

Abstract
This paper describes a new bioinformatic tool for use in psychiatric research, “SLEP” (Sullivan Lab Evidence Project). SLEP is a searchable archive of findings from psychiatric genetics that is freely available on the web for non-commercial use (http://slep.unc.edu). Via a simple interface, users can retrieve findings from genomewide linkage, genomewide association, and microarray studies for ADHD, autism, bipolar disorder, eating disorders, major depression, nicotine dependence, and schizophrenia. Findings can be save to disk or viewed via a genome browser.
Keywords: psychiatric genetics, genome-wide association, genome-wide linkage, microarray, database
The purpose of this paper is to describe a new bioinformatic tool for use in psychiatric research. The tool is called “SLEP” (Sullivan Lab Evidence Project) and is a searchable archive of findings from psychiatric genetics that is freely available on the web for non-commercial use (http://slep.unc.edu). SLEP is an example of “soft” bioinformatics: the use of existing genomic data in the service of a biological problem (“hard” bioinformatics is more concerned with novel algorithms or querying genomes in order to generate new hypotheses).
Motivation
The motivation for creating SLEP is that findings from psychiatric genetic studies are difficult to use and to contextualize. As a consequence, potentially useful and important findings may be lost. There are a relatively large number of genomewide linkage (GWL), genomewide association (GWA), and microarray (MA) studies and even a cursory summary of findings for one disorder (much less across several disorders) is quite challenging. For example, which linkage studies for what disorders provide support for NRG1? Or, what GWA studies implicate rs4680 (the widely-studies val-met SNP in COMT)? In a practical manner, these questions are very hard for most psychiatric geneticists to address and even more difficult for researchers from other disciplines who wish to make use of the accumulated results from genetic investigations of psychiatric disorders.
Focus on evidence
The intention of SLEP is to supply evidence in support of a particular gene, marker, or genomic region collated from unbiased empirical searches of the genome (i.e., genomewide linkage or genomewide association studies) or the transcriptome (i.e., microarray studies) for a set of core psychiatric disorders. As such, SLEP is not a meta-analytic tool. The reasons why SLEP is a qualitative and not a quantitative tool are threefold. First, meta-analysis requires access to the findings for all genetic markers investigated (and preferably to individual genotype and phenotype data). Until quite recently, these data were not routinely available and the predominant practice in the field was to publish only the findings for the best markers. Second, comparison of data across study types — e.g., GWL with GWA results — is complex and not readily handled in the absence of extensive data. Third, our aim was to create a way to explore and synthesize findings from the literature and the only practical solution was via a qualitative interface.
Results obtained from searches of the SLEP database will certainly contain false positive findings. This approach is appropriate if the literature is viewed as being populated by under-powered studies and if and “hits” from a SLEP search are viewed as “tentative knowledge” (Ioannidis 2006) requiring rigorous experimental confirmation. Even with this important limitation, SLEP can be of considerable utility in attempts to place an empirical finding in the context of prior studies.
Study technologies
The single greatest advantage of modern human genetic approaches to the etiology of complex traits is in their ability to uncover new and previously unsuspected etiological factors for a disease. They do this by proving an unbiased screen of the relevant search space (the genome and the transcriptome). Thus, the focus of SLEP is on three main study technologies — GWL, GWA, and MA studies. Study technologies available in SLEP are thus mixed — searches for DNA-level genomic variation along with searches for RNA-level changes in transcript levels predisposing or protecting against a disorder. However, these study types have in common their use of an unbiased exploration of the genomic or transcriptomic search space.
Publication identification and review
The disorders currently contained in SLEP are attention-deficit hyperactivity disorder (ADHD), autism (AUT), bipolar disorder (BIP), eating disorders (ED), major depressive disorder (MDD), nicotine dependence (ND), and schizophrenia (SCZ), and we hope to expand this list in the near future.
There were multiple steps involved in creating the SLEP database. (1) All relevant primary studies were identified via overlapping PubMed (Wheeler and others 2006) searches augmented by review of citation lists. All studies with a genomewide focus (excluding studies limited to a chromosomal region or candidate genes), sufficient descriptive information, and clear phenotype designations were included. (2) Where available, the most comprehensive quantitative meta-analysis for each disorder was also included. (3) Two reviewers independently abstracted information from each published report with any disagreements resolved by discussion. The data abstracted were of two types — study meta-data (study citation, technology, disorder, number of subjects, etc) and results (marker or gene name, statistical test, and p-value). (4) Given the lack of widespread adherence to presentation standards in this area, a number of decisions were required in regard to which publications and sets of results to include. (a) One report per sample was included. If there were multiple reports from the same sample, the most comprehensive report was abstracted. (b) One set of results was abstracted per report. As there were usually multiple choices, the a priori decisions for SLEP were to select:
The initial Stage 1 genomewide findings and not analyses of more markers (i.e., fine-mapping) or secondary analyses (e.g., stratified or sex-specific analyses). The intention here was to capture the initial, unbiased representation of the genome or transcriptome screen before complicated by secondary analyses or fine-mapping genotyping.
The narrowest phenotypic definition per sample was used. Many studies presented results for multiple phenotypic definitions (e.g., narrow, intermediate, and broad definitions of affection for SCZ) and the narrowest was generally the one that conformed most closely to DSM/ICD definitions of illness and are the most likely to be comparable across studies. The exceptions to this were for AUT (given our group’s interest in the broad AUT phenotype), eating disorders (because of a paucity of studies), and smoking behavior (due to phenotypic inconsistencies across studies).
If multiple sets of initial analyses were presented (e.g., singlepoint, multipoint, non-parametric linkage, parametric linkage, etc.), a single set was chosen with a preference for multipoint, non-parametric results for GWL studies and Cochran-Armitage trend test for GWA studies.
Filters were applied in order to select results for inclusion in the SLEP database. For GWL studies, markers with non-parametric LOD scores ≥ 1.5 were selected. For GWA studies, single nucleotide polymorphisms with p-values < 0.05 were included. To account for linkage disequilibrium, we used the “--clump” feature in PLINK (Purcell and others 2007) to compress individual SNP findings with p < 0.001 to genomic segments with high linkage disequilibrium patterns with reference to the appropriate HapMap panel (Frazer and others 2007). The focus on genomic segments that take into account linkage disequilibrium is a more useful portrayal of results and is considerably more compact. Finally, for MA studies, all transcripts with significant differences at the 0.05 level were selected.
Database creation
Once the findings were accurately entered, a custom SAS (SAS Institute Inc. 2004) program was used to create a series of data files. As part of this process, results specific to a marker were placed on the NCBI Build 35 human genome assembly (Wheeler and others 2006), also known as UCSC build hg17 (Hinrichs and others 2006). The data files were imported into a relational database which served as the back-end data system for the application. A custom, web-based user-interface was developed to enable users to search the findings based on a variety of criteria. The core application was built entirely upon an open-source framework (LAMP), written in PHP, backed by mySql, and employs AJAX technologies for seamless interaction between the user, the web browser and the database.
Signposts
A potentially useful feature of SLEP is the inclusion of genomic “signposts”. These are a conglomerate of findings from human genetics that can be searched along with the psychiatric genetics literature. These data attempt to provide information about the genome that might prove useful. We hope to include as much reliable genomewide data as possible. The signpost dataset includes:
Over 3,700 gene entries in the Online Mendelian Inheritance in Man (OMIM) (McKusick 2007) that can be mapped to hg17 (e.g., GABRA1 and juvenile myoclonic epilepsy).
A manually curated list of confirmed associations from complex human diseases (e.g., FTO- body mass index, PPARG-type 2 diabetes mellitus, and a copy number variant on 16p11.2-autism).
Genes for which a SNP is associated with expression differences in human cortex (Myers and others 2007).
Genes with evidence of imprinting (Luedi and others 2007)
Genes with evidence of selection from a genomic perspective (Sabeti and others 2007).
Genes with evidence of monoallelic expression from a genomic survey (Gimelbrant and others 2007).
Genes commonly mutated in colon and breast cancer from a genomic survey (Wood and others 2007).
Over 10,300 copy number variants and 77 inversions from the Database of Genomic Variants (Iafrate and others 2004).
Genomic features such as persistent sequencing gaps, heterochromatin, centromeres, telomeres, and the pseudo-autosomal regions (Hinrichs and others 2006).
Querying SLEP
The database can be queried in four ways — by gene name, SNP or microsatellite marker, chromosome band (e.g., 22q11), or chromosome region (e.g., chr1:12,000,000-13,500,000). The SLEP interface has one tab for each of these query methods. Gene name queries require standard HUGO gene names (http://www.genenames.org/cgibin/hgnc_search.pl) (Eyre and others 2006) and there is a lookup facility for common aliases. There are over 43,000 overlapping entries in the gene name database for all RefSeq (Pruitt and others 2005) and KnownGenes (Hsu and others 2006). The marker data base contains over 422,000 microsatellite markers (many known by several names) along with 11.9 million SNPs adapted from the TAMAL database (Hemminger and others 2006).
Users can modify their queries in several ways. First, the search can be for primary studies and/or meta-analyses. Second, the search can be for any or all of the psychiatric disorders currently in the SLEP database. Third, the user can select the technology used, i.e., GWL, GWA, and/or MA studies (for convenience, the signpost database is included here as well). Fourth, all searches can be widened by a specific number of kilobases or megabases in order to accommodate differences in localization for each technology. Linkage analysis is known to be imprecise and a linkage peak can be megabases from the true genomic variant. Association analysis is known to be more precise in outbred human populations but its precision is limited by local patterns of linkage disequilibrium. MA studies are, at least in theory, the most precise as an altered transcript should be exactly identified.
The computer algorithm for processing SLEP queries has the following steps:
All types of queries are converted chrN:start-end coordinate format with reference to the appropriate hg17 files.
The query is expanded by the number of bases specified by the user which may vary by methodology. The defaults are 10 megabases for GWL, 5 kilobases for GWA, 0 bases for MA, and 0 bases for signposts.
The search is performed. For the expanded genomic region specified by the user, SLEP identifies empirical findings that match all of the following criteria: the type of study selected (primary report and/or meta-analysis), the disorder selected (ADHD, AUT, BIP, EDs, MDD, ND, SCZ, and/or SCZ/BIP), and the study methodology (GWL, GWA, MA, and/or signposts).
Output
SLEP returns a listing of all study findings on the web page below the user query. Basic data about each hit are shown by default, and additional study metadata are optionally available (e.g., definition of affection, diagnostic criteria, sample size, etc.). The user can request that SLEP open a new web page with the study search and results displayed as custom annotation tracks on the UCSC genome browser in order to access the wealth of genomic data available there. Third, the results of a query can be downloaded as a comma-separated value file (.csv) in order to be imported into a spreadsheet or some other program.
Example
An example of a SLEP search is depicted in Figure 1. Bullet a shows the four ways in which SLEP can be searched. This example is a search by “Gene Name” for “comt” (bullet b). Clicking on either of the lookup options opens a window to assist in finding the correct standard gene name. Bullet c shows the three classes of options that can be used to fine-tune a search (checking a box means to include studies matching that criterion). The type of study can include primary reports and/or meta-analyses. Which psychiatric disorders included can be modified to suit the purpose of the search (here, all are selected). The technology used can be specified — i.e., GWL, GWA, or MA empirical studies along with the curated “signpost” list described above. Each of these can be given an optional but different number of bases by which to expand the query given the varying precision of these technologies.
Figure 1.
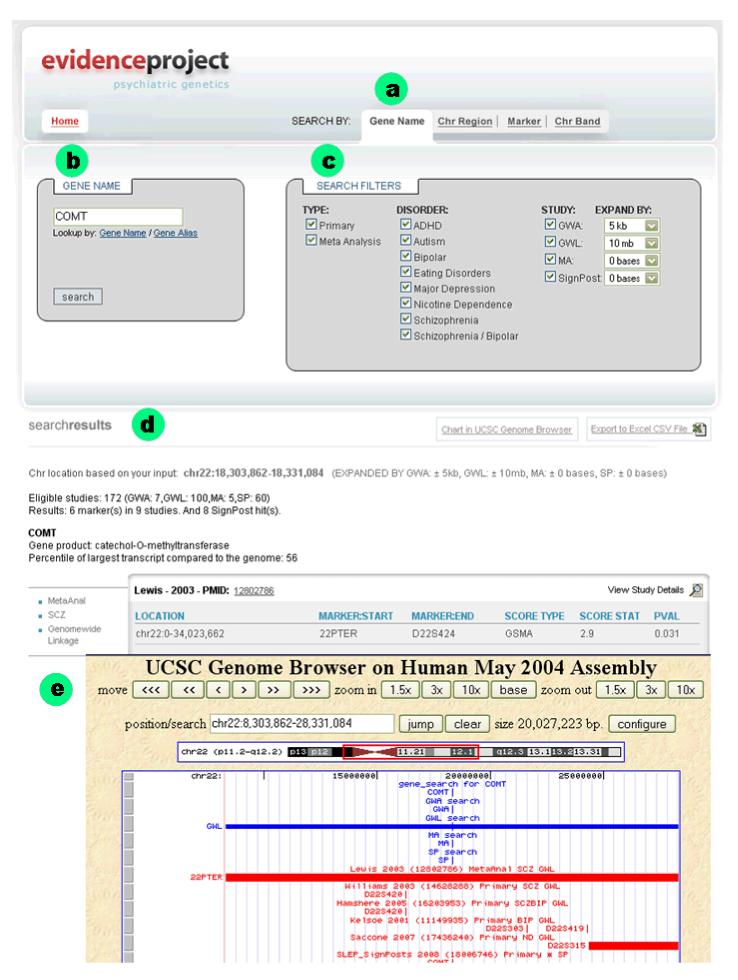
Example of a SLEP search for the gene “COMT”. See text for full description.
A portion of the search results are shown next to bullet d (in this instance, a 2003 SCZ GWL by Williams et al.). There are hyperlinks to download the search results to a comma-separate-value text file (.csv), to show more information about each study, and to view the results in the UCSC genome browser (bullet e). The topmost track in the UCSC browser shows the location of the user query followed by the expanded extent of GWL, GWA, MA, and “signpost” approaches. These are followed by additional tracks depicting the hits for the specific search from the SLEP database. All other tracks normally available on the UCSC genome browser can be viewed according to the user’s preference.
Conclusions
SLEP is a tool for searching and integrating findings from psychiatric genetics research. The user interface is simple, flexible, and powerful. The SLEP database will be updated approximately quarterly in order to integrate new findings from the upcoming generation of GWA studies. It must be emphasized that SLEP searches are likely to contain false positive findings. This is done by design given that limited power is a serious concern for most psychiatric genetic studies. Separating true positive from false positive findings is a complex empirical exercise. However, SLEP can be of considerable utility in attempts to place a new empirical finding in the context of prior studies.
ACKNOWLEDGEMENTS
HF was supported by K07 CA118412 (Anna Helena Furberg Barnes) from the National Cancer Institute.
REFERENCES
- Eyre T, Ducluzeau F, Sneddon T, Povey S, Bruford E, Lush M. The HUGO Gene Nomenclature Database, 2006 updates. Nucleic Acids Res. 2006;34:D319–21. doi: 10.1093/nar/gkj147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, Gibbs RA, Belmont JW, Boudreau A, Hardenbol P, Leal SM. and others. A second generation human haplotype map of over 3.1 million SNPs Nature 20074497164851–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimelbrant A, Hutchinson JN, Thompson BR, Chess A. Widespread monoallelic expression on human autosomes. Science. 2007;318(5853):1136–40. doi: 10.1126/science.1148910. [DOI] [PubMed] [Google Scholar]
- Hemminger BM, Saelim B, Sullivan PF. TAMAL: An integrated approach to choosing SNPs for genetic studies of human complex traits. Bioinformatics. 2006;22:626–7. doi: 10.1093/bioinformatics/btk025. [DOI] [PubMed] [Google Scholar]
- Hinrichs AS, Karolchik D, Baertsch R, Barber GP, Bejerano G, Clawson H, Diekhans M, Furey TS, Harte RA, Hsu F. and others. The UCSC Genome Browser Database: update 2006 Nucleic Acids Res 200634(Database issue)D590–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu F, Kent WJ, Clawson H, Kuhn RM, Diekhans M, Haussler D. The UCSC Known Genes. Bioinformatics. 2006;22(9):1036–46. doi: 10.1093/bioinformatics/btl048. [DOI] [PubMed] [Google Scholar]
- Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, Scherer SW, Lee C. Detection of large-scale variation in the human genome. Nat Genet. 2004;36(9):949–51. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- Ioannidis JP. Commentary: Grading the credibility of molecular evidence for complex diseases. Int J Epidemiol. 2006;35(3):572–8. doi: 10.1093/ije/dyl003. [DOI] [PubMed] [Google Scholar]
- Luedi PP, Dietrich FS, Weidman JR, Bosko JM, Jirtle RL, Hartemink AJ. Computational and experimental identification of novel human imprinted genes. Genome Res. 2007;17(12):1723–30. doi: 10.1101/gr.6584707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKusick VA. Mendelian Inheritance in Man and its online version, OMIM. Am J Hum Genet. 2007;80(4):588–604. doi: 10.1086/514346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers AJ, Gibbs JR, Webster JA, Rohrer K, Zhao A, Marlowe L, Kaleem M, Leung D, Bryden L, Nath P. and others. A survey of genetic human cortical gene expression Nat Genet 200739121494–9. [DOI] [PubMed] [Google Scholar]
- Pruitt KD, Tatusova T, Maglott DR.NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins Nucleic Acids Res 200533(Database issue)D501–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira M, Bender D, Maller J, de Bakker P, Daly M, Sham P. PLINK: a toolset for whole-genome association and population-based linkage analysis. American Journal of Human Genetics. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabeti PC, Varilly P, Fry B, Lohmueller J, Hostetter E, Cotsapas C, Xie X, Byrne EH, McCarroll SA, Gaudet R. and others. Genome-wide detection and characterization of positive selection in human populations Nature 20074497164913–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAS Institute Inc. SAS Institute, Inc. SAS/STAT® Software: Version 9. Cary, NC: 2004. [Google Scholar]
- Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K, Chetvernin V, Church DM, DiCuccio M, Edgar R, Federhen S. and others. Database resources of the National Center for Biotechnology Information Nucleic Acids Res 200634(Database issue)D173–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J. and others. The genomic landscapes of human breast and colorectal cancers Science 200731858531108–13. [DOI] [PubMed] [Google Scholar]