

Abstract
We have designed polypeptides combining selected lipophilic (LP) and hydrophilic (HP) sequences that assemble into amphiphilic (AP) α-helical bundles to reproduce key structure characteristics and functional elements of natural membrane proteins. The principal AP maquette (AP1) developed here joins 14 residues of a heme binding sequence from a structured diheme-four-α-helical bundle (HP1), with 24 residues of a membrane-spanning LP domain from the natural four-α-helical M2 channel of the influenza virus, through a flexible linking sequence (GGNG) to make a 42 amino acid peptide. The individual AP1 helices (without connecting loops) assemble in detergent into four-α-helical bundles as observed by analytical ultracentrifugation. The helices are oriented parallel as indicated by interactions typical of adjacent hemes. AP1 orients vectorially at non-polar/polar interfaces and readily incorporates into phospholipid vesicles with >97% efficiency, although most probably without vectorial bias. Mono- and diheme-AP1 in membranes enhance functional elements well established in related HP analogues. These include strong redox charge coupling of heme with interior glutamates and internal electric field effects eliciting a remarkable 160 mV splitting of the redox potentials of adjacent hemes that leads to differential heme binding affinities. The AP maquettes variants, AP2 and AP3, removed heme-ligating histidines from the HP domain and included heme-ligating histidines in LP domains by selecting the bH heme binding sequence from membrane-spanning d-helix of respiratory cytochrome bc1. These represent the first examples of AP maquettes with heme and bacteriochlorophyll binding sites located within the LP domains.
Keywords: amphiphilic, maquette, membrane protein, heme protein, de novo design, air-water interface, four-helix bundle, redox potential, charge-charge interaction, proton coupling to oxidation/reduction
The last two decades has seen progress toward reproducing structural motifs and functional elements drawn from Nature in simple, robust, de novo-designed peptide and protein models or maquettes. Amongst the first of these were water-insoluble, lipophilic (LP) maquettes (1-4) that drew inspiration mainly from channel formation and ion transport of natural membrane proteins. Early success came in the LS2 proton channel designed from homo-oligomeric α-helical segments that verified selective and transient proton transport across membranes under applied voltage (1). Later, ion transport selectivity of many natural channels has been reproduced by the stabilizing template-assembled synthetic protein (TASP) approach (2-4). More extensive work with water-soluble, hydrophilic (HP) maquettes took advantage of well-established specific ligation of metals, metal clusters and metallo-organic and organic cofactors to formulate design strategies to assemble the cofactor within three to four α-helices (5-21). These maquettes successfully established a variety of functional elements (11, 22-24) and simple redox activity (25-28).
The idea of bringing HP and LP protein segments together to form an amphiphilic (AP) maquette family (29) is a response to a requirement for vectorial performance across an interface (Figure 1). Vectorial orientation was conferred on early HP heme protein maquettes by introducing amphiphilic character via two palmitoylates or cholesterols that were conjugated to the loop region of each dihelix of the four helix bundle (30, 31); the clustering of these non-polar groups imposed a syn- (all helices parallel) topology. These lipo-proteins remained water-soluble but had sufficient amphiphilic character for the designed purpose of forming stable, oriented Langmuir monolayer films as established by X-ray reflectivity and interferometry (32, 33).
Figure 1.
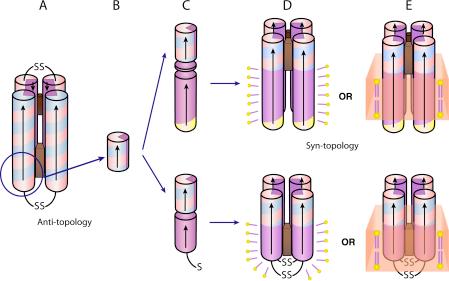
Schematic drawing of the AP family design and assembly. De novo designed maquettes are built from α-helices patterned along the helical axis with polar and non-polar residues: positive residues are colored blue, negative are red, polar uncharged are yellow and non-polar residues are purple. The incorporation of heme is displayed by a brown box. The HP domains of the AP maquettes described here are based on the structured diheme four-α-helical bundle HP1 that consists of two antiparallel dihelices (A). The HP1 dihelices are covalently linked by disulfide bond at the end of the flexible loops. AP1 (top) and AP3 (bottom) maquettes utilized only 14 residues close to the N-terminus and excluded the loops (blue circle). The abstracted HP domain (B) was linked to an LP domain either via flexible linker (AP1, top) or directly (AP3, bottom) (C). The polar/non-polar arrangement of the residues is designed (see also Figure 2) to drive assembly of four monomeric peptides to form a parallel four-α-helix bundle with two domains of sharply different polarity that confer amphiphilic character of the entire maquette. AP maquettes cannot be monodispersed and soluble in aqueous solution but they readily co-assemble with detergents to form micelles (D) or with lipids to form membranes (E).
The new AP maquettes described here are built from two distinct continuous HP and LP α-helical polypeptide domains following the general architecture of many natural membrane proteins. The HP-domains selected are variants of the recently presented singularly structured HP1 diheme-four-α-helical bundle (34), while the LP-domains are drawn from de novo designed LS2 channel (1), the native transmembrane segment of M2 channel from the influenza virus (35), and the heme binding helix d from the respiratory protein cytochrome bc1 (36). Monolayer structure determination for a representative AP protein, AP0, has established formation of well-ordered structures with the bundle axis perpendicular to the interface in Langmuir films at higher surface pressures (37).
In the present work we describe general engineering guidelines and assembly strategies of AP maquettes designed to accommodate porphyrin (heme) and chlorin (bacteriochlorophylls) cofactor binding sites within either the HP or LP domains. We focus on quantifying the courses of physical change during transformation from an overall HP maquette to related AP maquettes as seen by secondary structures, oligomeric states, properties after detergent solubilization or incorporation into membrane bilayers. Special attention is given to the way each maquette supports heme binding capacity and affinity, and how the heme oxidation-reduction and coupled reactions in AP maquettes compare to HP maquettes. Heme binding with ≤ 10−8 M affinity provides a sensitive probe of maquette structural integrity as it indicates histidines proximity and thereby availability for bis-axial ligation (22, 38, 39). Furthermore, heme provides an electrostatic probe of maquette interior by virtue of the formal cationic charge on the ferric form (bis-histidine ligated) that cannot be fully charge-compensated by proton or ion exchange (22, 39). This contrasts sharply with charge compensations possible with acid-base amino acid sidechains or with redox cofactor such as quinones or flavins. Thus equilibrium binding affinities of charged ferric heme to an α-helical maquette scaffold or ferric/ferrous oxidation-reduction potentials report environmental polarity and proximity/exposure to the aqueous solvent (38, 40). We have assessed the strength of the charge-charge coupling between two hemes by measuring binding affinity or redox potential of one heme alone and then in the presence of a second heme (22, 38), and the strength of heme redox charge coupling to proton exchange as seen in shifts in the pK values of interior glutamates (39). Such heme charge coupled field-induced events have allowed us to track the changes encountered as the design sequence takes us from HP maquettes surrounded by water, to the hydrophilic and then hydrophobic domains of AP maquettes dispersed in aqueous-detergent micelles and inserted across the aqueous-membrane interface of phospholipid bilayer vesicles.
MATERIALS AND METHODS
Peptide synthesis and purification
The peptides described in Table 1 were synthesized on Pioneer continuous flow solid phase synthesizer (Applied Biosystems) using standard Fmoc/tBu protection strategy on a Fmoc-PEG-PAL-PS resin (Applied Biosystems) at 0.1 mmol scale. The peptides were acetylated at their N-terminus (1:1 (v:v) acetic anhydride:pyridine for 20 min) and purified on a reversed phase C18 HPLC column (Vydac) using gradients of acetonitrile (Fisher) and water both containing 0.1% (v/v) trifluoroacetic acid (Sigma). The purity and molecular weight of the acetylated peptides were confirmed by matrix assisted laser desorption/ionization mass spectrometry (MALDI-MS) to be 4,733; and 3,755 Da for AP1, and AP3, respectively. The peptides were purified without difficulty with yields comparable to water-soluble peptides of similar length.
Table 1.
Primary amino acid sequences of HP1 maquette and current AP maquettes. Color code same as in the legend of Figure 1.
 |
Unless noted otherwise, all experiments were performed in buffered solutions containing 100 mM potassium chloride and 20 mM potassium phosphate (pH = 8.0). As will be shown in the results, the maquettes reported here assemble as tetramers and therefore the words maquette or bundle in the text refer to an assembly of four helices and the reported peptide concentrations are for the assembled four-helix bundles. Peptide concentrations were determined by absorbance spectroscopy at 280 nm assuming an extinction coefficient of 22,760 M−1 cm −1bundle−1 calculated from sequence by the Swiss institute of bioinformatics' EXPASY server (http://us.expasy.org/cgi-bin/protparam).
Peptide solubilization
Water insoluble AP peptides and the assembled maquettes need a detergent for solubilization in aqueous buffers. We have found that the AP maquette family members are readily solubilized by common detergents, including n-octylpentaoxyethylene (C8E5), n-octyl β-D-glucopyranoside (OG), or zwittergents (n-Alkyl-N,N-dimethyl-3-ammonio-1-propanesulfonates), near or above critical micellar concentration (CMC).
Heme binding
Although detergent concentration above the CMC is crucial for the peptide solubilization, the detergent micelles efficiently solubilize not only the protein but also the amphiphilic (but overall hydrophobic) heme and thereby interfere with the heme-bundle assembly. We have found, however, that in a narrow range of the detergent concentrations close to the CMC the protein remains soluble and still binds heme. Since the CMC of C8E5 and OG are within 0.15 − 0.3 % (w/v) and 0.55 − 0.7 % (w/v) respectively (41), a working protocol is to first solubilize the protein with detergent concentration several fold above CMC and then dilute the detergent concentration to 0.15% (w/v) for C8E5 and to 0.9% (w/v) for OG prior to heme addition.
Vesicle preparation
Vesicles were prepared from a 9:1 mixture of 1-palmitoyl-2-oleoyl-phosphatidylcholine (POPC) and 1-palmitoyl-2-oleoyl-phosphatidylserine (POPS) (Avanti, Alabaster, AL) by either dialysis or rehydration.
a) Solubilization/Dialysis
10 μmoles of the lipid mixture were dried under N2 and then overnight under vacuum. The dry lipids were co-dissolved with 45 nmoles of AP1 bundle (bundle:lipid mol ratio = 1:222) in 100 mM KCl, 50 mM potassium phosphate buffer, pH = 7.0, 5% (v/v) C8E5. The sample was dialyzed against the same buffer solution without detergent.
b) Rehydration/Sonication
10 μmoles of lipid mixture and 45−50 nmoles of AP1 bundle (bundle:lipid mol ratio = 1:200 to 1:222) were co-dissolved in chloroform:methanol 3:1. The protein/lipid solution was dried under N2 and then overnight under vacuum. The dry mixture was resuspended in buffered solution and then subjected to sonication in an ice bath with a Branson Sonifier Cell Disrupter, model 185, for ten cycles of 30 s sonication and 30 s rest which prevented the solution from overheating.
Analytical ultracentrifugation
a) Aggregation number
A Beckman XLA/I analytical ultracentrifuge was used to determine the molecular weight of the self-assembled complex of the apo- and holo- form of the maquette and to establish the efficiency of the peptide incorporation into vesicles. The molecular weight of the complex was assessed in two detergent solutions: 0.25% C8E5 and 0.9% OG. The partial specific volume of C8E5 (vbar ∼ 0.993 ml/g) is very close to that of the buffer (vbar = 0.995 ml/g at 20 °C) making the C8E5 contribution to the buoyant molecular weight of the peptide complex negligible and therefore the molecular weight can be calculated directly. The partial specific volume of OG is too low (vbar = 0.859 ml/g) to be matched by D2O (vbar = 0.899 ml/g). Therefore we have used the method of Noy et al. (42) and globally fitted sedimentation data at 6 different buffer densities with 0%, 16%, 32%, 48%, 64% and 80% D2O/H2O (v/v). This method not only determines the molecular weight of the protein complex but also resolves the average number of detergent molecules within the peptide-detergent complex. Sedimentation equilibrium experiments in C8E5 were performed at 20 °C and 40,000 RPM, whereas experiments in OG were done at 25 °C and utilized three different speeds: 35,000; 40,000; and 45,000 RPM. Data analysis made use of the theoretical partial specific volume of the apo-AP1, 0.765 ml/g (calculated from the peptide sequence using the amino acid residue parameters of Kharakoz (43) and the program “Sednterp” (44)). We used a partial specific volume of 0.82 ml/g for heme (45) and therefore the calculated partial specific volume of the holo- AP1 is 0.767 and 0.769 ml/g for heme1-AP1 and heme2-AP1, respectively.
b) Protein incorporation into vesicles
The Beckman XLA/I analytical ultracentrifuge was also used to determine the efficiency of protein incorporation into vesicles. Vesicles were prepared by rehydration as described above with buffer (100 mM KCl, 20 mM KPi, pH = 8.0) containing 50% D2O (density of 1.059 g/ml) and 24.5 μM four helix bundle. The sedimentation velocity experiments were performed at 25 °C and 30,000 RPM and scanned at 280 nm.
Characterization at the air-water interface
a) Pressure-area isotherm
Isotherms were measured using a Langmuir film balance (Lauda Filmbalance FW2, Sybron/Brinkmann, Westbury, NY) following a general procedure described earlier (46). AP1 was dissolved in methanol at protein concentrations of 4.12 mg/ml and 20 μl were spread at the air-water interface. A 15 minutes time interval was allowed between the spreading and compression cycle for complete solvent evaporation. The monolayer was compressed at 3 Å2/molecule/minute.
b) X-ray reflectivity
The X-ray reflectivity experiments were performed on beamline X-22B at the National Synchrotron Light Source (NSLS) at Brookhaven National Laboratory, Upton, New York using liquid surface spectrometer reported previously (47, 48). The AP1 monolayers were prepared as described above and compressed to the desired surface pressure (10, 20, 30, and 40 mN/m) that was then held constant to collect the X-ray reflectivity data. The normalized reflectivity data were analyzed by the box-refinement method, that requires no a priori assumptions and is therefore model-independent (49), to provide the monolayer electron density profiles.
Electron paramagnetic resonance (EPR)
EPR spectroscopy was performed using a Bruker (Karlsruhe, Germany) ESP300E spectrometer. Temperature of the sample was controlled by an Oxford ESR 900 continuous-flow liquid helium cryostat interfaced with an Oxford ITC4 temperature controller. Frequency was measured by a Hewlett-Packard 5350B frequency counter. The EPR parameters were: sample temperature, 77K; microwave frequency, 9.446 GHz; microwave power, 5.05 mW; modulation frequency, 100 kHz; modulation amplitude, 20.24 G; and time constant, 164 ms.
Circular dichroism spectroscopy
Peptide samples were measured in buffered aqueous solutions containing 0.9% OG. The concentration of the tetrameric bundle was 9 μM for the measurement without heme and 8 μM for the measurement with 1.0 heme per bundle. Circular dichroism (CD) spectra were recorded with an Aviv 62DS spectropolarimeter in a rectangular quartz cells with 1 mm path length. The temperature between each scan was increased by 5 °C by a Neslab CFT-22 re-circulating water bath. A time interval of at least 10 minutes was allowed between scans for temperature equilibration.
Heme redox midpoint potentials by potentiometry
Equilibrium redox potentiometry was used as a structurally related functional analysis of the oxidation-reduction of hemes bound to HP and AP maquettes.
a) Redox titrations
These were performed in an anaerobic cuvette equipped with a platinum electrode and a saturated potassium chloride calomel reference electrode (50). All the potentials reported are referred to the standard hydrogen electrode (SHE). Solution redox potentials were adjusted by microliter aliquots of freshly prepared solutions of sodium dithionite or potassium ferricyanide. Electrode-solution-heme redox mediation was facilitated by redox mediators as follows: 5 μM 1,2-naphthoquinone, 5 μM 1,4-naphthoquinone, 20 μM duroquinone, 10 μM pyocyanine, 5 μM indigotrisulphonate, 10 μM 2-hydroxy-1,4-naphthoquinone, 1 μM indigocarmine, 10 μM anthraquinone-2-sulphonate, and 5 μM benzyl viologen. After equilibration at each potential, the heme optical spectrum was recorded and the course of the reduction of heme was followed by the increase in the sharp α-band absorption at 559 nm relative to a 575 nm reference wavelength.
b) Heme redox titration data analysis
The data were analyzed with the Nernst equation using an n-value of 1.0 in the case of one heme incorporated into the bundle:
| (Eq. 1) |
or two n-values of 1.0 when two hemes were present:
| (Eq. 2) |
where R is the fraction of reduced heme, Eh is the solution redox potential vs. SHE, n is the number of electrons participating in the redox reaction, and Emi is the redox midpoint potential.
c) Heme redox coupling to proton exchange
Proton coupling to heme oxidation/reduction requires that an acid/base group or groups on the cofactor or nearby undergo a charge (electric field) induced shift in pK value depending on the heme oxidation state (34, 39), as described by this thermodynamic square:
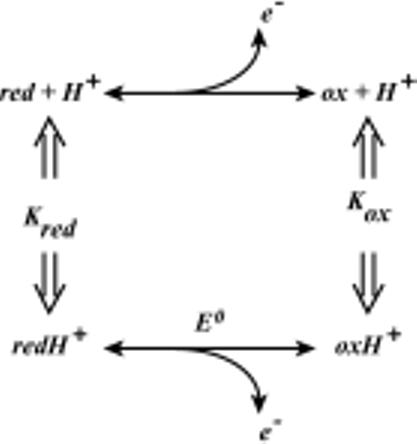
Oxidizing the cofactor increases its positive charge, which thereby reduces the proton affinity of the acid/base group(s). Hence, the pK of an acid/base group is generally lower when the cofactor is in the oxidized state. It follows that the redox midpoint potential Em value of the cofactor will in turn depend on the protonation state of the acid/base group, decreasing as the pH increases. This was evaluated by performing separate redox titrations at different pH values that span the range of the pK shift and analysis as shown in Eq. 3.
The heme oxidation/reduction when coupled to protonation of i independent acid/base groups is described according to
| (Eq. 3) |
where E0 is the redox midpoint potential at low pH, when all acid/base groups are protonated, [H+] is the proton concentration, Kri is the association constant for proton binding to the ith acid/base group in the reduced state of the heme, and K0i is the association constant for proton binding to the ith acid/base group in the oxidized state of the heme.
Heme charge-heme charge coupling
The presence of two ferric hemes in close proximity results in charge-charge repulsion that acts on the redox potentials and the dissociation constants. The difference between the Em1 and Em2 in Eq. 2 represents the charge-charge interaction energy (E = Em1 – Em2). When other factors are not involved (such as steric hindrance), this interaction energy and one Kd value can be used to estimate the unresolved binding constant for the first heme from E = RTln(Kd2/Kd1).
RESULTS
Design
The design and construction of the AP maquette family is intended to provide a platform in which to build function across an interface such as a membrane with non-polar core or a soft interface between polar and non-polar media. This requires a design that will establish structured assembly of stable maquette monolayers or co-assembly of the maquettes with other amphiphiles to form vesicles (Figure 1). Such assembly can be driven by a well-developed exterior amphiphilic pattern of polar and non-polar residues exposed along the axis of the four-α-helix bundle. However, the establishment of such an amphiphilic exterior must not conflict with the selection of other hydrophilic and hydrophobic amino acids that occupy the helix interfaces and interior along the axis of the assembled bundle. In order to ligate metal porphyrins (heme) and chlorin cofactors within the interior of AP bundle in both the externally polar HP and the nonpolar LP domains, we require a reasonably accurate prediction of the location of each residue, especially for the metal ligating histidine residues along the helix. For HP maquettes, such positioning of interior histidines in the hydrophobic interior is now relatively straightforward due to the wealth of information available on water soluble heme-binding maquettes (34, 51-53). However, designing maquettes with cofactor binding site in the hydrophobic domain has not been previously explored.
a) Polar plots
Selection of the primary sequence that assembles into an AP maquette has been aided by the use of amino acid polar plots of the kind shown in Figure 2. The figure emphasizes angular position of each residue on the polar plot which has been determined as θ = 360°/n where n is number of residues per turn. Since the value of n is not known without structural characterization, we have made use of n-values from either related protein structures or from peptide modeling assuming an ideal, straight α-helix with 3.6 residues per turn.
Figure 2.
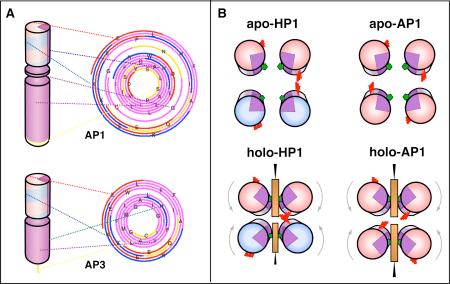
(A) Designing the amphiphilic pattern of polar and non-polar residues along the helical axis. The HP and LP domains of the AP maquettes have each their own amphiphilic exterior/interior patterns and specific interior residues (such as cofactor binding Histidines), which need to be aligned with respect to each other. The alignment was accomplished by plotting the sequence on polar graphs that have easily adjustable angle between the amino acids. The polar graphs were plotted with a 100° angle that is typical of an idealized α-helix. The colors reflect the type of amino acids as described in Figure 1. (B) Positions of heme-ligating histidines (green pentagons) and redox-coupled glutamates (red sticks) with respect to the bundle interior before and after heme heme addition. The figure illustrates the difference between the anti-topology of HP-1 and the syn-topology of AP1.
b) AP maquettes with a heme binding site within the HP domain
These prototype AP maquettes incorporated the heme binding sequence taken from uniquely structured HP1 maquette (Table 1) (34) and tested two different sequences for the LP domain (see AP0 and AP1 in Table 1). The first is based on heptad repeat as previously applied in de novo designed LS2 proton channel (1). The second sequence is based on a natural homo-oligomeric four-helix bundle protein, the transmembrane domain of M2 proton channel of the influenza virus (54). The length of the AP maquettes was selected with the intent to span a phospholipid bilayer which translates to LP domain comprising a polypeptide of more than 20 residues (55). In order to maintain the LP:HP ratio of 3:2 that is commonly found in membrane-forming amphiphiles (56, 57), the HP domain comprised a polypeptide of 14 residues. We have arranged the residues along the helical axis based on the crystal structure of HP apo-maquette (L31M) which revealed 3.58 residues per turn (52) and on the M2 proton channel structure with 3.56 residues per turn (35). Additionally, the AP1 design addressed the difference between the tilt angles in the domains that are being joined. The structure of the transmembrane M2 proton channel revealed −38° tilt angle between straight α-helices (35) whereas the structure of L31M revealed +5° tilt angle (52). To accommodate such a large difference in tilt angles, we have included a linking sequence between the HP and LP domains. The polar plots of amino acid sequences revealed that we need four amino acids long linking sequence to pattern the residues along the helical axis for assembly into four-helix bundle (Figure 2). Therefore the LP and HP domains of AP1 are connected by GGNG, where the glycine residues were selected for their flexibility to accommodate different tilts and the asparagine residue to cap the helix.
c) AP maquettes with a heme binding site within the LP domain
AP2 and AP3 were designed to accommodate cofactors in the hydrophobic domain in preparation for maquette development supporting electron transfer across a membrane or a hydrophobic interface. As no previous designs were available for cofactor binding to LP domain, we have analyzed native, hydrophobic, heme-binding sequences from 11 different cytochromes bc1. The core of all cytochromes bc1 contains a four helix bundle identifiable with bL and bH binding sites that ligate two hemes between four His residues on helices b and d. We have found that the sequences around the heme bL binding site are highly conserved and structurally more complicated because the diameter of the four-helix bundle increases toward the heme bL site to allow interactions with additional helical segments. On the other hand, the sequences around the heme bH binding site are less conserved and form a relatively straight α-helix (see PDB structure 1BE3 (36)). Therefore, we have selected residues 188 to 200 around the heme bH binding site from bovine mitochondrial cytochrome bc1 as our working template for the LP domain of AP2 and AP3 maquettes. The AP2 and AP3 maquettes differ in the HP domain. The HP domain of AP2 maquette has the same length of the HP1 except for the loop region, while the HP domain of AP3 maquette is identical to the HP domain of AP1 maquette with the exception of His residue that has been replaced by Phe (Table 1) to ensure that the desired ligation of cofactors to His residues occurs only in the LP domain. An 8.5° tilt angle of the four-helix bundle present in cytochrome bc1 (58) closely matches the 5° tilt angle of the HP maquette and therefore no flexible linking sequence needs to be integrated between the two domains. The sequence of the HP and LP domains were plotted on the polar graph as in AP1 design (Figure 2) assuming 102° per residue for the LP domain based on the analysis of cytochrome bc1 (58). The polar graph reveals that one additional residue must be inserted between the two domains to keep the hydrophobicity pattern along the helical axis consistent with assembly into a four-helix amphiphilic bundle. Phe was selected because its aromatic character allows this residue to preferentially partition at a polar-nonpolar interface (59, 60).
Co-assembly of AP maquettes with phospholipids vesicles
Analytical ultracentrifugation analysis of AP1 tested maquette incorporation into phospholipids membranes. The density of the suspending buffer was adjusted by D2O so that the centrifuged vesicles with incorporated protein float whereas the non-incorporated protein (monomeric or aggregated) sink. Sedimentation velocity experiments revealed very efficient incorporation of the AP1 maquettes into vesicles (Figure 3). Almost all of the AP1 protein moved rapidly upwards together with the lipids. The area under the absorbance profile of sinking non-incorporated protein contributes less than 3% to the initial absorbance and suggests incorporation efficiency above 97%.
Figure 3.
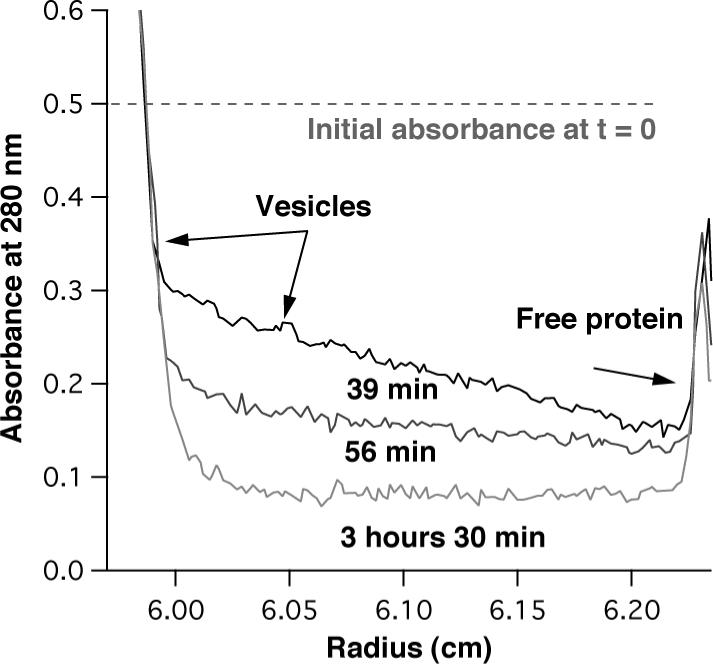
Peptide incorporation into vesicles determined from sedimentation velocity experiments. The vesicles with the protein float because of their lower density (ρ = 1.034 g/ml) with respect to the D2O containing buffer (ρ = 1.059 g/ml), whereas the free protein sediments (r = 1.3 g/ml).. The figure shows three traces from a velocity sedimentation experiment taken at 39, 56, and 210 minutes. Although approximately half of the vesicles floated to the top of the cell before the first spectrum has been taken (39 min), the traces shown clearly demonstrate the trend. The protein was monitored by the absorption of tryptophan at 280 nm, adjusted to an initial value of 0.5 which is depicted by the dashed line. Comparison of the area under the dashed line and the area under the protein peak (right side) provides an estimate of the efficiency of protein incorporation; in this typical example it was higher than 97%.
AP maquette monolayers at air-water interfaces
Molecular orientation of AP monolayers at a polar-nonpolar interface was examined with a Langmuir film balance. A film of AP1 spread at the air-water interface was compressed as shown by the π-A isotherm (24 °C) in Figure 4A. At low compression values and high molecular areas (850 − 650 Å2/molecule) the maquette behaves at the air-water interface as an ideal 2D gas (61) and, as demonstrated in Figure 4B, we can apply the 2D version of the gas law: π (A-A0) = kT/n, where π is surface pressure, A is the area per α-helix, A0 is the limiting area per α-helix, k is the Boltzmann constant, T is absolute temperature and n is the aggregation number. Plotting kT/π vs A gives a slope that corresponds to the aggregation number n. The n value of 1.04 ± 0.03 and the A0 of 552 ± 25 Å2 indicate that in the very low surface pressure regime, the individual polypeptide monomers do not assemble into a four-helix bundle. Figure 4C demonstrates how the compressibility of the film changes as the surface pressure is increased. The film is very compressible at low surface pressures up to ∼ 17 mN/m. The compressibility starts to decrease at about 360 Å2/helix (surface pressure of 18 mN/m) and continues to decrease until the monolayer reaches a pressure of 30 mN/m. The film becomes again very compressible at 40 mN/m, but this is most likely due to the monolayer collapse into the subphase.
Figure 4.
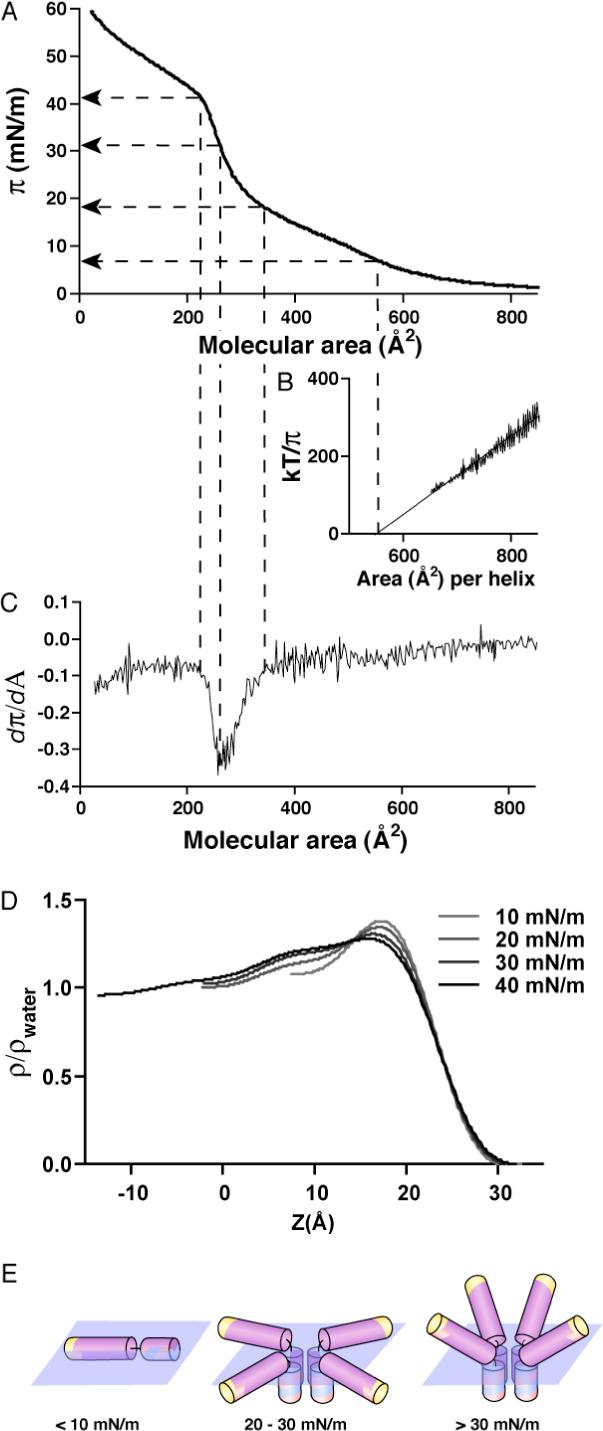
Orientation of AP1 at the air-water interface. (A) Pressure (π) - area (A) isotherm of apo-AP1 at 24 °C with water as a subphase. (B) kT/π versus A. The linear relationship indicates that the AP1 behaves as an ideal 2D gas at very low “2D concentration” (high molecular areas). The fit of the experimental points with linear equation kT/π = n(A - A0) yields n = 1.04 ± 0.03 and A0 = 552 ± 25 Å2. (B) Derivative of the π - A isotherm shown in (A) provide information on the changes in compressibility of the film. (C) Electron density profiles of the apo-AP1 monolayer at four different pressures (see methods and references). (D) Model of AP1 behavior based on the isotherm and the electron density profiles.
Electron density profiles for the Langmuir monolayers of AP1 were obtained from the Fresnel-normalized X-ray reflectivity and analyzed via the box-refinement method (Figure 4D) (49). The interpretation of these profiles is analogous to that described in our earlier studies of the alkylated di-helical peptide BBC16 (32) and the amphiphilic 4-helix bundle peptide AP0 (37). At the lowest pressure investigated of 10 mN/m, the single maximum in the electron density profile at the water/air interface arises from the profile of single α-helices aligned with their long axis lying in the plane of the interface. At somewhat higher pressures of 20−30 mN/m, the amplitude of this maximum decreases and a 20 Å long shoulder appears extending into the sub-phase, which increases in amplitude as the pressure increases. At the same time the compressibility of the film decreases (Figure 4C). The compressibility decrease together with the length of the shoulder, which corresponds to that of a 13−14 residue α-helix, suggest the formation of assembled HP domains into four-helix bundles with the helices perpendicular to the interface and non-assembled LP helices laying parallel to the interface. Upon further compression of the monolayer to 40 mN/m, an additional feature appears in the profile extending its length by ∼15 Å to a total length as large as ∼40 Å. This suggests that the LP helices start to orient with their long axis more perpendicular to the interface, as also consistent with the broadening of the peptide-air interface. The proposed pressure-dependent conformations of the AP1 maquettes with respect to the interface are illustrated in Figure 4E.
Characterization of AP maquettes in detergent micelles
The insolubility of AP maquettes in aqueous solution can be overcome by stabilizing the non-polar domain with detergent (see the Methods Section). Proteins solubilized in detergent micelles provide a broad selection of characterizations of polypeptide α-helical stability and oligomerization and cofactor binding.
a) Assembly of AP1 in detergent
The molecular weight of AP1 in C8E5 and OG was assessed by analytical ultracentrifugation and the aggregation number of AP1 was calculated as n = (MW – m*652)/4733, where m is the number of hemes added per tetramer (Table 2). The apo-AP1 assembles as a four-helix bundle in both OG and C8E5 detergents (n = 4.0 ± 0.1 and n = 3.6 ± 0.2, respectively). Heme1-AP1 and heme2-AP1 were analyzed in C8E5 at two different wavelengths: 280 nm for protein and 410 nm for heme. In the case of heme1-AP1, the sedimentation profiles for both protein and heme are again consistent with four-helix bundle (n= 3.9 ± 0.1 at 280 nm and 4.0 ± 0.0 at 410 nm). However, addition of second heme per four-helix bundle resulted in sedimentation profiles that deviated from ideal four-helix bundles (n= 3.4 ± 0.1 at 280 nm and 3.2 ± 0.1 at 410 nm). Moreover, the 410 nm sedimentation profile indicates that most of the second heme remained in its own micelle under these experimental conditions.
Table 2.
Aggregation number, n, of α-helical bundle and cofactor-bundle complex determined by analytical ultracentrifugation. The average number of detergent molecules per protein complex were obtained as previously described (42). The empty space indicates that aggregation number has not been measured.
| n (aggregation number) in C8E5 | n in OG | ||
|---|---|---|---|
| Wavelength | 280 nm | 410 nm | 280 nm |
| Apo-AP1 | 3.6 ± 0.2 | N/A | 4.0 ± 0.1 (ndet =52±2) |
| Heme1-AP1 | 3.9 ± 0.1 | 4.0 ± 0.0 | |
| Heme2-AP1 | 3.4 ± 0.1 | 3.2 ± 0.1 | |
b) Heme binding in the HP domain of AP1
The 14 amino acid length of HP domain of AP1, and AP3 is significantly shorter than the previously studied water-soluble HP maquettes which have ∼30 amino acid long helices. However, we have confirmed that these 14 residues are sufficient for tight heme binding in all studied constructs. Figure 5A shows heme binding to AP1 maquette. The absorbance of heme at 412 nm increases linearly with heme additions until one heme per bundle is bound. That means that the first dissociation constant (Kd1) is below the approximately 5 nM resolution level of our experimental assay. The linear relationship of the first 7 points was used to determine the extinction coefficient of the bound heme (εbound = 117,420 M−1cm−1) to reduce the number of unknowns in the fitting equation. The binding data were then fitted based on equilibrium binding to two independent binding sites. The least square fit yielded Kd2 = 218 ± 3 nM and εfree = 32897 ± 513 M−1cm−1. As will be presented later, the Kd2 value was combined with a measured redox charge interaction between the two hemes to provide an estimate of the Kd1 value for the first heme bound of 0.5 nM.
Figure 5.

Characterization of detergent solubilized AP1. (A) Heme binding: Binding titration of ferric hemin to AP1 bundle (0.476 μM) in 0.15% C8E5, 100 mM KCl, and 10 mM phosphate buffer, pH = 8.0. The dissociation constant was determined from the difference in absorbance at 412 nm for bound and free heme. The linear relationship of the first 7 points was used to determine the extinction coefficient of bound heme (εbound = 117,420 M−1cm−1). The line represents the least square fit for heme binding to two independent binding sites which yielded Kd2 = 218 ± 3 nM and εfree = 32,897 ± 513 M−1cm−1. (B) Electron paramagnetic resonance characterization of the heme-AP1 maquette solubilized in buffer containing 0.25% C8E5. The resonances gz,y,x of 2.94; 2.26; and 1.52 are characteristic of low spin ferric heme iron (III). The resonance at g ∼ 6.03 is typical of high spin ferric iron and the resonance at g value of 4.28 corresponds to non-heme iron. (C) Circular dichroism (CD) spectra at different temperatures for AP1 maquette in 0.9% OG without heme (left) and with one heme per four-helix bundle (right). The insets show the mean molar ellipticity at 222 nm, which results from the α-helical character of the maquette and it is plotted as a function of temperature.
c) Electron paramagnetic resonance (EPR) of AP1 heme
Figure 5B shows EPR spectrum of AP1 with 1 ferric heme bound per bundle. The resonances at g-values of 2.94; 2.26; and 1.52 are typical of a paramagnetic low-spin iron (III) and are almost identical to the resonances of the bis-His ligated iron in the hydrophilic maquettes (53) suggesting that the heme in AP1 is bis-His ligated in a similar way as in HP maquettes. The small increase in EPR signal at 6.03 corresponds to high spin heme and most likely results from small amount of detergent-solubilized heme present in the sample.
d) α-Helical stability of AP1 without and with heme
α-helical stability of both apo- and holo- AP1 measured by circular dichroism (CD) spectroscopy confirmed high α-helical character of the proteins (Figure 5C). The mean α-helical content, fH, is linearly dependent on the mean molar ellipticity at 222 nm, θ222: fH = (θ222 - θC)/(θH - θC), where θH and θC are the baseline ellipticities for the helix and random coil respectively (62). Using the empirical values of Luo and Baldwin (θH = 2220 − 53T and θC = (−44000 + 250T)(1−3/Nr), where T is temperature in °C and Nr number of amino acid residues) (63) yields 100% α-helical content at 25 °C for both apo- and holo- AP1. Increasing temperature to 90 °C linearly reduces the mean molar ellipticity at 222 nm (insets in Figure 5C) as expected for α-helical structures (63). The increase of the θ222 with temperature results mostly from the temperature dependence of baseline ellipticities (63) and minimal if any loss of α-helical content. High stability of an α-helical structure of the LP domain is expected considering the high energetic penalty for disrupting hydrogen bonds of α-helices in hydrophobic environment. The high α-helical stability of the HP domain can be explained by stabilization of the hydrophilic domain by the hydrophobic domain, which can occur because hydrogen bonding in α-helical secondary structure encourages further hydrogen bonding and continuation of the α-helical secondary structure. Moreover, many of the previously studied HP maquettes were also found to be extremely stable with kcal/mol (53).
Redox potentials of heme in HP1 (aqueous) and in the HP domain of AP1 in detergent micelles and lipid vesicles
a) Redox titrations of single heme
Figure 6 shows redox titrations of single Fe-protoporphyrin IX in two maquettes, HP1 and AP1, with identical sequence in the vicinity of the heme binding regions and, as revealed by visible and EPR spectra, with similar bis-histidine coordination. The titrations reveal a difference in the redox potentials that originates from the surrounding environment: the heme incorporated in water-soluble HP1 maquette (that has never been exposed to lipids nor detergents) titrates with a homogenous n-value of 1.0 and Em value of −202 mV; the heme incorporated into AP1 that has been solubilized by detergent (C8E5) titrates with >90% homogeneity (n-value of 1.0) and an Em8 value shifted substantially more positive (−90 mV) than in the HP1; and the heme incorporated into AP1 that has been inserted into bilayer membrane vesicles displays a homogenous n-value of 1.0 and a smaller positive shift in Em8 value to −175 mV. Thus heme binding to the AP1 maquettes appears acceptably homogeneous in both detergent micelles and lipid bilayers, and in comparison to their HP counterparts the redox midpoint potentials appear elevated.
Figure 6.

Redox potentials of heme bound to the four-α-helix maquettes in three different protein environments: (1) aqueous; heme2-HP1 (squares); (2) membrane; heme in the HP domain of heme-AP1 incorporated into vesicles (triangles); and (3) detergent; heme in the HP domain of heme-AP1 solubilized in 0.25% C8E5 (circles). All redox titrations were performed in 100 mM KCl, and 20 mM phosphate buffer, pH = 8.0.
b) Heme redox state-coupling to glutamate proton exchange
However, before going further in analyzing the Em8 value shifts it is necessary to address the strong proton coupling of the hemes to glutamates at positions (s +1+7t) where s is the position of heme ligating histidine (located in the bundle interior) and t = 0, 1 and 2 (2 only for longer HP sequences). The approximate heptad repeat of an α-helix locates all these glutamates at the interface between the bundle interior and exterior. Heme redox state-glutamate coupling is quantified in the Em/pH relationships described in the Methods section and shown in Figure 7. The dependency of Em on pH has been demonstrated for several HP maquettes, including 2 heme HP1. In the pH 4−7 range, these all show that one proton is exchanged by glutamate(s) per one-electron ferrous/ferric heme transition (∼ −0.06V/pH unit) and becomes pH independent under alkaline conditions. The fit to Eq. 3 with i=1 yields pKs of glutamate: 6.8 with heme ferrous and ∼ 4.2 with heme ferric. In contrast, AP1 in aqueous detergent micelles appears pH independent over the test pH range of 6 to 8. Apparently, detergent causes a dramatic decoupling of the heme redox transition from charge-compensating effects of the glutamate and proton exchange, presumably by interacting with these interface exposed glutamates.
Figure 7.

Proton exchange coupling to heme oxidation/reduction. The pH dependence of midpoint potentials has been demonstrated in natural proteins including bL heme from cytochrome bc1 (Rhodobacter sphaeroides) (open diamonds) and reproduced in water soluble HP maquettes, such as H10A24 maquette in solution (closed diamonds) (39), and uniquely structured HP1 maquette (closed triangles). The pH dependence of midpoint potentials is diminished when the protein is solubilized by detergent (red squares) but is restored upon incorporation of AP1 maquettes into lipid membranes (purple circles).
However, when AP1 is inserted into membrane vesicles dispersed in an aqueous buffer, the pH dependence of the Em value of the one heme AP1 over the test pH range is a full −0.06V/pH unit (Figure 7). When AP1 is assembled in bilayers, the internal arrangement of heme ligation and interaction with glutamate appears intact within its HP-domain. Indeed, it seems that bringing the heme binding domain close to the membrane phospholipids interface raises the ferrous heme pK of the glutamate above 8.
c) Heme-heme charge interactions
In the presence of two hemes per four-helix bundle, the heme-heme distances, and thereby heme-heme charge interactions, are dependent on the topology of the maquettes (22). The HP1 maquette represents a typical anti-topology (Figure 1) in which the two hemes bind in identical environments and are separated some 26 Å (Fe to Fe). Figure 8A shows a redox titration of HP1 with a single heme. The incorporation of a second heme lowers the Em value but both hemes titrate identically and display no splitting in the redox titration indicative of significant electric field acting over their distance (Figure 8B).
Figure 8.
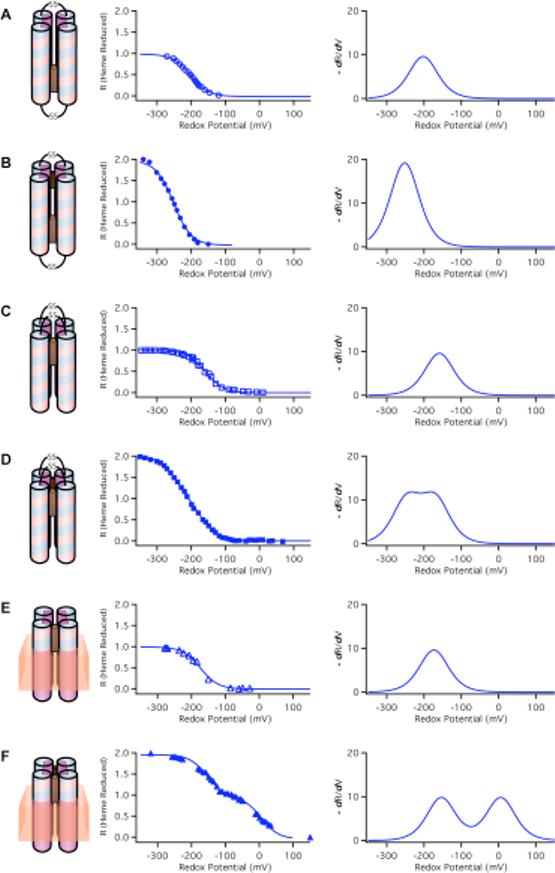
Comparison of redox properties of water soluble HP maquettes in anti-topology (A, B) and syn-topology (C, D) and amphiphilic AP maquettes incorporated into vesicles (E, F) with one (A, C, E) and two (B, D, F) hemes bound. On the left, the data are fitted by Nernst equation curve with n = 1.0 and on the right the same data are replotted as a derivative. (A) heme1-HP1, pH=8.0, −202 mV; (B) heme2-HP1, pH=8.0, Em=−252 mV (34); (C) heme1-H10A24, pH=8.5, Em=−159 mV (22); (D) heme2-H10A24, pH=8.5, Em1=−246 mV and Em2=−170 mV (22); (E) heme1-AP1 in vesicles, pH=8.0, Em=−173 mV; (F) heme2-AP1 in vesicles pH = 8.0, Em1=−154 mV and Em2=6 mV. The derivative of the fraction reduced vs redox potential curves clearly demonstrate the difference between single and split potentials as well as the large difference in the potential split between HP (D) and AP (F) family maquettes.
Maquettes with syn-topology ligate two hemes in close proximity, leading to an electrostatic interaction that adds to the heme-glutamate interaction (22). A typical example of syn-topology maquette is a prototype-H10A24 maquette that binds two hemes adjacent (Fe to Fe estimate 10−12 Å). The single Em value (Figure 8C) is split by 76 mV when two hemes are added (Figure 8D) (38). The split can be better discerned in the derivative plot of the Nernst curve.
Figures 8E and 8F compare the redox titrations of the syn-topology AP1 incorporated into membrane bilayer vesicles with one and two hemes added. The dramatic potential splitting is consistent with pure electric field effect: the heme with the more positive Em8 value (it could be either one) is titrated while under the influence of the field from the other, charged ferric heme. The heme with a lower Em8 is titrated in the presence of an uncharged ferrous heme and displays the value similar to that of a single heme in AP1. The splitting of 160 mV in the Em8 values of the otherwise similar hemes represents a significant increase in field strength and charge-charge interaction energy over that typically seen in water-soluble HP maquettes. This enhanced electric field effect is likely to be result of the exclusion of the polar aqueous phase by nearby phospholipids and tighter association of the tetrameric bundle.
Effect of heme-heme charge interactions on ferric heme binding
The 160 mV split in the Em8 values, corresponds to a 3.6 kcal/mol interaction energy that readily explains the widely differing binding strengths of the first and second ferric hemes bound to AP1 shown in Figure 5A. The second heme, bound against the repulsive force of the already present first heme, displayed a weakened dissociation constant (Kd2) of 218 ± 3 nM. The first heme dissociation constant (Kd1) was too tight to measure using the UV-VIS assay method for binding.
The interaction energy (E) and one Kd value can be used to estimate the unresolved binding constant for the first heme from E = RTln(Kd2/Kd1) which yields Kd1 = 0.5 nM. This charge-charge repulsion effect that acts on the dissociation constants of the first and second heme bound to AP1 provides compelling evidence that the two hemes in AP1 are ligated as designed in an overall syn-topology in very close proximity. This in turn implies that the minimal assembly unit of two-heme AP1 inside the membrane bilayer of the vesicles is a four-helix bundle.
Comparisons of heme and bacteriochlorophyll binding to HP1 and to the HP-domains and the LP-domains of AP maquettes
With the proviso that there is a higher repulsion between adjacent charged cofactors in the AP maquettes even in the HP-domains, we have found that the binding of cofactors to the maquettes with binding site within the HP domain of AP1 is qualitatively comparable to binding of cofactors to fully water soluble HP maquettes as indicated in Table 3. Both heme b and one heme a (with a farnesyl tail) bind tightly to the HP domain of AP1 as well as to all tested HP maquettes. By contrast, chlorophylls (Chls) and bacteriochlorophylls (BChls), and their metal substituted analogs (M-BChls) bind neither to HP maquettes nor to the HP domain of AP1. The high hydrophobicity of these cofactors results in their self-aggregation and precipitation in aqueous and polar environments. However, detergent micelles and the LP domains of AP3 and AP2 provide hydrophobic environments that significantly enhance BChl solubility and affinity. This new environment opens new possibilities for binding novel hydrophobic Chls and BChls with a variety of transition metal centers that will confer functional versatility to AP proteins.
Table 3.
Cofactor binding to HP1 and AP maquettes.*
|
Peptide |
Binding site in HP |
Binding site in LP |
|||
|---|---|---|---|---|---|
|
Cofactor |
HP1 |
AP0 |
AP1 |
AP2 |
AP3 |
| Heme b |
< 20 nM | < 50 nM* | 0.5 nM | < 50 nM | 430 ± 50 nM |
| < 20 nM |
< 50 nM* |
218 ± 3 nM |
< 50 nM |
- |
|
| Heme a |
++ | ++ | |||
| |
|
- |
|
- |
|
| Ni-BChl |
- | + | + | ||
| - |
|
|
- |
- |
|
| Zn-BChl | - | + | + | ||
| - | + | ||||
Accurate binding constants have been determined only for heme b. The binding of other cofactors has been evaluated qualitatively: “++” represents binding in the nM range; “+” in μM range; “-” is for above μM range or no binding; and empty space indicates that binding titration has not been performed.
Re-evaluation of binding titrations in reference (37) Ye, S. X., Strzalka, J. W., Discher, B. M., Noy, D., Zheng, S. Y., Dutton, P. L., and Blasie, J. K. (2004) Amphiphilic 4-helix bundles designed for biomolecular materials applications. Langmuir 20, 5897−5904.
Two examples of such novel complexes are shown in Figure 9 whereby AP3 incorporates either one 132-OH-Ni-BChl (Ni-BChl) or two 132-OH-Zn-BChls (Zn-BChls). The former is an extremely hydrophobic BChl derivative which requires detergent concentrations well above CMC (2% w:v Zwittergent 3−12) for solubility. Ni-BChl is especially useful as a reporter for the local coordination and solvation environment because its rich absorption spectra is well correlated with the type and number of axial ligands (64, 65). The absorption spectrum of unbound Ni-BChl (Figure 9, top) changes dramatically upon binding to AP3 and differ significantly from the respective spectra of imidazole ligated Ni-BChl. Particularly, the Qx absorption band of Ni-BChl shifts from 538 for unbound Ni-BChl to 584 to 609 nm upon imidazole ligation and AP3 binding, respectively. These bands are typical of non-, mono-, and bi-axially ligated Ni-BChl thus indicating that Ni-BChl is bis-His ligated in AP3. Ni-BChl binding affinity is very low compared to heme (Kd ∼ 200 mM) but is still about three orders of magnitude higher than imidazole binding affinity. The characterization of AP3-Ni-BChl is described in more detail in a subsequent manuscript (66).
Figure 9.
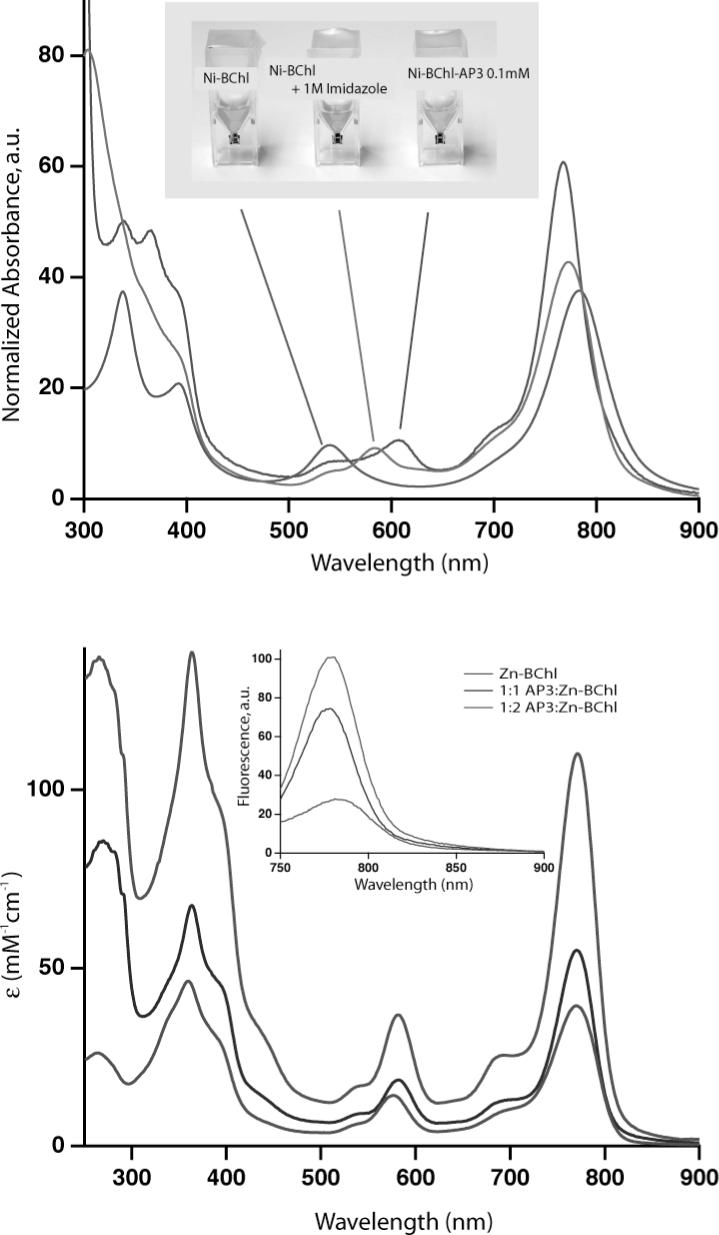
(A) Absorption spectra of Ni-BChl (purple), Ni-BChl with 1 M imidazole (green), and Ni-BChl-AP3 (blue). The Ni-BChl spectra are highly sensitive to Ni coordination chemistry and demonstrate that binding of Ni-BChl to AP3 changes the coordination state of Ni from 4- to mostly 6-coordinated, presumably due to bis-His ligation. (B) Molar absorption and fluorescence (inset) spectra of Zn-BChl (red), 1:1 Zn-BChl:AP3 (blue) and 2:1 Zn-BChl:AP3 (green) in 0.9% OG, pH 8.0. Excitation wavelength for the fluorescence spectra was 360 nm.
The importance of the second complex, AP3-Zn-BChl, lies in its photophysical properties. Unlike heme and Ni-BChl, both unbound Zn-BChl and its AP3 complex are strongly fluorescent. Typically, the excited state lifetimes of Zn-BChl in organic solvents are 2−3 nanoseconds (67) which is conveniently long to efficiently initiate photoinduced electron transfer reactions. The absorption spectrum of Zn-BChl (Figure 9, bottom) changes only slightly upon binding to AP3 yet its fluorescence emission spectrum changes significantly upon binding one or two pigments to AP3 (Figure 9, bottom inset). Particularly, the lowest energy fluorescence emission band intensifies and blue shits from 780 to 776 nm upon binding the first Zn-BChl, but the intensity is not doubled upon binding the second pigment. This self-quenching suggests mutual electronic interactions between the two bound pigments. Zn-BChl is readily soluble in OG at CMC (0.9% w:v) and can bind to AP3 under the same conditions used for heme binding. Nevertheless, AP3 affinity to Zn-BChl, though much higher than to Ni-BChl, is significantly lower than to heme (Kd ∼ 0.3 and 1 μM for the first and second Zn-BChl, respectively).
DISCUSSION
The work presented here started with four goals: (1) to design cofactor binding AP maquettes; (2) to co-assemble AP maquettes and cofactors with detergents and lipid bilayers; (3) to structurally characterize the maquettes at an air-water interface; and (4) to test whether the functional elements established in HP maquettes are preserved in the new, more hydrophobic, environment of AP maquettes. En route, we have examined the effect of detergent on structural and functional properties of the AP1 maquette and taken advantage of the ability to bind a larger variety of cofactors, increasing the range of redox potentials for future applications.
(1) Design strategies
The first challenge of AP maquettes not present in HP or LP maquettes was to propose a design that combines domains with different polarity patterns within one peptide in a way that the amino acids of the same polarity do not cross-react outside their domain and lead to a range of assembly/aggregation states. As no specific design principles were available for the AP maquettes, we approached this problem empirically by selecting different lengths of the HP and LP domains (AP1 and AP2) and by using partial sequences of natural membrane proteins that are mostly hydrophobic without clear hydrophilic patterns (AP1, AP2, and AP3). The second design challenge arises from the need for longer helices (> 35 amino acids) to both span a membrane and extend into aqueous phase. Longer helices are more difficult to design because the angular position of the amino acids with respect to the bundle interior/exterior becomes increasingly sensitive to number of residues per turn. This is illustrated by the structure of L31M maquette (52). Although the design of 30 amino acid long L31M has been based on a heptad repeat with 3.5 residues per turn (68), the maquette assembles with 3.58 residues per turn as revealed by the crystal structure (52). As a consequence, Leu 23, intended in the design to be located inside the bundle, is forced outside, in an energetically unfavorable aqueous environment. Since no specific design principles are available to build long helices with predetermined numbers of residues per turn, our designs relied on numbers previously determined for related proteins. While the successful assembly of the AP maquettes presented here proves the feasibility of AP designs, we are aware that much more systematic work will have to follow to establish general design principles for construction of AP maquettes.
(2) Heme properties as a structural tool
Although we have selected heme for its manifold functional activities in natural enzymes, heme proves to be also excellent probe for both the structural topology and quantitative assessment of the functionally important dielectric properties of the bundle interior. An interaction energy of 70−100 mV has been observed for adjacent hemes (Fe-Fe distance ∼12Å) in HP maquettes with syn-topology (see scheme in Figure 8D) whereas the charge interaction is undetectable in anti-topology (see scheme in Figure 8B) where hemes are distant (Fe-Fe distance ∼26Å) (34). The repulsive force between the two cationic ferric hemes is expressed in the difference in the dissociation constants (Kd) of the first and second ferric heme bound in the syn- but not the anti-topology (38). This repulsion has been demonstrated more dramatically in the reversible quantitative shift between syn- and anti-topologies in an HP maquette designed for the purpose (22).
The transition from HP to AP maquettes starts with HP1, a maquette with anti-topology that binds two distant, non-interacting hemes. Construction of AP1 used about a half of HP1 (including one heme ligation site) as the HP domain and the M2 channel of the influenza virus as the LP domain. This was expected to flip the relative orientation of the HP dihelices and impose a syn-topology to the a-helical bundle. The syn- assembly was verified both in detergent where two different binding constants were observed (Figure 5A) and in a membrane bilayers where two adjacent hemes revealed enhanced charge interaction (Figure 8F). Similarly, the AP3 maquette bound only one ferric heme, implying that the repulsive force between two ferric hemes increases the second Kd above our experimental concentrations as expected for syn-topology. In sharp contrast, the AP2 maquette in detergent bound two hemes with indistinguishable binding constants and redox mid-point potentials (see Table 3 and (69)), suggesting that the force driving the 28 residue HP domain into anti-topology can overcome the hydrophobic tetrameric association of the 14 residue LP sequence. The result is likely to be a complex topology with an anti- HP bundle in the middle, with detergent associated dimeric LP domains at either end.
Heme properties also report on the oligomerization state of the peptide. While oligomerization state of the HP maquettes is relatively easily assessed by size exclusion chromatography or analytical ultracentrifugation, the molecular weight of AP maquettes cannot be determined directly because the AP maquettes assemble only in the presence of detergents or membrane forming amphiphiles, such as lipids or amphiphilic copolymers. Although this obstacle has been overcome by density matching when dealing with detergents, very limited tools are available to obtain the oligomerization state in membranes (70). Therefore the heme spectral and redox properties provide invaluable information as they can report the minimal oligomerization number of the maquettes incorporated in membranes: bis-His ligation indicates that the peptide assembles at least as a dimer and charge-charge interaction implies at least a tetrameric assembly.
(3) Assembly at the air-water interface
Assembly of the maquettes at the air-water interface can provide means of structural characterization that is otherwise inaccessible. The AP1 maquettes are stable at the air-water interface, initially with density profiles that corresponds to maquettes lying parallel to the surface and in monomeric form. This implies that the tetrameric assembly that has been driven by hydrophobic sequestration is not strong enough to withstand the availability of both polar and non-polar media at the air-water interface. Upon compression from 10 to 30 mN/m, the length of the density profiles increases from ∼ 10 to ∼ 20 Å, suggesting that only HP domains of AP1 maquettes start to orient with respect to the surface, whereas the LP helices remain parallel to the plane of the interface. The final profile length at 40 mN/m (∼40 Å) does not match the total length of the AP1 peptide (∼ 66 Å), which means that the maquettes do not fully reach perpendicular orientation with respect to the surface. This behavior of AP1 is in contrast to that of AP0, which has the same total length as AP1, but contains no flexible linker between its HP and LP domains. Both domains of AP0 achieve an orientation with the long axis of the bundle perpendicular to the interface at 40mN/m. The failure of AP1 maquette to achieve completely perpendicular orientation is not surprising because the AP1 maquette has not been optimized for the assembly at the air-water interface. In addition to the flexible linker connecting the HP and LP domains, it also contains three polar residues at the end of the LP domain that extends to the air. These three residues originated from the sequence of the M2 proton channel of influenza virus and they have been kept to promote peptide assembly across a supporting lipid bilayer.
(4) Heme properties as a functional tool
The topologically anti- HP1, with two distant hemes and no heme-heme charge coupling, exhibits significant oxidation-reduction coupled shifts in the pK values as the glutamate is rotated into the interior as shown in Figure 2B (34). The pK of < 4.3 when heme is oxidized and formally positive moves to 6.8 when heme is reduced and formally uncharged. These pK shifts are comparable to bioenergetically functional pK shifts in natural proteins. Construction and assembly of AP1 in a membrane bilayer forces a syn-topology and the adjacent binding of two hemes together with the symmetric rotation of the glutamates into the maquette interior (Figure 2B). AP1 substantially elevates the pK on the glutamate when the heme is ferrous and formally uncharged from 6.8 to 8 or above. This reveals a stabilization of the neutral glutamic acid form of over 4 units (5.5 kcal/mole). Such extreme pK shift suggests that heme binds to membrane associated AP1 with sufficient affinity to drive rotation of glutamates into a bundle interior where they become more protected from water and/or the cofactor-bundle assembly is more stabilized by the hydrophobic LP domain structure and the supporting membrane. This pK is extremely redox sensitive, dropping below 5 when heme is oxidized, a substantial energetic change that spans much of the physiological pH range. Despite the fact that each of the ferric heme cations is stabilized by charge compensations that are mediated by the proton exchange on the glutamates, the remaining, destabilizing heme-heme charge interaction is 160mV (3.6 kcal/mole), much greater than the 70−100mV range displayed by syn-topology diheme HP maquettes. Thus, the diheme AP1 assembly in membranes is structurally strong enough to withstand the forces of these strong charge-charge interactions without loss of heme or flipping to an anti-topology.
Detergent micelles offer a ready analytical stage for the AP maquettes that includes some measurements that are less readily done or cannot be done when associated in membrane vesicles. But other measurements of the AP1-detergent micelles in dispersion suggest caution when considering detergents as a supporting medium alternative to AP maquettes in membranes. AP1 detergent micelles are assemblies of four helical units, bind two hemes with typical low spin heme character and display all the properties established with their HP analogues. Moreover, the finding that the cationic ferric hemes display different Kd values (<50nM and 218nM) is indicative of significant heme-heme charge interactions and a clear syn-topology as expected from the design. However, detergent appears to uncouple the heme oxidation-reduction from proton coupling, as shown by the rise of Em value with the loss of the ferric heme stabilization by the glutamate to assume a value independent of pH over the pH 6−8 test range. This is most likely a consequence of disruption of non-specific or environmental effects around the heme in the bundle when dispersed in detergent micelles, rather than an intrinsic and distinct difference between AP1 and the HP maquettes, because the coupling is fully restored upon placement of the maquette into lipid membrane (Figure 7).
The exploratory investigations done on the binding of hemes A and B and bacteriochlorophylls in AP3 has been mainly done in detergent micelle dispersions. The work suggests considerable promise, particularly in successfully binding the extremely hydrophobic and strongly self-associating heme A and bacteriochlorophylls. However, the intrinsically weak binding suggests the need for more work, including the development of new tools to assess the nature and strength of the interaction (66). Likewise the capacity to bind only one ferric heme in the LP domain of AP3 may signal the requirement for extensive measures to charge compensate for the ferric heme charge in a low dielectric medium.
It seems clear that the AP1 structure in the membrane supports redox and acid-base actions in its HP domain that are qualitatively very similar to, and that quantitatively match the energetic magnitude of those familiar in natural membrane proteins. Thus, AP1 offers a robust framework for designs directed to specific electron and proton transfer functions of complex substrate oxidation-reduction catalysis. Provision of selected HP and specifically designed LP domains should confer upon AP maquette constructions the ability to couple to HP redox activities with LP vectorial transmembrane action for the construction of light-activated and redox-driven transmembrane charge separation.
Abbreviations
- AP
amphiphilic
- BChl
bacteriochlorophyll
- C8E5
n-Octylpentaoxyethylene
- CD
circular dichroism
- CMC
critical micellar concentration
- OG
n-octyl β-D-glucopyranoside
- heme
Fe-protoporphyrin IX
- HP
hydrophilic
- LP
lipophilic
- POPC
1-palmitoyl-2-oleoyl-phosphatidylcholine
- POPS
1-palmitoyl-2-oleoyl-phosphatidylserine
- TASP
template-assembled synthetic proteins
Footnotes
This work has been supported by NIH grants GM48130, GM63388, and NSF grant DMR00−79909.
REFERENCES
- 1.Lear JD, Wasserman ZR, DeGrado WF. Synthetic Amphiphilic Peptide Models for Protein Ion Channels. Science. 1988;240:1177–1181. doi: 10.1126/science.2453923. [DOI] [PubMed] [Google Scholar]
- 2.Tomich JM, Grove A, Iwamoto T, Marrer S, Montal MS, Montal M. Biomembrane Electrochemistry. 1994. Design Principles and Chemical Synthesis of Oigomeric Channel Proteins; pp. 329–354. [Google Scholar]
- 3.Montal M. Molecular Mimicry in Channel-Protein Structure. Current Opinion in Structural Biology. 1995;5:501–506. doi: 10.1016/0959-440x(95)80035-2. [DOI] [PubMed] [Google Scholar]
- 4.Montal M. Design of Molecular Function - Channels of Communication. Annual Review of Biophysics and Biomolecular Structure. 1995;24:31–57. doi: 10.1146/annurev.bb.24.060195.000335. [DOI] [PubMed] [Google Scholar]
- 5.Hill RB, Raleigh DP, Lombardi A, DeGrado NF. De novo design of helical bundles as models for understanding protein folding and function. Accounts of Chemical Research. 2000;33:745–754. doi: 10.1021/ar970004h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cochran AG, Skelton NJ, Starovasnik MA. Tryptophan zippers: Stable, monomeric beta-hairpins. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:5578–5583. doi: 10.1073/pnas.091100898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuhlman B, Dantas G, Ireton GC, Varani G, Stoddard BL, Baker D. Design of a novel globular protein fold with atomic-level accuracy. Science. 2003;302:1364–1368. doi: 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
- 8.Lieberman M, Sasaki T. Iron(II) Organizes a Synthetic Peptide into 3-Helix Bundles. Journal of the American Chemical Society. 1991;113:1470–1471. [Google Scholar]
- 9.Ghadiri MR, Soares C, Choi C. Design of an Artificial 4-Helix Bundle Metalloprotein Via a Novel Ruthenium(II)-Assisted Self-Assembly Process. Journal of the American Chemical Society. 1992;114:4000–4002. [Google Scholar]
- 10.Gibney BR, Mulholland SE, Rabanal F, Dutton PL. Ferredoxin and ferredoxinheme maquettes. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:15041–15046. doi: 10.1073/pnas.93.26.15041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharp RE, Moser CC, Rabanal F, Dutton PL. Design, synthesis, and characterization of a photoactivatable flavocytochrome molecular maquette. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:10465–10470. doi: 10.1073/pnas.95.18.10465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dieckmann GR, McRorie DK, Lear JD, Sharp KA, DeGrado WF, Pecoraro VL. The role of protonation and metal chelation preferences in defining the properties of mercury-binding coiled coils. Journal of Molecular Biology. 1998;280:897–912. doi: 10.1006/jmbi.1998.1891. [DOI] [PubMed] [Google Scholar]
- 13.Marsh ENG, DeGrado WF. Noncovalent self-assembly of a heterotetrameric diiron protein. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:5150–5154. doi: 10.1073/pnas.052023199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Summa CM, Rosenblatt MM, Hong JK, Lear JD, DeGrado WF. Computational de novo design, and characterization of an A(2)B(2) diiron protein. Journal of Molecular Biology. 2002;321:923–938. doi: 10.1016/s0022-2836(02)00589-2. [DOI] [PubMed] [Google Scholar]
- 15.Calhoun JR, Kono H, Lahr S, Wang W, DeGrado WF, Saven JG. Computational design and characterization of a monomeric helical dinuclear metalloprotein. Journal of Molecular Biology. 2003;334:1101–1115. doi: 10.1016/j.jmb.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 16.Magistrato A, DeGrado WF, Laio A, Rothlisberger U, VandeVondele J, Klein ML. Characterization of the dizinc analogue of the synthetic diiron protein DF1 using ab initio and hybrid quantum/classical molecular dynamics simulations. Journal of Physical Chemistry B. 2003;107:4182–4188. [Google Scholar]
- 17.Maglio O, Nastri F, Pavone V, Lombardi A, DeGrado WF. Preorganization of molecular binding sites in designed diiron proteins. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:3772–3777. doi: 10.1073/pnas.0730771100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Williamson DA, Benson DR. Remarkable helix stabilization via edge-to-face tryptophan-porphyrin interactions in a peptide-sandwiched mesoheme. Chemical Communications. 1998:961–962. [Google Scholar]
- 19.Wei YN, Liu T, Sazinsky SL, Moffet DA, Pelczer I, Hecht MH. Stably folded de novo proteins from a designed combinatorial library. Protein Science. 2003;12:92–102. doi: 10.1110/ps.0228003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosenblatt MM, Wang JY, Suslick KS. De novo designed cyclic-peptide heme complexes. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:13140–13145. doi: 10.1073/pnas.2231273100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ghirlanda G, Osyczka A, Liu WX, Antolovich M, Smith KM, Dutton PL, Wand AJ, DeGrado WF. De novo design of a D-2-symmetrical protein that reproduces the diheme four-helix bundle in cytochrome bc1. Journal of the American Chemical Society. 2004;126:8141–8147. doi: 10.1021/ja039935g. [DOI] [PubMed] [Google Scholar]
- 22.Grosset AM, Gibney BR, Rabanal F, Moser CC, Dutton PL. Proof of principle in a de novo designed protein maquette: An allosterically regulated, charge-activated conformational switch in a tetra-alpha-helix bundle. Biochemistry. 2001;40:5474–5487. doi: 10.1021/bi002504f. [DOI] [PubMed] [Google Scholar]
- 23.Shifman JM, Gibney BR, Sharp RE, Dutton PL. Heme redox potential control in de novo designed four-alpha- helix bundle proteins. Biochemistry. 2000;39:14813–14821. doi: 10.1021/bi000927b. [DOI] [PubMed] [Google Scholar]
- 24.Topoglidis E, Discher BM, Moser CC, Dutton PL, Durrant JR. Functionalizing nanocrystalline metal oxide electrodes with robust synthetic redox proteins. ChemBioChem. 2003;4:1332–1339. doi: 10.1002/cbic.200300707. [DOI] [PubMed] [Google Scholar]
- 25.Moffet DA, Certain LK, Smith AJ, Kessel AJ, Beckwith KA, Hecht MH. Peroxidase activity in heme proteins derived from a designed combinatorial library. Journal of the American Chemical Society. 2000;122:7612–7613. [Google Scholar]
- 26.Wei YN, Hecht MH. Enzyme-like proteins from an unselected library of designed amino acid sequences. Protein Engineering Design & Selection. 2004;17:67–75. doi: 10.1093/protein/gzh007. [DOI] [PubMed] [Google Scholar]
- 27.Hecht MH, Das A, Go A, Bradley LH, Wei YN. De novo proteins from designed combinatorial libraries. Protein Science. 2004;13:1711–1723. doi: 10.1110/ps.04690804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaplan J, DeGrado WF. De novo design of catalytic proteins. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:11566–11570. doi: 10.1073/pnas.0404387101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Discher BM, Koder RL, Moser CC, Dutton PL. Hydrophillic to amphiphilic design in redox protein maquettes. Current Opinion in Chemical Biology. 2003;7:741–748. doi: 10.1016/j.cbpa.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 30.Chen XX, Moser CC, Pilloud DL, Dutton PL. Molecular orientation of Langmuir-Blodgett films of designed heme protein and lipoprotein maquettes. Journal of Physical Chemistry B. 1998;102:6425–6432. [Google Scholar]
- 31.Chen XX. Ph.D. thesis, Biochemistry and Biophysics. University of Pennsylvania; Philadelphia: 1999. [Google Scholar]
- 32.Strzalka J, Chen XX, Moser CC, Dutton PL, Ocko BM, Blasie JK. X-ray scattering studies of maquette peptide monolayers. 1. Reflectivity and grazing incidence diffraction at the air/water interface. Langmuir. 2000;16:10404–10418. [Google Scholar]
- 33.Strzalka J, Chen XX, Moser CC, Dutton PL, Bean JC, Blasie JK. X-ray scattering studies of maquette peptide monolayers. 2. Interferometry at the vapor/solid interface. Langmuir. 2001;17:1193–1199. [Google Scholar]
- 34.Huang SS, Koder RL, Lewis M, Wand AJ, Dutton PL. The HP-1 maquette: from an apoprotein to a structured hemoprotein designed to promote redox-coupled proton exchange. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:5536–5541. doi: 10.1073/pnas.0306676101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang JF, Kim S, Kovacs F, Cross TA. Structure of the transmembrane region of the M2 protein H+ channel. Protein Science. 2001;10:2241–2250. doi: 10.1110/ps.17901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Iwata S, Lee JW, Okada K, Lee JK, Iwata M, Rasmussen B, Link TA, Ramaswamy S, Jap BK. Complete structure of the 11-subunit bovine mitochondrial cytochrome bc1 complex. Science. 1998;281:64–71. doi: 10.1126/science.281.5373.64. [DOI] [PubMed] [Google Scholar]
- 37.Ye SX, Strzalka JW, Discher BM, Noy D, Zheng SY, Dutton PL, Blasie JK. Amphiphilic 4-helix bundles designed for biomolecular materials applications. Langmuir. 2004;20:5897–5904. doi: 10.1021/la0363884. [DOI] [PubMed] [Google Scholar]
- 38.Gibney BR, Huang SS, Skalicky JJ, Fuentes EJ, Wand AJ, Dutton PL. Hydrophobic modulation of heme properties in heme protein maquettes. Biochemistry. 2001;40:10550–10561. doi: 10.1021/bi002806h. [DOI] [PubMed] [Google Scholar]
- 39.Shifman JM, Moser CC, Kalsbeck WA, Bocian DF, Dutton PL. Functionalized de novo designed proteins: Mechanism of proton coupling to oxidation/reduction in heme protein maquettes. Biochemistry. 1998;37:16815–16827. doi: 10.1021/bi9816857. [DOI] [PubMed] [Google Scholar]
- 40.Larsen RW. Volume and thermodynamic profiles of CO-binding to Fe(II) protoporphyrin IX in detergent micelles. Inorganica Chimica Acta. 1999;288:74–81. [Google Scholar]
- 41.le Maire M, Champeil P, Moller JV. Interaction of membrane proteins and lipids with solubilizing detergents. Biochimica Et Biophysica Acta-Biomembranes. 2000;1508:86–111. doi: 10.1016/s0304-4157(00)00010-1. [DOI] [PubMed] [Google Scholar]
- 42.Noy D, Calhoun JR, Lear JD. Direct analysis of protein sedimentation equilibrium in detergent solutions without density matching. Analytical Biochemistry. 2003;320:185–192. doi: 10.1016/s0003-2697(03)00347-6. [DOI] [PubMed] [Google Scholar]
- 43.Kharakoz DP. Partial volumes and compressibilities of extended polypeptide chains in aqueous solution: Additivity scheme and implication of protein unfolding at normal and high pressure. Biochemistry. 1997;36:10276–10285. doi: 10.1021/bi961781c. [DOI] [PubMed] [Google Scholar]
- 44.Laue T, Shaw BD, Ridgeway TM, Pelletier SL. Computer-aided interpretation of analytical sedimentation data for proteins. The Royal Society of Chemistry; Cambridge, U.K.: 1992. [Google Scholar]
- 45.Mayer G, Anderka O, Ludwig B, Schubert D. Progressi n Clolloid and Polymer Science. Springer-Verlag Heidelberg; Heidelberg, Germany: 2002. The State of Association of the Cytochrome bc1 Complex from Paracoccus Dentrification in Solutions of dodecyl maltoside; pp. 77–83. [Google Scholar]
- 46.Popovic ZD, Kovacs GJ, Vincett PS, Alegria G, Dutton PL. Electric field dependence of recombination kinetics in reaction centers of photosynthetic bacteria. Chem. Phys. 1986;110:227–237. [Google Scholar]
- 47.Helm CA, Tippmannkrayer P, Mohwald H, Alsnielsen J, Kjaer K. Phases of Phosphatidyl Ethanolamine Monolayers Studied By Synchrotron X-Ray-Scattering. Biophysical Journal. 1991;60:1457–1476. doi: 10.1016/S0006-3495(91)82182-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Braslau A, Pershan PS, Swislow G, Ocko BM, Alsnielsen J. Capillary Waves On the Surface of Simple Liquids Measured By X- Ray Reflectivity. Physical Review A. 1988;38:2457–2470. doi: 10.1103/physreva.38.2457. [DOI] [PubMed] [Google Scholar]
- 49.Blasie JK, Zheng S, Strzalka J. Solution to the phase problem for specular x-ray or neutron reflectivity from thin films on liquid surfaces. Physical Review B. 2003;67 [Google Scholar]
- 50.Dutton PL. Methods in Enzymology, Biomembranes, Physical and Chemical Methods, Chapter 25, Part C, Biological Oxidation. 1978. Redox Potentiometry: Determination of Midpoint Potentials of Oxidation-Reduction Components in Biological Electron Transfer Systems; pp. 411–435. [DOI] [PubMed] [Google Scholar]
- 51.Gibney BR, Dutton PL. Advances in Inorganic Chemistry. Vol. 51. 2001. De novo design and synthesis of heme proteins; pp. 409–455. [Google Scholar]
- 52.Huang SS, Gibney BR, Stayrook SE, Dutton PL, Lewis M. X-ray structure of a Maquette scaffold. Journal of Molecular Biology. 2003;326:1219–1225. doi: 10.1016/s0022-2836(02)01441-9. [DOI] [PubMed] [Google Scholar]
- 53.Gibney BR, Dutton PL. Histidine placement in de novo-designed heme proteins. Protein Science. 1999;8:1888–1898. doi: 10.1110/ps.8.9.1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nishimura K, Kim SG, Zhang L, Cross TA. The closed state of a H+ channel helical bundle combining precise orientational and distance restraints from solid state NMR-1. Biochemistry. 2002;41:13170–13177. doi: 10.1021/bi0262799. [DOI] [PubMed] [Google Scholar]
- 55.Arkin IT, Brunger AT. Statistical analysis of predicted transmembrane alpha-helices. Biochimica Et Biophysica Acta-Protein Structure and Molecular Enzymology. 1998;1429:113–128. doi: 10.1016/s0167-4838(98)00225-8. [DOI] [PubMed] [Google Scholar]
- 56.Discher BM, Hammer DA, Bates FS, Discher DE. Polymer vesicles in various media. Current Opinion in Colloid & Interface Science. 2000;5:125–131. [Google Scholar]
- 57.Discher BM, Won YY, Ege DS, Lee JCM, Bates FS, Discher DE, Hammer DA. Polymersomes: Tough vesicles made from diblock copolymers. Science. 1999;284:1143–1146. doi: 10.1126/science.284.5417.1143. [DOI] [PubMed] [Google Scholar]
- 58.North B, Summa CM, Ghirlanda G, DeGrado WF. D-n-symmetrical tertiary templates for the design of tubular proteins. Journal of Molecular Biology. 2001;311:1081–1090. doi: 10.1006/jmbi.2001.4900. [DOI] [PubMed] [Google Scholar]
- 59.de Planque MRR, Boots JWP, Rijkers DTS, Liskamp RMJ, Greathouse DV, Killian JA. The effects of hydrophobic mismatch between phosphatidylcholine bilayers and transmembrane alpha-helical peptides depend on the nature of interfacially exposed aromatic and charged residues. Biochemistry. 2002;41:8396–8404. doi: 10.1021/bi0257686. [DOI] [PubMed] [Google Scholar]
- 60.Strandberg E, Morein S, Rijkers DTS, Liskamp RMJ, van der Wel PCA, Killian JA. Lipid dependence of membrane anchoring properties and snorkeling behavior of aromatic and charged residues in transmembrane peptides. Biochemistry. 2002;41:7190–7198. doi: 10.1021/bi012047i. [DOI] [PubMed] [Google Scholar]
- 61.Chen XX, Moser CC, Pilloud DL, Gibney BR, Dutton PL. Engineering oriented heme protein maquette monolayers through surface residue charge distribution patterns. Journal of Physical Chemistry B. 1999;103:9029–9037. [Google Scholar]
- 62.Rohl CA, Baldwin RL. Comparison of NH exchange and circular dichroism as techniques for measuring the parameters of the helix-coil transition in peptides. Biochemistry. 1997;36:8435–8442. doi: 10.1021/bi9706677. [DOI] [PubMed] [Google Scholar]
- 63.Luo PZ, Baldwin RL. Mechanism of helix induction by trifluoroethanol: A framework for extrapolating the helix-forming properties of peptides from trifluoroethanol/water mixtures back to water. Biochemistry. 1997;36:8413–8421. doi: 10.1021/bi9707133. [DOI] [PubMed] [Google Scholar]
- 64.Noy D, Yerushalmi R, Brumfeld V, Ashur I, Scheer H, Baldridge KK, Scherz A. Optical absorption and computational studies of [Ni]-bacteriochlorophyll-a. New insight into charge distribution between metal and ligands. Journal of the American Chemical Society. 2000;122:3937–3944. [Google Scholar]
- 65.Yerushalmi R, Noy D, Baldridge KK, Scherz A. Mutual control of axial and equatorial ligands: Model studies with [Ni]-bacteriochlorophyll-a. Journal of the American Chemical Society. 2002;124:8406–8415. doi: 10.1021/ja0121078. [DOI] [PubMed] [Google Scholar]
- 66.Noy D, Discher BM, Rubtsov I, Hochstraesser R, Dutton PL. Design of Amphiphilic Protein Maquettes: Enhancing Maquette Functionality through Binding of Extremely Hydrophobic Cofactors to Lipophilic Domains. Biochemistry. 2005 doi: 10.1021/bi050696e. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Teuchner K, Stiel H, Leupold D, Scherz A, Noy D, Simonin I, Hartwich G, Scheer H. Fluorescence and excited state absorption in modified pigments of bacterial photosynthesis - A comparative study of metal-substituted bacteriochlorophylls a. Journal of Luminescence. 1997;72-4:612–614. [Google Scholar]
- 68.Robertson DE, Farid RS, Moser CC, Urbauer JL, Mulholland SE, Pidikiti R, Lear JD, Wand AJ, DeGrado WF, Dutton PL. Design and Synthesis of Multi-Heme Proteins. Nature. 1994;368:425–431. doi: 10.1038/368425a0. [DOI] [PubMed] [Google Scholar]
- 69.Ye S, Discher BM, Strzalka J, Xu T, Wu SP, Noy D, Kuzmenko I, Gog T, Therien MJ, Dutton PL, Blasie JK. Amphiphilic 4-helix bundle peptides designed for light-induced electron transfer across a soft interface. Nano Letters. 2005 doi: 10.1021/nl050542k. in press. [DOI] [PubMed] [Google Scholar]
- 70.Cristian L, Lear JD, DeGrado WF. Use of thiol-disulfide equilibria to measure the energetics of assembly of transmembrane helices in phospholipid bilayers. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:14772–14777. doi: 10.1073/pnas.2536751100. [DOI] [PMC free article] [PubMed] [Google Scholar]