

Abstract
Pharmacogenetics is the study of the role of inheritance in variation in drug response phenotypes. Those phenotypes can range from life-threatening adverse drugs reactions at one end of the spectrum to equally serious lack of therapeutic efficacy at the other. Over the past half century, pharmacogenetics has—like all of medical genetics—evolved from a discipline with a focus on monogenetic traits to become pharmacogenomics, with a genome-wide perspective. This article will briefly review recent examples of the application of genome-wide techniques to clinical pharmacogenomic studies and to pharmacogenomic model systems that vary from cell line-based model systems to yeast gene deletion libraries. Functional validation of candidate genes and the use of genome-wide techniques to gain mechanistic insights will be emphasized for the establishment of biological plausibility and as essential follow-up steps after the identification of candidate genes.
INTRODUCTION
Pharmacogenetics is the study of the role of inheritance in variation in drug response (1,2). Although many factors can contribute to variation in response to drug therapy—factors that include age, sex, diet and the effects of other drugs—it is clear that inheritance can also play a major role (1,2). Pharmacogenetics has now ‘matured’ to the point that the United States Food and Drug Administration (FDA) has held a series of public hearings on the incorporation of pharmacogenetic information into drug labeling (www.fda.org). Most of these mature examples of the clinical relevance of pharmacogenetics involve monogenic variation in drug metabolism. Those examples include thiopurine S-methyltransferase (TPMT) polymorphisms and variation in the metabolism and effect of the thiopurine drugs that are used to treat childhood leukemia (3); UGT1A1 and the metabolism of the anticancer drug irinotecan (4); cytochrome P450 (CYP) 2C9 and the metabolism of the anticoagulant warfarin (5) and CYP2D6 and the metabolic ‘activation’ of the breast cancer drug tamoxifen (6,7). Drug metabolism alters ‘pharmacokinetics’ factors that influence the concentration of drug that reaches its target. Equally important genetic variation can also occur in the target itself or signaling downstream of the target, so-called ‘pharmacodynamic’ factors. The past half century has witnessed increasing understanding of this type of monogenic pharmacogenetic trait (1,2). However, the recent development of genome-wide techniques has made it possible to move beyond known genes in known pathways to identify unanticipated candidate genes that might contribute to risk for the occurrence of adverse drug reactions or lack of the desired drug effect. Although the application of genome-wide techniques to perform ‘pharmacogenomic’ studies is only in its infancy, subsequent paragraphs will highlight examples of genome-wide studies in pharmacogenomics. These examples will include both clinical studies involving patients and studies performed with pharmacogenomic model systems. Model systems are being used increasingly for candidate gene identification and hypothesis generation, for the functional validation of candidates and to explore pharmacogenomic mechanisms. As a result, there are currently more examples of the application of genome-wide techniques to pharmacogenomic model systems than of clinical studies—a situation that will undoubtedly change rapidly in the future.
GENOME-WIDE CLINICAL PHARMACOGENOMICS
Striking examples of the clinical relevance of monogenic pharmacogenetic traits have been known for decades (1,2). However, the application of this information in clinical practice has been limited. That is true in spite of the fact that, for example, subjects homozygous for the most common TPMT variant allele, TPMT*3A, are at greatly increased risk for life-threatening myelosuppression (bone marrow suppression), when treated with ‘standard’ doses of thiopurine drugs for diseases such as childhood leukemia (3). Conversely, breast cancer patients who are homozygous for CYP2D6 variant alleles that result in the absence of enzyme activity are at increased risk for breast cancer recurrence, presumably because they lack the ability to convert tamoxifen to its active metabolites, 4-hydroxytamoxifen and 4-hydroxy-N-desmethyltamoxifen (endoxifen) (6,7). The fact that the clinical application of these monogenic traits has been so slow raises a serious question with regard to the practical therapeutic use of data obtained with genome-wide association studies (GWAS). That is because studies of risk alleles for diseases such as diabetes and breast cancer have shown that, although GWAS can identify novel genes that contribute to risk, the odds ratios (ORs) are generally relatively small (8–11). The practical clinical utility of that type of information, even in a public health setting, remains controversial (12). Pharmacogenomic information would presumably be used by physicians to make practical decisions with regard to drug selection and dosage in an individual patient. It is questionable whether ORs as quantitatively small as most of those that have been reported for disease risk will prove to be useful for clinical application to individual patients. However, it has been speculated that the application of genome-wide techniques might result in the identification of drug response genes having major effects—particularly for adverse drug reactions. Currently, so few clinically relevant examples of genome-wide studies of drug response phenotypes have been reported that it is impossible to answer that question. However, one example was reported recently which suggests that the application of genome-wide techniques might make it possible to identify polymorphisms of practical value for therapeutic decision making.
HMG-CoA reductase inhibitors, the ‘statins’, are among the best-selling drugs worldwide. These agents are generally safe and effective. However, they do have side effects. The most serious is myopathy, which can be life-threatening in a small number of patients (13). A very recent study used a GWAS to identify genes that might contribute to risk for statin-induced myopathy (14). Specifically, the 6031 patients in the SEARCH trial who were treated with an 80 mg dose of the commonly used statin, simvastatin, were surveyed to identify patients who had suffered serious myopathy. Eighty-five myopathy patients with adequate DNA for genotyping were identified, a number that usually would be considered inadequate for use in a GWAS based on power calculations for OR values like those observed in studies of disease pathophysiology. However, these investigators persevered and performed a GWAS with these 85 patients and 90-matched controls from the same group of patients treated with simvastatin. A single nucleotide polymorphism (SNP) in the transporter gene SLCO1B1, encoding the organic anion-transporting polypeptide OATP1B1, had a P-value of 4 × 10−9 with an OR of 16.9 for subjects homozygous for the variant allele, and 4.5 for heterozygous subjects (Fig. 1A). The authors then replicated their finding in an independent study that included 10 269 patients who had been treated with 40 mg of simvastatin (15). The replication study showed an OR value of 2.6 per copy of the variant allele. The authors estimated that more than 60% of statin-induced myopathy cases in their study could be attributed to this single variant allele (Fig. 1B).
Figure 1.
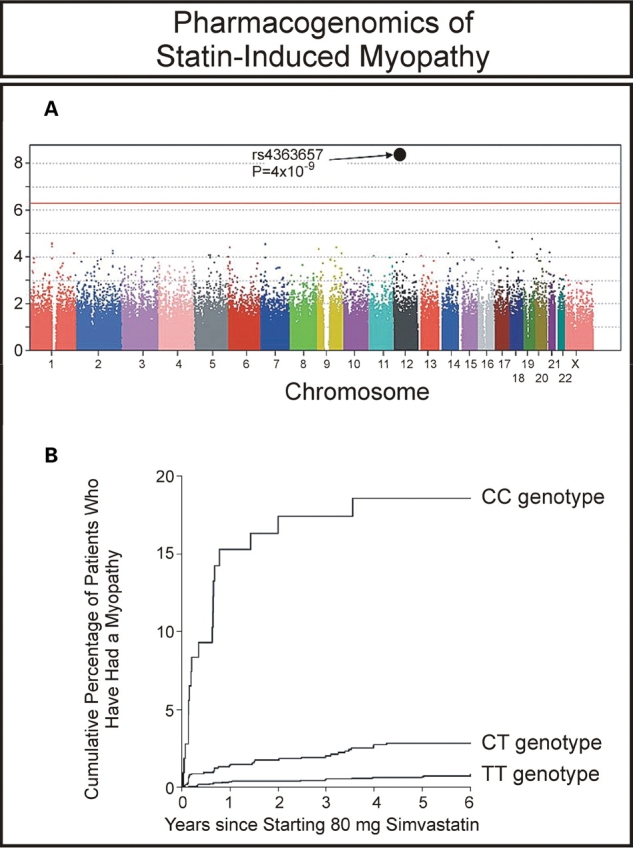
GWAS of statin-induced myopathy. (A) Association between myopathy and each SNP assayed in the GWAS. (B) Estimated cumulative risk of myopathy associated with 80 mg of simvastatin daily. (Modified from the SEARCH Collaborations Group (14) with permission of the Massachusetts Medical Society.)
Whether this example will prove to be representative of pharmacogenomic traits associated with adverse drug reactions remains to be seen, pending the application of genome-wide techniques to study many drug reaction phenotypes. However, the size required, as well as the expense and difficulty associated with gaining access to clinical samples suitable for this type of study, highlights the need for the development of genome-wide model systems that can be used to screen for candidate genes that play a role in drug sensitivity or resistance. That is particularly true for drugs with a narrow therapeutic index—drugs for which the toxic dose is similar to the therapeutic dose—or drugs like the statins for which adverse reactions can be life-threatening. It is for that reason that more genome-wide pharmacogenomic data are currently available for model systems than for clinical pharmacogenomics studies. Subsequent paragraphs will briefly outline the current status of the application of genome-wide techniques to pharmacogenomic model systems.
GENOME-WIDE MODEL SYSTEM PHARMACOGENOMICS
In vitro model systems such as cell lines from large numbers of individuals represent an attractive and cost-effective approach that has been used to identify genes associated with variation in drug response by the application of genome-wide techniques. Observations made with such model systems can be pursued by functional validation and by replication studies performed with clinical samples, as outlined schematically in Figure 2. These cell line model systems can be used to perform pharmacogenomic studies using a variety of phenotypes in a setting that is much more highly controlled than the clinical environment. An additional advantage is that many of these model systems use cell lines and associated genomic data that are publicly available. That is an important advantage because all too often more time is spent negotiating access to clinical samples than in actually performing the study and analyzing the data. Finally, unlike the situation with clinical studies, information obtained with cell lines is cumulative, i.e. as new genomic and phenotypic assays are developed, they can be applied to the same ‘patients’, i.e. the same cell lines. Therefore, in vitro cell line model systems are well suited for the generation of pharmacogenomic hypotheses during the ‘discovery phase’, followed by both functional validation—as outlined subsequently—and by the translation of laboratory-based observations into the clinic (Fig. 2). Conversely, these same in vitro cell line systems can also be used for the functional validation of significant ‘hits’ observed in the course of clinical genome-wide studies.
Figure 2.
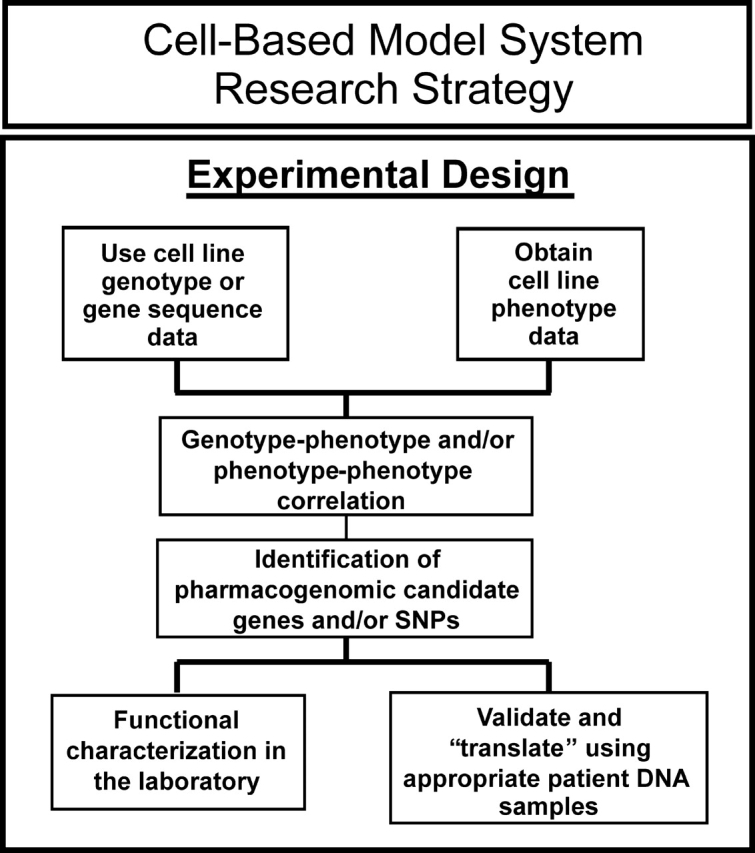
Diagramatic outline of the use of a cell line-based model system to identify and both functionally and clinically validate pharmacogenomic candidate genes.
The in vitro cell line model systems that have been used most often in pharmacogenomic studies include HapMap and Human Variation Panel lymphoblastoid cell lines—both of which are publicly available from the Coriell Institute. These are Epstein–Barr virus (EBV)-transformed lymphocytes immortalized from hundreds of individuals of different ethnic groups. This approach focuses on common variation in germline DNA and can be contrasted with the use of tumor cell lines such as the NCI-60 cells in which cell lines of multiple tumor types—but obtained from only a few individuals each—are studied (16–18). The samples used in the original International HapMap Project included four populations (19–22), and the Human Variation Panel cell lines were collected in the U.S. from unrelated individuals of different ethnicities (http://ccr.coriell.org/ Sections/Collections/ NIGMS/Populations.aspx?PgId=177&coll=GM). A great deal of data, including genome-wide SNP and expression array data, are available for most of these cell lines (22–30). Therefore, the genome-wide information necessary to perform a GWAS are publicly available for these cell line-based model systems. Studies performed with HapMap samples have demonstrated that the variation in gene expression or in drug response phenotypes such as cytotoxicity in these cells is regulated, in part, by inheritance (26). A variety of drug response phenotypes can be tested, with cytotoxicity being the most common endpoint that has been studied.
The major goal of pharmacogenomic studies is to identify genetic variation that might be responsible for individual differences in drug efficacy and/or risk for adverse drug reactions. Although classic examples of pharmacogenetics—as mentioned previously—most often involve only one or two genes and only a few SNPs (1,2), many drug response phenotypes may also be influenced by multiple genes. Therefore, the use of GWAS could potentially identify candidate genes that lie outside of our current range of knowledge. Those studies could also provide novel insight into mechanisms of drug action. For example, Huang et al. (29,31), Shukla et al. (32) and Duan et al. (33) have used HapMap cell lines to perform GWAS that included SNPs, expression array data and cytotoxicity phenotypes to study the antineoplastic drugs cisplatin, daunorubicin and etopside. These investigators identified a series of SNPs that were associated with drug-induced cytotoxicity as a result of their influence on gene expression. Li et al. (30) used nearly 200 Human Variation Panel cell lines to identify genes with expression levels that were significantly associated with sensitivity to two antineoplastic cytidine analogues, gemcitabine and AraC. Specifically, Li et al. (30) studied the association between gene expression and cytotoxicity and identified two top candidate genes, one within the known ‘metabolic pathway’ for these drugs and the other outside of current knowledge that were each significantly associated with gemcitabine and AraC cytotoxicity. These studies provide examples of the way in which cell line model systems can be used for pharmacogenomic hypotheses generation.
Although cell line-based model systems have already proved useful in identifying pharmacogenomic candidate genes, there are also significant potential limitations associated with their use. First, these lymphoblastoid cells are not derived from tumor tissue and they are not tumor cell lines. Second, <50% of human genes are expressed in lymphoblastoid cells (34), and the EBV transformation required to immortalize these cells is known to influence sensitivity to some drugs (35,36). Therefore, like any genome-wide association data, the results obtained with these in vitro systems must be functionally validated to provide biological plausibility and—ultimately—these results must be replicated in clinical studies. For example, in the study performed by Li et al. (30), the authors functionally validated the two top candidate genes that they identified by using siRNA ‘knockdown’ performed with tumor cell lines, followed by cytotoxicity and other functional assays. Figure 3A shows an example of this functional validation in which Li et al. (30) demonstrated that knockdown of a nucleotidase candidate gene (NT5C3) identified using the Human Variation Panel cell lines shifted dose–response curves for AraC in cancer cell lines to the left, as anticipated. These investigators also showed that expression of NT5C3 mRNA was inversely correlated with the active phosphorylated metabolite levels for AraC in lymphoblastoid cells (Fig. 3B). Although no data are currently available to demonstrate the success of using novel candidates identified with these cell lines to predict clinical drug response in patients, this approach could significantly narrow the list of candidate genes or SNPs to be tested during clinical studies.
Figure 3.
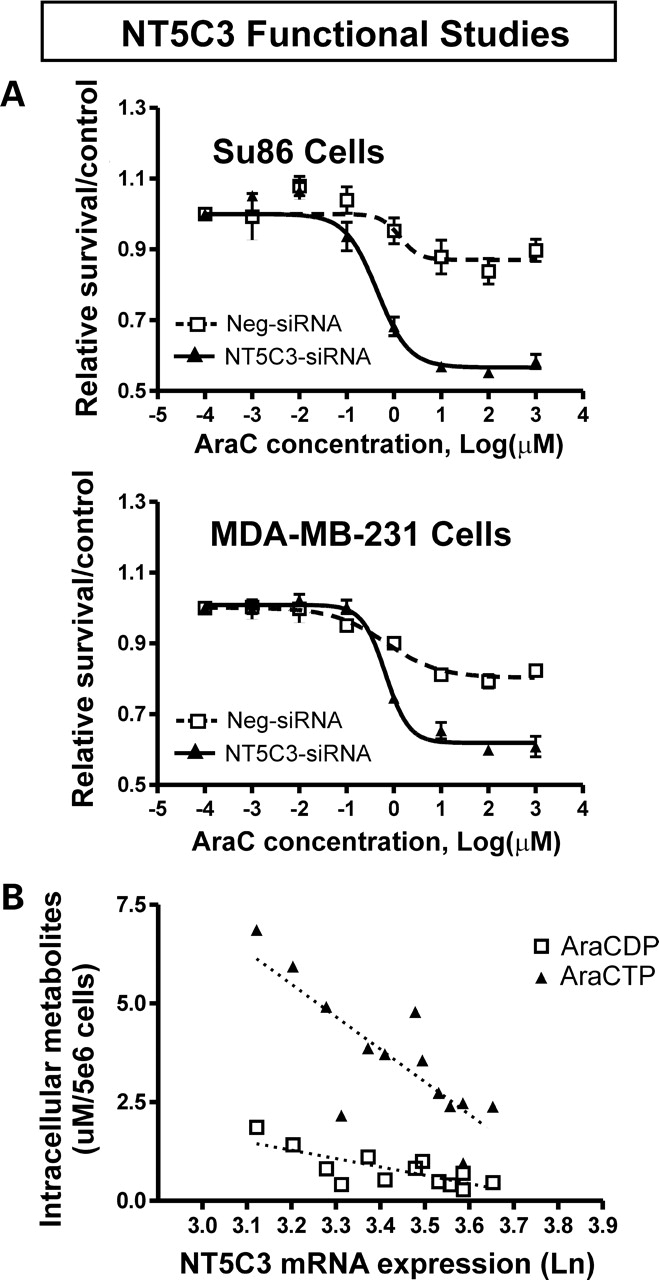
Functional validation of a candidate gene identified using the lymphoblastoid cell line model system. (A) siRNA knockdown of NT5C3 in cancer cell lines shifts the dose–response curves for AraC to the left, as anticipated. (B) Inverse correlation between NT5C3 mRNA levels and levels of AraC active metabolites in lymphoblastoid cells. (Modified from Li et al. (30) with permission of the American Association for Cancer Research.)
Yeast gene deletion libraries represent another ‘genome-wide’ model system that has been applied in pharmacogenomic studies. For example, a yeast gene deletion library was used to study mechanisms responsible for the striking functional effects of the common TPMT*3A variant allele that is associated with life-threatening myelosuppression in subjects treated with standard doses of thiopurine drugs (3). TPMT*3A includes two nonsynonymous SNPs—and is associated with lack of detectable TPMT protein in human tissues. These two SNPs appear to result in misfolding of the TPMT*3A variant allozyme, with resultant aggregation and accelerated proteasome-mediated degradation (37–39). The recent use of a yeast library in which 4667 nonessential yeast genes had been deleted resulted in the identification of 24 genes that participated in the degradation of TPMT*3A—including a series of genes encoding proteins required for autophagy (40). Autophagy had never previously been shown to be a mechanism in pharmacogenomics, but the application of this ‘genome-wide’ approach made it possible to move beyond candidate gene identification to study mechanisms responsible for the effect of a common variant allele of clinical pharmacogenomic importance.
CONCLUSIONS
Pharmacogenetics is a discipline with a history that extends back for a half century (1,2). During the past decade, pharmacogenetics has evolved into pharmacogenomics by the application of genome-wide techniques to study drug response phenotypes. However, even the very limited data currently available already make it possible to predict that these techniques—when applied to both clinical samples and model systems—will result in a dramatic expansion of our understanding of the role of genetics in variation in risk for either adverse drug reactions or lack of the desired therapeutic effect of the powerful drugs that are used in the twenty-first century medicine.
FUNDING
Supported in part by NIH grants K22 CA130828 (L.W.), R01 GM28157 (R.M.W.), R01 GM35720 (R.M.W.), R01 CA132780 (R.M.W.), U01 GM61388 (The Pharmacogenetics Research Network) (L.W. and R.M.W.), an ASPET-Astellas Award (L.W.) and a PhRMA Foundation ‘Center of Excellence in Clinical Pharmacology’ Award (L.W. and R.M.W.).
ACKNOWLEDGEMENTS
We thank Ms. Luanne Wussow for her assistance with the preparation of this manuscript.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Weinshilboum R. Inheritance and drug response. N. Engl. J. Med. 2003;348:529–537. doi: 10.1056/NEJMra020021. [DOI] [PubMed] [Google Scholar]
- 2.Weinshilboum R.M., Wang L. Pharmacogenetics and pharmacogenomics: development, science, and translation. Annu. Rev. Genomics Hum. Genet. 2006;7:223–245. doi: 10.1146/annurev.genom.6.080604.162315. [DOI] [PubMed] [Google Scholar]
- 3.Wang L., Weinshilboum R. Thiopurine S-methyltransferase pharmacogenetics: insights, challenges and future directions. Oncogene. 2006;25:1629–1638. doi: 10.1038/sj.onc.1209372. [DOI] [PubMed] [Google Scholar]
- 4.Innocenti F., Ratain M.J. ‘Irinogenetics’ and UGT1A: from genotypes to haplotypes. Clin. Pharmacol. Ther. 2004;75:495–500. doi: 10.1016/j.clpt.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 5.Aithal G.P., Day C.P., Kesteven P.J., Daly A.K. Association of polymorphisms in the cytochrome P450 CYP2C9 with warfarin dose requirement and risk of bleeding complications. Lancet. 1999;353:717–719. doi: 10.1016/S0140-6736(98)04474-2. [DOI] [PubMed] [Google Scholar]
- 6.Goetz M.P., Knox S.K., Suman V.J., Rae J.M., Safgren S.L., Ames M.M., Visscher D.W., Reynolds C., Couch F.J., Lingle W.L., et al. The impact of cytochrome P450 2D6 metabolism in women receiving adjuvant tamoxifen. Breast Cancer Res. Treat. 2007;101:113–121. doi: 10.1007/s10549-006-9428-0. [DOI] [PubMed] [Google Scholar]
- 7.Weinshilboum R. Pharmacogenomics of endocrine therapy in breast cancer. In: Bernstein L.M., Santen R.J., editors. Innovative Endocrinology of Cancer, Chapter 14. Landes Biosciences; 2008. pp. 220–231. Austin, TX. [DOI] [PubMed] [Google Scholar]
- 8.Saxena R., Voight B.F., Lyssenko V., Burtt N.P., de Bakker P.I., Chen H., Roix J.J., et al. Diabetes Genetics Initiative of Broad Institute of Harvard and MIT, Lund University, Novartis Institutes of BioMedical Research. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316:1331–1336. doi: 10.1126/science.1142358. [DOI] [PubMed] [Google Scholar]
- 9.Easton D.F., Pooley K.A., Dunning A.M., Pharoah P.D., Thompson D., Ballinger D.G., Struewing J.P., Morrison J., Field H., Luben R., et al. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature. 2007;447:1087–1093. doi: 10.1038/nature05887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zeggini E., Weedon M.N., Lindgren C.M., Frayling T.M., Elliott K.S., Lango H., Timpson N.J., Perry J., Rayner N.W., Freathy R.M., et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science. 2007;316:1336–1341. doi: 10.1126/science.1142364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scott L.J., Mohlke K.L., Bonnycastle L.L., Willer C.J., Li Y., Duren W.L., Erdos M.R., Stringham H.M., Chines P.S., Jackson A.U., et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341–1345. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kathiresan S., Melander O., Anevski D., Guiducci C., Burtt N.P., Roos C., Hirschhorn J.N., Berglund G., Hedblad B., Groop L., et al. Polymorphisms associated with cholesterol and risk of cardiovascular events. N. Engl. J. Med. 2008;358:1240–1249. doi: 10.1056/NEJMoa0706728. [DOI] [PubMed] [Google Scholar]
- 13.Thompson P.D., Clarkson P., Karas R.H. Statin-associated myopathy. J. Am. Med. Assoc. 2003;289:1681–1690. doi: 10.1001/jama.289.13.1681. [DOI] [PubMed] [Google Scholar]
- 14.The SEARCH Collaborative Group. SLCO1B1 variants and statin-induced myopathy—a genomewide study. The SEARCH Collaborative Group. N. Engl. J. Med. 2008;359:789–799. doi: 10.1056/NEJMoa0801936. [DOI] [PubMed] [Google Scholar]
- 15.Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high- risk individuals: a randomised placebo-controlled trial. Lancet. 2002;360:7–22. [Google Scholar]
- 16.Shoemaker R.H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer. 2006;6:813–823. doi: 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]
- 17.Weinstein J.N., Pommier Y. Transcriptomic analysis of the NCI-60 cancer cell lines. C. R. Biol. 2003;326:909–920. doi: 10.1016/j.crvi.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 18.Weinstein J.N. Biochemistry. A postgenomic visual icon. Science. 2008;319:1772–1773. doi: 10.1126/science.1151888. [DOI] [PubMed] [Google Scholar]
- 19.The International HapMap Consortium. The international HapMap project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 20.International HapMap Consortium. Integrating ethics and science in the international HapMap project. Nat. Rev. Genet. 2004;5:467–475. doi: 10.1038/nrg1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.International HapMap Consortium. A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frazer K.A., Ballinger D.G., Cox D.R., Hinds D.A., Stuve L.L., Gibbs R.A., Belmont J.W., Boudreau A., Hardenbol P., Leal S.M., et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thorisson G.A., Smith A.V., Krishnan L., Stein L.D. The international HapMap project web site. Genome Res. 2005;15:1592–1593. doi: 10.1101/gr.4413105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Redon R., Ishikawa S., Fitch K.R., Feuk L., Perry G.H., Andrews T.D., Fiegler H., Shapero M.H., Carson A.R., Chen W., et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stranger B.E., Forrest M.S., Dunning M., Ingle C.E., Beazley C., Thorne N., Redon R., Bird C.P., de Grassi A., Lee C., et al. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science. 2007;315:848–853. doi: 10.1126/science.1136678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spielman R.S., Bastone L.A., Burdick J.T., Morley M., Ewens W.J., Cheung V.G. Common genetic variants account for differences in gene expression among ethnic groups. Nat. Genet. 2007;39:226–231. doi: 10.1038/ng1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Storey J.D., Madeoy J., Strout J.L., Wurfel M., Ronald J., Akey J.M. Gene-expression variation within and among human populations. Am. J. Hum. Genet. 2007;80:502–509. doi: 10.1086/512017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stranger B.E., Nica A.C., Forrest M.S., Dimas A., Bird C.P., Beazley C., Ingle C.E., Dunning M., Flicek P., Koller D., et al. Population genomics of human gene expression. Nat. Genet. 2007;39:1217–1224. doi: 10.1038/ng2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang R.S., Duan S., Bleibel W.K., Kistner E.O., Zhang W., Clark T.A., Chen T.X., Schweitzer A.C., Blume J.E., Cox N.J., et al. A genome-wide approach to identify genetic variants that contribute to etoposide-induced cytotoxicity. Proc. Natl. Acad. Sci. USA. 2007;104:9758–9763. doi: 10.1073/pnas.0703736104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li L., Fridley B.L., Kalari K.R., Jenkins G.D., Batzler A., Safgren S.L., Hildebrandt M.A.T., Ames M.M., Schaid D.J., Wang L. Gemcitabine and cytosine arabinoside cytotoxicity: association with lymphoblastoid cell expression. Cancer Res. 2008;68:7050–7088. doi: 10.1158/0008-5472.CAN-08-0405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang R.S., Duan S., Kistner E.O., Bleibel W.K., Delaney S.M., Fackenthal D.L., Das S., Dolan M.E. Genetic variants contributing to daunorubicin-induced cytotoxicity. Cancer Res. 2008;68:3161–3168. doi: 10.1158/0008-5472.CAN-07-6381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shukla S.J., Duan S., Badner J.A., Wu X., Dolan M.E. Susceptibility loci involved in cisplatin-induced cytotoxicity and apoptosis. Pharmacogenet. Genomics. 2008;18:253–262. doi: 10.1097/FPC.0b013e3282f5e605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duan S., Bleibel W.K., Huang R.S., Shukla S.J., Wu X., Badner J.A., Dolan M.E. Mapping genes that contribute to daunorubicin-induced cytotoxicity. Cancer Res. 2007;67:5425–5433. doi: 10.1158/0008-5472.CAN-06-4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheung V.G., Conlin L.K., Weber T.M., Arcaro M., Jen K.Y., Morley M., Spielman R.S. Natural variation in human gene expression assessed in lymphoblastoid cells. Nat. Genet. 2003;33:422–425. doi: 10.1038/ng1094. [DOI] [PubMed] [Google Scholar]
- 35.Liu M.T., Chen Y.R., Chen S.C., Hu C.Y., Lin C.S., Chang Y.T., Wang W.B., Chen J.Y. Epstein–Barr virus latent membrane protein 1 induces micronucleus formation, represses DNA repair and enhances sensitivity to DNA-damaging agents in human epithelial cells. Oncogene. 2004;23:2531–2539. doi: 10.1038/sj.onc.1207375. [DOI] [PubMed] [Google Scholar]
- 36.Feng W.H., Hong G., Delecluse H.J., Kenney S.C. Lytic induction therapy for Epstein–Barr virus-positive B-cell lymphomas. J. Virol. 2004;78:1893–1902. doi: 10.1128/JVI.78.4.1893-1902.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tai H.L., Fessing M.Y., Bonten E.J., Yanishevsky Y., d’Azzo A., Krynetski E.Y., Evans W.E. Enhanced proteasomal degradation of mutant human thiopurine S-methyltransferase (TPMT) in mammalian cells: mechanism for TPMT protein deficiency inherited by TPMT*2, TPMT*3A, TPMT*3B or TPMT*3C. Pharmacogenetics. 1999;9:641–650. doi: 10.1097/01213011-199910000-00011. [DOI] [PubMed] [Google Scholar]
- 38.Wang L., Nguyen T.V., McLaughlin R.W., Sikkink L.A., Ramirez-Alvarado M., Weinshilboum R.M. Human thiopurine S-methyltransferase pharmacogenetics: variant allozyme misfolding and aggresome formation. Proc. Natl. Acad. Sci. USA. 2005;102:9394–9399. doi: 10.1073/pnas.0502352102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang L., Sullivan W., Toft D., Weinshilboum R. Thiopurine S-methyltransferase pharmacogenetics: chaperone protein association and allozyme degradation. Pharmacogenetics. 2003;13:555–564. doi: 10.1097/01.fpc.0000054124.14659.99. [DOI] [PubMed] [Google Scholar]
- 40.Li F., Wang L., Burgess R.J., Weinshilboum R.M. Thiopurine S-methyltransferase pharmacogenetics: autophagy as a mechanism for variant allozyme degradation. Pharmacogenet Genomics. 2008 doi: 10.1097/FPC.0b013e328313e03f. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]