

Abstract
ValG is a glycosyltransferase (GT) that is responsible for the glucosylation of validoxylamine A to validamycin A. To explore the potential utilization of ValG as a tool for the production of validamycin analogs, a number of nucleotidyldiphosphate-sugars were evaluated as alternative substrates for ValG. The results indicated that in addition to its natural substrate, UDP-glucose, ValG also efficiently utilized UDP-galactose as sugar donor and resulted in the production of an unnatural compound 4″-epi-validamycin A. The new compound demonstrated a moderate growth inhibitory activity against the plant fungal pathogen Rhizoctonia solani (=Pellicularia sasakii). A comparative analysis of ValG with its homologous proteins revealed that ValG contains an unusual DTG motif, in place of the DXD motif proposed for metal ion binding and/or NDP-sugar binding and commonly found in other glycosyltransferases. Site-directed mutagenesis of the DTG motif of ValG to DCD altered its preferences for metal ion binding, but did not seem to affect its substrate specificity.
Combinatorial biocatalysis is an emerging technology in the field of drug discovery and development. Some of the approaches include the use of enzymatic, chemoenzymatic, and microbial transformations to generate libraries of new chemical entities from lead compounds.1-3 Since many bioactive natural products contain sugar moieties, which play a critical role in their pharmacological activities,4, 5 glycosylation processes of natural products have become a prime subject of investigation.6
Glycosyltransferases (GTs) catalyze the transfer of a sugar moiety from a nucleotidyldiphosphate (NDP)-sugar to an acceptor, which could be a growing oligosaccharide, a lipid, a protein, or a small molecule. Based on tertiary structure analysis, GTs have been divided into two superfamilies, known as GT-A and GT-B.7 Members of the GT-A superfamily contain two dissimilar domains, one involved in the recognition of the NDP-sugar and the other in the recognition of the acceptor molecule. Most of the GTs in the Leloir pathway that reside in the Golgi apparatus and the endoplasmic reticulum belong to this family. The GT-B superfamily is remarkably diverse and contains members that are extremely promiscuous to their NDP-sugar donors.8, 9 The GT-B family consists of most of prokaryotic enzymes that glycosylate secondary metabolites to produce active natural products, as well as some glycosyltranferases from the primary pathways.
Validamycin A (2), a commercially used agricultural antifungal antibiotic, is a microbial-derived pseudotrisaccharide, which contains a core aminocyclitol moiety, validoxylamine A (1), and glucose. The core aminocyclitol moiety is derived from two units of 2-epi-5-epi-valiolone, each of which is a cyclization product of the C7-sugar phosphate, sedoheptulose 7-phosphate. The glucose attachment is critical for the intake of the drug by the fungal cells through the common oligosaccharide transport system. Inside the cells, the compound is hydrolyzed to 1, and serves as a competitive inhibitor of trehalase.10 In fungi, trehalose is commonly used as a storage carbohydrate, which can be hydrolyzed by trehalase to two molecules of glucose for energy supply and other physiological purposes. Therefore, inhibition of the trehalase activity is detrimental to fungal growth.
As part of our ongoing study on the biosynthesis of validamycin, we have identified the complete biosynthetic gene cluster in Streptomyces hygroscopicus subsp. jinggangensis 5008.11 Among the proteins believed to be directly involved in the biosynthesis, ValG has been characterized both in vivo and in vitro as a glycosyltransferase that catalyzes the conversion of 1 to 2 using UDP-glucose as the sugar donor (Fig. 1).11 Interestingly, ValG belongs to the GT-A family of glycosyltransferases, even though it functions in secondary metabolism. ValG is also unique in that it has an unusual DTG motif instead of the DXD motif common in closely related proteins. While generally there is no significant identity between different GT families, the acidic DXD motif is highly conserved in almost all GTs,12 and is predicted to participate in the coordination of a divalent metal ion (most commonly Mn2+) and/or in the binding of the NDP-sugar. Mutagenesis of a mannosyltransferase has showed that altering either of these aspartates completely eliminates the enzymatic activity without causing the protein to misfold or denature.13 In this study, we investigated the utilization of ValG as a tool for generating analogs of validamycin by testing a number of commercially available NDP-sugars as substrates. The involvement of the DTG sequence of ValG in its catalytic activity or substrate specificity was explored by replacing the DTG sequence with a DCD motif.
Figure 1.
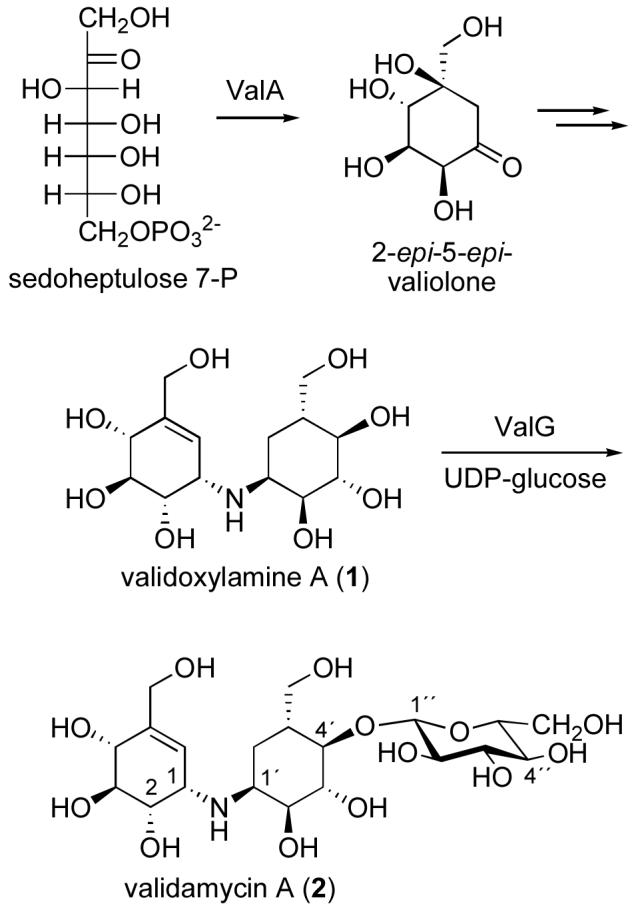
Biosynthesis of validamycin A.
Results and Discussion
Enzymatic assays of ValG with different NDP-sugars
To explore the possibility of employing ValG as a tool for generating analogs of validamycin, a number of commercially available NDP-sugars were tested as sugar donors. The recombinant histidine-tagged protein was prepared heterologously in E. coli BL21Gold(DE3)pLysS and purified on a BD TALON™ affinity column. The enzyme was incubated with validoxylamine A as a sugar acceptor and either UDP-glucose, UDP-galactose, UDP-N-acetylglucosamine, UDP-glucuronic acid, or GDP-mannose as a sugar donor. The reactions were carried out in the presence of Mg2+ for 1-6 hours and monitored by TLC. UDP-glucose is the natural sugar donor of ValG, which converts validoxylamine A (1) to validamycin A (2), and was used in this study as a positive control to validate the activity of the enzyme. The results revealed that in addition to UDP-glucose, ValG also utilizes UDP-galactose as substrate to produce a new validamycin analog, 4″-epi-validamycin A (3) (Fig. 2), whereas incubations with UDP-N-acetylglucosamine, UDP-glucuronic acid, or GDP-mannose did not give any products at a detectable level.
Figure 2.

Bioconversion of 1 to 3 using the glycosyltransferase ValG.
Interestingly, in contrast to the conversion of 1 to 2, a complete conversion of 1 to 3 can only be achieved when a purified ValG was used. When a cell-free extract of E. coli harboring valG was used for the enzyme reaction, only part of 1 was converted to 3 after 3 hours, and most of the product was hydrolyzed back to 1 after 6 hours (as monitored by TLC and MS). We hypothesized that this hydrolysis was due to an endogenous β-galactosidase activity of E. coli origin. Therefore, both 2 and 3 were incubated individually with either cell-free extract of E. coli harboring the valG gene or cell-free extract of E. coli harboring the pRSET-B vector. The reactions were carried out with or without an addition of uridyldiphosphate (UDP). Validoxylamine A (1) was not produced during any of the incubations with 2, whereas a significant amount of 1 was produced in incubations with 3. This activity was not dependent on the presence of additional UDP (data not shown).
Chemical Structure Determination of 4″-epi-Validamycin A
For a complete characterization of 4″-epi-validamycin A (3), a large-scale enzyme reaction of purified ValG with validoxylamine A (1) and UDP-galactose was carried out and the product was purified by ion-exchange chromatography. High-resolution mass spectrum of 3 revealed a pseudo-molecular ion m/z 498 [M+H]+, which is identical to that of validamycin A (2). However, the 1H and 13C NMR spectra of 3 are slightly different from those of 2.14 In particular, the C-4″ proton of 3 appeared as a small doublet (J = 3 Hz), as opposed to the large doublet (J = 12 Hz) of 2 (Fig. 3), which confirmed the identity of a galactosyl moiety in 3. On the other hand, the coupling constant of the anomeric proton (H-1″) of 3 is 7.5 Hz, which is consistent with that of 2, indicating a β-glycosidyl linkage of galactose. Furthermore, a correlation between H-1″ (δH 4.42 ppm) with C-4′ (δC 83.8 ppm) in the HMBC analysis provides evidence that the sugar moiety attaches at the C-4′ position of 3, which is consistent with the regioselectivity of ValG.11 Complete structure elucidation and assignments of the protons and carbons were based on direct comparisons of the 1H- and 13C NMR spectroscopic data with those for 2, as well as 2D NMR, including 1H-1H COSY, HSQC, and HMBC.
Figure 3.
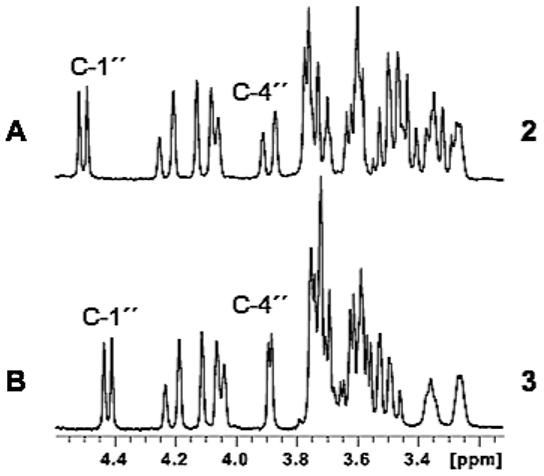
Partial 1H NMR spectra of validamycin A (2) and 4″-epi-validamycin A (3).
Fungal growth inhibitory assay of 4″-epi-validamycin A
It has been reported that 1 has a stronger in vitro inhibitory activity against trehalase than 2. However, 2 is more active in vivo, as it is efficiently taken up by the fungal cells and then hydrolyzed by an intracellular β-glucosidase to give the active pharmacophore 1.15 Given that 3 is readily hydrolyzed by the E. coli enzymes to give 1, it was anticipated that 3 might be comparably or more active than 2, if a similar β-galactosidase activity is present in the fungal cells. To compare their fungal growth inhibitory activity, 2 and 3 were tested against Pellicularia sasakii using an agar plug assay.16 Agar plugs with diameter of 5 mm and containing P. sasakii mycelia were placed in the center of agar plates that contain 1 mg of each compound and the growth of the pathogen was monitored after two days. Active compounds were determined based on their ability to inhibit the expansion of the fungal mycelia on the Petri dishes. Consistent with the reported results, validamycin A (2) demonstrated a potent fungal growth inhibitory activity (Fig. 4A), which validated the sensitivity of the assay. While 3 demonstrated a moderate growth inhibitory activity against P. sasakii, it is unexpectedly less active than 2 (Fig. 4A and 4B). On the other hand, the negative control (Fig. 4C) showed extensive fungal mycelial expansion. The lower activity of 3 may be due to a number of reasons, including less efficient uptake by the fungal cells and/or less effective intracellular hydrolysis by the fungal enzyme.
Figure 4.
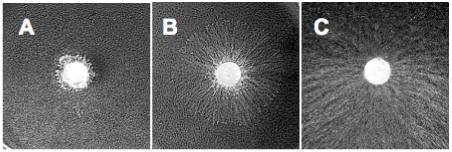
Fungal growth inhibitory assays of validamycin A (A), 4″-epi-validamycin A (B), and the negative control - water (C).
Site-directed Mutagenesis of ValG
BLAST analysis of ValG showed that it is a member of the glycosyltransferase family 2 (GT-2), classified by the CAZy system. This family is the largest and evolutionarily the most ancient of the inverting enzyme families with over 8,000 members to date and it utilizes a vast array of potential sugar donors and acceptors. The SpsA from Bacillus subtilis is one of the few well studied glycosyltransferases with crystal structures in this most widespread family.17 The N-terminal domains of this family often display a much higher similarity since they bind a common nucleotidyl moiety, and the C-terminal domains show enormous diversity probably because of the variety of sugar acceptors. Alignment of ValG showed that the N-terminal domain (amino acid residues 12-110) has high homology with the sugar donor-binding domain of SpsA, and the C-terminal domain shows very low similarity to other proteins.
Structurally, ValG belongs to the GT-A superfamily. A general feature of all enzymes of the GT-A fold family is the presence of a common DXD motif, which interacts with a divalent metal ion and/or an NDP-sugar. In the DXD motif, the third aspartate residue binds the divalent metal ion, typically Mn2+, and the middle amino acid residue binds the hydroxyl groups on the ribose moiety of the nucleotide moiety. Alignments of the amino acid sequence of ValG with its homologous proteins revealed that instead of having the common DXD motif at the position 99-101, ValG contains a DTG sequence. Using Hydrophobic Cluster Analysis (HCA),18 which is commonly used for predicting structural similarities in protein sequences with low levels of identity, we were able to predict that the DTG sequence in ValG is indeed located at the DXD motif position in other glycosyltransferases. To investigate the role of this DTG sequence, as opposed to the common DXD motif, the D99/T100/G101 motif was replaced with DCD residues by site-directed mutagenesis. An overlapping PCR method was used for this experiment and the mutated gene was confirmed by DNA sequencing. The original DNA sequence ACTGGT encoding threonine (T) and glycine (G) was replaced with TGCGAC for cysteine (C) and aspartic acid (D).
Effects of DTG → DCD mutation on ValG catalytic activity
Recombinant wild-type and mutant forms of ValG were produced in E. coli BL21 Gold(DE3)pLysS, purified by nickel columns, and tested for their catalytic activity using the suite of different sugar donors described above. While the enzyme maintained its catalytic activity with UDP-glucose and UDP-galactose (Fig. 5), no other NDP-sugars were recognized as alternative substrates. On the other hand, compared to the native ValG, the mutated enzyme appeared to have different preferences toward divalent metal ions.
Figure 5.

(A) SDS-PAGE of ValG and ValG mutant. M, protein marker; I, cell-free extract of E. coli harboring valG; II, purified ValG; III, cell-free extract of E. coli harboring mutated valG; IV, purified ValG mutant. (B) TLC analysis (nPrOH:AcOH:H2O 4:1:1) of ValG reactions. a, validoxylamine A; b, glycosylation product of validoxylamine A with UDP-galactose; c, glycosylation product of validoxylamine A with UDP-glucose.
A comparative study between the native and mutated ValG revealed that the native DTG-bearing ValG efficiently used a number of divalent metal ions, e.g., Mn2+, Mg2+, Co2+, and Ca2+, as co-factor, whereas the DCD-bearing ValG could only effectively use Mn2+, and to some extent Co2+, as co-factor (Fig. 6). The result is consistent with the nature of other DXD-containing glycosyltransferases, which mostly require Mn2+ for their catalytic activities. The DTG sequence of ValG may provide greater co-factor flexibility for the enzyme; however, it is not clear as to what extent this would benefit the host bacteria in relation to validamycin A biosynthesis.
Figure 6.
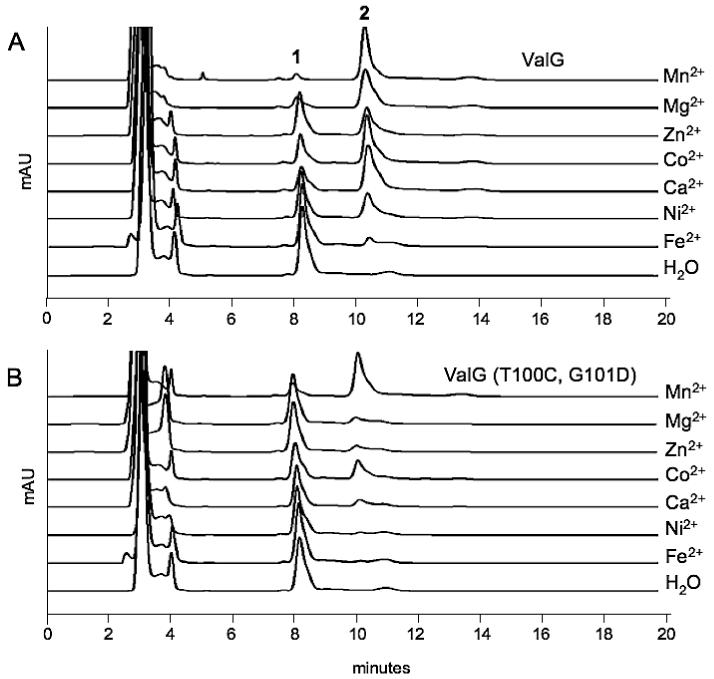
HPLC analyses of the reaction products of ValG (A) and mutated ValG (B) with different divalent metal ions.
Experimental Section
General Experiment Procedures
[α]D was recorded on a P-1010 polarimeter (JASCO Corporation). 1H and 13C NMR were recorded on a Bruker DPX300 NMR spectrometer. Low-resolution electrospray ionization (ESI) mass spectra were recorded on a ThermoFinnigan Liquid Chromatograph-Ion Trap Mass Spectrometer. High-resolution electrospray mass spectra were recorded on Waters/Micromass LCT spectrometer. Reactions were monitored by TLC (silica gel 60 F254, Merck) with detection by use of Ce(SO4)2 solution. Column chromatography was performed on Dowex 50Wx8-200 (Fluka). All chemicals were purchased from Aldrich or Sigma and were used without further purification unless otherwise noted. Reversed-phase preparative HPLC was carried out on a C18 column (Atlantis™ C18 5 mm column, Waters) with CH3CN-KH2PO4 buffer (pH 6.92, 1mM) (3:97) as mobile phase at 0.6 mL/min. Cloning and overexpression of the valG gene and purification of the recombinant His6-tagged ValG were carried out as described previously.11
Enzyme Assays
The enzyme assays were carried out at 30 °C for 3-12 h in a 100 μL volume of 25 mM Tris-HCl (pH 7.5), 10 mM MgCl2, 20 mM NH4Cl, 15 mM UDP-sugar, 10 mM validoxylamine A, and 50 μL of purified ValG solution (1 mg/mL) or 50 μL of the cell-free extract of E. coli BL21(DE3)pLysS containing ValG. The reaction progress was monitored by TLC analysis. For a scale-up experiment, 5.71 mg of validoxylamine A was used. To terminate the reaction, EtOH was added to precipitate the protein. The supernatant was subjected to ion-exchange (Dowex 50Wx8-200 [H+ form]) column chromatography. The column was washed with H2O and then eluted with 0.5 M aq. NH4OH. The elution of the product was monitored by TLC. Fractions containing the desired product [Rf value 0.15 (nPrOH/AcOH/H2O=4:1:1)] were pooled and lyophilized. The product was subsequently subjected to preparative HPLC to give 3.6 mg of pure 4″-epi-validamycin A (3). 3: white powder, [α]23D +49.2 (c = 0.07, H2O); 1H NMR (D2O, 300MHz) δ 6.01 (1H, d, J=5 Hz, H-6), 4.42 (1H, d, J=7.5 Hz, H-1″), 4.21 (1H, d, J=14 Hz, H-7b), 4.08 (1H, d, J=14 Hz, H-7a), 4.05 (1H, brd, J=7.0 Hz, H-4), 3.89 (1H, d, J=3 Hz, H-4″), 3.76-3.68 (6H, overlap, H-2′, H-7′a,b, H-5″, H-6″a,b), 3.60 (1H, brd, J=3 Hz, H-3″), 3.58-3.56 (3H, overlap, H-2, H-3, H-2″), 3.52 (1H, d, J=2.5 Hz, H-3′), 3.48 (1H, brd, J=2.5 Hz, H-4′), 3.34 (1H, m, H-1), 3.25 (1H, m, H-1′), 2.07 (1H, m, H-5′), 1.91 (1H, brd, J=15 Hz, H-6′b), 1.29 (1H, brd, J=15 Hz, H-6á); 13C NMR (D2O, 75 MHz) δ 139.1 (C-5), 123.1 (C-6), 103.3 (C-1″), 83.8 (C-4′), 75.3 (C-5″), 73.6 (C-3), 73.1 (C-2′), 72.7 (C-3″), 72.6 (C-2), 71.4 (C-4), 71.2 (C-3′), 69.4 (C-2″), 68.5 (C-4″), 61.7 (C-7′), 61.5 (C-7), 60.9 (C-6″), 53.7 (C-1′), 52.3 (C-1), 37.4 (C-5′), 26.8 (C-6′); HRESIMS m/z 498.2169 [M+H]+ (calcd for C20H36NO13, 498.2187).
Site-directed Mutagenesis ofvalG
The mutagenesis in valG was introduced by an overlapping PCR method. For the first step, two fragments of valG were amplified separately by PCR with Platinum Pfx DNA polymerase (Invitrogen) using the cosmid clone 17F2 as template. A 323 bp DNA encoding the N-terminal fragment of valG (fragment 1) was amplified using primers ValGPM-1F2 (5′-AAAGGATCCACATATGCCCGGTGCGCATCCC-3′, the BamHI and NdeI sites are in italics) and ValGPM-1R2 (5′-TACTGCGGCCCCGCCAGGACGTCGCAGTCGAGGAAGGCCAGGAGTG-3′, the introduced mutation sites are underlined). Similarly, a 992 bp DNA encoding the C-terminal fragment of valG (fragment 2) was amplified using primers ValGPM-2F2 (5′-CACTCCTGGCCTTCCTCGACTGCGACGTCCTGGCGGGGCCGCAGTA-3′, the mutation sites are underlined) and ValGPM-2R2 (5′-AAAGAATTCAGTCACCGCGAAGAGAC-3′, the EcoRI site is in italics). For the second step, the whole modified valG was amplified using fragments 1 and 2 as templates with primers ValGPM-1F2 and ValGPM-2R2. Gel-purified PCR products were digested with BamHI and EcoRI, and subsequently ligated into BamHI/EcoRI-digested pRSET-B. The constructs were transferred into E. coli DH10B and plated on LB agar plates containing 100 μg/mL ampicillin. The expected mutation was confirmed by sequencing.
Enzyme assays with different metal ions
Enzyme assays of mutated ValG with different metal ions were carried out at 30 °C for 3 h in a 100 μL volume of 25 mM Tris-HCl (pH 7.5), 1 mM divalent metal ion, 20 mM NH4Cl, 15 mM UDP-sugars, 10 mM validoxylamine A, and 50 μL of protein solution (0.1 mg/mL). Subsequently, 100 μL of MeOH was added into the reaction and the mixture was centrifuged for 5 min at 12,000 rpm, and the supernatant was transferred into a clean Eppendorf tube and dried. The product was then dissolved in 100 μL of H2O for HPLC analysis.
Bioassay
A 1% agar solution (14 mL) was mixed with the compounds (dissolved in 1 mL H2O) and plated into Petri dishes. PDA agar plugs with mycelia of Pellicularia sasakii were placed in the center of the plates as the indicator strain for bioassay of the compounds. The plates were incubated at 30 °C for 2 days.
Supplementary Material
Acknowledgment
The authors thank P. M. Flatt for the critical reading of this manuscript. This work was supported by grants from the National Institutes of Health (R01 AI061528), the Herman Frasch Foundation, and in part, by the Mass Spectrometry Facilities and Services Core of the Environmental Health Sciences Center, Oregon State University, grant number P30 ES00210, National Institute of Environmental Health Sciences, National Institutes of Health. HX was in part supported by the Exchange Program from China Scholarship Council.
References and Notes
- (1).Michels PC, Khmelnitsky YL, Dordick JS, Clark DS. Trends. Biotechnol. 1998;16:210–215. doi: 10.1016/s0167-7799(98)01190-1. [DOI] [PubMed] [Google Scholar]
- (2).Altreuter DH, Clark DS. Curr. Opin. Biotechnol. 1999;10:130–136. doi: 10.1016/s0958-1669(99)80022-6. [DOI] [PubMed] [Google Scholar]
- (3).Woodley JM. Trends Biotechnol. 2008 Apr 22; [Epub ahead of print] doi:10.1016/j.tibtech.2008.03.004. [Google Scholar]
- (4).Weymouth-Wilson AC. Nat. Prod. Rep. 1997;14:99–110. doi: 10.1039/np9971400099. [DOI] [PubMed] [Google Scholar]
- (5).Dembitsky VM. Chem. Biodivers. 2004;1:673–781. doi: 10.1002/cbdv.200490060. [DOI] [PubMed] [Google Scholar]
- (6).Luzhetskyy A, Mendez C, Salas JA, Bechthold A. Curr. Top. Med. Chem. 2008;8:680–709. doi: 10.2174/156802608784221514. [DOI] [PubMed] [Google Scholar]
- (7).Bourne Y, Henrissat B. Curr. Opin. Struct. Biol. 2001;11:593–600. doi: 10.1016/s0959-440x(00)00253-0. [DOI] [PubMed] [Google Scholar]
- (8).Jiang J, Biggins JB, Thorson JS. Angew. Chem. Int. Ed. Engl. 2001;40:1502–1505. [PubMed] [Google Scholar]
- (9).Barton WA, Biggins JB, Jiang J, Thorson JS, Nikolov DB. Proc. Natl. Acad. Sci. U.S.A. 2002;99:13397–13402. doi: 10.1073/pnas.192468299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Qian X, Li Z, Liu Z, Song G, Li Z. Carbohydr. Res. 2001;336:79–82. doi: 10.1016/s0008-6215(01)00126-4. [DOI] [PubMed] [Google Scholar]
- (11).Bai L, Li L, Xu H, Minagawa K, Yu Y, Zhang Y, Zhou X, Floss HG, Mahmud T, Deng Z. Chem. Biol. 2006;13:387–397. doi: 10.1016/j.chembiol.2006.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Breton C, Imberty A. Curr. Opin. Struct. Biol. 1999;9:563–571. doi: 10.1016/s0959-440x(99)00006-8. [DOI] [PubMed] [Google Scholar]
- (13).Wiggins CA, Munro S. Proc. Natl. Acad. Sci. U.S.A. 1998;95:7945–7950. doi: 10.1073/pnas.95.14.7945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Jin WZ, Rinehart KL, Jr., Toyokuni T. J. Antibiot. 1987;40:329–339. doi: 10.7164/antibiotics.40.329. [DOI] [PubMed] [Google Scholar]
- (15).Asano N, Yamaguchi T, Kameda Y, Matsui K. J. Antibiot. 1987;40:526–532. doi: 10.7164/antibiotics.40.526. [DOI] [PubMed] [Google Scholar]
- (16).Minagawa K, Zhang Y, Ito T, Bai L, Deng Z, Mahmud T. ChemBioChem. 2007;8:632–641. doi: 10.1002/cbic.200600528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Tarbouriech N, Charnock SJ, Davies GJ. J. Mol. Biol. 2001;314:655–661. doi: 10.1006/jmbi.2001.5159. [DOI] [PubMed] [Google Scholar]
- (18).Gaboriaud C, Bissery V, Benchetrit T, Mornon JP. FEBS Lett. 1987;224:149–155. doi: 10.1016/0014-5793(87)80439-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.