

Abstract
This study was conducted to examine whether the renin-angiotensin system contributes to hyperthyroidism-induced cardiac hypertrophy without involving the sympathetic nervous system. Sprague-Dawley rats were divided into control-innervated, control-denervated, hyperthyroid-innervated, and hyperthyroid-denervated groups using intraperitoneal injections of thyroxine and 6-hydroxydopamine. After 8 wk, the heart-to-body weight ratio increased in hyperthyroid groups (63%), and this increase was only partially inhibited by sympathetic denervation. Radioimmunoassays and reverse transcription-polymerase chain reaction revealed increased cardiac levels of renin (33%) and angiotensin II (53%) and enhanced cardiac expression of renin mRNA (225%) in the hyperthyroid groups. These increases were unaffected by sympathetic denervation or 24-h bilateral nephrectomy. In addition, losartan and nicardipine decreased systolic blood pressure to the same extent, but only losartan caused regression of thyroxine-induced cardiac hypertrophy. These results suggest that thyroid hormone activates the cardiac renin-angiotensin system without involving the sympathetic nervous system or the circulating renin-angiotensin system; the activated renin-angiotensin system contributes to cardiac hypertrophy in hyperthyroidism.
Keywords: chemical sympathectomy, polymerase chain reaction, tissue renin, angiotensin II type 1 receptor antagonist
CARDIAC HYPERTROPHY IS a serious complication of hyperthyroidism (31). It was previously believed that the enhanced hemodynamics produced by increased sympathetic nerve activity were a major factor in cardiac hypertrophy induced by hyperthyroidism (15). Increased sympathetic nerve activity also augments plasma renin activity (PRA) and thus activates the circulating renin-angiotensin system (RAS) (15). Some evidence indicates that the circulating RAS may be involved in the development of cardiac hypertrophy (29). Angiotensin II exerts a direct physiological effect on the cardiovascular system through specific receptors on the cardiomyocyte plasma membrane that are coupled to guanine nucleotide-binding proteins (1). The administration of angiotensin-converting enzyme inhibitors is clinically efficacious in reducing cardiac hypertrophy (9). However, more recent reports have shown that sympathetic nerve activity is elevated in hyper- and hypothyroidism (27), yet cardiac hypertrophy is only recognized in hyperthyroidism (16). These data suggest that hyperthyroidism-induced cardiac hypertrophy can be caused by factors independent of the sympathetic nervous system.
Furthermore, although hyperthyroidism is associated with cardiac hypertrophy (16), hyperthyroidism frequently demonstrates normal PRA (15); thus a factor other than the circulating RAS may be involved in hyperthyroidism-induced cardiac hypertrophy. Recent in vitro studies suggest that thyroid hormone itself regulates the expression of the tissue renin gene (12, 20). The expression of the renin gene in the mouse submandibular gland has been shown to be stimulated by thyroid hormone (20). In a pituitary cell line, thyroid hormone regulates promoter activity of the renin gene (12). Therefore, we hypothesized that thyroid hormone directly enhances cardiac expression of the renin gene and the cardiac RAS without involving the sympathetic nervous system, thereby causing cardiac hypertrophy in hyperthyroidism. To evaluate this hypothesis, the effects of sympathetic denervation, bilateral nephrectomy, and treatment with losartan or nicardipine on the circulating and cardiac RAS and the heart-to-body weight ratio were investigated in hyperthyroid rats.
METHODS
Preparation of Animals
Male Sprague-Dawley rats (Charles River Japan, Kanagawa, Japan) weighing 150−200 g were used in the present study. They received standard laboratory food containing 110 μmol/g sodium (Oriental Yeast, Tokyo, Japan), with tap water ad libitum. They were individually caged with a 12:12-h light-dark cycle. Body weight (BW) was checked daily. For the first series of this study, 20 rats were divided into control (Cont) and hyperthyroid (Hyper) groups by daily intraperitoneal injections of saline vehicle or thyroxine (0.1 μg/g) for 8 wk as previously described (22). These groups were subdivided into sympathetic innervated (IN) and sympathetic denervated (DX) groups by intraperitoneal injections of saline vehicle or 6-hydroxydopamine (100 μg/g) on days 1, 3, 27, and 47 as previously described (22). Systolic blood pressure (BP) and heart rate (HR) were measured weekly by tail-cuff plethysmography. All rats were decapitated at 8 wk. Blood was collected into tubes with and without EDTA, separated into plasma and serum by centrifugation at 4°C and stored at −20°C. After blood was collected, the heart and an aliquot of femoral muscle were immediately removed, washed in water free of ribonuclease, weighed, frozen in liquid nitrogen, and stored at −20°C until assayed.
Nephrectomy Study
The second series of this study was performed to examine whether the changes in cardiac levels of renin and angiotensin II were caused by changes in circulating levels of renin and angiotensin II. Twenty rats were divided into four groups (Cont-IN, Cont-DX, Hyper-IN, and Hyper-DX) in the same manner as the first series, then were bilaterally nephrectomized 24 h before decapitation. Samples were taken as described above.
Losartan and Nicardipine Study
In the third series of this study, 15 hyperthyroid rats were prepared and chemically denervated as described above. These rats were then treated with daily intraperitoneal administration of saline vehicle, losartan (5 μg/g), or nicardipine (10 μg/g) and were decapitated at 8 wk.
Hormone Measurements in Serum and Plasma
Serum levels of free 3,3’,5triiodothyronine (Ts) were determined with a commercially available radioimmunoassay (RIA) kit according to the manufacturer's instructions (Amarex-MAB free T3, Ortho-Clinical Diagnostics, Tokyo, Japan). PRA was determined with a commercially available RIA kit according to the manufacturer's instructions (Renin-Riabead, Dainabot, Tokyo, Japan). Plasma levels of angiotensin II were determined by a combined solid-phase extraction (SPE)-high-performance liquid chromatography (HPLC)-RIA method, as previously described (22).
SPE of angiotensin peptides
After a mixed inhibitor solution (5 mM EDTA, 10 μM pepstain, 20 μM enalapril, and 1.25 mM 1, 10-phenanthroline, final concentration) was added to 1 ml of plasma, the sample was immediately applied to an octadecasilyl-silica SPE column (Sep-Pak Plus Cl8 cartridge, Millipore, Bedford, MA) that had been moistened with 3 ml of methanol followed by 10 ml of 0.1 M HCl. After the sample was washed with 10 ml of 0.1 M HCl, it was eluted with a mixture of methanol-distilled water-trifluoroacetic acid (80: 19.1:0.l volume ratio). The eluate was evaporated to dryness in a vacuum centrifuge and resuspended in 1 ml of a buffer that contained 0.1 M tris(hydroxymethyl)aminomethane (Tris) acetate, 2.6 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, and 0.1% bovine serum albumin. These processes remove contaminating proteins, salts, and lipids that affect measurement of angiotensin II.
Separation of angiotensin peptides by HPLC
HPLC was performed with a reverse-phase column (Nucleosil C18 column, Alltech Associates, Deerfield, IL) to separate angiotensin II from other angiotensin peptides. Mobile phase A consisted of 0.085% orthophosphoric acid and 0.02% sodium azide. Mobile phase B consisted of methanol. The gradient was isocratic with phase A 65%-phase B 35% from 0 to 9 min followed by a gradient of mobile phase B to 55% over 9 min at 45°C. Fractions were collected and neutralized to pH 7.4. The sample was lyophilized, reconstituted in an RIA buffer containing 50 mM Tris·HCl (pH 7.4) and 0.3% bovine serum albumin, and measured directly by RIA. Retention times of angiotensin I, II, and III were 15.5, 7.5, and 5.8 min, respectively.
Quantification of angiotensin II by RIA
A competitive protein-binding RIA was performed with a commercially available kit (angiotensin II RIA kit, Nichols Institute Diagnostics, San Juan Capistrano, CA). Cross-reactivity of the RIA antibody with angiotensin I, Asp1-Ile5-angiotensin II, Asn1-Va15-angiotensin II, Sar1-Ile5-angiotensin II, and angiotensin III is 0.1, 100,30,0.02, and 67%, respectively.
The reconstituted samples were incubated with rabbit antiangiotensin II antiserum for 6 h at 4°C and then incubated with 125I-labeled angiotensin II for 18 h at 4°C. Antibody-bound angiotensin II was separated from free angiotensin II using donkey anti-rabbit coated cellulose in suspension. After an incubation for 20 min at 20°C and centrifugation at 5,000 revolutions/min for 15 min at 4°C the unbound angiotensin II was measured in a gamma counter for 3 min. A standard curve was prepared using the dose-response relationship with standard samples, and the concentration of angiotensin II in the test samples was read from the curve.
Hormone Measurements in Cardiac Tissue
Frozen hearts were divided into four chambers. One-fourth of each chamber was used in the following measurements.
The first piece of each chamber was used to measure cardiac levels of renin as previously described (22). In brief, the heart was thawed and homogenized with the Polytron (Kinematica, Littau, Switzerland) in 10 ml of a buffer containing 2.6 mM EDTA, 1.6 mM dimercaprol, 3.4 mM 8-hydroxyquinoline sulfate, 0.2 mM phenylmethylsulfonyl fluoride, and 5 mM ammonium acetate. The homogenate was frozen and thawed four times and spun at 5,000 revolutions/min for 30 min at 4°C and the supernatant was removed. An aliquot of the supernatant was diluted 1:10. In addition, 0.5 ml of plasma obtained from the nephrectomized male rats was added to the same volume of the diluted solution as a substrate for the enzymatic reaction. Renin activity of the sample was determined as previously described (18) using the Renin-Riabead. Cardiac renin was calculated by the following formula: cardiac level (ng angiotensin I·h−l·g heart−l) = renin activity (ng angiotensin I·h−l·ml−1) × dilution rate (10 × 2 = 20) × volume of the buffer (10 ml)/weight of the aliquot of the heart assayed (g).
The second piece of each chamber was used for determination of cardiac angiotensin II by a combined SPE-HPLC-RIA method (22). In brief, the heart was thawed and homogenized with the Polytron in 10 ml of a buffer that contained 0.1 M HCl that inactivates endogenous tissue protease. The homogenate was centrifuged at 5,000 revolutions/min for 30 min at 4°C and 1 ml of the supernatant was immediately applied to the Sep-Pak Plus C18 cartridge. The concentration of angiotensin II in the sample was determined in the same manner as angiotensin II measurement in plasma described above. Cardiac angiotensin II was calculated from the following formula: cardiac level (pg/g heart) = angiotensin II concentration (pg/ml) × volume of the buffer (10 ml)/weight of the aliquot of the heart assayed (g). As a control for angiotensin II in muscular tissue, the skeletal muscle level of angiotensin II was measured in the same manner using the femoral muscle.
The third piece of each chamber was used to determine cardiac norepinephrine as previously described (22). In brief, the heart was thawed and homogenized with the Polytron in 10 ml of a cooled phosphate buffer containing 0.4 M perchloric acid and 5 mM reduced glutathione. The homogenate was spun at 5,000 revolutions/min for 30 min at 0°C and the supernatant was removed. The concentration of norepinephrine in the sample was determined by a kit for HPLC according to the manufacturer's instructions (Cat-a-Kit assay system, Amersham, Buckinghamshire, UK). Cardiac norepinephrine was calculated from the following formula: cardiac level (rig/g heart) = norepinephrine concentration (rig/ml) × volume of the buffer (10 ml)/weight of the aliquot of the heart assayed (g).
Reverse Transcription-Polymerase Chain Reaction
The fourth piece of each chamber was used for extraction of total RNA, according to the manufacturer's instructions for using the total RNA separator kit (Clontech, Palo Alto, CA). The extracted RNA was suspended in water that was free of ribonuclease and quantified by measuring the absorbance at 260 nm.
Total RNA from each heart was reverse transcribed using the GeneAmp RNA polymerase chain reaction (PCR) core kit (Perkin-Elmer Cetus, Norwalk, CT). Each sample contained 0.5 pg total RNA, 100 nmol MgC12, 1,000 nmol KCl, 200 nmol Tris·HCl (pH 8.3), 20 nmol of each dNTP (dATP, dTTP, dGTP, dCTP), 20 units ribonuclease inhibitor, 50 pmol random hexamers, and 50 units murine leukemia virus reverse transcriptase in a final volume of 20 μl. After being incubated at 42°C for 15 min, the samples were heated for 5 min at 99°C to finish the reactions and were then stored at 5°C until assayed.
Oligonucleotide primers were constructed from the published cDNA sequences of renin (32) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (35). Because treatments had no effect on GAPDH mRNA expression (Table I), GAPDH was used as an internal standard. The sequences of the renin primers were 5’-TGCCACCTTGTTGTGTGAGG-3’ (sense) that corresponded to bases 851−870 (from exon 7) of the cloned full-length sequence and 5’-ACCCGATGCGATTGTTATGCCG-3’ (antisense) that annealed to bases 1203−1224 (from exon 9). The sequences of the GAPDH primers were 5’-TCCCTCAAGATTGTCAGCAA-3’ (sense) that corresponded to bases 492−511 of the cloned full-length sequence and 5’-AGATCCACAACGGATACATT-3’ (antisense) that annealed to bases 780−799. The predicted sizes of the amplified renin and GAPDH cDNA products were 374 and 308 bp, respectively. The sense primers in each reaction were radiolabeled using [γ-32P]ATP (Amersham) and T4 polynucleotide kinase as described in the kination kit (Toyobo, Osaka, Japan).
Table 1.
Changes in parameters produced by thyroxine treatment and sympathetic denervation
| Control |
Hyperthyroid |
|||
|---|---|---|---|---|
| Parameter | IN | DX | IN | DX |
| Serum free T3, ng/l | 2.5 ± 0.1 | 2.4 ± 0.2 | 6.6 ± 0.3* | 6.5 ± 1.5* |
| Cardiac norepi-nephrine, ng/g | 122 ± 14 | 18 ± 3† | 102 ± 5 | 15 ± 2† |
| HW/BW, mg/g | 3.5 ± 0.2 | 3.6 ± 0.3 | 5.7 ± 0.4* | 4.8 ± 0.4*† |
| Systolic blood pressure, mmHg | 138 ± 2 | 125 ± 2† | 144 ± 1* | 131 ± 8† |
| Heart rate, beats/min | 424 ±40 | 409 ±17 | 499 ±20* | 455±5*† |
| PRA, μg·h−1·l−1 | 7 ± 1 | 5 ± 1† | 28 ± 5* | 18 ± 3*† |
| Cardiac renin, ng·h−1·g−1 | 12 ± 1 | 11 ± 1 | 16 ± 3* | 15 ± 1* |
| GAPDH, densitometric units | 1,183 ± 31 | 1,238 ± 27 | 1,201 ± 42 | 1,252 ± 37 |
| Renin/GAPDH, %ratio | 12 ± 2 | 11 ± 2 | 39 ± 5* | 34 ± 4* |
| Plasma ANG II, ng/l | 44 ± 6 | 48 ± 8 | 86 ± 13* | 64 ± 11 |
| Skeletal muscle ANG II, pg/g | 100 ± 9 | 115 ± 9 | 105 ± 11 | 113 ± 17 |
| Cardiac ANG II, pg/g | 100 ± 16 | 89 ± 13 | 153 ± 18* | 182 ± 26* |
Values are means ± SE; n = 5 experiments. IN, sympathetic innervated group; DX, sympathetic denervated group; T3, 3,3′,5-triiodothyronine; HW/BW, ratio of heart weight to body weight; PRA, plasma renin activity; GAPDH, glyceraldehyde-3-phosphate dehydro-genase; ANG II, angiotensin II.
P < 0.05 vs. respective control group,
P < 0.05 vs. respective sympathetic innervated group.
Five microliters of the reverse transcription (RT) mixture were used for amplification; 25 nmol MgCla, 1,000 nmol KCl, 200 nmol Tris·HCl (pH 8.3), 3.75 pmol and 106 counts/min of both sense primers, 3.75 pmol both antisense primers, and 0.625 units AmpliTaq DNA polymerase were added to each sample as described in the GeneAmp RNA PCR core kit. To minimize nonspecific amplification, a “hot start” procedure was used in which PCR samples were placed in a thermocycler (Perkin Elmer Cetus) that was prewarmed to 94°C. After 2 min, PCR was performed for 25−45 cycles using a 30-s denaturation at 94°C 60-s annealing at 57°C and 75-s extension at 72°C. A 5-min extension at 72°C was added. After completion of RT-PCR, DNA was electrophoresed on an 8% (wt/vol) polyacrylamide gel. Gels were dried on filter paper and then exposed to the BAS 2000 imaging plate (Fuji Film, Tokyo, Japan) for 1 min and quantified with the BAS 2000 laser image analyzer (Fuji Film) (19). Renin mRNA expression was evaluated as the renin-to-GAPDH ratio.
Statistical Analysis
Results are presented as means ± SE. Comparison of multiple groups used one-way factorial analysis of variance with post hoc Scheffé F-test to evaluate the significance of the differences between groups. A level of probability (P) <o.05 was considered a statistically significant difference.
RESULTS
Effects of Administration of Thyroxine and 6-Hydroxydopamine
Serum free T3 rose significantly after the intraperitoneal administration of thyroxine in Hyper vs. Cont groups (Table 1). Cardiac norepinephrine decreased significantly after the intraperitoneal administration of 6-hydroxydopamine in DX as compared with IN.
Effects of RT on Amplification of Renin and GAPDH mRNAs
Two clear bands were detected with RT, and the bands had the predicted size of 374 bp for renin and 308 bp for GAPDH. When the PCR procedure was performed without RT, these bands were not observed, and no other bands were present. This indicated that the 374- and 308-bp bands originated from mRNA, not from genomic DNA.
Relationship Between PCR Cycle Number and Quantity of Amplified Products for Renin and GAPDH mRNAs
The cycle dependency of the radioactivity of the RT-PCR product was evaluated (Fig. 1). A linear relationship between the number of PCR cycles and the amount of PCR product (renin and GAPDH) was obtained from cycles 33 to 38. Thirty-five cycles of PCR were selected for analysis.
Fig. 1.
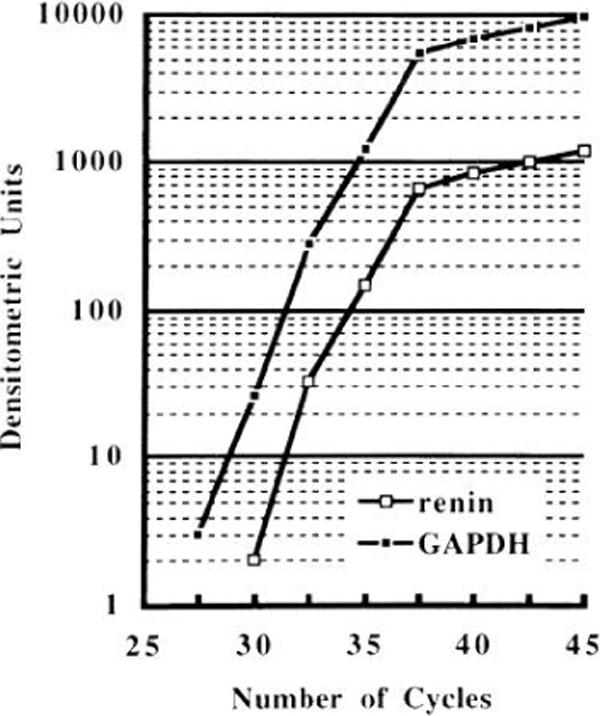
Cycle-dependent amplification of renin and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNAs in semiquantitative reverse transcription-polymerase chain reaction. A linear relationship between numbers of cycles and amount of product (renin and GAPDH) was obtained between cycles 33 and 38. Thus 35 cycles were chosen for analysis.
Hemodynamic Changes Produced by Thyroxine Treatment and Sympathetic Denervation
The ratio of the heart weight to BW (HWIBW) was significantly increased in Hyper-IN compared with Cont-IN (Table 1). HW/BW was significantly reduced in Hyper-DX as compared with Hyper-IN. HWBW was significantly increased in Hyper-DX vs. Cont-DX. There was a significant increase in systolic BP in Hyper-IN vs. Cont-IN. Systolic BP was significantly reduced in Hyper-DX vs. Hyper-IN and in Cont-DX vs. Cont-IN. HR was significantly increased in Hyper-IN vs. Cont-IN. HR was significantly reduced in Hyper-DX vs. Hyper-IN. HR was significantly increased in Hyper-DX vs. Cont-DX.
Changes in Circulating and Cardiac RAS
PRA was significantly increased in Hyper-IN as compared with Cont-IN (Table 1). PRA was significantly reduced in Hyper-DX vs. Hyper-IN and in Cont-DX vs. Cont-IN. PRA was significantly increased in Hyper-DX vs. Cont-DX. Cardiac renin was significantly increased in Hyper-IN vs. Cont-IN. Sympathetic denervation did not affect cardiac renin. Cardiac renin was significantly increased in Hyper-DX as compared with Cont-DX. Figure 2 and Table 1 show the changes in the renin-to-GAPDH ratio induced by thyroxine and sympathetic denervation. Although the densitometric unit of GAPDH was similar across the four groups, thyroxine administration significantly increased the renin-to-GAPDH ratio by approximately threefold. These increases were not significantly affected by sympathetic denervation. Plasma angiotensin II was significantly increased in Hyper-IN vs. Cont-IN (Table 1, and sympathetic denervation had no effect on plasma angiotensin II. Although angiotensin II in femoral muscle did not change in each group, cardiac angiotensin II was significantly increased in Hyper-IN vs. Cont-IN. Sympathetic denervation did not affect cardiac angiotensin II. Cardiac angiotensin II was significantly increased in Hyper-DX vs. Cont-DX.
Fig. 2.
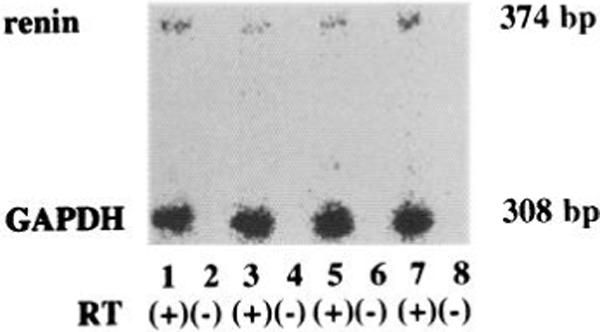
Semiquantitative polymerase chain reaction of renin GAPDH mRNAs in each group. As repeated 3 times for each sample, 15 experiments were performed in the following manner: lane 1, control (Cent)-innervated (IN) with reverse transcription (RT); lane 2, Cont-IN without RT, lane 3, Cont-denervated (DX) with RT; lane 4, Cont-DX without RT; lane 5, hyperthyroid (Hyper)-IN with RT, lane 6, Hyper-IN without RT; lane 7, Hyper-DX with RT; lane 8, Hyper-DX without RT.
Effect of Nephrectomy
Table 2 shows the effects of nephrectomy with and without thyroxine administration. Bilateral nephrectomy 24 h before decapitation significantly reduced PRA below the measurable level in all four groups. Plasma angiotensin II also significantly decreased with nephrectomy and did not differ among the four groups. However, cardiac levels of renin and angiotensin II and the renin-to-GAPDH ratio were still significantly greater in the nephrectomized Hyper group than in the nephrectomized Cont group.
Table 2.
Changes in parameters in 24-h nephrectomized rats
| Control |
Hyperthyroid |
|||
|---|---|---|---|---|
| Parameter | IN | DX | IN | DX |
| PRA, μg · h−1 · l−1 | 0.2 ± 0.1 | 0.2 ± 0.1 | 0.2 ± 0.1 | 0.2 ± 0.1 |
| Cardiac renin, ng · h−1 · g−1 | 9 ± 1 | 8 ± 1 | 14 ± 2* | 13 ± 1* |
| Renin/GAPDH, %ratio | 10 ± 2 | 10 ± 2 | 36 ± 4* | 33 ± 3* |
| Plasma ANG II, ng/l | 18 ± 3 | 17 ± 2 | 24 ± 4 | 22 ± 3 |
| Cardiac ANG II, pg/g | 74 ± 16 | 63 ±16 | 127 ± 17* | 136 ± 20* |
Values are means ± SE; n = 5 experiments. Definitions are as in Table 1.
P < 0.05 vs. respective control group.
Effects of Losartan and Nicardipine
Table 3 depicts the effects of vehicle, losartan, and nicardipine in hyperthyroid rats with sympathetic denervation. Vehicle treatment did not influence hyperthyroid rats with chemical denervation, and all parameters were similar to those observed in Hyper-DX in Table 1. Losartan or nicardipine did not alter serum free T3 or HR compared with vehicle treatment. Losartan and nicardipine significantly decreased systolic BP to the same extent; however, only losartan significantly decreased the HWIBW compared with vehicle. PRA was significantly increased with losartan but was not altered with nicardipine. Cardiac renin and the renin-to-GAPDH ratio were similar among the three groups. Plasma angiotensin II was significantly increased with losartan but not with nicardipine. Losartan significantly decreased cardiac angiotensin II, which was unaffected by nicardipine compared with vehicle treatment.
Table 3.
Influences of losartan or nicardipine treatment in hyperthyroid rats with sympathetic denervation
| Treatment | Vehicle | Losartan | Nicardipine |
|---|---|---|---|
| Serum free T3, ng/l | 6.7 ± 0.8 | 6,7 ± 6.1 | 6.8 ±0.2 |
| HW/BW, mg/g | 4.7 ±0.5 | 3.3 ± 0.2* | 4.4 ±0.2 |
| Systolic blood pressure, mmHg | 135 ±6 | 117 ±1* | 115 ±4* |
| Heart rate, beats/min | 447 ±5 | 438 ± 42 | 449 ±9 |
| PRA, μg · h−1 · l−1 | 14 ±3 | 30 ±1* | 12 ± 2 |
| Cardiac renin, ng · h−1 · g−1 | 16 ± 2 | 15 ± 1 | 16 ± 1 |
| Renin/GAPDH, %ratio | 33 ± 4 | 33 ± 3 | 31 ± 5 |
| Plasma ANG II, ng/l | 68 ± 9 | 193 ± 13* | 72 ± 4 |
| Cardiac ANG II, pg/g | 186 ± 9 | 144 ± 8* | 184 ± 11 |
Values are means ± SE; n = 5 experiments. Definitions are as in Table 1.
P < 0.05 vs. vehicle treatment.
DISCUSSION
Administration of thyroxine for 8 wk significantly increased serum free T3, HR, PRA, and HW/BW. Thus thyroxine administration was effective at producing hyperthyroidism in this model. Administration of 6-hydroxydopamine significantly reduced cardiac norepinephrine after 8 wk, indicating the effectiveness of sympathetic denervation of the heart.
It was previously thought that cardiac hypertrophy induced by hyperthyroidism resulted from increased sympathetic nervous activity and an increase in BP (15). The present data agree with these mechanisms, since the increase in HW/BW by thyroxine was partially inhibited by denervation associated with complete inhibition of the increase in BP, and this effect persisted. However, if cardiac hypertrophy induced by hyperthyroidism resulted only from increased sympathetic nervous activity and an increase in BP, sympathetic denervation associated with normalization of BP would completely inhibit the increase in HW/BW produced by thyroxine administration. The present study showed that HW/BW in Hyper-DX was significantly increased vs. Cont-DX. Therefore, other factors must be involved.
Thyroxine administration significantly increased HW/BW, BP, HR, PRA, plasma angiotensin II, cardiac renin, and angiotensin II, and the renin-to-GAPDH ratio in Hyper-IN vs. Cont-IN. In Hyper-DX, the increases in cardiac renin, cardiac angiotensin II, and the renin-to-GAPDH ratio were not inhibited by sympathetic denervation. These results indicate that thyroid hormone activates the cardiac RAS independent of the sympathetic nervous system through enhancement of the expression of renin mRNA. Some studies have shown that activation of the cardiac RAS produced cardiac hypertrophy (28). Activation of the cardiac RAS has been shown to provoke cardiac hypertrophy through enhancement of the expression of the sarcoplasmic reticulum Ca2+-ATPase mRNA (4). The increases in cardiac renin, cardiac angiotensin II, and the renin-to-GAPDH ratio observed in the present study suggest that activation of the cardiac RAS results in increases in HWIBW.
It was proposed 30 years ago that thyroid hormone caused cardiac hypertrophy through a mechanism that was independent of the sympathetic nervous system (5, 33). Segal (30) reported that thyroid hormone induced uptake of calcium by the heart as an acute effect, which would then increase myocardial contractility. Ojamaa et al. (25) demonstrated that thyroid hormone upregulated the expression of the myosin heavy chain genes. Thomas et al. (34) showed that insulin-like growth factor I produced cardiac hypertrophy, as well as endocrine and cardiac paracrine effects, during hyperthyroidism. Current information suggests that these possible mediators are also induced by angiotensin II. Angiotensin II increases the intracellular concentration of calcium through voltage-dependent and receptor-dependent calcium channels (29) and enhances the expression of the myosin heavy chain genes in the heart (11) and insulin-like growth factor I mRNA (23). These studies suggest that thyroid hormone can induce these mediators through activation of the RAS. Activation of the cardiac RAS may be the first step in a mechanism independent of the sympathetic nervous system that leads to cardiac hypertrophy induced by hyperthyroidism.
Some investigators have suggested that increased cardiac renin may be secondary to the uptake of circulating renin (6, 37). However, other researchers have shown that cardiac renin may be endogenously produced (3, 8, 10). We have shown that cardiac levels of renin were increased with thyroxine treatment and were accompanied by enhanced expression of renin mRNA, even when nephrectomy drastically reduced the circulating levels of renin. It is therefore strongly suggested that the increase in cardiac renin observed in hyperthyroidism results from a local increase in cardiac production of renin.
We then examined hyperthyroid rats with sympathetic denervation to determine the involvement of the RAS in hyperthyroidism-induced cardiac hypertrophy. Losartan treatment significantly suppressed the development of thyroxine-induced cardiac hypertrophy. In contrast, nicardipine, which reduced systolic BP to the same extent as losartan, failed to diminish the increased HW/BW observed in vehicle-treated hyperthyroid rats with sympathetic denervation. These results suggested that losartan improves hyperthyroidism-induced cardiac hypertrophy independently of its effects on hemodynamics and on the sympathetic nervous system. Therefore, the activated RAS observed in hyperthyroid rats may be important in the development of cardiac hypertrophy. The thyroid hormone-induced RAS activation included sympathetic nervous system-independent and circulating renin-independent increases in cardiac levels of renin and the renin gene. The increased cardiac level of renin can trigger cardiac RAS activation in hyperthyroidism, such that thyroid hormone contributes to the development of cardiac hypertrophy. However, it has been reported that thyroid hormone upregulates expression of angiotensinogen (17) and angiotensin-converting enzyme (24). Because the present study focused on renin, a rate-limiting enzyme in the RAS cascade (7, 14), the expression of angiotensinogen or angiotensin-converting enzyme in the heart was not determined. Therefore, we cannot exclude the possibility that thyroid hormone may also upregulate cardiac expression of angiotensinogen and angiotensin-converting enzyme. Such upregulations would enhance the cardiac RAS in cooperation with the enhanced expression of cardiac renin observed in the present study.
A previous study provided opposing evidence as to whether the RAS has a role in thyroid hormone-induced cardiac hypertrophy (2). Captopril decreased left ventricular systolic and diastolic pressures but did not change heart rate, dP/dt, and left ventricular-to-body weight ratio in animals treated for 10 days with thyroxine (2). In the present study, 8-wk treatment with losartan significantly inhibited the development of cardiac hypertrophy induced by thyroxine treatment. This discrepancy may result from differences in the treatment period. The RAS may be essential for maintenance rather than the initiation of hyperthyroidism-induced cardiac hypertrophy. Alternatively, the discrepancy may be based on the difference in pharmacological effects between angiotensin-converting enzyme inhibi tor and angiotensin II receptor antagonist. Recent studies elucidated that angiotensin-converting enzyme inhibitor-independent angiotensin II synthesis exists in the heart (26,36). It is therefore possible that chronic administration of angiotensin-converting enzyme inhibi tor fails to completely inhibit cardiac angiotensin II synthesis. In contrast, the angiotensin II receptor antagonist blocks the elects of angiotensin II on the heart regardless of its synthetic pathway. In the pres ent study, losartan treatment significantly increased PRA and plasma angiotensin II, consistent with the pharmacological effects of angiotensin II type 1 recep tor antagonists (13), and losartan also significantly decreased cardiac angiotensin II. These results indi cate that losartan effectively antagonized circulating and cardiac angiotensin II and thereby could decrease the heart-to-body weight ratio.
Zierhut and Zimmer (38) have provided evidence that the β-adrenergic blocker metoprolol had no effect on cardiac hypertrophy in T3-treated rats. Bedotto et al. (2) demonstrated that propranolol treatment failed to decrease the left ventricular-to-body weight ratio in thyroxine-treated rats. These studies suggest that the sympathetic nervous system has little involvement in developing cardiac hypertrophy in hyperthyroid rats. In contrast, Klein (21) reported that propranolol treat ment prevented cardiac hypertrophy in thyroxine-treated rats. In the present study, thyroxine treatment induced cardiac hypertrophy without involving the sympathetic nervous system, and simultaneous sympa thetic denervation attenuated the development of thy roxine-induced cardiac hypertrophy. Thus the sympathetic nervous system may not be essential but does act as a modulator of the development of hyperthyroidism-induced cardiac hypertrophy. The complex interaction between thyroxine and the sympathetic nervous system remains to be fully characterized.
Skeletal muscle angiotensin II was also measured as a control in the present study. Although no difference in skeletal muscle angiotensin II was observed, cardiac angiotensin II levels were significantly different among the four groups. This result indicated that the effects of thyroid hormone on angiotensin II were specific for the heart, excluding the possibility that the increased plasma angiotensin II diffusely influenced the systemic muscular tissues.
In conclusion, the present study demonstrated that 8-wk treatment with thyroxine caused cardiac hypertrophy in rats independent of the sympathetic nervous system, although the sympathetic nervous system modulated the development of cardiac hypertrophy. The sympathetic nervous system-independent cardiac hypertrophy caused by thyroid hormone accompanied RAS activation that included increases in cardiac re nin, cardiac angiotensin II, and cardiac renin mRNA expression. However, complete inhibition of PRA with bilateral nephrectomy did not influence the thyroid hormone-induced increase in cardiac renin, suggesting that thyroid hormone directly enhanced cardiac renin expression without involving circulating renin. Addi tional experiments revealed that losartan, but not nicardipine, improved thyroid hormone-induced car diac hypertrophy. It appears that the RAS has an essential role in the development of cardiac hypertro phy in our hyperthyroid model. Thyroid hormone-induced enhancement of cardiac renin expression may be one trigger of the RAS activation that leads to cardiac hypertrophy.
Acknowledgments
We thank Dr. Lisa M. Harrison-Bernard for critical review of this manuscript.
This work was supported in part by research grants from the Japan Health Sciences Foundation (Tokyo, Japan) and Ministry of Education, Science, and Culture, Japan, Grant 08770511.
REFERENCES
- 1.Baker KM, Singer HA. Identification and characterization of guinea pig angiotensin II ventricular and atria1 receptors: coupling to inositol phosphate production. Circ. Res. 1988;62:896–904. doi: 10.1161/01.res.62.5.896. [DOI] [PubMed] [Google Scholar]
- 2.Bedotto JB, Gay RG, Graham SD, Morkin E, Goldman S. Cardiac hypertrophy induced by thyroid hormone is independent of loading conditions and beta adrenoceptor blockade. J. Pharmacol. Exp. Ther. 1989;248:632–636. [PubMed] [Google Scholar]
- 3.Boer PH, Ruzicka M, Lear W, Harmsen E, Rosenthal J, Leenen FH. Stretch-mediated activation of cardiac renin gene. Am. J. Physiol. 1994;267:Hl630–H1636. doi: 10.1152/ajpheart.1994.267.4.H1630. (Heart Circ. Physiol. 36)
- 4.Bruckschlegel G, Holmer SR, K Jandeleit, D Grimm, F Muders, Kromer EP, Riegger GA, H Schunkert. Blockade of the renin-angiotensin system in cardiac pressure-overload hypertrophy in rats. Hypertension. 1995;25:250–259. doi: 10.1161/01.hyp.25.2.250. [DOI] [PubMed] [Google Scholar]
- 5.Buccino RA, Spann J, Jr., Pool PE, Sonnenblick EH, Braunwald E. Influence of the thyroid state on the intrinsic contractile properties and energy stores of the myocardium. J. Clin. Inuest. 1967;46:1669–1682. doi: 10.1172/JCI105658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Danser AH, van-Kats JP, Admiraal PJ, Derkx FH, Lamers JM, Verdouw PD, Saxena PR, Schalekamp MA. Cardiac renin and angiotensins. Uptake from plasma vs. in situ synthesis. Hypertension. 1994;24:37–48. doi: 10.1161/01.hyp.24.1.37. [DOI] [PubMed] [Google Scholar]
- 7.Davis JO, Freeman RH. Mechanisms regulating renin release. Physiol. Reu. 1976;56:1–56. doi: 10.1152/physrev.1976.56.1.1. [DOI] [PubMed] [Google Scholar]
- 8.Dostal DE, Rothblum KN, Chernin MI, Cooper GR, Baker KM. Intracardiac detection of angiotensinogen and renin: a localized renin-angiotensin system in neonatal rat heart. Am. J. Physiol. 1992;263:C838–C850. doi: 10.1152/ajpcell.1992.263.4.C838. (Cell Physiol. 32)
- 9.Dunn FG, Oigman W, Ventura H0, Messerli FH, Kobrin I, Frohlich ED. Enalapril improves systemic and renal hemodynamics and allows regression of left ventricular mass in essential hypertension. Am. J. Cardiol. 1984;53:105–108. doi: 10.1016/0002-9149(84)90692-1. [DOI] [PubMed] [Google Scholar]
- 10.Dzau VJ, Re R. Tissue angiotensin system in cardiovascular medicine. A paradigm shift? Circulation. 1994;89:493–498. doi: 10.1161/01.cir.89.1.493. [DOI] [PubMed] [Google Scholar]
- 11.Geenen DL, Malhotra A, Scheuer J. Angiotensin II increases cardiac protein synthesis in adult rat heart. Am. J. Physiol. 1993;265:H238–H243. doi: 10.1152/ajpheart.1993.265.1.H238. (Heart Circ. Physiol. 34)
- 12.Gilbert MT, Sun J, Yan Y, Oddoux C, Lazarus A, Tansey WP, Lavin TN, Catanzaro DF. Renin gene promoter activity in GC cells is regulated by CAMP and thyroid hormone through Pit-l-dependent mechanisms. J. Biol. Chem. 1994;269:28049–28054. [PubMed] [Google Scholar]
- 13.Goa KL, Wagstaff AJ. Losartan potassium-a review of its pharmacology, clinical efficacy and tolerability in the management of hypertension. Drugs. 1996;51:820–845. doi: 10.2165/00003495-199651050-00008. [DOI] [PubMed] [Google Scholar]
- 14.Hackenthal E, Paul M, Ganten D, Taugner R. Morphology, physiology, and molecular biology of renin secretion. Physiol. Rev. 199O;70:1067–1116. doi: 10.1152/physrev.1990.70.4.1067. [DOI] [PubMed] [Google Scholar]
- 15.Hauger-Klevene JH, Brown H, Zavaleta J. Plasma renin activity in hyper- and hypothyroidism: effect of adrenergic blocking agents. J. Clin. Endocrinol. Metab. 1972;34:625–629. doi: 10.1210/jcem-34-4-625. [DOI] [PubMed] [Google Scholar]
- 16.Heron MI, Rakusan K. Geometry of coronary capillaries in hyperthyroid and hypothyroid rat heart. Am. J. Physiol. 1994;267:H1024–H1031. doi: 10.1152/ajpheart.1994.267.3.H1024. (Heart Circ. Physiol. 36)
- 17.Hong-Brown LQ, Deschepper CF. Effects of thyroid hormones on angiotensinogen gene expression in rat liver, brain, and cultured cells. Endocrinology. 1992;130:1231–1237. doi: 10.1210/endo.130.3.1537289. [DOI] [PubMed] [Google Scholar]
- 18.Ichihara A, Suzuki H, Murakami M, Naitoh M, Matsumoto A, Saruta T. Interactions between angiotensin II and norepinephrine on renin release by juxtaglomerular cells. Eur. J. Endocrinol. 1995;133:569–577. doi: 10.1530/eje.0.1330569. [DOI] [PubMed] [Google Scholar]
- 19.Itohara S, Farr AG, Lafaille JJ, Bonneville M, Takagaki Y, Haas W, Tonegawa S. Homing of a gamma delta thymocyte subset with homogeneous T-cell receptors to mucosal epithelia. Nature. 199O;343:754–757. doi: 10.1038/343754a0. [DOI] [PubMed] [Google Scholar]
- 20.Karen P, Morris BJ. Stimulation by thyroid hormone of renin mRNA in mouse submandibular gland. Am. J. Physiol. 1986;251:E290–E293. doi: 10.1152/ajpendo.1986.251.3.E290. (Endocrinol. Metab. 14)
- 21.Klein I. Thyroxine-induced cardiac hypertrophy: time course of development and inhibition by propranolol. Endocrinology. 1988;123:203–210. doi: 10.1210/endo-123-1-203. [DOI] [PubMed] [Google Scholar]
- 22.Kobori H, Ichihara A, Suzuki H, Miyashita Y, Hayashi M, Saruta T. Thyroid hormone stimulates renin synthesis in rats without involving the sympathetic nervous system. Am. J. Physiol. 1997;272:E227–E232. doi: 10.1152/ajpendo.1997.272.2.E227. (Endocrinol. Metab. 35)
- 23.Lowe W, Jr., Yorek MA, Teasdale RM. Ligands that activate protein kinase-C differ in their ability to regulate basic fibroblast growth factor and insulin-like growth factor-I messenger ribonucleic acid levels. Endocrinology. 1993;132:1593–1602. doi: 10.1210/endo.132.4.8462457. [DOI] [PubMed] [Google Scholar]
- 24.Michel B, Grima M, Coquard C, Welsch C, Barthelmebs M, Imbs JL. Effects of triiodothyronine and dexamethasone on plasma and tissue angiotensin converting enzyme in the rat. Fundam. Clin. Pharm. 1994;8:366–372. doi: 10.1111/j.1472-8206.1994.tb00814.x. [DOI] [PubMed] [Google Scholar]
- 25.Ojamaa K, Samarel AM, Kupfer JM, Hong C, Klein I. Thyroid hormone effects on cardiac gene expression independent of cardiac growth and protein synthesis. Am. J. Physiol. 1992;263:E534–E540. doi: 10.1152/ajpendo.1992.263.3.E534. (Endocrinol. Metab. 26)
- 26.Phillips MI, Speakman EA, Kimura B. Levels of angiotensin and molecular biology of the tissue renin angiotensin systems. Regul. Pept. 1993;43:1–20. doi: 10.1016/0167-0115(93)90403-u. [DOI] [PubMed] [Google Scholar]
- 27.Polikar R, Kennedy B, Ziegler M, O'Connor DT, Smith J, Nicod P. Plasma norepinephrine kinetics, dopa-mine-beta-hydroxylase, and chromogranin-A, in hypothyroid patients before and after replacement therapy. J. Clin. Endocrinol. Metab. 1990;70:277–281. doi: 10.1210/jcem-70-1-277. [DOI] [PubMed] [Google Scholar]
- 28.Ruzicka M, Leenen FH. Renin-angiotensin system and minoxidil-induced cardiac hypertrophy in rats. Am. J. Physiol. 1993;265:H1551–H1556. doi: 10.1152/ajpheart.1993.265.5.H1551. (Heart Circ. Physiol. 34)
- 29.Sadoshima J, Qiu Z, Morgan JP, Izumo S. Angiotensin II and other hypertrophic stimuli mediated by G protein-coupled receptors activate tyrosine kinase, mitogen-activated protein kinase, and 90-kD S6 kinase in cardiac myocytes. The critical role of Ca2+-dependent signaling. Circ. Res. 1995;76:1–15. doi: 10.1161/01.res.76.1.1. [DOI] [PubMed] [Google Scholar]
- 30.Segal J. Calcium is the first messenger for the action of thyroid hormone at the level of the plasma membrane: first evidence for an acute effect of thyroid hormone on calcium uptake in the heart. Endocrinology. 199O;126:2693–2702. doi: 10.1210/endo-126-5-2693. [DOI] [PubMed] [Google Scholar]
- 31.Shirani J, Barron MM, Pierre-Louis ML, Roberts WC. Congestive heart failure, dilated cardiac ventricles, and sudden death in hyperthyroidism. Am. J. Cardiol. 1993;72:365–368. doi: 10.1016/0002-9149(93)90691-5. [DOI] [PubMed] [Google Scholar]
- 32.Tada M, Fukamizu A, Seo MS, Takahashi S, Murakami K. Nucleotide sequence of rat renin cDNA. Nucleic Acids Res. 1988;16:3576. doi: 10.1093/nar/16.8.3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taylor RR, Covell JW, Ross J., Jr Influence of the thyroid state on left ventricular tension-velocity relations in the intact, sedated dog. J. Clin. Invest. 1969;48:775–784. doi: 10.1172/JCI106035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thomas MR, Miell JP, Taylor AM, Ross RJ, Arnao JR, Jewitt DE, McGregor AM. Endocrine and cardiac paracrine actions of insulin-like growth factor-I (IGF-I) during thyroid dysfunction in the rat: is IGF-I implicated in the mechanism of heart weight/body weight change during abnormal thyroid function? J. Mol. Endocrinol. 1993;10:313–323. doi: 10.1677/jme.0.0100313. [DOI] [PubMed] [Google Scholar]
- 35.Tso JY, Sun XH, Kao TH, Reece KS, Wu R. Isolation and characterization of rat and human glyceraldehyde-3-phosphate dehydrogenase cDNAs: genomic complexity and molecular evolution of the gene. Nucleic Acids Res. 1985;13:2485–2502. doi: 10.1093/nar/13.7.2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Urata H, Kinoshita A, Misono KS, Bumpus FM, Husain A. Identification of a highly specific chymase as the major angiotensin II-forming enzyme in the human heart. J. Biol. Chem. 199O;265:22348–22357. [PubMed] [Google Scholar]
- 37.Von Lutterotti N, Catanzaro DF, Sealey JE, Laragh JH. Renin is not synthesized by cardiac and extrarenal vascular tissues. A review of experimental evidence. Circulation. 1994;89:458–470. doi: 10.1161/01.cir.89.1.458. [DOI] [PubMed] [Google Scholar]
- 38.Zierhut W, Zimmer HG. Triiodothyronine-induced changes in function, metabolism and weight of the rat heart: effects of alphaand beta-adrenergic blockade. Basic Res. Cardiol. 1989;84:359–370. doi: 10.1007/BF02650870. [DOI] [PubMed] [Google Scholar]