

Abstract
Human topoisomerase IB (hTopo) forms a covalent phosphotyrosyl linkage with the DNA backbone, and controls genomic DNA topology by relaxing DNA supercoils during the processes of DNA replication, transcription, chromosome condensation and decondensation. The essential role of hTopo in these processes has made it a preeminent anticancer drug target. We have screened a small library of arylstibonic acids for their effects on plasmid supercoil relaxation catalyzed by hTopo. Despite the similar structures of the library compounds, some compounds were found to be effective competitive inhibitors, and others, nonessential activators. Some arylstibonic acids show selectivity in their action against hTopo and the related enzyme from poxvirus (vTopo). Structure-activity relationships and structural modeling suggest that competitive inhibition may result from positioning of the negatively charged stibonic acid and carboxylate groups of the inhibitors into DNA phosphate binding pockets on hTopo. The hTopo activators act by a surprising allosteric mechanism without interfering with DNA binding or binding of the widely used hTopo poison camptothecin. Arylstibonic acid competitive inhibitors may become useful small molecules for elucidating the cellular functions of hTopo.
The human type I DNA topoisomerase (hTopo) has long been a validated target for anticancer drug therapy [1]. The mechanism of one widely used hTopo drug, camptothecin (CPT), is a classic example of uncompetitive, mechanism-based inhibition [2]. When hTopo cleaves DNA to form a covalent phosphotyrosyl linkage between its nucleophilic active site tyrosine and the DNA phosphodiester backbone, CPT intercalates into the gap and sterically prevents the expelled 5′ DNA hydroxyl from reconnecting to the phosphate [3–5]. Thus, the topoisomerase is trapped in its 3′ covalent linkage with the DNA and the strand break persists. The persistence of the complex presumably leads to double strand DNA breaks when a replication fork or transcription complex encounters the obstruction. Because of this unique mechanism of drug action, which does not inhibit DNA cleavage, camptothecin is usually called a “poison” rather than an inhibitor of hTopo [6, 7].
Although camptothecin analogues show potent killing of rapidly proliferating cells, drug resistance inevitably develops in patients [8, 9], and mechanistically distinct drugs that effectively target hTopo would be of interest. Recently we reported the discovery of several novel inhibitors of the closely related poxvirus topoisomerase (vTopo) [10]. One of the most potent compounds against vTopo was the arylstibonic acid NSC 13778 shown in Table 1. This compound has a novel structure and mode of action in which the covalently bound enzyme is trapped in a nonproductive state that is unable to turnover [10]. NSC 13778 does not intercalate into the DNA base stack, indicating that it exerts its uncompetitive inhibitory effects by an entirely different mechanism than CPT. Interestingly, 13778 showed high selectivity for vTopo as compared to hTopo. The activity and selectivity of this arylstibonic acid compound for vTopo prompted us to further interrogate a 37 member library of related arylstibonic acids in search of compounds that had activity against hTopo. Here we report the surprising inhibitory and activating properties of several arylstibonic acids contained in this library and their selectivities for the human and poxviral enzymes.
Table 1.
Chemical Structures of Arylstibonic Acids
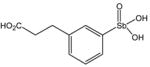
P6949 |
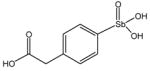
P6971 |
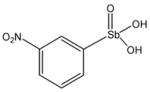
13743 |
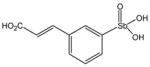
13778 |
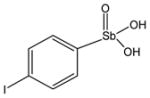
P6952 |
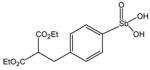
P6972 |
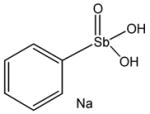
13744 |
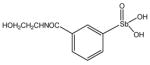
13782 |
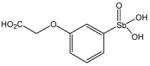
P6953 |
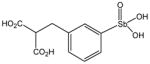
P6981 |
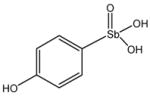
13745 |
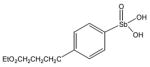
13793 |
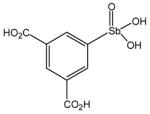
P6954 |
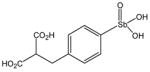
P6982 |
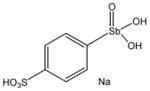
13746 |
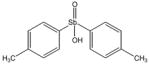
13794 |
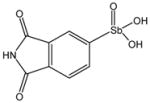
P6964 |
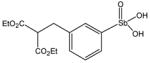
P6983 |
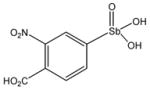
13755 |
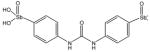
15408 |
|
P6965 |
13730 |
13759 |
15591 |
P6966 |
13731 |
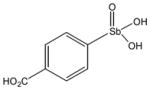
13760 |
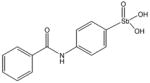
15596 |
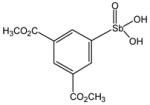
P6967 |
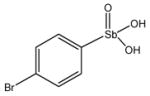
13740 |
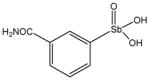
13765 |
|
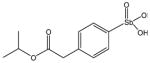
P6968 |
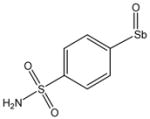
13741 |
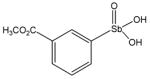
13771 |
|
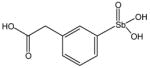
P6970 |
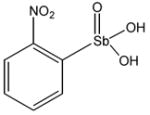
13742 |
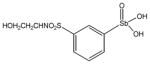
13776 |
Materials and Methods
Materials
The 70 kDa fragment of recombinant human topoisomerase I and vaccinia topoisomerase I were expressed and purified as previously described [11, 12]. The 37member library of arylstibonic acids was obtained from the National Cancer Institute Developmental Therapeutics Program. pUC19 plasmid DNA was purified from Escherichia coli strain DH5α using a Qiagen plasmid maxi kit. The sequence of the 22-mer oligonucleotide containing the hTopo I preferred cleavage sequence is 5′-AAAAAGACTTGGAAAAATTTTT-FAM-3′ (where cleavage occurs after the ACTT sequence) and 3′–TTTTTCTGAACCTTTTTAAAAA-5′ where FAM is 6-carboxyfluorescein. The oligonucleotides were HPLC purified using UNO Q-12 column (Bio-Rad) and then desalted using a disposable gel filtration PD-10 column. The purity of oligonucleotides was confirmed using electrophoresis through a 19% denaturing polyacrylamide gel containing 7 M urea.
Initial Compound Screening
To initially identify active compounds in the 37 member library, we performed a screen of hTopo and vTopo using 5 μM of each compound. For hTopo, a supercoil relaxation assay was performed in 40 μl of reaction buffer (10 mM Tris·HCl, pH 7.5, 150 mM KCl, 1 mM EDTA, 1 mM DTT, 100 μg/ml BSA and 0.01% Brij 35) supercoiled pUC19 (14 nM) and 1 nM hTopo. For hTopo, reaction mixtures were incubated for 15 min at room temperature and were then quenched with 2 × loading buffer containing 1% SDS, 10% glycerol and Tris-glycine. For vTopo, relaxation assays were performed in 30 μl of reaction buffer (50 mM Tris·HCl, pH 7.5, 100 mM NaCl, 1 mM DTT, 20 μg/ml BSA and 0.01% Brij 35) with 14 nM of supercoiled pUC19 and 1 nM vTopo. Reactions were incubated for either 20 or 30 min before quenching. The DNA was resolved on a 1% agarose gel at 125 V for 90 min in Tris-glycine running buffer (50 mM Tris and 160 mM glycine). The gels were stained with ethidium bromide (0.8 μg/ml) and the fluorescence of the DNA bands was imaged using a GelDoc 2000 imaging system (Bio-Rad). Standard curves were performed to ensure that the fluorescence signal was linear in the DNA concentration range used in the assay. The bands corresponding to supercoiled and relaxed DNA in each lane were quantified by integration using Quantity One software and corrected for background (Bio-Rad). The % inhibition was calculated as 100 × {1 − [%relaxation in presence of compound/% relaxation in absence of compound]}. For compounds that displayed activation the % activation was calculated as 100 × [% relaxation in presence of compound/% relaxation in absence of compound] – 1}. Compounds that displayed greater than 30% inhibition or activation under the screening conditions were investigated further.
Determination of Compound Potency
To further characterize the inhibitor and activator compounds the concentration dependence of their activities were determined. In general, the supercoil relaxation time courses performed as described above were restricted to the observed linear range (< 50% consumption of substrate). To estimate the concentrations that gave rise to half-maximal inhibition (C0.5 ) the inhibition data were fitted to a single site binding equation with a variable cooperativity term (n) using GraphPad Prism software (eq 1):
| (1) |
To characterize the most potent activator of supercoil relaxation, P6966, the concentration dependence was measured by determining the initial rates in the presence and absence of P6966 using DNA concentrations in the range 3.5 to 26 nM. The activation is reported as the ratio vA/vo, where vA and vo are the rates in the presence and absence of activator.
Electrophoretic Mobility Shift Assay (EMSA)
To test the compounds ability to prevent hTopo from binding to DNA, 10 nM of the 3′ FAM 22/22-mer duplex DNA substrate and 20 nM of hTopo were incubated in 20 μl of binding buffer (10 mM Tris·HCl, pH 7.5, 10 mM KCl, 1 mM EDTA, 1 mM DTT, 100 μg/ml BSA and 0.01% Brij 35) in the absence and presence of the compounds at 8 °C for 15 min. Samples were immediately loaded onto the 6% polyacrylamide gel and separated by electrophoresis at 50 V for 90 min in the running buffer (22.5 mM Tris-boric acid buffer and 0.5 mM EDTA). The bands corresponding to the hTopo-DNA complex and free DNA were imaged using a Typhoon 9210 instrument and Image Quant software (Amersham Biosciences). The fraction DNA bound to hTopo was calculated as (fluorescence complex)/(fluorescence complex + fluorescence free DNA).
Inhibition of hTopo Religation
The 3′ FAM 22/22-mer oligoduplex substrate (10 nM) was incubated with 20 nM of hTopo either with or without compound and with or without CPT for 15 min at room temperature in binding buffer. Reactions were stopped by adding sodium dodecyl sulfate (SDS, final concentration 1%) to trap the covalently bound enzyme on the DNA. The samples were run through a 19% denaturing polyacrylamide gel containing 7 M urea to separate 3′ FAM 12 mer from 3′ FAM-22 mer. Imaging and quantification were performed as described above. The fraction of the total DNA that was covalently bound to hTopo was calculated as (3′ FAM 12 mer fluorescence)/(fluorescence 3′ FAM-22 + 3′ FAM-12).
Results
Arylstibonic Acid Library Screening Against hTopo
The 37 member arylstibonic acid library (Table 1) was obtained from the National Cancer Institute and the compounds were screened for their activity using a standard hTopo DNA supercoil relaxation assay. The initial screening was carried out using 5 μM concentrations of each compound and the activity results are summarized as black bars in Figure. 1. As a reference compound for the screen, we also included the known hTopo poison camptothecin (CPT). This preliminary screen uncovered four compounds that inhibited hTopo activity by greater than 40% (P6954, P6964, P6982, and 13759). This level of inhibition is similar to the effect of 5 μM CPT (Fig.1). Surprisingly, two substituted aryl stibonic acids were found to activate rather than inhibit hTopo (P6966 and 13778). The activation by 13778 is especially intriguing given that this compound has been previously characterized as a potent inhibitor of vTopo I [10]. Although the observed activation by these compounds is only 30% under the screening conditions, these levels are only lower limits to the activating potencies because the reactions in the presence of the activators were already complete at the time of quench. A complete analysis of the potent activating properties of P6966 is reported below.
Figure 1.

Inhibitory and activating properties of arylstibonic acids on supercoil relaxation by hTopo I (black bars) and vTopo (open bars). Screening reactions contained 5 μM compound, 14 nM pUC19 plasmid DNA and 1 nM hTopo or vTopo (see Methods).
We brought forward seven of the compounds for further testing in the DNA supercoil relaxation assay (P6954, P6964, P6966, P6982, 13759, 13765 and 13778). At least ten different compound concentrations in the range 0.1 to 20 μM were used to determine the concentration dependences of their activities, and representative data for three compounds P6982, P6966, 13759 and CPT are shown in Fig. 2. The most potent inhibitor, P6982, fully inhibited hTopo in this concentration range with half-maximal inhibition occurring at C0.5 = 3.4 ± 0.3 μM (Fig. 2A and Table 2). This inhibitory potency is similar to CPT (C0.5 = 1.2 ± 0.5 μM) (Fig. 2B and Table 2). Rather than inhibition, compound P6966 showed saturable activation in the micromolar concentration range (Fig. 2C), while 13759 showed activation at low concentrations, and inhibition at higher concentrations (Fig. 2D). The other compounds (13765 and 13778) also displayed a similar concentration dependent activation and inhibition (not shown). Because of the inherent difficulties in analyzing compounds that show both activation and inhibition, we only characterized the pure activator P6966.
Figure 2.

DNA supercoil relaxation by hTopo in the presence of various concentrations of compounds. pUC19 (14 nM) was incubated with 1 nM hTopo at room temperature for 15 min with increasing amounts of compounds (A) P6982, (B) CPT, (C) P6966, and (D) 13759. The first lane is a negative control without hTopo and compound. Abbreviations: R, relaxed DNA; SC, supercoiled DNA.
Table 2.
Potencies of Selected Arylstibonic Acids and Camptothecina
| compound | structure |
C0.5 (μM) |
|
|---|---|---|---|
| hTopob | vTopo | ||
| camptothecin |
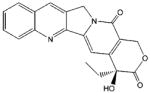
|
1.2 ± 0.5
(n = 1) |
~100c |
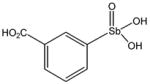
| P6954 |
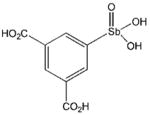
|
6.6 ± 0.4
(n = 3.6 ± 0.7) |
1.4 ± 0.6
(n = 1) |
| P6964 |
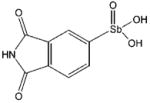
|
6.5 ± 0.7
(n = 3.9 ± 1.5) |
≥ 6 |
| P6982 |
|
3.4 ± 0.3
(n = 2.0 ± 0.4) |
≥ 5 |
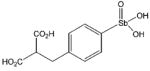
| 13759 |
|
~ 6 d | 0.21 ± 0.07
(n = 1) |
Reactions were carried out in the presence of 14 nM pUC19 plasmid DNA substrate and 1 nM hTopo or vTopo.
Values in parentheses are Hill coefficients.
ref #
This compound shows activation and inhibition. The value is the apparent concentration for 50% inhibition.
P6966 Activates hTopo
To further investigate the activating properties of P6966, we measured the initial rates of DNA supercoil relaxation at three different concentrations of the DNA substrate and four concentrations of P6966 in the range 1 to 16 μM (Fig. 3). Given the limited sensitivity of the supercoil relaxation assay, we were unable to extend rate measurements to DNA concentrations below 2 nM. However, we estimate the Km for supercoiled DNA to be around 1 nM based on extrapolation of initial rate data to lower concentrations and assuming a standard hyperbolic concentration dependence (not shown). This limitation prevents us from accurately measuring the activating effects of P6966 on the kinetic parameter kcat/Km. Nevertheless, we determined the velocity ratio (vA/vo) in the presence (vA) and absence (vo) of activator as a function of both substrate and P6966 concentration (Fig. 3B). This analysis clearly shows that activation is apparent when hTopo is present in a mixture of its free and bound forms ([S] = 3.5 nM), as well as its fully bound form ([S] ≫ Km), and suggests that P6966 is an activating ligand (A) that binds to both the free enzyme and enzyme-substrate complex. The maximal observed activation was vA/vo = 6.5 at 3.5 nM substrate and 16 μM activator (Fig. 3B).
Figure 3.
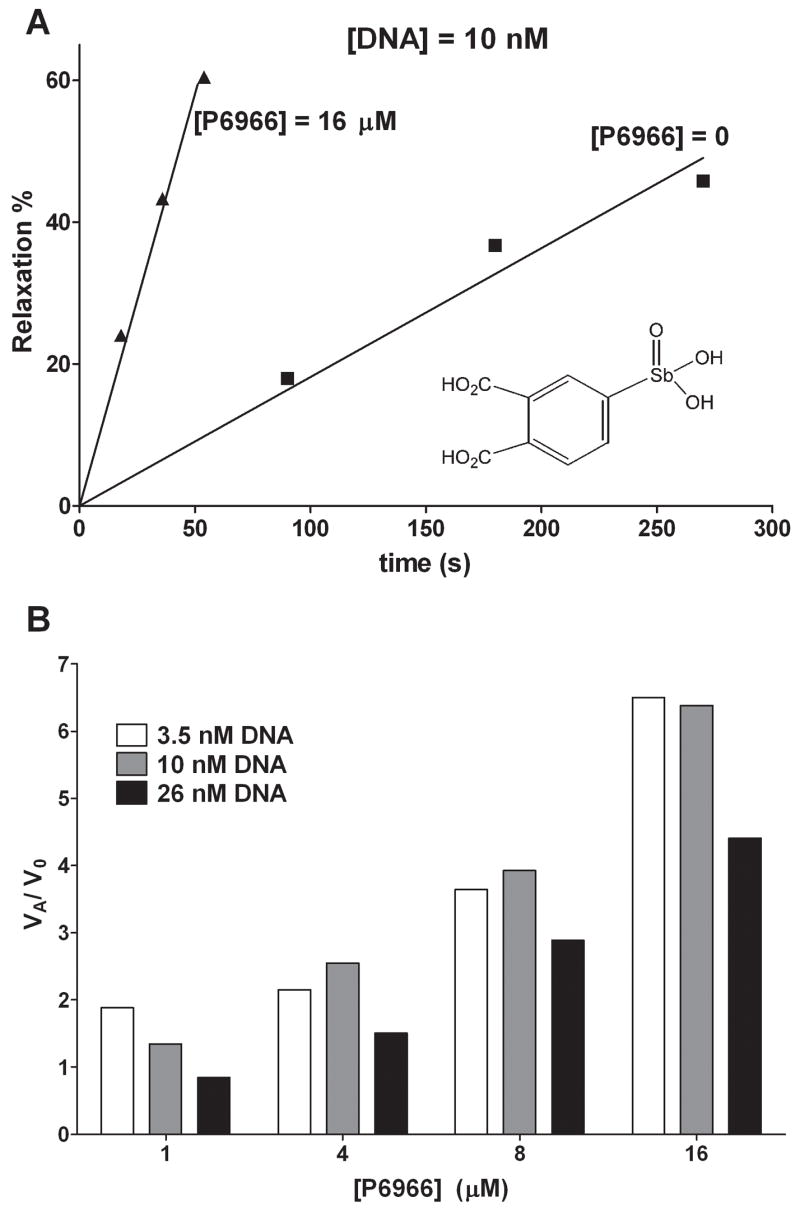
Activation by compound P6966. (A) Percentage relaxation as a function of reaction time with 10 nM of DNA substrate and 1 nM hTopo in the presence and absence of 16 μM P6966. (B) Relative reaction rate (vi/vo) of DNA supercoil relaxation with 1, 4, 8 and 16 μM of P6966 at 3.5, 10 and 26 nM DNA substrate and 1 nM hTopo.
Activity of Arylstibonic Acids with vTopo
Given that compound 13778 was first identified as a vTopo inhibitor [10], and this compound is an activator for hTopo, we were interested in further exploring the differential effects of this library on both enzymes. An initial screen was performed on vTopo using 1 and 5 μM concentrations of each compound. The vTopo inhibition profile at 5 μM compound is shown with open bars in Figure 1 for comparison with hTopo. Although ten compounds inhibited vTopo by ≥40% at 5 μM concentrations, only three of these were significantly inhibitory at 1 μM (P6954, 13759, 13778). Aside from the already characterized compound 13778, the most notable observations are (1) P6966 activates hTopo but has no effect on vTopo activity (Figs. 1 and 3), and (2) 13759 potently inhibits vTopo (Fig. 4), but activates hTopo at low concentrations and inhibits at higher concentrations (Figure 2D). The other weakly inhibitory compounds had similar potencies with both enzymes and were not investigated further (13755 and 13793). Complete concentration dependences of vTopo inhibition by the most potent new compounds P6954 and 13759 are shown in Figure 4 and the C0.5 values for inhibition are reported in Table 2.
Figure 4.
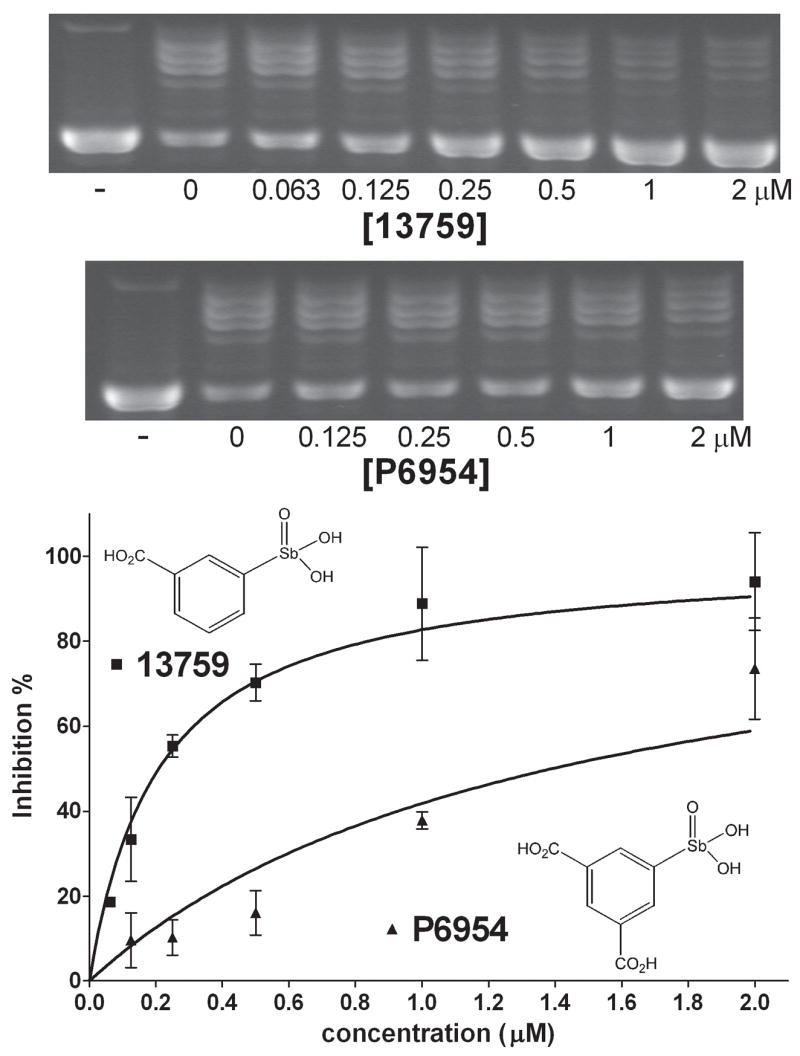
Concentration dependence of inhibition for the most potent arylstibonic acid vTopo inhibitors (13759, P6954) using 14 nM pUC19 DNA. Concentrations that produced half-maximal inhibition are reported in Table 2.
Effects of Compounds on Noncovalent and Covalent DNA Binding by hTopo
We investigated whether the compounds interfered with noncovalent DNA binding by hTopo using an electrophoretic mobility shift assay (EMSA)(Fig. 5). Inhibitors P6964 and P6982 prevented DNA binding by hTopo as judged by the disappearance of the slower mobility species corresponding to the hTopo-DNA complex (Fig. 5). In contrast, noncovalent DNA binding by hTopo was not diminished by either the activator P6966 or camptothecin, a known uncompetitive inhibitor that stabilizes the covalent adduct.
Figure 5.
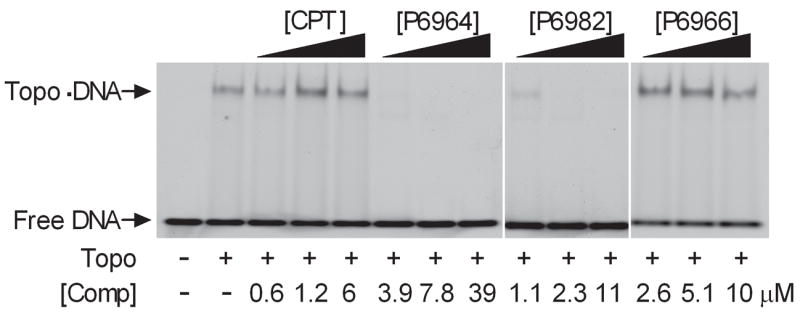
Effects of arylstibonic acid compounds on noncovalent DNA binding of hTopo (10 nM DNA). The hTopo bound and free DNA bands were resolved on 6% native polyacrylamide gel.
We then assessed whether the arylstibonic acids altered the equilibrium for DNA strand cleavage. In this assay, a 3′-end labeled oligonucleotide substrate is used where a labeled 12 mer strand is generated when hTopo reversibly cleaves the labeled strand at the preferred cleavage site (Fig. 6A). The amount of covalent species at equilibrium is then assessed by the rapid addition of sodium dodecylsulfate to the reaction, which irreversibly traps the enzyme in the covalent state. The cleaved 12-mer and uncleaved 22-mer DNAs are then separated by denaturing polyacrylamide gel electrophoresis (Fig. 6B). As expected, no detectable covalent complex is detected with hTopo alone, but the addition of camptothecin results in a concentration dependent accumulation of the 12-mer strand (Fig. 6B). We then assayed inhibitors P6964 and P6982 and the activator P6966 in the absence and presence of 5 μM CPT (Fig. 6 B). None of the compounds had any stimulatory effect on the formation of the covalent complex in the absence of CPT, and the inhibitors P6964 and P6982 decreased the amount of complex that was produced in the presence of 5 μM CPT. This indicates that these inhibitors bind competitively with respect to DNA, and shift the equilibrium towards free DNA and the EI complex. In contrast, the activator P6966 had no effect on the level of covalent complex induced by CPT, indicating that this compound is an allosteric activator that does not affect DNA binding or the equilibrium concentration of the covalent complex.
Figure 6.

Effects of CPT and arylstibonic acid on the DNA cleavage equilibrium. (A) hTopo forms a reversible phosphotyrosyl linkage with DNA. Upon trapping the covalent complex with SDS, a FAM-labeled 12-mer DNA strand is generated which is in direct proportion to the amount of covalent complex formed. (B) Separations of 22-mer DNA and 12-mer cleavage product by denaturing PAGE. The additions to each reaction are indicated. A DNA concentration of 10 nM was used.
Discussion
Structure-Activity Relationships
The discrete structural differences between the arylstibonic acid library members allows the inference of functional groups that are required for inhibition and activation of hTopo and vTopo (Figure 7). For hTopo, potent activation requires the presence of negatively charged carboxylate groups at both the meta and para positions of the phenyl ring relative to the stibonic acid moiety (i.e. P6966). This conclusion is supported by the findings that (1) placement of a single carboxylate group at the meta position results in combined activation and inhibition, and furthermore, esterification of this group abolishes activity (compare 13759 and 13771), (2) placement of a single carboxylate group at the para position results in loss of compound activity (i.e. 13760), (3) a propenoic acid group at the meta position confers both activation and inhibition (i.e. 13778), and (4) placement of a charged carboxylate group at both meta positions abolishes activation and leads to weak inhibition (Table 2, P6954). We construe that for P6966, the negatively charged substituent at the meta position is required for activation, and the second carboxylate at the para position serves to thwart binding to an inhibitory binding site. Thus without the para substituent, the other compounds activate and then inhibit as the compound concentration increases, or alternatively, simply act as a weak inhibitors.
Figure 7.

Structure-activity relationships for arylstibonate activators and inhibitors of hTopo and vTopo (see text).
Although the inhibitors of hTopo follow less obvious structure-activity trends as compared to the activators (Figure 7), the two most potent inhibitors have essential carboxylate side chains that superimpose in three-dimensional space and could occupy the same inhibitory binding site on hTopo (P6982 and P6954). The importance of the anionic dicarboxylate functional group at the para position of P6982 is exemplified by the lack of activity of the esterified version (P6972), and the absence of activity of compound P6981, which has the same dicarboxylate group located at the meta position. The weaker binding inhibitor P6964 has a pyrrole-2, 5-dione ring system and may bind in a different binding mode from the other two.
Specificity of Aryl stibonic Acids for hTopo and vTopo
Although several weak binding arylstibonic acids inhibit both vTopo and hTopo, interesting specificities are observed for other arylstibonic acids. As reported previously, the arylstibonic acid 13778 strongly inhibited vTopo in the nanomolar concentration range, but concentrations higher than 5 μM were required to inhibit hTopo, resulting in a specificity for vTopo of approximately 102-fold [10]. Here a more detailed study has revealed that 13778 actually stimulates hTopo at lower concentrations, and inhibits at higher concentrations. Like 13778, the other inhibitor of vTopo found in this work (13759) has a carboxylate group at the meta position relative to the stibonic acid group, suggesting a common binding site for these analogues (Fig. 7). Also reminiscent of 13778, the potent vTopo inhibitor 13759 is both an activator and weak inhibitor of hTopo, with a selectivity for inhibition of vTopo of around 50-fold. The varied effects and selectivities of the compounds contained in this fairly small directed library suggest that further diversification around the arylstibonic acid scaffold would be fruitful.
Possible Mechanisms of Inhibition and Activation
Since antimony is a group V-A element of the periodic table, stibonate resembles both phosphate and arsenate in its electronic properties [13]. However, certain properties of Sb(V) may make it an especially potent element in the inhibition of macromolecules that bind to DNA. These properties include its lower electronegativity which would tend to increase the Sb-O bond length as compared to P-O. Increased bond lengths would be expected to lead to greater negative charge density on the stibonate oxygen atoms and would enhance coulombic interactions with cationic enzyme groups [13]. Another feature of the most potent arylstibonic acid inhibitors discovered here is the combined presence of the stibonate group and at least one carboxylate group (Table 2, Figure 7). We imagined that these functional groups might be positioned at an appropriate distance and orientation to mimic the negatively charged phosphate ester groups in B DNA, and therefore, competitively occupy phosphate binding sites on the enzyme. Both hTopo and vTopo bind circumferentially to B DNA and interact with phosphates on both DNA strands for 6 to 10 base pairs around the cleavage site [3, 14, 15]. To test this binding model, we attempted to align the antimony and carboxylate moieties of the best hTopo inhibitor P6982 with DNA extracted from the crystal structure of hTopo bound to DNA (Fig. 8)(pdb code 1A35). We found that the stibonate and carboxylate moieties of P6982 could not be superimposed with the intrastrand phosphates of DNA, but that the oxygen atoms could be positioned to nearly superimpose the cross strand phosphate oxygens (Fig. 8). In contrast, the stibonate and carboxylate moieties of the vTopo inhibitors 13759 and 13778 very nicely superimpose the adjacent intrastrand phosphates of B DNA (Figure 8). Thus, the antimonate and carboxylate moieties of these aryl stibonic acids are positioned appropriately to serve as mimics of both intra- and interstrand phosphates in B DNA, which may be the mechanistic basis for their effects on hTopo and vTopo activity. Occupation of several phosphate binding sites on the protein target provides a reasonable explanation for the Hill coefficients in the range n ~ 2 to 4 for binding of P6982, P6964 and P6954 to hTopo (Table 2). In contrast, inhibition of vTopo by P6954 and 13759 involves binding to only one site (n = 1, Table 2). Finally, it is significant to point out that although the inhibitor P6982 binds with a modest C0.5 of about 3 μM under our standard reaction conditions, the DNA substrate binds competitively with an affinity in the low nanomolar range. Thus, the true dissociation constant for P6982 must be at least an order of magnitude lower than the observed value, which exemplifies the inherent difficulty of developing effective inhibitors that bind competitively with DNA.
Figure 8.

Stibonate and carboxylate moieties of the arylstibonic acids may mimic both intra- and interstrand phosphates in B DNA and affect hTopo and vTopo DNA binding or catalysis. The stibonate and carboxylate moieties of hTopo inhibitor P6982 cannot be superimposed with the intrastrand phosphate of DNA. However, its carboxylate and stibonate oxygen atoms nearly superimpose the cross strand phosphate oxygens. In contrast, vTopo inhibitors 13759 and 13778 superimpose the intrastrand phosphates of B DNA. The models were generated by extracting the given nucleotides from the bound DNA in the crystallographic model (pdb code 1A35) and then superimposing the carboxylate and stibonate moieties with either the inter- or intrastrand DNA phosphates.
The observed activation of hTopo by P6966 requires a binding site different from the competitive inhibitors of DNA substrate binding. Little can be said with respect to the nature of the activation mechanism of P6966 except that the rate limiting step during catalytic turnover must be accelerated. Based on studies of vTopo, it is likely that turnover of hTopo is limited by product release [11, 16], which may be the step targeted by P6966. The interesting finding that a ligand of hTopo serves as an activator, suggests an allosteric mechanism where the effects of ligand binding are transmitted over a distance to the enzyme-DNA interaction surface. This finding suggests that it may also be possible to identify new hTopo poisons that also operate allosterically rather than by direct intercalation at the site of DNA cleavage. Arylstibonic acids could serve as a useful scaffold for this purpose.
Arylstibonates as Potential Tools to Elucidate hTopo Functions in Cells
It has been problematic to study the biological functions of hTopo because its genetic knockout is embryonic lethal in flies and mice [17, 18]. Recently, small interfering RNA (siRNA) cell lines have been designed that provide a knock down of hTopo protein levels by 80% [19], and have shown that hTopo plays a role in a maintaining genomic stability, the transcription of specific genes, and modulating the response to a variety of chemotherapeutic agents. The ability of P6982 to reverse the formation of CPT induced covalent complexes in vitro indicates that small molecules may be designed that could selectively, rapidly and reversibly knock down the enzymatic activity of hTopo in cells, providing an additional useful tool to elucidate its function. However, P6982 itself may not be an ideal candidate for this purpose because of its potential off-target toxic effects, and the observation that this inhibitor did not reverse covalent complexes in the context of chromatin. However, more potent arylstibonic acids related to P6982 may provide such tools.
Acknowledgments
We thank the Developmental Therapeutics Program at the National Cancer Institute for the small molecule libraries. This work was supported by NIH grants GM56834 to J.T.S and GM49156 to J.J.C. and in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract N01-CO-12400, and in part by the Developmental Therapeutics Program in the Division of Cancer Treatment and Diagnosis of the National Cancer Institute. The content of the publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does the mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Liu LF, Desai SD, Li TK, Mao Y, Sun M, Sim SP. Ann N Y Acad Sci. 2000;922:1–10. doi: 10.1111/j.1749-6632.2000.tb07020.x. [DOI] [PubMed] [Google Scholar]
- 2.Chrencik JE, Staker BL, Burgin AB, Pourquier P, Pommier Y, Stewart L, Redinbo MR. J Mol Biol. 2004;339:773–784. doi: 10.1016/j.jmb.2004.03.077. [DOI] [PubMed] [Google Scholar]
- 3.Redinbo MR, Stewart L, Kuhn P, Champoux JJ, Hol WG. Science. 1998;279:1504–113. doi: 10.1126/science.279.5356.1504. [DOI] [PubMed] [Google Scholar]
- 4.Stewart L, Redinbo MR, Qiu X, Hol WG, Champoux JJ. Science. 1998;279:1534–1541. doi: 10.1126/science.279.5356.1534. [DOI] [PubMed] [Google Scholar]
- 5.Staker BL, Hjerrild K, Feese MD, Behnke CA, Burgin AB, Jr, Stewart L. Proc Natl Acad Sci U S A. 2002;99:15387–15392. doi: 10.1073/pnas.242259599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Champoux JJ. Ann N Y Acad Sci. 2000;922:56–64. doi: 10.1111/j.1749-6632.2000.tb07025.x. [DOI] [PubMed] [Google Scholar]
- 7.Pommier Y. Nat Rev Cancer. 2006;6:789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- 8.Pommier Y, Pourquier P, Urasaki Y, Wu J, Laco GS. Drug Resist Updat. 1999;2:307–318. doi: 10.1054/drup.1999.0102. [DOI] [PubMed] [Google Scholar]
- 9.Pourquier P, Pommier Y. Bull Cancer Spec. 1998;99:5–10. [PubMed] [Google Scholar]
- 10.Bond A, Reichert Z, Stivers JT. Mol Pharmacol. 2006;69:547–557. doi: 10.1124/mol.105.019067. [DOI] [PubMed] [Google Scholar]
- 11.Kwon K, Stivers JT. J Biol Chem. 2002;277:345–352. doi: 10.1074/jbc.M109449200. [DOI] [PubMed] [Google Scholar]
- 12.Stewart L, Champoux JJ. Methods Mol Biol. 1999;94:223–234. doi: 10.1385/1-59259-259-7:223. [DOI] [PubMed] [Google Scholar]
- 13.Seiple LA, Cardellina JH, Akee R, Stivers JT. Mol Pharmacol. 2008;73:669–677. doi: 10.1124/mol.107.042622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tian L, Claeboe CD, Hecht SM, Shuman S. Structure. 2004;12:31–40. doi: 10.1016/j.str.2003.11.025. [DOI] [PubMed] [Google Scholar]
- 15.Perry K, Hwang Y, Bushman FD, Van Duyne GD. Mol Cell. 2006;23:343–354. doi: 10.1016/j.molcel.2006.06.015. [DOI] [PubMed] [Google Scholar]
- 16.Kwon K, Nagarajan R, Stivers JT. Biochemistry. 2004;43:14994–15004. doi: 10.1021/bi048801s. [DOI] [PubMed] [Google Scholar]
- 17.Morham SG, Kluckman KD, Voulomanos N, Smithies O. Mol Cell Biol. 1996;16:6804–6809. doi: 10.1128/mcb.16.12.6804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee MP, Brown SD, Chen A, Hsieh TS. Proc Natl Acad Sci U S A. 1993;90:6656–6660. doi: 10.1073/pnas.90.14.6656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miao ZH, Player A, Shankavaram U, Wang YH, Zimonjic DB, Lorenzi PL, Liao ZY, Liu H, Shimura T, Zhang HL, Meng LH, Zhang YW, Kawasaki ES, Popescu NC, Aladjem MI, Goldstein DJ, Weinstein JN, Pommier Y. Cancer Res. 2007;67:8752–8761. doi: 10.1158/0008-5472.CAN-06-4554. [DOI] [PubMed] [Google Scholar]