

Abstract
Recent studies have indicated that both endothelin (ET) and angiotensin (Ang) II stimulate oxidative stress, which contributes to the development of hypertension. Here, we examined the effects of Ang II type 1 (AT1) receptor blockade on reactive oxygen species (ROS) formation in ET-dependent hypertension. Chronic ET-1 infusion (2.5 pmol/kg/min, i.v., n=7) into rats for 14 days increased systolic blood pressure from 113±1 to 141±2 mmHg. ET-1-infused rats showed greater plasma renin activity (8.1±0.8 Ang I/ml/h), and greater Ang I (122±28 fmol/ml) and Ang II levels (94±13 fmol/ml) than vehicle (0.9% NaCl)-infused rats (3.1±0.6 Ang I/ml/h, 45±8 and 47±7 fmol/ml, respectively, n=6). Angiotensin converting enzyme and AT1 receptor expression in aortic tissues were similar between the vehicle- and ET-1-infused rats. Vascular superoxide anion (O2-) production and plasma thiobarbituric acid-reactive substance (TBARS) levels were greater in ET-1-infused rats (27±1 counts per minutes [CPM]/mg dry tissue weight and 8.9±0.8 μmol/l, respectively) than vehicle-infused rats (16±1 CPM/mg and 5.1±0.1 μmol/l, respectively). The ET-1-induced hypertension was prevented by simultaneous treatment with a new AT1 receptor antagonist, olmesartan (0.01% in chow, 117±5 mmHg, n=7), or hydralazine (15 mg/kg/day in drinking water, 118±4 mmHg, n=6). Olmesartan prevented ET-1-induced increases in vascular O2- production (15±1 CPM/mg) and plasma TBARS (5.0±0.1μmol/l). Vascular O2- production and plasma TBARS were also decreased by hydralazine (21±1 CPM/mg and 7.0±0.3 μmol/l, respectively), but these levels were significantly higher than in vehicle-infused rats. These data suggest that ET-dependent hypertension is associated with augmentation of Ang II levels and ROS formation. The combined effects of the elevations in circulating ET-1 and Ang II, as well as the associated ROS production, may contribute to the development of hypertension induced by chronic ET-1 infusion.
Keywords: endothelin, angiotensin II, angiotensin II type 1 receptor, reactive oxygen species, olmesartan
Introduction
The renin-angiotensin system (RAS) and the endothelin (ET) system entail potent vasopressor mechanisms that contribute to the development of hypertension and cardiovascular injury (1, 2). Many studies have indicated that angiotensin (Ang) II exerts its hypertensive effects via interaction with the ET system (1, 3-6). For example, it has been shown that ET mediates some of the vasoconstrictor actions of Ang II in the peripheral vasculature (3). Furthermore, ET treatment augments the pressor effects of an acutely administered subpressor dose of Ang II (4). The hypertension associated with chronic infusion of Ang II can be markedly attenuated by ETA receptor blockade (5, 6). Although these observations indicate potential interactions between the RAS and the ET system, there has been little investigation into the specific role of the RAS in the development of ET-dependent hypertension. In hypertensive uremic rats, ET-1 levels in blood vessels and glomeruli were reduced by AT1 receptor blockade with losartan (7, 8). It was also shown that losartan reduced constrictor responses to ET-1 in aortic rings from spontaneously hypertensive rats (9). Chronic infusion of a subpressor dose of Ang II in combination with a subpressor dose of ET-1 induced hypertension, indicating a synergistic interaction between Ang II and ET-1 (10). In addition, treatment with an angiotensin converting enzyme (ACE) inhibitor attenuated the reductions in renal blood flow and urinary sodium excretion induced by acute administration of ET-1 (11). Similarly, the hypertension induced by chronic infusion of ET-1 was prevented by an ACE inhibitor (12).
Ang II stimulates superoxide anion (O2-) production through NAD(P)H oxidase-dependent pathways (13, 14), and this may at least partially contribute to the development of Ang II-dependent hypertension (13, 15). Recent studies have also indicated a role for ET in reactive oxygen species (ROS) formation (16-21). In vitro studies have shown that ET-1 increases ROS formation in endothelial cells (16), vascular smooth muscle cells (17) and rat aortic rings (18). Treatment with ET antagonists has been shown to reduce ROS levels in streptozotocin-induced diabetic rats (19) and hypertensive rats (18, 20). Li et al. (18, 21) showed that both the ET-1 level and the vascular NAD(P)H oxidase activity are increased in deoxycorticosterone acetate (DOCA)-salt hypertensive rats, and that NAD(P)H oxidase-derived O2- augments ET-1-induced venous vasoconstriction in these animals. Recent studies also have shown that thiobarbituric acid-reactive substance (TBARS) levels in renal tissues and urinary 8-isoprostaglandin F2α excretion are significantly increased in ET-induced hypertensive rats, and that treatment with tempol, a superoxide dismutase mimetic, normalizes the hypertension and these oxidative stress markers in these animals (17). These observations indicate that the formation of ROS plays a critical role in mediating the hypertension induced by chronic elevations in ET.
The aim of the present study was to determine whether the RAS was involved in the development of ET-dependent hypertension and ROS formation. Therefore, we investigated the status of oxidative stress and the RAS, including the plasma renin activity (PRA), Ang I, Ang II and angiotensinogen (AGT) levels as well as vascular ACE and AT1 receptor expression in ET-1-infused hypertensive rats. We also examined the effects of olmesartan, a potent new AT1 receptor antagonist (22, 23), on blood pressure and oxidative stress in ET-1-infused rats.
Methods
Animal Preparation
All surgical and experimental procedures were performed according to the guidelines for the care and use of animals established by the Kagawa Medical University. Male Sprague-Dawley rats (Clea Japan, Tokyo, Japan), 5-6 weeks of age, were selected at random to receive a continuous infusion of ET-1 (n=7) or vehicle (0.9% NaCl, n=6). ET-1 (Peptide Institute Inc., Minoh, Japan) was dissolved in 0.9% NaCl and infused into the jugular vein at a rate of 2.5 pmol/kg/min for a period of 14 days. Rats were anesthetized with sodium pentobarbital (50 mg/kg, i.p.), and the vascular catheter (PE-60) was tunneled subcutaneously to an osmotic minipump (model 2ML2; Alza Co., Cupertino, USA) implanted subcutaneously at the dorsum of the neck. ET-1 was also infused into rats treated with a new AT1 receptor antagonist, olmesartan (CS-866; Sankyo Co. Ltd., Tokyo, Japan; 0.01% in chow, approximately 6 mg/kg/day, n=7), or a nonspecific antihypertensive agent, hydralazine (Wako Co., Osaka, Japan; 15 mg/kg/day in drinking water, n=6). The doses of ET-1, olmesartan and hydralazine were determined based on previous studies in rats (24-26).
Systolic blood pressure (SBP) was measured in conscious rats by tail-cuff plethysmography (BP-98A; Softron Co., Tokyo, Japan) at 0, 7 and 14 days. Blood and tissue samples were harvested at the end of the experimental periods. After decapitation, the trunk blood was collected into chilled tubes containing an inhibitor mixture (5 mmol/l EDTA+20μmol/l enalaprilat+1.25 mmol/l 1,10-o-phenanthroline+10μmol/l pepstatin A), and processed for measurements of the plasma Ang I and Ang II concentrations as well as the AGT protein level (27-29). Blood was also collected into chilled tubes containing 5 mmol/l EDTA for measuring PRA (28, 29) and TBARS (28, 30). The aorta was quickly removed followed by the perivascular tissues. Five millimeter ring segments of the aorta were used to measure the O2- production (15, 28), and the remaining aorta samples were snap-frozen in liquid nitrogen and stored at -80 °C until processing for protein extraction.
Plasma AGT Levels and the Expression of ACE and AT1 Receptors in Aortic Tissues
Plasma AGT protein levels were determined by Western blotting analysis as described previously (27, 28). Briefly, protein samples were separated by 8% polyacrylamide gel electrophoresis and then transferred to a nitrocellulose membrane. The membrane was incubated with the primary antibody (sheep anti-rat AGT, 1:5,000), followed by incubation with a horseradish peroxidase-conjugated secondary antibody (donkey anti-sheep IgG, 1:30,000; Sigma Chemical Co., St. Louis, USA). All values were normalized by arbitrarily setting the integrated densitometric values (IDV) of vehicle-infused rats to 1.0.
The levels of ACE and AT1 receptor protein expression in aortic tissues were determined by Western blotting analyses (28, 29). Briefly, protein samples were separated by 8% polyacrylamide gel electrophoresis and then transferred to a nitrocellulose membrane. The membrane was reacted with a polyclonal anti-ACE antibody (1:200; Santa Cruz Biotechnology Inc., Santa Cruz, USA) or a polyclonal anti-AT1 receptor antibody (1:1,000; Santa Cruz Biotechnology Inc.), followed by incubation with a horseradish peroxidase-conjugated secondary antibody (for ACE: donkey anti-goat immunoglobulin G [IgG], 1:1,000, Santa Cruz Biotechnology Inc.; for AT1 receptors: goat anti-rabbit IgG, 1:4,000, Cell Signaling Technology, Beverly, USA). To check for equal loading, membranes were re-probed with an antibody against β-actin. All values were normalized by arbitrarily setting the IDV of vehicle-infused rats to 1.0.
Measurement of Vascular O2- Production
O2- production in aortic segments was determined with the use of lucigenin chemiluminescence, as described previously (15, 28). Briefly, 5-mm aortic rings were placed in bicarbonate buffer for 30 min at 37 °C. After the equilibration, the rings were rinsed with prewarmed (37 °C) modified Krebs-HEPES buffer. Rings were placed in 1 ml Krebs-HEPES buffer containing lucigenin (20μmol/l) and equilibrated in the dark for 10 min at 37 °C. The chemiluminescence was then recorded every 30 s for 15 min with a luminescence reader (BLR-301; Aloka Co., Ltd., Tokyo, Japan). Thereafter, a cell-permeant, nonenzymatic O2- scavenger, Tiron (20 mmol/l) was added to quench the O2--dependent chemiluminescence (31), and 18 more cycles were read. The final 10 values, which appeared to be maximally reduced, were averaged. The differences between the values obtained before and after Tiron addition were calculated and defined as the Tiron-quenchable lucigenin chemiluminescence. Lucigenin chemiluminescence was expressed as counts per minute (CPM) per milligram of dry tissue weight.
Analytical Procedures
We determined the degree of lipid peroxidation using a biochemical assay of the TBARS level in plasma as described previously (28, 30). PRA, Ang I and Ang II levels were measured by radioimmunoassays as previously reported (27-29).
Statistical Analysis
The values are presented the as means±SEM. Statistical comparisons of differences were performed using one way or two way ANOVA for repeated measures combined with Newman-Keuls post hoc test. Values of p<0.05 were considered statistically significant.
Results
Blood Pressure
The temporal profiles of SBP are depicted in Fig. 1. SBP was identical among the 4 groups at the beginning of the protocol. SBP was significantly elevated in ET-1-infused rats at week 2 compared with that in vehicle-infused rats (141±2 vs. 113±1 mmHg). Concurrent administration of olmesartan or hydralazine prevented ET-1-induced hypertension (117±5 mmHg and 118±4 mmHg, respectively).
Fig. 1.
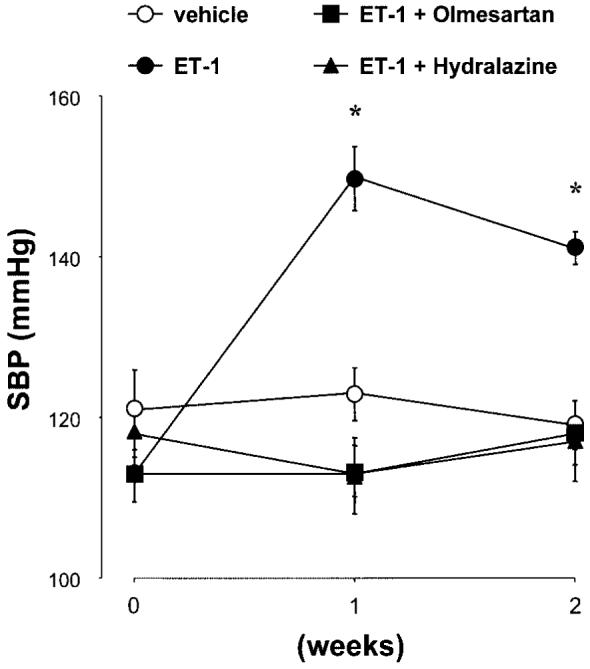
Temporal profiles of systolic blood pressure (SBP). SBP was similar among the 4 groups at the beginning of the protocol. Endothelin (ET)-1 infusion for 2 weeks significantly increased SBP. Concurrent administration of olmesartan or hydralazine prevented the development of hypertension in ET-1-infused rats. *p<0.05 vs. week 0.
The RAS in ET-Induced Hypertensive Rats
Consistent with previous studies (27), Western blotting analysis of the plasma proteins using a specific anti-AGT polyclonal antibody showed 2 bands at 52 and 64 kDa (slightly glycosylated and highly glycosylated forms of circulating AGT), with the greatest abundance at 52 kDa. Figure 2 shows a representative Western blot of plasma AGT. Densitometric analysis of the immunoreactive bands showed that ET-1 infusion had no effect on the expression of plasma AGT. On the other hand, concurrent administration of olmesartan to ET-1-infused rats significantly increased both forms of plasma AGT protein compared with the levels in vehicle-infused rats (1.7±0.2-, 1.8±0.2- and 1.5±0.2-fold for 52 kDa, 64 kDa and total AGT, respectively). Similarly, treatment with hydralazine also increased both forms of plasma AGT protein (1.5±0.2-, 1.6±0.1- and 1.6±0.1-fold for 52 kDa, 64 kDa and total AGT, respectively).
Fig. 2.

Western blotting analysis of plasma angiotensinogen (AGT) expression. Densitometric analysis of the immunoreactive bands shows that chronic endothelin (ET)-1 infusion had no effect on the expression of plasma AGT. Concurrent administration of olmesartan or hydralazine to ET-1-infused rats significantly increased the expression of both forms of plasma AGT. All values were normalized by arbitrarily setting the densitometry of vehicle-treated rats to 1.0, as described in the Materials and Methods. *p<0.05 vs. vehicle-infused rats.
Compared with vehicle-infused rats, PRA was significantly increased in ET-1-infused rats (3.1±0.6 vs. 8.1±0.8 Ang I/ml/h). Treatment with olmesartan caused a further increase in PRA in ET-1-infused rats (16.6±0.8 Ang I/ml/h). On the other hand, concurrent administration of hydralazine to ET-1-infused rats did not alter PRA (10.7±1.7 Ang I/ml/h, Fig. 3A). In ET-1-infused rats, the plasma Ang I level was significantly elevated compared with that in vehicle-infused rats (122±28 vs. 45±8 fmol/ml, Fig. 3B). Similarly, the plasma Ang II levels was higher in ET-1-infused rats (94±13 fmol/ml) than in vehicle-infused rats (47±7 fmol/ml, Fig. 3C). Concurrent administration of olmesartan to ET-1-infused rats further increased the plasma Ang I and Ang II levels (471±70 and 354±42 fmol/ml, respectively). The plasma Ang I and Ang II levels in hydralazine-treated ET-1-infused rats (153±20 and 118±13 fmol/ml, respectively) were higher than those in vehicle-infused rats, but these levels were not different from those in rats treated with ET-1 alone.
Fig. 3.
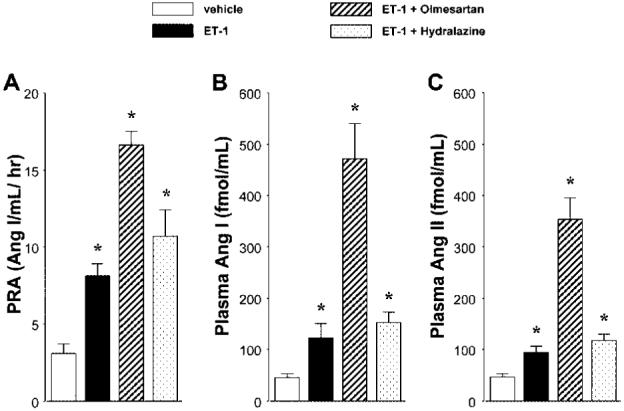
A: Plasma renin activity (PRA), B: plasma angiotensin I (Ang I) and C: plasma angiotensin II (Ang II) levels are shown. PRA, plasma Ang I and Ang II levels were significantly elevated in endothelin (ET)-1-infused hypertensive rats. Concurrent administration of olmesartan to ET-1-infused rats further increased these levels. PRA, Ang I and Ang II levels were significantly higher in hydralazine-treated ET-1-infused rats than in vehicle-infused rats, but these levels were not different from those in rats treated with ET-1 alone. *p<0.05 vs. vehicle-infused rats.
Western blotting analyses showed that ACE and AT1 receptor expression levels in aortic tissue were similar between vehicle- and ET-1-infused rats. Furthermore, neither olmesartan nor hydralazine altered vascular ACE or AT1 receptor expression in ET-1-infused rats (Fig. 4).
Fig. 4.
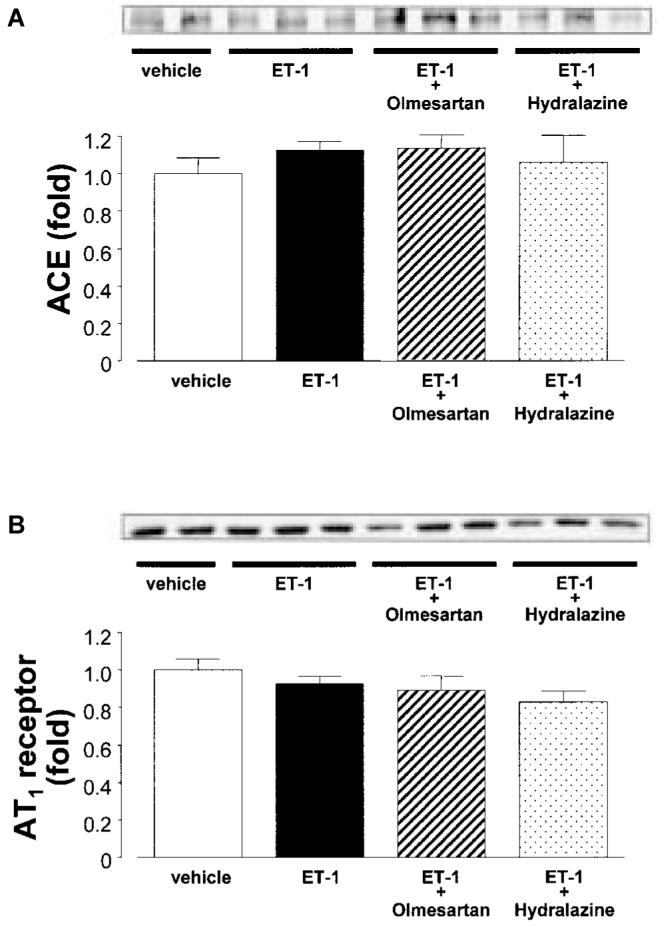
A: Western blotting analyses for A: protein expression of angiotensin converting enzyme (ACE) and B: AT1 receptors in aortic tissues. The expression of ACE and AT1 receptor in aortic tissues were similar between vehicle- and ET-1-infused rats. Furthermore, neither olmesartan nor hydralazine altered vascular ACE and AT1 receptor expression in ET-1-infused rats. To check for equal loading, membranes were re-probed with an antibody against β-actin. All values were normalized by arbitrarily setting the densitometry of vehicle-treated rats to 1.0, as described in the Materials and Methods.
Plasma TBARS Levels
ET-1-infused rats showed a significantly higher plasma TBARS level compared with vehicle-infused rats (8.9±0.8 vs. 5.1±0.1μmol/l, Fig. 5A). Concurrent administration of olmesartan prevented the increase in the plasma TBARS levels in ET-infused rats (5.0±0.1μmol/l). On the other hand, hydralazine did not alter the plasma TBARS level in ET-infused rats (7.0±0.3μmol/l), but this level was significantly higher than that in vehicle-infused rats. Furthermore, the TBARS level in hydralazine-treated ET-1-infused rats was significantly higher than that in olmesartan-treated ET-1-infused rats (p<0.05).
Fig. 5.
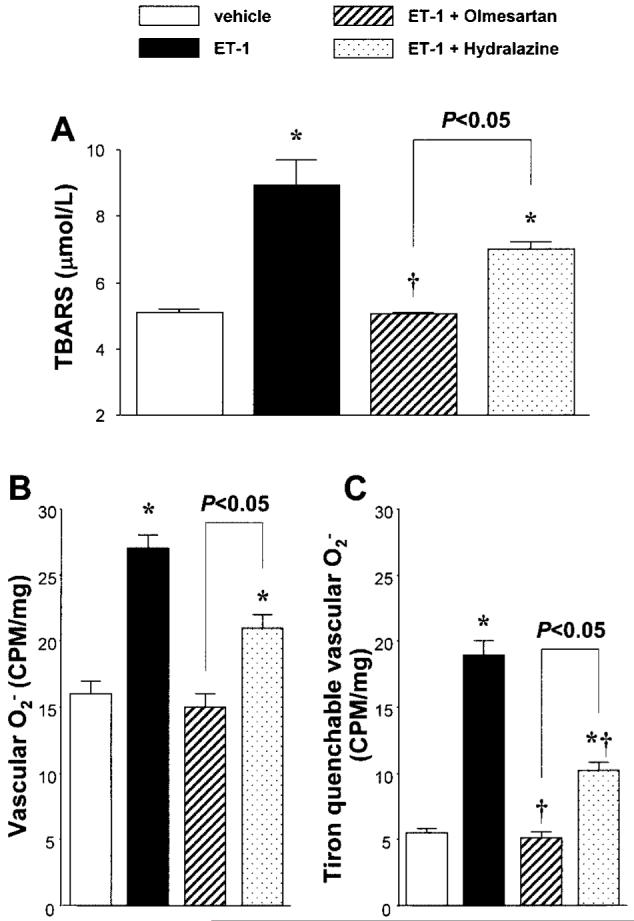
A: Plasma thiobarbituric acid-reactive substance (TBARS) levels. B: Vascular superoxide anion (O2-) production assessed by lucigenin chemiluminescence in aortic segments. C: The differences between the values obtained before and after Tiron (20 mmol/l) addition were calculated and defined as the Tiron-quenchable lucigenin chemiluminescence. Lucigenin chemiluminescence was expressed as counts per minute (CPM) per milligram of dry tissue weight. *p<0.05 vs. vehicle-infused rats. †p<0.05: endothelin (ET)-1-infused rats vs. ET-1-infused rats+olmesartan or hydralazine.
Vascular O2- Production
Lucigenin chemiluminescence from aortic segments of vehicle-infused rats averaged 16±1 CPM/mg dry tissue weight. Compared with vehicle-infused rats, ET-1-infused rats showed higher lucigenin chemiluminescence (27±1 CPM/mg dry tissue weight, p<0.05, Fig. 5B). Olmesartan prevented the ET-1-induced increase in lucigenin chemiluminescence (15±1 CPM/mg dry tissue weight). Similarly, hydralazine treatment of ET-1-infused rats also decreased lucigenin chemiluminescence (21±2 CPM/mg dry tissue weight, n=6). However, this level was significantly higher than that in vehicle-infused rats (p<0.05). Furthermore, lucigenin chemiluminescence from aortic segments of hydralazine-treated ET-1-infused rats was significantly higher than that of olmesartan-treated ET-1-infused rats (p<0.05).
Figure 5C shows the Tiron-quenchable lucigenin chemiluminescence from aortic segments. Compared with vehicle infused rats, ET-1-infused rats showed higher Tiron-quenchable lucigenin chemiluminescence (5.5±0.3 vs. 18.9±1.1 CPM/mg dry tissue weight, p<0.05). The increased Tironquenchable lucigenin chemiluminescence in ET-1-infused rats was normalized by treatment with olmesartan (5.1±0.1 CPM/mg dry tissue weight). Similarly, hydralazine administration to ET-1-infused rats decreased the Tiron-quenchable lucigenin chemiluminescence (10.2±0.6 CPM/mg dry tissue weight, n=6). On the basis of group comparisons, however, this level was significantly higher than those in vehicle-infused rats and olmesartan-treated ET-1-infused rats (p<0.05, respectively).
Discussion
In vitro and in vivo studies have indicated that Ang II is a potent stimulator of ET-1 production (1, 32, 33). It has been shown that Ang II can turn on the transcription of the ppET-1 gene in vascular smooth muscle cells (32), and that chronic administration of Ang II increased renal ET-1 production in rats (33). On the other hand, the effects of the ET system on the RAS activity remain unclear. Matsumura et al. (34) showed that ET-1 decreased renin secretion from kidney cortical slices. However, ET-1 had no consistent effects on basal renin secretion from juxtaglomerular cells (35). In vivo studies have also shown that acute infusion of ET-1 significantly decreased (36) or significantly increased (37) renin secretion. In the present study, we investigated the status of the RAS in rats treated with chronic ET-1 infusion. The results showed that the plasma Ang II level was significantly increased by chronic infusion of ET-1, while the vascular ACE and AT1 receptor expression remained constant. Elmarakbly et al. (38) reported that AT1 receptor blockade with candesartan had no effect on the increase in blood pressure induced by acute infusion of ET-1. However, the present study has shown that a new AT1 receptor antagonist, olmesartan (22, 23), prevents the hypertension induced by chronic infusion of ET-1. These observations suggest that the RAS system is maintained or activated when ET-1 is infused chronically. Since AT1 receptor blockade prevents ET-induced hypertension, activation of the RAS might be involved, at least in part, in the development of ET-dependent hypertension.
We observed that PRA was significantly increased in ET-1-infused hypertensive rats. These results are consistent with previous observations that acute hypertension induced by systemic infusion of ET is associated with an elevation in PRA (37). We also found that the plasma AGT levels were maintained in ET-1-infused hypertensive rats. These data indicate that increases in PRA, rather than increases in the AGT level, are responsible for the elevated Ang II level in ET-1-induced hypertensive rats. However, the mechanisms by which chronic infusion of ET-1 increases PRA remain to be elucidated. Acute administration of ET-1 constricts the renal afferent arterioles (39, 40), and decreases the renal blood flow (11) and glomerular filtration rate (38). Furthermore, significantly increased renal vascular resistance has been observed in ET-1-infused hypertensive rats (17). Therefore, the possibility exists that sustained renal vasoconstriction, a decrease in the glomerular filtration rate, or the subsequent renal damage induced by chronic elevation of the ET-1 level contributes to the increases in renin release. Other possibilities cannot be ruled out and need to be examined further.
Recent studies have indicated that ET-induced vasoconstriction may be mediated, at least partially, by increased ROS production (17-21). The present study showed that vascular O2- production and the plasma TBARS level were significantly increased in ET-1-infused hypertensive rats. These observations are in accordance with those of previous studies showing that the TBARS content in the kidney and urinary excretion of 8-isoprostaglandin F2α were increased in ET-1-infused hypertensive rats (17). However, the mechanisms by which ET-1 stimulates ROS production are not clear. It is well known that Ang II stimulates O2- production through NAD(P)H oxidase activation (13, 14). Recently, it has also been shown that ET-1 can stimulate O2- production via the NAD(P)H oxidase pathway (18, 21). Furthermore, ET-1 increases O2- production and the expression of gp91phox, an essential component of NAD(P)H oxidase, in endothelial cells (16). ET-1 is a potent stimulator of protein kinase C (PKC) (40), which is also involved in NAD(P)H oxidase activity and O2- production (41). Therefore, it is also possible that ET-1 activates NAD(P)H oxidase through a PKC-dependent pathway. In the present study, we have demonstrated that ET-dependent hypertension is associated with augmentation of Ang II and ROS levels. Interestingly, AT1 receptor blockade with olmesartan prevents the ET-1-induced hypertension and ROS formation. Although multiple mechanisms may contribute to the ET-induced ROS formation, it can be speculated that the augmented Ang II level also participates in the ROS formation induced by chronic infusion of ET.
In the present study, we also examined the effects of a non-specific vasodilator, hydralazine. We observed that treatment with hydralazine did not alter the plasma TBARS level, but did significantly decrease vascular O2- in ET-1-infused rats. This observation is consistent with previous observations that hydralazine did not change the plasma TBARS level but did reduce NAD(P)H oxidase-mediated vascular O2- production in Ang II-infused rats (42). These data raise two possibilities: first, hydralazine has antioxidant properties; and second, reductions in the ROS levels are a consequence of the antihypertensive effect of hydralazine. The possibility of antioxidative effects of hydralazine cannot be addressed from the present data. However, the vascular O2- level may not be simply changed by alterations in blood pressure, since previous studies have shown that vascular O2- release was not altered in norepinephrine-infused hypertensive rats (43).
In conclusion, the present study demonstrates that ET-1-induced hypertension is associated with augmentation of Ang II and ROS levels in rats. In addition, AT1 receptor blockade with olmesartan prevents the development of ET-1-induced hypertension and oxidative stress. These data suggest that the combined effects of the elevations in circulating ET-1 and Ang II levels, as well as the associated ROS production, contribute to the development of ET-induced hypertension.
Acknowledgements
The polyclonal antibody against rat AGT was generously provided by Conrad Sernia, Ph.D. (University of Queensland, Australia). We are also grateful to Mss. Kayoko Miyata, Kanako Ogura, Junko Osada (Kagawa Medical University) and Eri Hiramoto (Okayama University Medical School) for their excellent technical assistance, and to Sankyo Co., Ltd. (Tokyo, Japan) for supplying olmesartan.
This work was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Science, Sports and Culture of Japan (Y.A., A.N.), and by grants from the Salt Sciences Research Foundation (A.N., No. 04C2), the Health Excellence Found of the Louisiana Board of Regents (H.K.), the Mitsui Life Social Welfare Foundation (A.N.), Japan Research Foundation for Clinical Pharmacology (A.N.), and the National Kidney Foundation (H.K.).
References
- 1.Schiffrin EL. Endothelin and endothelin antagonists in hypertension. J Hypertens. 1998;16:1891–1895. doi: 10.1097/00004872-199816121-00007. [DOI] [PubMed] [Google Scholar]
- 2.Minami S, Yamano S, Yamamoto Y, et al. Associations of plasma endothelin concentration with carotid atherosclerosis and asymptomatic cerebrovascular lesions in patients with essential hypertension. Hypertens Res. 2001;24:663–670. doi: 10.1291/hypres.24.663. [DOI] [PubMed] [Google Scholar]
- 3.Chen L, McNeill JR, Wilson TW, Gopalakrishnan V. Heterogeneity in vascular smooth muscle responsiveness to angiotensin II: role of endothelin. Hypertension. 1995;26:83–88. doi: 10.1161/01.hyp.26.1.83. [DOI] [PubMed] [Google Scholar]
- 4.Balakrishnan SM, Wang HD, Gopalakrishnan V, Wilson TW, McNeill JR. Effect of an endothelin antagonist on hemodynamic responses to angiotensin II. Hypertension. 1996;28:806–809. doi: 10.1161/01.hyp.28.5.806. [DOI] [PubMed] [Google Scholar]
- 5.Ballew JR, Fink GD. Role of ET(A) receptors in experimental ANG II-induced hypertension in rats. Am J Physiol Regul Integr Comp Physiol. 2001;281:R150–R154. doi: 10.1152/ajpregu.2001.281.1.R150. [DOI] [PubMed] [Google Scholar]
- 6.Rajagopalan S, Laursen JB, Borthayre A, et al. Role for endothelin-1 in angiotensin II-mediated hypertension. Hypertension. 1997;30:29–34. doi: 10.1161/01.hyp.30.1.29. [DOI] [PubMed] [Google Scholar]
- 7.Lariviere R, Lebel M, Kingma I, Grose JH, Boucher D. Effects of losartan and captopril on endothelin-1 production in blood vessels and glomeruli of rats with reduced renal mass. Am J Hypertens. 1998;11:989–997. doi: 10.1016/s0895-7061(98)00088-0. [DOI] [PubMed] [Google Scholar]
- 8.Dumont Y, D’Amours M, Lebel M, Lariviere R. Blood pressure-independent effect of angiotensin AT1 receptor blockade on renal endothelin-1 production in hypertensive uremic rats. J Hypertens. 2001;19:1479–1487. doi: 10.1097/00004872-200108000-00017. [DOI] [PubMed] [Google Scholar]
- 9.Maeso R, Rodrigo E, Munoz-Garcia R, et al. Losartan reduces constrictor responses to endothelin-1 and the thromboxane A2 analogue in aortic rings from spontaneously hypertensive rats: role of nitric oxide. J Hypertens. 1997;15:1677–1684. doi: 10.1097/00004872-199715120-00072. [DOI] [PubMed] [Google Scholar]
- 10.Yoshida K, Yasujima M, Kohzuki M, Kanazawa M, Yoshinaga K, Abe K. Endothelin-1 augments pressor response to angiotensin II infusion in rats. Hypertension. 1992;20:292–297. doi: 10.1161/01.hyp.20.3.292. [DOI] [PubMed] [Google Scholar]
- 11.Cao LQ, Banks RO. Cardiorenal actions of endothelin, part I: effects of converting enzyme inhibition. Life Sci. 1990;46:577–583. doi: 10.1016/0024-3205(90)90125-b. [DOI] [PubMed] [Google Scholar]
- 12.Mortensen LH, Fink GD. Captopril prevents chronic hypertension produced by infusion of endothelin-1 in rats. Hypertension. 1992;19:676–680. doi: 10.1161/01.hyp.19.6.676. [DOI] [PubMed] [Google Scholar]
- 13.Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res. 2000;86:494–501. doi: 10.1161/01.res.86.5.494. [DOI] [PubMed] [Google Scholar]
- 14.Ying CJ, Xu JW, Ikeda K, Takahashi K, Nara Y, Yamori Y. Tea polyphenols regulate nicotinamide adenine dinucleotide phosphate oxidase subunit expression and ameliorate angiotensin II-induced hyperpermeability in endothelial cells. Hypertens Res. 2003;26:823–828. doi: 10.1291/hypres.26.823. [DOI] [PubMed] [Google Scholar]
- 15.Nishiyama A, Fukui T, Fujisawa Y, et al. Systemic and regional hemodynamic responses to tempol in angiotensin II-infused hypertensive rats. Hypertension. 2001;37:77–83. doi: 10.1161/01.hyp.37.1.77. [DOI] [PubMed] [Google Scholar]
- 16.Duerrschmidt N, Wippich N, Goettsch W, Broemme HJ, Morawietz H. Endothelin-1 induces NAD(P)H oxidase in human endothelial cells. Biochem Biophys Res Commun. 2000;269:713–717. doi: 10.1006/bbrc.2000.2354. [DOI] [PubMed] [Google Scholar]
- 17.Sedeek MH, Llinas MT, Drummond H, et al. Role of reactive oxygen species in endothelin-induced hypertension. Hypertension. 2003;42:806–810. doi: 10.1161/01.HYP.0000084372.91932.BA. [DOI] [PubMed] [Google Scholar]
- 18.Li L, Fink GD, Watts SW, et al. Endothelin-1 increases vascular superoxide via endothelin(A)-NADPH oxidase pathway in low-renin hypertension. Circulation. 2003;107:1053–1058. doi: 10.1161/01.cir.0000051459.74466.46. [DOI] [PubMed] [Google Scholar]
- 19.Kanie N, Kamata K. Effects of chronic administration of the novel endothelin antagonist J-104132 on endothelial dysfunction in streptozotocin-induced diabetic rat. Br J Pharmacol. 2002;135:1935–1942. doi: 10.1038/sj.bjp.0704659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Callera GE, Touyz RM, Teixeira SA, et al. ETA receptor blockade decreases vascular superoxide generation in DOCA-salt hypertension. Hypertension. 2003;42:811–817. doi: 10.1161/01.HYP.0000088363.65943.6C. [DOI] [PubMed] [Google Scholar]
- 21.Li L, Watts SW, Banes AK, Galligan JJ, Fink GD, Chen AF. NADPH oxidase-derived superoxide augments endothelin-1-induced venoconstriction in mineralocorticoid hypertension. Hypertension. 2003;42:316–321. doi: 10.1161/01.HYP.0000084853.47326.F2. [DOI] [PubMed] [Google Scholar]
- 22.Ichikawa S, Takayama Y. Long-term effects of olmesartan, an Ang II receptor antagonist, on blood pressure and the renin-angiotensin-aldosterone system in hypertensive patients. Hypertens Res. 2001;24:641–646. doi: 10.1291/hypres.24.641. [DOI] [PubMed] [Google Scholar]
- 23.Mizuno M, Sada T, Ikeda M, et al. Pharmacology of CS-866, a novel nonpeptide angiotensin II receptor antagonist. Eur J Pharmacol. 1995;285:181–188. doi: 10.1016/0014-2999(95)00401-6. [DOI] [PubMed] [Google Scholar]
- 24.Potter GS, Johnson RJ, Fink GD. Role of endothelin in hypertension of experimental chronic renal failure. Hypertension. 1997;30:1578–1584. doi: 10.1161/01.hyp.30.6.1578. [DOI] [PubMed] [Google Scholar]
- 25.Mizuno M, Sada T, Koike H. Renoprotective effects of blockade of angiotensin II AT1 receptors in an animal model of type 2 diabetes. Hypertens Res. 2002;25:271–278. doi: 10.1291/hypres.25.271. [DOI] [PubMed] [Google Scholar]
- 26.Casellas D, Benahmed S, Artuso A, Jover B. Candesartan and progression of preglomerular lesions in NG-nitro-l-arginine methyl ester hypertensive rats. J Am Soc Nephrol. 1999;10:S230–S233. [PubMed] [Google Scholar]
- 27.Kobori H, Nishiyama A, Harrison-Bernard LM, Navar LG. Urinary angiotensinogen as an indicator of intrarenal angiotensin status in hypertension. Hypertension. 2003;41:42–49. doi: 10.1161/01.hyp.0000050102.90932.cf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nishiyama A, Kobori H, Fukui T, et al. Role of angiotensin II and reactive oxygen species in cyclosporine A-dependent hypertension. Hypertension. 2003;42:754–760. doi: 10.1161/01.HYP.0000085195.38870.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nishiyama A, Yoshizumi M, Rahman M, et al. Effects of AT1 receptor blockade on renal injury and mitogen-activated protein activity in Dahl salt-sensitive rats. Kidney Int. 2004;65:972–981. doi: 10.1111/j.1523-1755.2004.00476.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nishiyama A, Yoshizumi M, Hitomi H, et al. The SOD mimetic tempol ameliorates glomerular injury and reduces mitogen-activated protein kinase activity in Dahl salt-sensitive rats. J Am Soc Nephrol. 2004;15:306–315. doi: 10.1097/01.asn.0000108523.02100.e0. [DOI] [PubMed] [Google Scholar]
- 31.Rahman M, Kimura S, Nishiyama A, Hitomi H, Zhang G-X, Abe Y. Angiotensin II stimulates superoxide production via both angiotensin AT1a and AT1b receptors in mouse aorta and heart. Eur J Pharmacol. 2004;485:243–249. doi: 10.1016/j.ejphar.2003.11.074. [DOI] [PubMed] [Google Scholar]
- 32.Sung CP, Arleth AJ, Storer BL, Ohlstein EH. Angiotensin type 1 receptors mediate smooth muscle proliferation and endothelin biosynthesis in rat vascular smooth muscle. J Pharmacol Exp Ther. 1994;271:429–437. [PubMed] [Google Scholar]
- 33.Sasser JM, Pollock JS, Pollock DM. Renal endothelin in chronic angiotensin II hypertension. Am J Physiol Regul Integr Comp Physiol. 2002;283:R243–R248. doi: 10.1152/ajpregu.00086.2002. [DOI] [PubMed] [Google Scholar]
- 34.Matsumura Y, Nakase K, Ikegawa R, Hayashi K, Ohyama T, Morimoto S. The endothelium-derived vasoconstrictor peptide endothelin inhibits renin release in vitro. Life Sci. 1989;44:149–157. doi: 10.1016/0024-3205(89)90533-x. [DOI] [PubMed] [Google Scholar]
- 35.Ritthaler T, Scholz H, Ackermann M, Riegger G, Kurtz A, Kramer BK. Effects of endothelins on renin secretion from isolated mouse renal juxtaglomerular cells. Am J Physiol. 1995;268:F39–F45. doi: 10.1152/ajprenal.1995.268.1.F39. [DOI] [PubMed] [Google Scholar]
- 36.Matsumura Y, Hisaki K, Ohyama T, Hayashi K, Morimoto S. Effects of endothelin on renal function and renin secretion in anesthetized rats. Eur J Pharmacol. 1989;166:577–580. doi: 10.1016/0014-2999(89)90380-4. [DOI] [PubMed] [Google Scholar]
- 37.Goetz KL, Wang BC, Madwed JB, Zhu JL, Leadley RJ., Jr Cardiovascular, renal, and endocrine responses to intravenous endothelin in conscious dogs. Am J Physiol. 1988;255:R1064–R1068. doi: 10.1152/ajpregu.1988.255.6.R1064. [DOI] [PubMed] [Google Scholar]
- 38.Elmarakby AA, Morsing P, Pollock DM. Enalapril attenuates endothelin-1-induced hypertension via increased kinin survival. Am J Physiol Heart Circ Physiol. 2003;284:H1899–H1903. doi: 10.1152/ajpheart.00027.2003. [DOI] [PubMed] [Google Scholar]
- 39.Loutzenhiser R, Epstein M, Hayashi K, Horton C. Direct visualization of effects of endothelin on the renal microvasculature. Am J Physiol. 1990;258:F61–F68. doi: 10.1152/ajprenal.1990.258.1.F61. [DOI] [PubMed] [Google Scholar]
- 40.Takenaka T, Forster H, Epstein M. Protein kinase C and calcium channel activation as determinants of renal vasoconstriction by angiotensin II and endothelin. Circ Res. 1993;73:743–750. doi: 10.1161/01.res.73.4.743. [DOI] [PubMed] [Google Scholar]
- 41.Heitzer T, Wenzel U, Hink U, et al. Increased NAD(P)H oxidase-mediated superoxide production in renovascular hypertension: evidence for an involvement of protein kinase C. Kidney Int. 1999;55:252–260. doi: 10.1046/j.1523-1755.1999.00229.x. [DOI] [PubMed] [Google Scholar]
- 42.Virdis A, Neves MF, Amiri F, Viel E, Touyz RM, Schiffrin EL. Spironolactone improves angiotensin-induced vascular changes and oxidative stress. Hypertension. 2002;40:504–510. doi: 10.1161/01.hyp.0000034738.79310.06. [DOI] [PubMed] [Google Scholar]
- 43.Rajagopalan S, Kurz S, Munzel T, et al. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest. 1996;97:1916–1923. doi: 10.1172/JCI118623. [DOI] [PMC free article] [PubMed] [Google Scholar]