholar]
- 245.Demeure MJ, Klonoff DC, Karam JH, et al. Insulinomas associated with multiple endocrine neoplasia type I: The need for a different surgical approach. Surgery. 1991;110:998–1005. [PubMed] [Google Scholar]
- 246.Norton JA, Fang TD, Jensen RT. Surgery for gastrinoma and insulinoma in multiple endocrine neoplasia type 1. J Natl Compr Canc Netw. 2006;4:148–53. doi: 10.6004/jnccn.2006.0015. [DOI] [PubMed] [Google Scholar]
- 247.Jadoul M, Koppeschaar HP, Bax MA, et al. Insulinomas in MEN-I patients: early detection and treatment of insulinomas in patients with the multiple endocrine neoplasia syndrome type-I. Neth J Med. 1990;37:95–102. [PubMed] [Google Scholar]
- 248.Thompson NW. Current concepts in the surgical management of multiple endocrine neoplasia type 1 pancreatic-duodenal disease. Results in the treatment of 40 patients with Zollinger-Ellison syndrome, hypoglycaemia or both. J Intern Med. 1998;243:495–500. doi: 10.1046/j.1365-2796.1998.00307.x. [DOI] [PubMed] [Google Scholar]
- 249.Norton JA, Jensen RT. Unresolved surgical issues in the management of patients with the Zollinger-Ellison syndrome. World J Surg. 1991;15:151–9. doi: 10.1007/BF01658992. [DOI] [PubMed] [Google Scholar]
- 250.Metz DC, Strader DB, Orbuch M, et al. Use of omeprazole in Zollinger-Ellison: A prospective nine-year study of efficacy and safety. Aliment Pharmacol Ther. 1993;7:597–610. doi: 10.1111/j.1365-2036.1993.tb00140.x. [DOI] [PubMed] [Google Scholar]
- 251.Jensen RT, Collen MJ, McArthur KE, et al. Comparison of the effectiveness of ranitidine and cimetidine in inhibiting acid secretion in patients with gastric acid hypersecretory states. Am J Med. 1984;77(5B):90–105. [PubMed] [Google Scholar]
- 252.Maton PN, Lack EE, Collen MJ, et al. The effect of Zollinger-Ellison syndrome and omeprazole therapy on gastric oxyntic endocrine cells. Gastroenterology. 1990;99:943–50. doi: 10.1016/0016-5085(90)90611-4. [DOI] [PubMed] [Google Scholar]
- 253.Raufman JP, Collins SM, Pandol SJ, et al. Reliability of symptoms in assessing control of gastric acid secretion in patients with Zollinger-Ellison syndrome. Gastroenterology. 1983;84:108–13. [PubMed] [Google Scholar]
- 254.Lew EA, Pisegna JR, Starr JA, et al. Intravenous pantoprazole rapidly controls gastric acid hypersecretion in patients with Zollinger-Ellison syndrome. Gastroenterology. 2000;118:696–704. doi: 10.1016/s0016-5085(00)70139-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Vinayek R, Frucht H, London JF, et al. Intravenous omeprazole in patients with Zollinger-Ellison syndrome undergoing surgery. Gastroenterology. 1990;99:10–6. doi: 10.1016/0016-5085(90)91223-s. [DOI] [PubMed] [Google Scholar]
- 256.Vinayek R, Hahne WF, Euler AR, et al. Parenteral control of gastric hypersecretion in patients with Zollinger-Ellison syndrome. Dig Dis Sci. 1993;38:1857–65. doi: 10.1007/BF01296110. [DOI] [PubMed] [Google Scholar]
- 257.Saeed ZA, Norton JA, Frank WO, et al. Parenteral antisecretory drug therapy in patients with Zollinger- Ellison syndrome. Gastroenterology. 1989;96:1393–402. doi: 10.1016/0016-5085(89)90504-0. [DOI] [PubMed] [Google Scholar]
- 258.Fraker DL, Norton JA, Saeed ZA, et al. A prospective study of perioperative and postoperative control of acid hypersecretion in patients with Zollinger-Ellison syndrome. Surgery. 1988;104:1054–63. [PubMed] [Google Scholar]
- 259.Metz DC, Forsmark C, Lew EA, et al. Replacement of oral proton pump inhibitors with intravenous pantoprazole to effectively control gastric acid hypersecretion in patients with Zollinger-Ellison syndrome. Am J Gastroenterol. 2001;96:3274–80. doi: 10.1111/j.1572-0241.2001.05325.x. [DOI] [PubMed] [Google Scholar]
- 260.de Herder WW, Niederle B, Scoazec JY, et al. Well-differentiated pancreatic tumor/carcinoma: insulinoma. Neuroendocrinology. 2006;84:183–8. doi: 10.1159/000098010. [DOI] [PubMed] [Google Scholar]
- 261.Vezzosi D, Bennet A, Rochaix P, et al. Octreotide in insulinoma patients: efficacy on hypoglycemia, relationships with Octreoscan scintigraphy and immunostaining with anti-sst2A and anti-sst5 antibodies. Eur J Endocrinol. 2005;152:757–67. doi: 10.1530/eje.1.01901. [DOI] [PubMed] [Google Scholar]
- 262.Healy ML, Dawson SJ, Murray RM, et al. Severe hypoglycaemia after long-acting octreotide in a patient with an unrecognized malignant insulinoma. Intern Med J. 2007;37:406–9. doi: 10.1111/j.1445-5994.2007.01371.x. [DOI] [PubMed] [Google Scholar]
- 263.O’Toole D, Salazar R, Falconi M, et al. Rare functioning pancreatic endocrine tumors. Neuroendocrinology. 2006;84:189–95. doi: 10.1159/000098011. [DOI] [PubMed] [Google Scholar]
- 264.Jensen RT. Endocrine tumors of the pancreas. In: Yamada T, Alpers DH, Kaplowitz N, Laine L, Owyang C, Powell DW, editors. Textbook of Gastroenterology. 4. Philadelphia: Lippincott Williams and Wilkins; 2003. pp. 2108–46. [Google Scholar]
- 265.Akerstrom G, Hessman O, Skogseid B. Timing and extent of surgery in symptomatic and asymptomatic neuroendocrine tumors of the pancreas in MEN1. Langenbecks Arch Surg. 2002;386:558–69. doi: 10.1007/s00423-001-0274-6. [DOI] [PubMed] [Google Scholar]
- 266.Botsios D, Vasiliadis K, Tsalis K, et al. Management of nonfunctioning pancreatic endocrine tumors in the context of multiple endocrine neoplasia type 1 syndrome. J Gastrointestin Liver Dis. 2007;16:257–62. [PubMed] [Google Scholar]
- 267.Malagelada JR, Edis AJ, Adson MA, et al. Medical and surgical options in the management of patients with gastrinoma. Gastroenterology. 1983;84:1524–32. [PubMed] [Google Scholar]
- 268.Stabile BE, Morrow DJ, Passaro E., Jr The gastrinoma triangle: operative implications. Am J Surg. 1984;147:25–31. doi: 10.1016/0002-9610(84)90029-1. [DOI] [PubMed] [Google Scholar]
- 269.Bartsch DK, Langer P, Rothmund M. Surgical aspects of gastrinoma in multiple endocrine neoplasia type 1. Wien Klin Wochenschr. 2007;119:602–8. doi: 10.1007/s00508-007-0883-3. [DOI] [PubMed] [Google Scholar]
- 270.Skogseid B, Oberg K, Eriksson B, et al. Surgery for asymptomatic pancreatic lesion in multiple endocrine neoplasia type 1. World J Surg. 1996;20:872–7. doi: 10.1007/s002689900133. [DOI] [PubMed] [Google Scholar]
- 271.Thompson NW. Multiple endocrine neoplasia type I. Surgical therapy. Cancer Treat Res. 1997;89:407–19. doi: 10.1007/978-1-4615-6355-6_19. [DOI] [PubMed] [Google Scholar]
- 272.Terris B, Scoazeck JY, Rubbia L, et al. Expression of vascular endothelial growth factor in digestive neuroendocrine tumours. Histopathology. 1998;32:133–8. doi: 10.1046/j.1365-2559.1998.00321.x. [DOI] [PubMed] [Google Scholar]
- 273.Triponez F, Cadiot G. Non-functioning tumours of the pancreas in MEN1 patients. J Gastrointestin Liver Dis. 2007;16:295–6. [PubMed] [Google Scholar]
- 274.Beghelli S, Pelosi g, Zamboni G, et al. Pancreatic endocrine tumours: evidence for a tumour suppressor pathogenesis and for a tumour suppressor gene on chromosome 17p. J Pathol. 1998;186:41–50. doi: 10.1002/(SICI)1096-9896(199809)186:1<41::AID-PATH172>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 275.vanHeerden JA, Smith SL, Miller LJ. Management of the Zollinger-Ellison syndrome in patients with multiple endocrine neoplasia type 1. Surgery. 1986;100:971–7. [PubMed] [Google Scholar]
- 276.Thompson NW, Pasieka J, Fukuuchi A. Duodenal gastrinomas, duodenotomy, and duodenal exploration in the surgical management of Zollinger-Ellison syndrome. World J Surg. 1993;17:455–62. doi: 10.1007/BF01655104. [DOI] [PubMed] [Google Scholar]
- 277.Jaskowiak NT, Fraker DL, Alexander HR, et al. Is reoperation for gastrinoma excision in Zollinger-Ellison syndrome (ZES) indicated? Surgery. 1996;120:1057–63. doi: 10.1016/s0039-6060(96)80055-9. [DOI] [PubMed] [Google Scholar]
- 278.Fendrich V, Langer P, Celik I, et al. An Aggressive Surgical Approach Leads to Long-term Survival in Patients With Pancreatic Endocrine Tumors. Ann Surg. 2006;244:845–53. doi: 10.1097/01.sla.0000246951.21252.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Simon D, Starke A, Goretzki PE, et al. Reoperative surgery for organic hyperinsulinism: indications and operative strategy. World J Surg. 1998;22:666–72. doi: 10.1007/s002689900450. [DOI] [PubMed] [Google Scholar]
- 280.Sugg SL, Norton JA, Fraker DL, et al. A prospective study of intraoperative methods to diagnose and resect duodenal gastrinomas. Ann Surg. 1993;218:138–44. doi: 10.1097/00000658-199308000-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Norton JA, Doppman JL, Collen MJ, et al. Prospective study of gastrinoma localization and resection in patients with Zollinger-Ellison syndrome. Ann Surg. 1986;204:468–79. doi: 10.1097/00000658-198610000-00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Jensen RT, Fraker DL. Zollinger-Ellison syndrome: Advances in treatment of the gastric hypersecretion and the gastrinoma. J Am Med Assoc. 1994;271:1–7. doi: 10.1001/jama.271.18.1429. [DOI] [PubMed] [Google Scholar]
- 283.Stadil F. Treatment of gastrinomas with pancreaticoduodenectomy. In: Mignon M, Jensen RT, editors. Endocrine Tumors of the Pancreas: Recent advances in research and management. Series: Frontiers in Gastrointestinal Research. Vol. 23. Basel, Switzerland: S. Karger; 1995. pp. 333–41. [Google Scholar]
- 284.Phan GQ, Yeo CJ, Cameron JL, et al. Pancreaticoduodenectomy for selected periampullary neuroendocrine tumors: Fifty patients. Surgery. 1997;122:989–97. doi: 10.1016/s0039-6060(97)90200-2. [DOI] [PubMed] [Google Scholar]
- 285.Mignon M, Cadiot G. Diagnostic and therapeutic criteria in patients with Zollinger-Ellison syndrome and multiple endocrine neoplasia type 1. J Intern Med. 1998;243:489–94. doi: 10.1046/j.1365-2796.1998.00287.x. [DOI] [PubMed] [Google Scholar]
- 286.Akerstrom G, Hellman P. Surgery on neuroendocrine tumours. Best Pract Res Clin Endocrinol Metab. 2007;21:87–109. doi: 10.1016/j.beem.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 287.Lee CH, Peng FK, Lui WY. The clinical aspect of retained gastric antrum. Arch Surg. 1986;121:1181–6. doi: 10.1001/archsurg.1986.01400100093018. [DOI] [PubMed] [Google Scholar]
- 288.Clark OH, Ajani J, Benson AB, III, et al. Neuroendocrine tumors. J Natl Compr Canc Netw. 2006;4:102–38. doi: 10.6004/jnccn.2006.0013. [DOI] [PubMed] [Google Scholar]
- 289.McCallum RW, Parameswaran V, Burgess JR. Multiple endocrine neoplasia type 1 (MEN 1) is associated with an increased prevalence of diabetes mellitus and impaired fasting glucose. Clin Endocrinol (Oxf) 2006;65:163–8. doi: 10.1111/j.1365-2265.2006.02563.x. [DOI] [PubMed] [Google Scholar]
- 290.Norton JA. Surgery for primary pancreatic neuroendocrine tumors. J Gastrointest Surg. 2006;10:327–31. doi: 10.1016/j.gassur.2005.08.023. [DOI] [PubMed] [Google Scholar]
- 291.Norton JA. Surgical treatment of neuroendocrine metastases. Best Pract Res Clin Gastroenterol. 2005;19:577–83. doi: 10.1016/j.bpg.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 292.Wiedenmann B, Jensen RT, Mignon M, et al. Preoperative diagnosis and surgical manangement of neuroendocrine gastroenteropancreatic tumors: general recommendations by a consensus workshop. World J Surg. 1998;22:309–18. doi: 10.1007/s002689900387. [DOI] [PubMed] [Google Scholar]
- 293.Kaltsas GA, Besser GM, Grossman AB. The diagnosis and medical management of advanced neuroendocrine tumors. Endocr Rev. 2004;25:458–511. doi: 10.1210/er.2003-0014. [DOI] [PubMed] [Google Scholar]
- 294.Virgolini I, Traub-Weidinger T, Decristoforo C. Nuclear medicine in the detection and management of pancreatic islet-cell tumours. Best Pract Res Clin Endocrinol Metab. 2005;19:213–27. doi: 10.1016/j.beem.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 295.Vilar E, Salazar R, Perez-Garcia J, et al. Chemotherapy and role of the proliferation marker Ki-67 in digestive neuroendocrine tumors. Endocr Relat Cancer. 2007;14:221–32. doi: 10.1677/ERC-06-0074. [DOI] [PubMed] [Google Scholar]
- 296.Shuin T, Yamasaki I, Tamura K, et al. Von Hippel-Lindau disease: molecular pathological basis, clinical criteria, genetic testing, clinical features of tumors and treatment. Jpn J Clin Oncol. 2006;36:337–43. doi: 10.1093/jjco/hyl052. [DOI] [PubMed] [Google Scholar]
- 297.Hammel PR, Vilgrain V, Terris B, et al. Pancreatic involvement in von Hippel-Lindau disease. Gastroenterology. 2000;119:1087–95. doi: 10.1053/gast.2000.18143. [DOI] [PubMed] [Google Scholar]
- 298.Marcos HB, Libutti SK, Alexander HR, et al. Neuroendocrine tumors of the pancreas in von Hippel-Lindau disease: spectrum of appearances at CT and MR imaging with histopathologic comparison. Radiology. 2002;225:751–8. doi: 10.1148/radiol.2253011297. [DOI] [PubMed] [Google Scholar]
- 299.Libutti SK, Choyke PL, Alexander HR, et al. Clinical and genetic analysis of patients with pancreatic neuroendocrine tumors associated with von Hippel-Lindau disease. Surgery. 2000;128:1022–7. doi: 10.1067/msy.2000.110239. [DOI] [PubMed] [Google Scholar]
- 300.Binkovitz LA, Johnson CD, Stephens DH. Islet cell tumors in von Hippel-Lindau disease: increased prevalence and relationship to the multiple endocrine neoplasias. AJR Am J Roentgenol. 1990;155:501–5. doi: 10.2214/ajr.155.3.1974734. [DOI] [PubMed] [Google Scholar]
- 301.Yamasaki I, Nishimori I, Ashida S, et al. Clinical characteristics of pancreatic neuroendocrine tumors in Japanese patients with von Hippel-Lindau disease. Pancreas. 2006;33:382–5. doi: 10.1097/01.mpa.0000240604.26312.e4. [DOI] [PubMed] [Google Scholar]
- 302.Delman KA, Shapiro SE, Jonasch EW, et al. Abdominal visceral lesions in von Hippel-Lindau disease: incidence and clinical behavior of pancreatic and adrenal lesions at a single center. World J Surg. 2006;30:665–9. doi: 10.1007/s00268-005-0359-4. [DOI] [PubMed] [Google Scholar]
- 303.Girelli R, Bassi C, Falconi M, et al. Pancreatic cystic manifestations in von Hippel-Lindau disease. Int J Pancreatol. 1997;22:101–9. doi: 10.1007/BF02787467. [DOI] [PubMed] [Google Scholar]
- 304.Chetty R, Kennedy M, Ezzat S, et al. Pancreatic endocrine pathology in von Hippel-Lindau disease: an expanding spectrum of lesions. Endocr Pathol. 2004;15:141–8. doi: 10.1385/ep:15:2:141. [DOI] [PubMed] [Google Scholar]
- 305.Mukhopadhyay B, Sahdev A, Monson JP, et al. Pancreatic lesions in von Hippel-Lindau disease. Clin Endocrinol (Oxf) 2002;57:603–8. doi: 10.1046/j.1365-2265.2002.01637.x. [DOI] [PubMed] [Google Scholar]
- 306.Libutti SK, Choyke PL, Bartlett DL, et al. Pancreatic neuroendocrine tumors associated with von Hippel Lindau disease: diagnostic and management recomendations. Surgery. 1998;124:1153–9. doi: 10.1067/msy.1998.91823. [DOI] [PubMed] [Google Scholar]
- 307.Lubensky IA, Pack S, Ault D, et al. Multiple neuroendocrine tumors of the pancreas in von Hippel-Lindau disease patients. Am J Pathol. 1998;153:223–31. doi: 10.1016/S0002-9440(10)65563-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Ling H, Cybulla M, Schaefer O, et al. When to look for Von Hippel-Lindau disease in gastroenteropancreatic neuroendocrine tumors? Neuroendocrinology. 2004;80 Suppl 1:39–46. doi: 10.1159/000080740. [DOI] [PubMed] [Google Scholar]
- 309.Neumann HP, Dinkel E, Brambs H, et al. Pancreatic lesions in the von Hippel-Lindau syndrome. Gastroenterology. 1991;101:465–71. doi: 10.1016/0016-5085(91)90026-h. [DOI] [PubMed] [Google Scholar]
- 310.Hough DM, Stephens DH, Johnson CD, et al. Pancreatic lesions in von Hippel-Lindau disease: prevalence, clinical significance, and CT findings. AJR Am J Roentgenol. 1994;162:1091–4. doi: 10.2214/ajr.162.5.8165988. [DOI] [PubMed] [Google Scholar]
- 311.Lott ST, Chandler DS, Curley SA, et al. High frequency loss of heterozygosity in von Hippel-Lindau (VHL)-associated and sporadic pancreatic islet cell tumors: evidence for a stepwise mechanism for malignant conversion in VHL tumorigenesis. Cancer Res. 2002;62:1952–5. [PubMed] [Google Scholar]
- 312.Blansfield JA, Choyke L, Morita SY, et al. Clinical, genetic and radiographic analysis of 108 patients with von Hippel-Lindau disease (VHL) manifested by pancreatic neuroendocrine neoplasms (PNETs) Surgery. 2007;142:814–8. doi: 10.1016/j.surg.2007.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Maddock IR, Moran A, Maher ER, et al. A genetic register for von Hippel-Lindau disease. J Med Genet. 1996;33:120–7. doi: 10.1136/jmg.33.2.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Niemela M, Lemeta S, Summanen P, et al. Long-term prognosis of haemangioblastoma of the CNS: impact of von Hippel-Lindau disease. Acta Neurochir (Wien) 1999;141:1147–56. doi: 10.1007/s007010050412. [DOI] [PubMed] [Google Scholar]
- 315.Maher ER, Yates JR, Harries R, et al. Clinical features and natural history of von Hippel-Lindau disease. Q J Med. 1990;77:1151–63. doi: 10.1093/qjmed/77.2.1151. [DOI] [PubMed] [Google Scholar]
- 316.Ferner RE. Neurofibromatosis 1 and neurofibromatosis 2: a twenty first century perspective. Lancet Neurol. 2007;6:340–51. doi: 10.1016/S1474-4422(07)70075-3. [DOI] [PubMed] [Google Scholar]
- 317.Feldkamp MM, Gutmann DH, Guha A. Neurofibromatosis type 1: piecing the puzzle together. Can J Neurol Sci. 1998;25:181–91. doi: 10.1017/s0317167100033990. [DOI] [PubMed] [Google Scholar]
- 318.Trovo-Marqui AB, Tajara EH. Neurofibromin: a general outlook. Clin Genet. 2006;70:1–13. doi: 10.1111/j.1399-0004.2006.00639.x. [DOI] [PubMed] [Google Scholar]
- 319.Xu GF, O’Connell P, Viskochil D, et al. The neurofibromatosis type 1 gene encodes a protein relatd to GAP. Cell. 1990;62:599–608. doi: 10.1016/0092-8674(90)90024-9. [DOI] [PubMed] [Google Scholar]
- 320.Dayal Y, Tallberg KA, Nunnemacher G, et al. Duodenal carcinoids in patients with and without neurofibromatosis. A comparative study. Am J Surg Pathol. 1986;10:348–57. doi: 10.1097/00000478-198605000-00007. [DOI] [PubMed] [Google Scholar]
- 321.Soga J, Yakuwa Y. Somatostatinoma/inhibitory syndrome: a statistical evaluation of 173 reported cases as compared to other pancreatic endocrinomas. J Exp Clin Cancer Res. 1999;18:13–22. [PubMed] [Google Scholar]
- 322.Mao C, Shah A, Hanson DJ, et al. Von Recklinghausen’s disease associated with duodenal somatostatinoma: contrast of duodenal versus pancreatic somatostatinomas. J Surg Oncol. 1995;59:67–73. doi: 10.1002/jso.2930590116. [DOI] [PubMed] [Google Scholar]
- 323.van Basten JP, van Hoek B, de Bruine A, et al. Ampullary carcinoid and neurofibromatosis: case report and review of the literature. Neth J Med. 1994;44:202–6. [PubMed] [Google Scholar]
- 324.Clements WM, Martin SP, Stemmerman G, et al. Ampullary carcinoid tumors: rationale for an aggressive surgical approach. J Gastrointest Surg. 2003;7:773–6. doi: 10.1016/s1091-255x(03)00114-8. [DOI] [PubMed] [Google Scholar]
- 325.Lee WS, Koh YS, Kim JC, et al. Zollinger-Ellison syndrome associated with neurofibromatosis type 1: a case report. BMC Cancer. 2005;5:85. doi: 10.1186/1471-2407-5-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Chagnon JP, Barge J, Henin D, et al. [Recklinghausen’s disease with digestive localizations associated with gastric acid hypersecretion suggesting Zollinger-Ellison syndrome] Gastroenterol Clin Biol. 1985;9(1):65–9. [PubMed] [Google Scholar]
- 327.Fung JW, Lam KS. Neurofibromatosis and insulinoma. Postgrad Med J. 1995;71:485–6. doi: 10.1136/pgmj.71.838.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Perren A, Wiesli P, Schmid S, et al. Pancreatic endocrine tumors are a rare manifestation of the neurofibromatosis type 1 phenotype: molecular analysis of a malignant insulinoma in a NF-1 patient. Am J Surg Pathol. 2006;30:1047–51. doi: 10.1097/00000478-200608000-00018. [DOI] [PubMed] [Google Scholar]
- 329.Coskey RL, Tranquada RE. Insulinoma and multiple neurofibromatasis: report of a case. Metabolism. 1965;13:312–8. doi: 10.1016/0026-0495(64)90058-7. [DOI] [PubMed] [Google Scholar]
- 330.Fujisawa T, Osuga T, Maeda M, et al. Malignant endocrine tumor of the pancreas associated with von Recklinghausen’s disease. J Gastroenterol. 2002;37:59–67. doi: 10.1007/s535-002-8135-x. [DOI] [PubMed] [Google Scholar]
- 331.Miettinen M, Fetsch JF, Sobin LH, et al. Gastrointestinal stromal tumors in patients with neurofibromatosis 1: a clinicopathologic and molecular genetic study of 45 cases. Am J Surg Pathol. 2006;30:90–6. doi: 10.1097/01.pas.0000176433.81079.bd. [DOI] [PubMed] [Google Scholar]
- 332.Bettini R, Falconi M, Crippa S, et al. Ampullary somatostatinomas and jejunal gastrointestinal stromal tumor in a patient with Von Recklinghausen’s disease. World J Gastroenterol. 2007;13:2761–3. doi: 10.3748/wjg.v13.i19.2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Hatzitheoklitos E, Buchler MW, Friess H, et al. Carcinoid of the ampulla of Vater. Clinical characteristics and morphologic features. Cancer. 1994;73:1580–8. doi: 10.1002/1097-0142(19940315)73:6<1580::aid-cncr2820730608>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 334.Burke AP, Federspiel BH, Sobin LH, et al. Carcinoids of the duodenum. A histologic and immunohistochemical study of 65 tumors. Am J Surg Pathol. 1989;13:828–37. doi: 10.1097/00000478-198910000-00002. [DOI] [PubMed] [Google Scholar]
- 335.Mayoral W, Salcedo J, Al-Kawas F. Ampullary carcinoid tumor presenting as acute pancreatitis in a patient with von Recklinghausen’s disease: case report and review of the literature. Endoscopy. 2003;35:854–7. doi: 10.1055/s-2003-42613. [DOI] [PubMed] [Google Scholar]
- 336.Klein A, Clemens J, Cameron J. Periampullary neoplasms in von Recklinghausen’s disease. Surgery. 1989;106:815–9. [PubMed] [Google Scholar]
- 337.Witzigmann H, Loracher C, Geissler F, et al. Neuroendocrine tumours of the duodenum. Clinical aspects, pathomorphology and therapy. Langenbecks Arch Surg. 2002;386:525–33. doi: 10.1007/s00423-001-0260-z. [DOI] [PubMed] [Google Scholar]
- 338.Capella C, Riva C, Rindi G, et al. Endocrine tumors of the duodenum and upper jejunum. A study of 33 cases with clinico-pathological characteristics and hormone content. Hepatogastroenterology. 1990;37:247–52. [PubMed] [Google Scholar]
- 339.Rasmussen SA, Yang Q, Friedman JM. Mortality in neurofibromatosis 1: an analysis using U.S. death certificates. Am J Hum Genet. 2001;68:1110–8. doi: 10.1086/320121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Yohay K. Neurofibromatosis types 1 and 2. Neurologist. 2006;12:86–93. doi: 10.1097/01.nrl.0000195830.22432.a5. [DOI] [PubMed] [Google Scholar]
- 341.Khosrotehrani K, Bastuji-Garin S, Zeller J, et al. Clinical risk factors for mortality in patients with neurofibromatosis 1: a cohort study of 378 patients. Arch Dermatol. 2003;139:187–91. doi: 10.1001/archderm.139.2.187. [DOI] [PubMed] [Google Scholar]
- 342.Makhlouf HR, Burke AP, Sobin LH. Carcinoid tumors of the ampulla of Vater: a comparison with duodenal carcinoid tumors. Cancer. 1999;85:1241–9. doi: 10.1002/(sici)1097-0142(19990315)85:6<1241::aid-cncr5>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 343.Hartel M, Wente MN, Sido B, et al. Carcinoid of the ampulla of Vater. J Gastroenterol Hepatol. 2005;20:676–81. doi: 10.1111/j.1440-1746.2005.03744.x. [DOI] [PubMed] [Google Scholar]
- 344.Gilani N, Ramirez FC. Endoscopic resection of an ampullary carcinoid presenting with upper gastrointestinal bleeding: a case report and review of the literature. World J Gastroenterol. 2007;13:1268–70. doi: 10.3748/wjg.v13.i8.1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Leung AK, Robson WL. Tuberous sclerosis complex: a review. J Pediatr Health Care. 2007;21:108–14. doi: 10.1016/j.pedhc.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 346.Schwartz RA, Fernandez G, Kotulska K, et al. Tuberous sclerosis complex: advances in diagnosis, genetics, and management. J Am Acad Dermatol. 2007;57:189–202. doi: 10.1016/j.jaad.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 347.Schwarzkopf G, Pfisterer J. Metastasizing gastrinoma and tuberous sclerosis complex. Zentralbl Pathol. 1994;139:477–81. [PubMed] [Google Scholar]
- 348.Kim H, Kerr A, Morehouse H. The association between tuberous sclerosis and insulinoma. AJNR Am J Neuroradiol. 1995;16:1543–4. [PMC free article] [PubMed] [Google Scholar]
- 349.Eledrisi MS, Stuart CA, Alshanti M. Insulinoma in a patient with tuberous sclerosis: is there an association? Endocr Pract. 2002;8:109–12. doi: 10.4158/EP.8.2.109. [DOI] [PubMed] [Google Scholar]
- 350.Gutman A, Leffkowitz M. Tuberous sclerosis associated with spontaneous hypoglycemia. Brit Med J. 1959;2:1065–8. doi: 10.1136/bmj.2.5159.1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Davoren PM, Epstein MT. Insulinoma complicating tuberous sclerosis. J Neurol Neurosurg Psychiatry. 1992;55:1209. doi: 10.1136/jnnp.55.12.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Boubaddi NE, Imbert Y, Tissot B, et al. Secreting insulinoma and Bourneville’s tuberous sclerosis. Gastroenterol Clin Biol. 1997;21:343. [PubMed] [Google Scholar]
- 353.Verhoef S, van Diemen-Steenvoorde R, Akkersdijk WL, et al. Malignant pancreatic tumour within the spectrum of tuberous sclerosis complex in childhood. Eur J Pediatr. 1999;158:284–7. doi: 10.1007/s004310051073. [DOI] [PubMed] [Google Scholar]
- 354.Merritt JL, Davis DM, Pittelkow MR, et al. Extensive acrochordons and pancreatic islet-cell tumors in tuberous sclerosis associated with TSC2 mutations. Am J Med Genet A. 2006;140:1669–72. doi: 10.1002/ajmg.a.31351. [DOI] [PubMed] [Google Scholar]
- 355.Francalanci P, Domedi-Camassei F, Purificato C, et al. Malignant pancreatic endocrine tumor in a child with tuberous sclerosis. Am J Surg Pathol. 2003;27:1386–9. doi: 10.1097/00000478-200310000-00012. [DOI] [PubMed] [Google Scholar]
- 356.Ilgren EB, Westmoreland D. Tuberous sclerosis: unusual associations in four cases. J Clin Pathol. 1984;37:272–8. doi: 10.1136/jcp.37.3.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Eberle F, Grun R. Multiple endocrine neoplasia, type I (MEN I) Ergeb Inn Med Kinderheilkd. 1981;46:76–149. [PubMed] [Google Scholar]
- 358.Shepherd JJ, Challis DR, Davies PF, et al. Multiple endocrine neoplasm, type 1: Gastrinomas, pancreatic neoplasms, microcarcinoids, the Zollinger-Ellison syndrome, lymph nodes, and hepatic metastases. Arch Surg. 1993;128:1133–42. doi: 10.1001/archsurg.1993.01420220053007. [DOI] [PubMed] [Google Scholar]
- 359.Alexander HR, Jensen RT. Pancreatic Endocrine Tumors. In: De Vita VT, Hellman S, Rosenberg SA, editors. Cancer: Principles and Practice of Oncology. 7. Philadelphia: Lippincott Williams and Wilkins, Co.; 2005. pp. 1548–58. [Google Scholar]
- 360.Hausman MS, Jr, Thompson NW, Gauger PG, et al. The surgical management of MEN-1 pancreatoduodenal neuroendocrine disease. Surgery. 2004;136:1205–11. doi: 10.1016/j.surg.2004.06.049. [DOI] [PubMed] [Google Scholar]
- 361.Mignon M, Cadiot G, Rigaud D, et al. Management of islet cell tumors in patients with multiple endocrine neoplasia type 1. In: Mignon M, Jensen RT, editors. Endocrine Tumors of the Pancreas: Recent advances in research and management. Series: Frontiers in Gastrointestinal Research. Vol. 23. Basel, Switzerland: S. Karger; 1995. pp. 342–59. [Google Scholar]
- 362.Cherner JA, Sawyers JL. Benefit of resection of metastatic gastrinoma in multiple endocrine neoplasia type I. Gastroenterology. 1992;102:1049–53. doi: 10.1016/0016-5085(92)90196-6. [DOI] [PubMed] [Google Scholar]
- 363.Kisker O, Bastian D, Bartsch D, et al. Localization, malignant potential, and surgical management of gastrinomas. World J Surg. 1998;22:651–8. doi: 10.1007/s002689900448. [DOI] [PubMed] [Google Scholar]
- 364.Jordan PH., Jr A personal experience with pancreatic and duodenal neuroendocrine tumors. J Am Coll Surg. 1999;189:470–82. doi: 10.1016/s1072-7515(99)00162-3. [DOI] [PubMed] [Google Scholar]
- 365.Kato M, Imamura M, Hosotani R, et al. Curative resection of microgastrinomas based on the intraoperative secretin test. World J Surg. 2000;24:1425–30. doi: 10.1007/s002680010235. [DOI] [PubMed] [Google Scholar]
- 366.Thodiyil PA, El-Masry NS, Williamson RC. Achieving eugastrinaemia in Zollinger-Ellison syndrome: resection or enucleation? Dig Surg. 2001;18:118–23. doi: 10.1159/000050111. [DOI] [PubMed] [Google Scholar]
- 367.Gauger PG, Thompson NW. Early surgical intervention and strategy in patients with multiple endocrine neoplasia type 1. Best Practice & Res Clin Endocrinol Metabol. 2001;15:213–23. doi: 10.1053/beem.2001.0136. [DOI] [PubMed] [Google Scholar]
- 368.Waddell WR, Coppinger WR, Loughry RW. Pancreaticoduodenectomy for Zollinger-Ellison syndrome. Ann Surg. 1968;168(4):641–54. doi: 10.1097/00000658-196810000-00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Lind T, Olbe L. Long term follow up of patients with Zollinger-Ellison syndrome (ZES) Acta Chir Scand. 1989;155:383–8. [PubMed] [Google Scholar]
- 370.Delcore R, Friesen SR. Role of pancreatoduodenectomy in the management of primary duodenal wall gastrinomas in patients with Zollinger-Ellison syndrome. Surgery. 1992;112:1016–22. [PubMed] [Google Scholar]
- 371.Schroder W, Holscher AH, Beckurts T, et al. Duodenal microgastrinomas associated with Zollinger-Ellison syndrome. Hepato-Gastroenterol. 1996;43:1465–9. [PubMed] [Google Scholar]
- 372.Sarmiento JM, Farnell MB, Que FG, et al. Pancreaticoduodenectomy for islet cell tumors of the head of the pancreas: long-term survival analysis. World J Surg. 2002;26:1267–71. doi: 10.1007/s00268-002-6714-9. [DOI] [PubMed] [Google Scholar]
- 373.Fernandez-Cruz L, Martinez I, Cesar-Borges G, et al. Laparoscopic surgery in patients with sporadic and multiple insulinomas associated with multiple endocrine neoplasia type 1. J Gastrointest Surg. 2005;9:381–8. doi: 10.1016/j.gassur.2004.06.009. [DOI] [PubMed] [Google Scholar]