

Abstract
Microglia are the resident immune cells of the CNS, which are important for preserving neural tissue functions, but may also contribute to neurodegeneration. Activation of these cells in infection, inflammation, or trauma leads to the release of various toxic molecules, including reactive oxygen species (ROS) and the excitatory amino acid glutamate. In this study we used an electrophysiological approach and a D-[3H]aspartate (glutamate) release assay to explore the ROS-dependent regulation of glutamate-permeable volume-regulated anion channels (VRACs). Exposure of rat microglia to hypoosmotic media stimulated Cl− currents and D-[3H]aspartate release, both of which were inhibited by the selective VRAC blocker DCPIB. Exogenously applied H2O2 potently increased swelling-activated glutamate release. Stimulation of microglia with zymosan triggered production of endogenous ROS and strongly enhanced glutamate release via VRAC in swollen cells. The effects of zymosan were attenuated by the ROS scavenger MnTMPyP, and by two inhibitors of NADPH oxidase (NOX) diphenyliodonium and thioridazine. However, zymosan-stimulated glutamate release was insensitive to other NOX blockers, apocynin and AEBSF. This pharmacological profile pointed to the potential involvement of apocynin-insensitive NOX4. Using RT-PCR we confirmed that NOX4 is expressed in rat microglial cells, along with NOX1 and NOX2. To check for potential involvement of phagocytic NOX2 we stimulated this isoform using protein kinase C (PKC) activator PMA, or inhibited it with the broad spectrum PKC blocker Gö6983. Both agents potently modulated endogenous ROS production by NOX2, but not VRAC activity. Taken together, these data suggest that the anion channel VRAC may contribute to microglial glutamate release, and that its activity is regulated by endogenous ROS originating from NOX4.
Keywords: microglial cells, VRAC, glutamate release, reactive oxygen species, NOX4
Introduction
Microglia are the resident immunocompetent cells of the central nervous system. In the adult brain, microglial cells are typically found in a resting state, in which they survey their local environment for any signals indicating traumatic injury, infection, or inflammation (Kreutzberg 1996; Farber and Kettenmann 2005). Upon exposure to invading pathogens, neuronal debris, elevated extracellular ATP levels, or pro-inflammatory cytokines and chemokines, microglia rapidly convert to an activated state. Once activated, they produce both cytotoxic and neuroprotective molecules (Kreutzberg 1996; Nakajima and Kohsaka 2001). The most prevalent microglial toxins are IL-1β, TNFα, reactive oxygen species (ROS), and reactive nitrogen species (RNS) (Nakajima and Kohsaka 2001; Block et al. 2007). Although ROS and RNS production is an intrinsic part of the CNS response to infections, trauma, neurodegeneration, or brain tumors, it frequently results in bystander neurotoxicity. ROS and RNS, including those produced by microglia, are thought to play an important role in numerous neurodegenerative processes (Nakajima and Kohsaka 2001; Halliwell 2006; Block et al. 2007). The primary source of ROS production in microglia are NADPH oxidases, multi-subunit heme-containing enzymes that catalyze the reduction of molecular oxygen to superoxide anion (O2−) using NADH or NADPH as a substrate (Bedard and Krause 2007).
A less recognized mediator of microglial neurotoxicity is the amino acid glutamate, which serves as the main excitatory neurotransmitter in the CNS. Several previous studies demonstrated that activated microglia release glutamate in quantities sufficient to induce neuronal death via overactivation of ionotropic glutamate receptors (Piani et al. 1991; Piani et al. 1992; Barger and Basile 2001; Takeuchi et al. 2006). Piani and co-workers suggested that glutamate, rather than ROS and/or inflammatory cytokines, is the key factor contributing to microglial toxicity (Piani et al. 1992). Two glutamate transporting pathways that have been proposed to mediate glutamate release from microglia are the cystine-glutamate antiporter and connexin hemichannels (Piani and Fontana 1994; Barger and Basile 2001; Takeuchi et al. 2006). The cystine-glutamate antiporter (system Xc-) exchanges one intracellular glutamate for extracellular cystine, a homodimer of cysteine, which is used for the synthesis of the antioxidant glutathione (McBean 2002). This transporter is upregulated in immune cells in response to oxidative stress in order to maintain sufficient levels of glutathione, and is abundantly expressed in activated microglia (Sato et al. 2001; Qin et al. 2006). However, Takeuchi et al. (2006) recently found that connexin hemichannels, rather than the cystine-glutamate antiporter, largely mediate microglial glutamate release and excitotoxic neuronal damage in a TNFα toxicity model. Additionally, in astrocyte cultures, it has been demonstrated that glutamate release in response to hypoosmotic swelling and chemical ischemia is mediated by two anion channels: volume-regulated anion channel (VRAC) and Gd3+-sensitive maxi-anion channel (Liu et al. 2006).
VRACs are ubiquitously expressed Cl− channels whose molecular identity remains unknown (Strange et al. 1996; Nilius and Droogmans 2003; Okada 2006). In addition to inorganic anions, VRACs are permeable to several amino acids, including the excitatory amino acids glutamate and aspartate (Kimelberg et al. 1990; Banderali and Roy 1992; Jackson et al. 1994; Abdullaev et al. 2006). Although it has not been demonstrated in microglia, VRACs may contribute to pathological glutamate release, as seen in ischemia and other neurological disorders (Kimelberg 1995; Kimelberg and Mongin 1998; Mongin and Kimelberg 2005). The presence of VRACs in mammalian cells is typically determined by measuring increases in Cl− conductance in response to cell swelling using electrophysiology. Besides sensitivity to cell volume changes, the key biophysical characteristics of VRACs include intermediate conductance, moderate outward rectification, Eisenman's type I anion permeability sequence (SCN− > I− >NO3− > Cl− > F− > gluconate), requirement for cytosolic ATP, and time-dependent inactivation at large positive potentials (Strange et al. 1996; Okada 1997; Nilius et al. 1997).
Several studies demonstrated that microglial cell functions are strongly regulated by changes in the expression and activity of K+, H+, and Cl− channels (reviewed in Eder 2005). Volume- and/or membrane stretch-sensitive Cl− channels, resembling VRACs, were found in cultured microglial cells and have been proposed to participate in morphological transitions, migration, proliferation, and phagocytosis in this cell type (Schlichter et al. 1996; Eder et al. 1998; Ducharme et al. 2007). Although the primary physiological role for VRACs is regulation of cellular volume, they are also activated during other cellular processes, such as apoptosis, cell motility, and proliferation (Okada et al. 2001; Lang et al. 2000; Stutzin and Hoffmann 2006). Several recent studies have found that VRACs may be activated or positively modulated by ROS, particularly H2O2 (Shimizu et al. 2004; Varela et al. 2004; Haskew-Layton et al. 2005). In ventricular myocytes, endogenous ROS have been demonstrated to regulate stretch-activated and swelling-activated Cl− currents, both likely mediated by VRAC (Browe and Baumgarten 2004; Ren et al. 2008). Since microglia robustly generate ROS as part of their immunological responses, they represent an ideal cell type for studying the impact of endogenously produced ROS on VRAC activity and VRAC-mediated release of organic osmolytes. In the present work we focused on the VRAC-mediated release of the excitatory amino acids, glutamate and aspartate, because of the significant physiological and pathological impact of these neurotransmitters on neuronal signaling and viability in the brain.
Methods
Materials
4-(2-Aaminoethyl) benzenesulfonyl fluoride hydrochloride (AEBSF), apocynin, dipehnyliodonium chloride (DPI), nitroblue tetrazolium chloride (NBT), thioridazine, zymosan A (S. cerevisiae), and H2O2 (30%), were purchased from Sigma-Aldrich. Gö6983, Mn(III)tetrakis(1-methyl-4-pyridyl)porphyrin pentachloride (MnTMPyP), and phorbol 12-myristate 13-acetate (PMA) were acquired from EMD-Calbiochem (La Jolla, CA, USA). 4-[(2-Butyl-6,7-dichloro-2-cyclopentyl-2,3-dihydro-1-oxo-1H-inden-5-yl)oxy]butanoic (DCPIB) acid was from Tocris Cookson (Ellisville, MO, USA).
Cell culture preparation
Microglial cells were purified from mixed glial cultures, which were prepared from the cortices of neonatal Sprague-Dawley rat according to the procedure approved by the Albany Medical College Animal Care and Use Committee. After the isolation, the cortical tissue was enzymatically dissociated using three enzymatic extractions with protease Dispase II (Sigma-Aldrich, St. Louis, MO, USA). The first extraction was discarded and DNAse I (Sigma, St. Louis, MO, USA) was added. Dissociated cortical cells were plated in T-75 flasks and grown for two-three weeks in minimal essential medium (MEM) plus 10% fetal bovine serum (FBS) and additionally supplemented with 50 units/mL penicillin and streptomycin (all cell culture reagents were from Invitrogen, Carlsbad, CA, USA) in a humidified 5% CO2/95% air atmosphere at 37°C. Culture medium was replaced bi-weekly, and the penicillin and streptomycin were no longer added to the media after ten days of cultivation. This procedure yields a confluent mixed glial culture largely consisting of astrocytes and microglia. For purifying microglia, mixed glial cultures were shaken for approximately two to five minutes on a titer plate shaker. Floating microglial were collected, sedimented by a brief centrifugation, resuspended in Opti-MEM supplemented with 2% B-27 serum-free supplement minus antioxidants, and plated onto 18 mm2 glass coverslips (Caroline Biological Supply Co., Burlington, NC, USA) or cell culture dishes coated with poly-d-lysine (Sigma, St. Louis, MO, USA). Cells were maintained in Opti-MEM/B-27 for at least 12 hours before being used in subsequent experiments.
Immunocytochemistry
To verify the purity our microglia cultures, we stained the isolated microglial cells for OX-42 (CD11b), a complement receptor that within the CNS is expressed solely by microglia. Briefly, microglial cells plated onto 18 mm2 glass coverslips were fixed in 4% paraformaldehyde for 20 minutes at room temperature (22°C). The cells were then blocked in 10% normal goat serum (Invitrogen) for 30 minutes at room temperature. Blocked cells were incubated with an OX-42 mouse anti-rat monoclonal antibody (1:500; BD Pharmingen, San Diego, CA, USA) for two hours at room temperature, washed with physiological phosphate saline, and incubated with an Alexa488-conjugated goat anti-mouse secondary antibody (1:1,000 Molecular Probes, Carlsbad, CA, USA) for 45 minutes at room temperature. After completion of immunohisto-chemical staining microglia were additionally counterstained with DAPI (100 ng/mL; Sigma) for five minutes at room temperature. Images were analyzed with an Olympus Optical Provis AX70 microscope equipped with conventional fluorescence filters (Cy2/Alexa488: 460-500 nm excitation, 510-560 nm emission; DAPI/Hoechst: 375-400 nm excitation, 450-475 nm emission). Fluorescent images were captured with a high-resolution camera (Sony, DKC-ST5) interfaced with Northern Eclipse (Empix Imaging, Mississaugau, ON), Photoshop (Adobe, San Jose, CA), and Neurolucinda (MicroBrightField, Colchester, VT) software. This procedure revealed that >98% cells were positive for OX-42 (CD11b) and therefore are of microglial origin.
Whole-cell patch clamp recordings
Microglial cells were plated on poly-d-lysine-treated glass coverslips at low density in MEM supplemented with heat-inactivated horse serum (HIHS, Invitrogen). After three hours they were transferred into B-27-supplemented Opti-MEM as described above. Adhered cells were moved into electrophysiological media described below. Whole-cell recordings were performed at room temperature as described previously (Kubo and Okada 1992; Abdullaev et al. 2006). Patch electrodes were fabricated from borosilicate glass capillaries using a micropipette puller (P-97, Sutter Instruments, Novato, CA), with a resistance of 3-3.5 MΩ when filled with pipette solution. Series resistance was ≤15 MΩ. Currents were recorded using an Axopatch 200B amplifier (Axon Instruments, Foster City, CA). pCLAMP software (version 9.2, Axon Instruments) was used for command pulse control, data acquisition, and analysis. Series resistance was compensated in all experiments. Current signals were filtered at 2 kHz using a four-pole Bessel filter and digitized at 4 kHz. The time course of current development was monitored by applying every 15 seconds alternating one-second step pulses from a holding potential of 0 to test pulses ±40 mV. After attaining steady-state activation of Cl− currents, their biophysical properties were determined by applying 2-s step pulses from 0 mV to test potentials of -100 to +100 mV in 20-mV increments. Short prepulse to -100 mV were applied before each test pulse to assure complete activation of VRAC channels. The isoosmotic external solution contained (in mM): 110 CsCl, 2 CaCl2, 1 MgSO4, 5 glucose, 10 HEPES, and 60 mannitol (pH 7.4, 290 mosM). The hypoosmotic solution was made by omitting mannitol from isotonic solution and had an osmolarity of 230 mosM. The pipette solution contained (in mM): 110 CsCl, 1 MgSO4, 1 Na2-ATP, 0.3 Na2-GTP, 15 Na-HEPES, 10 HEPES, and 1 EGTA (pH 7.3, 255 mosM). The osmolarity of the pipette solution was set lower than that of the isotonic bath solution in order to prevent spontaneous cell swelling after attaining the whole-cell mode (Worrell et al. 1989).
Excitatory amino acid efflux assay
The release of excitatory amino acids from primary microglia was measured using a non-metabolized L-glutamate/L-aspartate analog, D-[3H]aspartate, as described elsewhere (Mongin and Kimelberg 2002). In brief, primary microglia plated on 18 mm2 glass coverslips were loaded for 3 hours with 8 μCi/mL D-[3H]aspartate in MEM plus 10% HIHS. Loaded microglial cells were washed of extracellular isotope and serum-containing medium with HEPES-buffered solution, and then placed in a Lucite perfusion chamber. Cells were suffused with isoosmotic or hypoosmotic media at 1.2 mL/min. The isoosmotic medium contained (in mM) 135 NaCl, 3.8 KCl, 1.2 MgSO4, 1.3 CaCl2, 1.2 KH2PO4, 10 d-glucose, and 10 HEPES (pH 7.4). The hypoosmotic medium was prepared by reducing NaCl concentration to 85 mM, which translates into a 30% reduction in the medium osmolarity. Suffused media fractions were collected at one minute intervals. Isotope (D-[3H]aspartate) content in each fraction was determined using a PerkinElmer Tri-Carb 1900TR liquid scintillation analyzer (PerkinElmer, Downers Grove, IL, USA) after addition of four mLs of Ecoscint A scintillation cocktail (National Diagnostics, Atlanta, GA, USA). The isotope remaining in the cells was extracted with a solution containing 2% sodium dodecylsulfate plus 8 mM EDTA. Percent fractional isotope release for each time point was determined by dividing radioactivity released in each 1-minute interval by the radioactivity remaining in the cells at this time point, as retroactively calculated using a custom computer program.
Nitroblue tetrazolium (NBT) assay
The quantitation of O2− production in primary microglia was performed using a nitroblue tetrazolium assay as described elsewhere (Choi et al. 2006). In brief, primary microglial cells grown on 24-well culture plates were incubated with nitroblue tetrazolium chloride and various compounds that promote and/or inhibit the production of O2− for 10 or 30 minutes at 37°C. Cells were then rinsed with warm PBS and fixed in 100% methanol. Reduced formazan particles, contained inside cells, were dissolved with 2M KOH in DMSO, and the absorbance was read at 620 nm on a BioTek ELx800 absorbance microplate reader (BioTek Winooski, VT, USA).
Total RNA isolation and RT-PCR
Total RNA was isolated from primary rat microglia using the RNAqueous-4PCR kit (Ambion, Austin, TX, USA) according to the manufacturer's protocol. The concentration and purity of the isolated microglial RNA was determined using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). One μg of isolated RNA was then converted into cDNA using an iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA), adhering to the manufacturer's instructions, in a Mastercycler thermocycler (Eppendorf, Westbury, NY, USA). One μl of the cDNA product was used for PCR amplification of the NOX1-4 and GAPDH transcripts, using rat specific primers included in Table 1.
Table 1.
Species specific primers for RT-PCR analysis of NOX isoform expression in microglia
| NOX isoform | PCR primers | Predicted product (bp) | |
|---|---|---|---|
| Sense | Antisense | ||
| NOX1 | AAGCCATTGGATCACAACCT | GGCTTCTACTGTAGCGTTCA | 115 |
| NOX2 | CATAAGATGGTAGCTTGGAT | GAGTGCTACTGAATATGGGT | 123 |
| NOX3 | AAGCCATTGGATCACAACCT | GGCTTCTACTGTAGCGTTCA | 108 |
| NOX4 | GCCTAGGATTGTGTTTGAGC | CGAAGGTAAGCCAGGACTGT | 100 |
Statistical analysis
The statistical analyses of the data were performed using either Student's t-test or one-way ANOVA followed by Tukey's post-hoc analysis for multiple comparisons. Results of the excitatory amino acid efflux assays were separately analyzed for the maximal release values under hypo-osmotic conditions, or integral release values during the whole duration of the hypoosmotic exposure. Because statistical significances of the data were very similar for both parameters, we presented only comparisons of maximal releases in the text and figures. Origin 6.0 (OriginLab, Northampton, MA) and Prism 5 (GraphPad, San Diego, CA) were used for statistical analysis.
Results
Characterization of swelling-activated, glutamate-permeable pathway in primary rat microglia
In our initial experiments, we tested for the functional expression of a volume-sensitive Cl− permeability pathway in primary rat microglial cells using a whole-cell patch clamp electrophysiological approach. We hypothesized that microglia, as the majority of other cells, express VRACs. To activate VRACs, we induced cell swelling by exposing microglia to hypoosmotic medium (20.7% reduction in medium osmolarity), which elicited slowly developing, outwardly rectifying Cl− currents (Fig. 1a). The average current density in five successfully patched cells was 40.8 ±5.7 pA/pF at +40 mV. This was in the same order of magnitude as the VRAC current densities registered under identical experimental conditions in primary rat astrocytes (19.7 ±3.3 pA/pF, Abdullaev et al., 2006). The registered currents were similar to those defined for VRAC in other cell types in several respects. Similar to what is shown in Fig. 1b, in all five cells, swelling-activated Cl− currents were outwardly rectifying and showed time-dependent inactivation at +100 mV, although it was less pronounced than in other cell types. Importantly, these Cl− currents were completely and reversibly inhibited by DCPIB, a pharmacological compound that discriminates VRACs from other Cl− channels (Decher et al. 2001; Abdullaev et al. 2006).
Fig. 1. Cultured primary rat microglial cells express volume-regulated anion channels, which are permeable to excitatory amino acids.

(a) Representative electrophysiological recording of Cl− currents in a single cultured microglial cell exposed to hypoosmotic medium. Cells were held at 0 mV and alternative one-second pulses to ±40 mV were applied to measure activation of Cl− currents every 15 seconds. The VRAC blocker DCPIB (20 μM) was applied after Cl− currents reached steady-state level. (b) Cl− current responses to step pulses from -100 to +100 mV in 20 mV increments applied after swelling-activated Cl− currents reached steady-state level. Representative traces of electrophysiological recordings in 5 cells. (c) Effect of the VRAC blocker DCPIB (20 μM) on hypoosmotic medium-induced D-[3H]aspartate release from microglia. Cells were loaded for 3 hours with D-[3H]aspartate. Extracellular isotope was then washed and cells were placed in a Lucite perfusion chamber. Data represent the mean values ±SE of six experiments. *** p < 0.001, DCPIB vs. control hypoosmotic medium (Hypo).
To determine whether VRACs expressed in primary microglia are permeable to glutamate, we employed an excitatory amino acid efflux assay. Primary microglia were loaded with D-[3H]aspartate, a non-hydrolyzable analog of glutamate, and then suffused with isoosmotic or hypoosmotic media. The exposure of microglia to hypoosmotic medium (30% reduction in medium osmolarity) resulted in a significant release of D-[3H]aspartate, and this effect was potently suppressed by 20 μM DCPIB, suggesting that microglial VRACs are permeable to glutamate (Fig. 1c).
Exogenous H2O2 enhances the release of glutamate from primary microglia via VRAC
It has been demonstrated in various cell types, including astrocytes, that VRACs may be activated or modulated by the reactive oxygen species hydrogen peroxide (H2O2) (Shimizu et al. 2004; Varela et al. 2004; Haskew-Layton et al. 2005; Ren et al. 2008). To address the question of whether H2O2 impacts VRAC activity in microglia, we exposed microglia to exogenous H2O2 under isoosmotic and hypoosmotic conditions. In non-swollen cells, 500 μM H2O2 produced a small gradual increase in D-[3H]aspartate release levels (Fig. 2a, ∼2-fold increase above basal levels, p<0.05, repeated measures ANOVA). However, when applied in conjunction with hypoosmotic medium, H2O2 significantly potentiated the swelling-activated D-[3H]aspartate release from microglia by ∼70% (Fig. 2a). The effect of H2O2 was completely inhibited by DCPIB, suggesting that VRAC is the source (Fig. 2a). DCPIB on its own had no antioxidant properties, as verified in ROS assays in microglia and macrophages (data not shown). We further explored which concentrations of H2O2 are most effective in regulating VRAC activity. We found a biphasic effect of H2O2 with the maximal stimulation at 100 μM (Fig. 2b). At higher H2O2 concentrations, potentiation of excitatory amino acid release was diminished, which may suggest a redox-dependent inactivation of VRAC itself or its regulatory mechanism(s) (Fig. 2b). To assure that the effect of H2O2 is mediated by increases in VRAC activity, we additionally performed several electrophysiological experiments in which H2O2 was applied to microglia under isoosmotic or hypoosmotic conditions. Similar to glutamate release results, 100 μM H2O2 did not increase basal Cl− currents in non swollen cells (n=3, data not shown). However, when 100 μM H2O2 was applied in conjunction with hypoosmotic medium, after swelling-activated Cl− currents reached steady-state levels, there was additional 54% increase in current density (from 45.9 ±2.6 to 70.7±3.9 pA/pF, n=3, p=0.006).
Fig. 2. Exogenous reactive oxygen species H2O2 potentiate the excitatory amino acid release from primary microglia via VRAC.

(a) Primary microglia were exposed to H2O2 (500 μM) 10 minutes prior to and during the 10-minute suffusion with hypoosmotic medium in the presence or absence of the VRAC blocker DCPIB (20 μM). Data represent the mean values ±SE of six experiments. **p<0.01, H2O2 vs. Hypo, and p<0.001, H2O2 vs. H2O2+DCPIB. (b) The effect of varying concentrations of H2O2 on the release of D-[3H]aspartate from microglia under hypoosmotic conditions. These experiments were conducted in a similar fashion as in (a). For clarity only maximal D-[3H]aspartate release data are shown. Mean values ±SE of five to ten experiments in each group are presented. *p<0.05, **p<0.01, ***p<0.001, H2O2 vs. hypoosmotic control; one-way ANOVA with Tukey's post-hoc analysis.
Zymosan stimulates the endogenous production of O2- and enhances microglial VRAC activity in a ROS-dependent manner
To determine whether endogenous ROS can modulate VRAC activity, we stimulated microglia with zymosan (500 μg/mL). Zymosan is a desiccated preparation of yeast cell wall commonly employed to stimulate the activity of the ROS-producing enzyme NADPH oxidase (NOX) in immune cells (DeChatelet et al. 1975). We measured endogenous ROS production using a nitroblue tetrazolium (NBT) assay, which detects O2− via quantification of NBT reduction to formazan (Choi et al. 2006). Stimulation of microglia with zymosan strongly increased NBT reduction (Fig. 3a). The effect of zymosan was evident at 10 minutes, but roughly tripled after a 30-minute incubation (data not shown). In all the subsequent experiments we used a 30-minute incubation to increase assay sensitivity. Several previous studies found increased ROS production upon application of hypoosmotic media (Lambert 2003; Ortenblad et al. 2003; Varela et al. 2004). In our microglial experiments we failed to register changes in NBT reduction under hypoosmotic conditions; also, hypoosmotic medium did not increase the zymosan-induced ROS production (data not shown). The effect of zymosan was blocked by DPI (10 μM), a potent NOX inhibitor (Bedard and Krause 2007) (Fig. 3a). This suggests that zymosan stimulates the production of ROS in microglia via NOX activation.
Fig. 3. Zymosan stimulates the endogenous production of superoxide anion (O2−) and enhances microglial excitatory amino acid release.

(a) Effects of zymosan and the NOX inhibitor DPI on microglial production of O2−. Primary microglial cells were exposed to hypoosmotic media additionally containing zymosan (500 μg/mL) and/or DPI (10 μM) for 30 minutes at 37°C. O2- production was measured as the reduction of NBT (see Materials and methods for details). ***p<0.001 Zymosan vs. Control; ###p<0.001 Zymosan vs. Zymosan+DPI. (b) Effect of zymosan on microglial D-[3H]aspartate release. Microglia were exposed to zymosan (500 μg/mL) for 10 minutes prior to and during 10-minute application of hypoosmotic medium. Arrow indicates a transient increase in D-[3H]aspartate release upon zymosan application under isoosmotic conditions. In a separate set of experiments, zymosan was applied in combination with 20 μM DCPIB. The mean values ±SE of six experiments are presented. ***p<0.001 Hypo vs. Zymosan. ###p<0.001 Zymosan+DCPIB vs. Hypo or Zymosan alone. (c) Effects of zymosan (500 μg/mL) on hypoosmotic medium-induced D-[3H]aspartate release in the presence or absence of DPI (10 μM). The design of these experiments was similar to those presented in (b); DPI was present 30 min before and during application of hypoosmotic medium. For clarity only maximal hypoosmotic release values are presented. Data are the mean values ±SE of 5-6 experiments. ***p<0.001, Zymosan vs. Hypo; ###p<0.001, Zymosan vs. Zymosan + DPI.
We further tested zymosan in the excitatory amino acid release assay. Zymosan was applied for 10 minutes prior to and during the 10-min hypoosmotic stimulation. In non-swollen cells, zymosan caused a small and transient increase in D-[3H]aspartate release (Fig. 3b, 2.4-fold increase above basal levels, p=0.021, marked by arrow). In swollen microglia, zymosan stimulation potentiated hypoosmotic d-[3H] aspartate release by ∼two-fold (Fig. 3b). As in the case of exogenous H2O2 (see Fig. 2a), 20 μM DCPIB suppressed zymosan-induced excitatory amino acid efflux both under isoosmotic and hypoosmotic conditions (Fig. 3b), confirming VRAC contribution. The zymosan-enhanced release of D-[3H]aspartate was also blocked with 10 μM DPI, under both isoosmotic and hypoosmotic conditions (Fig. 3c), suggesting NOX involvement.
Because the effect of zymosan in non-swollen cells was very small, in all the subsequent experiments we applied zymosan during exposure to hypoosmotic medium only. Such application produced a consistently stronger stimulation of volume-sensitive excitatory amino acid release (increased from ∼2-fold to ∼4-fold, compare Fig. 3c to Fig. 4b).
Fig. 4. Endogenous ROS production mediates zymosan effect on microglial excitatory amino acid release via VRAC.

(a) The effect of the ROS scavenger MnTMPyP on intracellular O2− levels in microglia. Primary microglial cells were exposed for 30 minutes to hypoosmotic media containing zymosan (500 μg/mL) and various concentrations of MnTMPyP. O2− production was measured as the reduction of NBT. Representative of two experiments. Data are the mean values ±SE of three independent measurements. *p<0.05, **p<0.01, p<0.001 vs. Hypo; #p<0.05, p<Zymosan+MnTMPyP vs. Zymosan control. (b) The effect of MnTMPyP (20 μM) on the zymosan-induced enhancement of hypoosmotic D-[3H]aspartate release. The mean values ± SE of five experiments are shown. **p<0.01, ***p<0.001 vs. Hypo; ##p<0.01, Zymo.+MnTMPyP vs. Zymosan. In these experiments one-way ANOVA with Tukey's post-hoc analysis was performed after logarithmic transformation of the data.
To further confirm that the effects of zymosan on VRAC activity are mediated by ROS, rather than by activation of ROS-independent intracellular signaling pathway(s), we applied MnTMPyP, which is a cell permeable superoxide dismutase (SOD) and catalase mimetic (Day et al. 1997). One-to-30 μM MnTMPyP dose-dependently reduced NBT-detectable levels of O2− in microglia stimulated with zymosan (Fig. 4a). However, even at the highest MnTMPyP concentration used, inhibition of ROS production incomplete (Fig. 4a). We have chosen 20 μM MnTMPyP concentration for the subsequent experiments in order to reduce risk of toxicity and assure specificity of the effects. 20 μM MnTMPyP significantly attenuated the zymosan-enhanced release of D-[3H]aspartate under hypoosmotic conditions (Fig. 4b), strongly suggesting that the effects of zymosan are mediated by ROS.
To further implicate NOX as the source of ROS involved in the regulation of VRAC activity in microglia, we employed apocynin, another commonly used inhibitor of NOX, which is more selective than DPI (Stolk et al. 1994). We used the concentration of 2 mM, because at lower concentrations apocynin showed limited efficacy in NBT assays (data not shown). Two mM apocynin decreased the zymosan-stimulated production of O2- in microglia by 75% (Fig. 5a) but, surprisingly, was completely ineffective against the zymosan-enhanced release of d-[3H] aspartate (Fig. 5b). Similarly, AEBSF, a protease inhibitor that is frequently used to block the NOX2 isoform (Bedard and Krause 2007), prevented the zymosan-activated ROS production, but was ineffective against zymosan-stimulated glutamate release (Supplementary Fig. 1).
Fig. 5. The NOX blocker apocynin potently suppresses the zymosan-induced ROS production, but not the zymosan effect on VRAC activity.

(a) The effects of apocynin on zymosan-induced O2- production in microglia. Microglial cells were exposed to hypoosmotic media additionally containing zymosan (500 μg/mL) and/or apocynin (2 mM). Apocynin was included in the media 30 minutes before and during 30-minute incubation with zymosan and NBT. Data are the mean values ±SE of four experiments. ***p<0.001, vs. Hypo; ###p<0.001 Zymosan vs. Zymo. +Apocynin. (b) The effect of zymosan on hypoosmotic medium-induced D-[3H]aspartate release in the presence or absence of 2 mM apocynin. The mean values ±SE of five experiments are shown. ***p<0.001 vs. Hypo. In these experiments one-way ANOVA with Tukey's post-hoc analysis was performed after logarithmic transformation of the data.
In an attempt to resolve the incongruity between results with DPI, apocynin and AEBSF, we tested thioridazine, another NOX inhibitor. Thioridazine is a phenothiazine derivative, which blocks dopaminergic receptors, as well as all NOX isoforms via the mechanism similar to DPI (Serrander et al. 2007). In our experiments, thioridazine completely blocked the zymosan-stimulated production of O2− at 30 μM (Fig. 6a), and effectively prevented the zymosan-enhanced release of D-[3H]aspartate from swollen microglia (Fig. 6b).
Fig. 6. The NOX inhibitor thioridazine blocks zymosan-enhanced ROS production and excitatory amino acid release.

(a) The effect of various concentration of thioridazine on zymosan-induced production of O2− in microglia exposed to hypoosmotic medium. Thioridazine was applied for 30 minutes in conjunction with 500 μg/mL zymosan and NBT. The mean values ±SE of three independent measurements in a representative experiment are shown. *p<0.05, ***p<0.001, vs. Hypo, ###p<0.001 vs. Zymosan control. (b) The effect of 20 μM thioridazine on zymosan-induced enhancement of hypoosmotic D-[3H]aspartate release. The data are the mean values ±SE of five experiments. ***p<0.001 vs. Hypo. ##p<0.01 Zymozan vs. Zymo.+Thioridazine.
Expression of different NOX isoforms in microglia
Because apocynin and AEBSF were unexpectedly ineffective in attenuating the zymosan-enhanced release of D-[3H]aspartate, we conducted an analysis of literature on NOX pharmacology. Five NOX isoforms (NOX1-5) are expressed in humans, while only four NOX isoforms (NOX1-4) have been identified in rat (Kawahara et al. 2007). Apocynin and AEBSF block NOX1-3, but are ineffective against NOX4, while DPI and thioridazine effectively inhibit all NOX isoforms (Bedard and Krause 2007; Serrander et al. 2007). Therefore, our data suggest that NOX4 is involved in the ROS-mediated effect of zymosan on VRAC.
To determine which NOX isoforms are expressed in primary rat microglia, we probed for the mRNAs expression of all four NOX rat isoforms (NOX1-4). Using RT-PCR, we found that primary rat microglia express high levels of NOX2 along with lower levels of NOX1 and NOX4, but not NOX3 (Fig. 7). The expression of NOX4 fits well with the pharmacological profile of zymosan-enhanced D-[3H]aspartate release, suggesting that NOX4, and not NOX2, may be the source of ROS that positively regulates VRAC activity.
Fig. 7. NOX1, NOX2, and NOX4 isoforms of NAD(P)H oxidase are expressed in primary microglial cells.
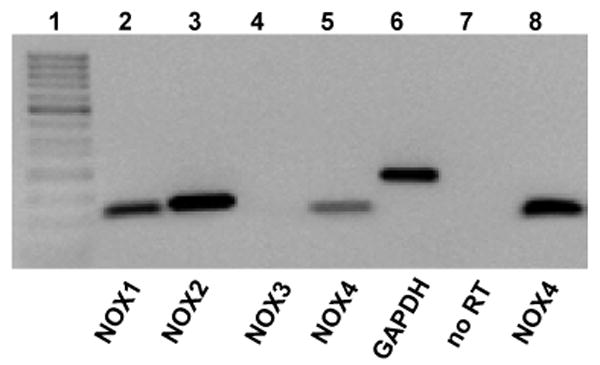
This figure shows a representative negative image of ethidium bromide staining of PCR products for transcripts of various NOX isoforms. PCR was performed after reverse transcriptase (RT) processing of the total RNA samples isolated from primary rat microglia. Total RNA was treated with DNAse in order to remove genomic DNA contamination. Lanes 2-6 show RT-PCR products for rat NOX1-4 and GAPDH in microglial samples, as indicated. NOX3 was not expressed in microglia. In the reaction presented in lane 7, PCR was performed using NOX4 primers, but the RT product was omitted. Lane 8 shows NOX4 RT-PCR product from primary rat endothelial cells (positive control). Lane 1 contains 50-bp MW ladder.
Activation and inhibition of NOX2 modulates O2− production without having any impact on microglial VRAC activity
NOX2 is considered to be the main NOX isoform expressed in microglia and other phagocytic cells; it serves as a prominent source of ROS in immunological reactions (Lambeth 2004), and, thus, is expected to be activated in response to zymosan. Our pharmacological data imply that NOX4, and not NOX2, is the source of ROS which impacts VRAC activity. NOX1-3, but not NOX4, require protein kinase activity for the phosphorylation-dependent assembly of an active NOX complex (Lambeth 2004; Bedard and Krause 2007). Therefore, we pharmacologically manipulated PKC activity in an attempt to discern whether ROS production by NOX2 (and perhaps NOX1, which is also expressed in rat microglia) regulates VRAC.
First, we interrupted the generation of ROS via NOX1/2 by inhibiting PKC with the broad-spectrum PKC blocker Gö6983 (Gschwendt et al. 1996). One μM Gö6983 completely blocked the zymosan-stimulated production of O2- (Fig. 8a) but had no effect on the zymosan-enhanced release of d-[3H] aspartate (Fig. 8b).
Fig. 8. The PKC inhibitor Gö6983 completely prevents NOX2-medited O2− production but not the zymosan-enhanced release of excitatory amino acids via VRAC.

(a) Effect of Gö6983 on the zymosan-induced of O2- production (reduction of NBT). Microglial cells were exposed for 30 minutes to hypoosmotic medium additionally containing zymosan (500 μg/mL) and the broad-spectrum PKC inhibitor Gö6983 (1 μM). Data are the mean values ±SE of three independent measurements in a representative of three experiments. **p<0.01, ***p<0.001 vs. Hypo; ###p<0.001, Zymosan vs Zymosan+Gö6983. (b) The effect of zymosan on hypoosmotic D-[3H]aspartate release in the presence or absence of 1 μM Gö6983. Data are the mean values ±SE of six to nine experiments. ***p<0.001,. vs. Hypo.
We then took an opposite approach by stimulating PKC directly using phorbol 12-myristate 13-acetate (PMA), which has been demonstrated to fully activate NOX2, as well as NOX1 and NOX3 (Bedard and Krause 2007). 500 nM PMA was more effective in stimulating microglial O2− production than zymosan (Fig. 9a). However, at the same concentration, PMA failed to enhance the VRAC-mediated release of glutamate in microglia (Fig. 9b).
Fig. 9. PKC-dependent stimulation of NOX2 enhances O2− production without impacting the VRAC-mediated release of excitatory amino acids.

(a) Effects of PMA (500 nM) or zymosan (500 μg/mL) on microglial O2− production measured as the reduction of NBT. Data are the mean values ±SE of four independent measurements in a representative of three experiments. ***p<0.001 vs. Hypo. (b) PMA (500 nM) had no effect on hypoosmotic D-[3H]aspartate release from primary microglia. Data are the mean values ±SE of six experiments
Together, the experiments presented in Figs. 8 and 9 strongly suggest that NOX2, the major microglial NOX isoform, and possibly NOX1, are not a critical source of ROS that regulate VRAC activity in zymosan-treated cells.
Discussion
In the present work, we found that in microglia, the immunocompetent cells of the CNS, both exogenous and endogenously produced reactive oxygen species strongly enhance excitatory amino acid release via volume-regulated anion channels (VRACs). To stimulate endogenous ROS production by NADPH oxidases, we challenged microglial cells with zymosan, thereby mimicking pathogenic infection. The surprising finding of this study is that the major phagocytic NOX isoform, NOX2, although activated by zymosan, does not contribute to VRAC regulation. Instead, such regulation is likely mediated by NOX4, which—until the present study—has not been identified in microglial cells.
VRACs contribute to glutamate release from microglia
Microglial activation in response to pathogenic infection or trauma is important for the preservation of neural tissue (Kreutzberg 1996; Nakajima and Kohsaka 2001; Block and Hong 2005). When activated, microglia produce and release various inflammatory cytokines, reactive oxygen and nitrogen species, and glutamate (Nakajima and Kohsaka 2001; Block and Hong 2005; Block et al. 2007). Glutamate is the primary excitatory neurotransmitter in the CNS, which may induce excitotoxic tissue damage if its extracellular levels are not properly regulated (Choi 1988; Choi 1992). One of several potential pathways for glutamate release are VRACs (Kimelberg and Mongin 1998; Mongin and Kimelberg 2005; Malarkey and Parpura 2008). In microglial cells, swelling-activated anion channels, which resemble VRAC, have been identified and suggested to play important roles in volume regulation, cell proliferation, and phagocytosis (Schlichter et al. 1996; Ducharme et al. 2007). Importantly, microglial swelling-activated Cl− channels are permeable to glutamate and aspartate with permeability values, relative to Cl−, of ∼0.18 and ∼0.22, respectively (Ducharme et al. 2007). However, Ducharme et al. (2007) have found that microglial swelling-activated anion channels are not inactivated at positive potentials and have much smaller unitary conductance of ∼1-3 pS in comparison to a prototypical VRAC. Conversely, our electrophysiological and pharmacological data suggest that rat microglia express classical VRACs. Several characteristics that support such a notion include: (i) outward rectification of the swelling induced Cl− currents, (ii) time-dependent current inactivation at positive potentials, and (iii) the sensitivity of swelling-activated Cl− currents to the selective VRAC blocker DCPIB. DCPIB has been found to discriminate VRACs from other cloned to-date Cl− channels (Decher et al. 2001). Additional studies are needed to determine if the method of cell preparation or differences in recording solutions explain variation between present electrophysiological findings and those in the work of Schlichter and co-workers (Ducharme et al. 2007). Along with our electrophysiological data, the inhibitory effect of DCPIB on amino acid release suggests that microglial VRACs serve as a major route for glutamate release, and in such a way may profoundly impact neural tissue function. This may especially be the case under pathological conditions associated with cell swelling or in the processes of microglial migration, proliferation, and phagocytosis, which likely involve enhanced VRAC activity (Schlichter et al. 1996; Eder 2005; Ducharme et al. 2007).
Microglial VRACs are potently regulated by exogenous and endogenous ROS
The major objective of this study was to determine if ROS may trigger glutamate release from microglial cells via glutamate-permeable VRACs. In several cell types, it has been demonstrated that ROS activate or positively modulate VRACs (Browe and Baumgarten 2004; Shimizu et al. 2004; Varela et al. 2004; Haskew-Layton et al. 2005; Ren et al. 2008), as well as similar volume-sensitive taurine permeability pathway (Ortenblad et al. 2003; Lambert 2003). Because activated microglia abundantly generate ROS, these cells represent an ideal subject for studying redox regulation of VRACs, and the functional impact of such regulation on cell physiology and function. In our experiments, application of exogenous H2O2 enhanced hypoosmotic medium-induced D-[3H]aspartate release and VRAC Cl− currents via a DCPIB-sensitive pathway, acting synergistically with cell swelling. Consistent with our previous findings in astrocytes (Haskew-Layton et al. 2005), H2O2 was essentially ineffective in non-swollen cells. Induction of endogenous ROS production with zymosan produced limited VRAC activation in non-swollen cells, and potently increased glutamate release by 2-5-fold from microglial cells exposed to hypoosmotic media. These data suggest that ROS preferentially modulate VRACs only when these channels are already active. The magnitude of D-[3H]aspartate release from non-swollen cells exposed to H2O2 or zymosan is too small to have physiological significance. Under isoosmotic conditions set in our experiments, VRACs are inactive. Therefore, we subject microglial cells to large hypotonic stimuli in order to activate the volume-sensitive Cl− currents and glutamate fluxes. Such hypoosmotic gradients do not exist in the brain, even in pathology. However, microglial VRACs may be activated under various physiological and pathological conditions without changes in extracellular osmolarity. Such a notion is supported by the observations that nonselective VRAC blockers inhibit proliferation, phagocytosis and shape transitions in microglia (Schlichter et al. 1996; Eder et al. 1998; Ducharme et al. 2007).
Zymosan is commonly used to stimulate ROS production in immune cells, including microglia, and this effect is mediated via activation of NADPH oxidases (Colton and Gilbert 1987; Sankarapandi et al. 1998). In our experiments, the effects of zymosan on D-[3H]aspartate release via VRACs were inhibited either by the antioxidant MnTMPyP or the NOX blockers DPI and thioridazine, which strongly suggests that they are mediated by ROS originating form NOX.
Differential impact of NOX isoforms on regulation of microglial VRAC by ROS
The paradoxical finding of the present work was insensitivity of zymosan-induced VRAC activation to the NOX inhibitors apocynin and AEBSF. Apocynin, DPI, AEBSF, and thioridazine all potently suppressed zymosan-induced superoxide production. However, only DPI and thioridazine inhibited VRAC-mediated glutamate release from zymosan-treated cells. This discrepancy may be explained by suggesting that the effect of zymosan on VRAC involves atypical NOX4. NOX1-3 isoforms require phosphorylation-dependent assembly of several cytosolic subunits in order to be catalytically active, while NOX4 activity does not require the same proteins and is regulated via alternative mechanisms (Bedard and Krause 2007, see Fig. 10). Apocynin and AEBSF suppress the activity of NOX2 (and related NOX1 and 3) by inhibiting the assembly of regulatory subunits (Stolk et al. 1994; Bedard and Krause 2007). However, these compounds are ineffective inhibitors of NOX4 (Serrander et al. 2007). DPI and thioridazine, on the other hand, inhibit all NOX isoforms, including NOX4, by interrupting electron flow in the catalytic subunit (Bedard and Krause 2007; Serrander et al. 2007). Although DPI and thioridazine are not specific for NOXs, and also block other electron-transferring enzymes, they are the only potent NOX4 inhibitors available to-date.
Fig. 10. Schematic representation of the hypothetical mechanisms contributing to the effects of zymosan on production of ROS and VRAC activity in rat microglial cells.
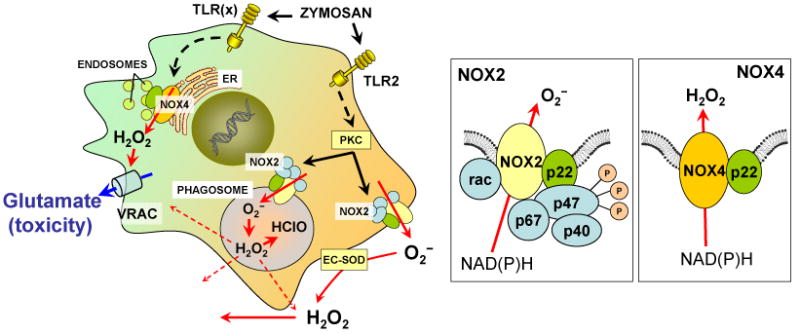
Zymosan activates TLR2 and perhaps another unidentified subclass of the TLR receptors. TLR2 activation stimulates cytosolic PKC via several additional steps (not shown). PKC phosphorylates regulatory cytosolic subunit p47phox (see NOX2 box on the right). Phosphorylation of p47phox prompts assembly of the enzymatically active NOX2 complex, which includes membrane proteins NOX2 (gp91phox) and p22phox, as well as cytosolic p47phox, p67phox, p40phox, and the small GTPase rac. An active NOX2 complex resides in plasmalemmal and phagocytic membranes and produces O2−, which is released to the extracellular space or into phagosome. An alternative signaling pathway causes activation of NOX4 via unidentified enzymatic steps. NOX4 is likely localized in endoplasmic reticulum and/or endosomes and does not require cytosolic regulatory subunits for its activity (see NOX4 box on the right). NOX4 produces predominantly H2O2, rather than O2−. The difference in the location of the ROS production and/or the nature of generated ROS likely determines differential contribution of NOX4 and NOX2 to the activation of VRAC by zymosan. Abbreviations: ER, endoplasmic reticulum; ES-SOD, extracellular superoxide dismutase; NOX, NAD(P)H oxidase, PKC, protein kinase C, TLR, Toll-like receptors. See text for additional details.
NOX4 is a recently discovered enzyme, whose expression in microglial cells has not been reported until the present study (Bedard and Krause 2007). Taken together, the presence of NOX4 messenger RNA and our pharmacological data strongly support the hypothesis of NOX4 involvement in regulation of VRAC. A similar idea has been previously put forth by Lambert and colleagues, who suggest that NOX4 is critical for activation of swelling-sensitive taurine release pathway in mouse NIH 3T3 fibroblasts (Friis et al. 2008).
It should be noted, however, that NOX4 is not a major NOX isoform in microglia, nor is it known to be activated by zymosan. In microglia, zymosan stimulates generation of ROS via activation of phagocytic NOX2 (Sankarapandi et al. 1998). It was not clear to us why NOX2-derived ROS were not contributing to VRAC activation by zymosan. In order to address this question, we stimulated NOX2 by treating microglial cells with PKC activator PMA, thereby promoting the assembly of catalytically active NOX2 complex (Bedard and Krause 2007). PMA potently enhanced generation of O2−, but it did not affect the VRAC activity. Conversely, the potent PKC inhibitor Gö6983 completely suppressed O2− generation in zymosan-treated cells, but did not affect VRAC activity. These data unequivocally prove that NOX2-derived ROS are not critical for VRAC regulation by zymosan under our experimental conditions.
The surprising lack of effect of NOX2-derived ROS may be explained by the location of ROS production and/or by the peculiarity of NOX4 biochemistry. As shown in Fig. 10, NOX2 is predominantly present in plasmalemmal and phagocytic membranes and secretes superoxide to the extracellular space or into the phagosome (Bedard and Krause 2007). In contrast, NOX4 is thought to be predominantly associated with the endoplasmic reticulum and/or endosomes and generates disproportionally higher amounts of intracellular H2O2 compared to other NOXs (Martyn et al. 2006; Bedard and Krause 2007; Serrander et al. 2007). Therefore, NOX4 may create sufficiently high local levels of H2O2 that stimulate VRACs. Our attempts to quantify intracellular H2O2 production in zymosan-treated cells using a fluorescent probe 5-chloromethyl-2′7′-dichlorofluorescein diacetate (CM-H2DCFDA) were unsuccessful because of the turbidity of zymosan solutions.
Pathological significance VRAC-mediated glutamate release from microglial
Microglia activation is a common feature of many neurological disorders. Although necessary for preservation of neural tissue, microglial responses may have deleterious effects, which are largely related to the excessive production of ROS (Kreutzberg 1996; Nakajima and Kohsaka 2001; Block and Hong 2005). Genetic deletion of the ROS-producing enzyme NOX2 or its regulatory subunits, or overexpression of ROS-scavenging enzymes, are protective in animal models of cerebral ischemia, Alzheimer's and Parkinson's diseases, and methamphetamine neurotoxicity (Yang et al. 1994; Walder et al. 1997; Deng and Cadet 2000; Zhang et al. 2004; Park et al. 2005). Tissue damage in the same animal models can also be prevented by antagonists of the NMDA subtype of glutamate receptors (Sonsalla et al. 1989; Zeevalk et al. 1994; Lipton 1999). Therefore, oxidative stress and glutamate toxicity are likely related processes. It is well known how glutamate promotes oxidative and nitrosative stress (Beckman and Koppenol 1996; Lipton 1999). Whether the opposite is true, i.e., if ROS production may lead to enhanced glutamate release, is less clear. Previous studies implicated the cystine/glutamate antiporter and connexin hemichannels in the ROS- and inflammation-induced glutamate release (Piani and Fontana 1994; Barger and Basile 2001; Takeuchi et al. 2006; Fogal et al. 2007). Our present and published work suggests that, in microglia, astrocytes and possibly other types of neural cells, oxidative stress may promote pathological glutamate release via the glutamate-permeable anion channel VRAC, and perhaps other permeability pathways (Haskew-Layton et al. 2005; Liu et al. 2006). Although our present in vitro data point to a key role for NOX4, in intact tissue NOX2 may be as important. A recent report by Z. Ren et al. (2008) implicated NOX2 in regulation of VRAC activity by angiotensin II in cardiac myocytes. NOX2-derived superoxide is converted extracellularly and intracellularly to H2O2, which in intact tissue is capable of accumulating in pathological quantities, regardless of the initial source (Halliwell 2006). In our experimental paradigm, NOX2-generated ROS may be consumed in phagosome or removed with perfusion, while NOX4-derived H2O2 may reach high intracellular levels (see Fig. 10). Overall, our study proposes one possible mechanism that links microglial ROS production to elevated glutamate release, which involves the anion channel VRAC. Such release may contribute to glutamate-associated neuronal damage seen in various models of neural pathologies.
Acknowledgments
We appreciate contributions of Aabha A. Shah and Marko Trauzzi, to the preliminary experiments for this study. We thank Dr. Alena Rudkouskaya for assistance in microglial work, Dr. Philip J. Albrecht for help with immunocytochemical experiments, and Patrick Bryant and Annemarie Tyrell for their expert advice in molecular biology. This study was supported in part by AMC grants 201-465005, 201-311053 to A.A. Mongin. T.J. Harrigan was supported by NIH (NIDA) training grant T32 DA007307.
Abbreviations used
- AEBSF
4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride (NOX2 inhibitor)
- DCPIB
4-[(2-Butyl-6,7-dichloro-2-cyclopentyl-2,3-dihydro-1-oxo-1H-inden-5-yl)oxy]butanoic acid (selective VRAC blocker)
- DPI
dipehnyliodonium chloride (NOX inhibitor)
- MEM
minimal essential medium
- FBS
fetal bovine serum
- NOX
NADPH oxidase
- PKC
protein kinase C
- ROS
reactive oxygen species
- VRAC
volume-regulated anion channel
References
- Abdullaev IF, Rudkouskaya A, Schools GP, Kimelberg HK, Mongin AA. Pharmacological comparison of swelling-activated excitatory amino acid release and Cl- currents in rat cultured astrocytes. J Physiol. 2006;572:677–689. doi: 10.1113/jphysiol.2005.103820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banderali U, Roy G. Anion channels for amino-acids in Mdck cells. Am J Physiol. 1992;263:C1200–C1207. doi: 10.1152/ajpcell.1992.263.6.C1200. [DOI] [PubMed] [Google Scholar]
- Barger SW, Basile AS. Activation of microglia by secreted amyloid precursor protein evokes release of glutamate by cystine exchange and attenuates synaptic function. J Neurochem. 2001;76:846–854. doi: 10.1046/j.1471-4159.2001.00075.x. [DOI] [PubMed] [Google Scholar]
- Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol. 1996;271:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- Block ML, Hong JS. Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol. 2005;76:77–98. doi: 10.1016/j.pneurobio.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Browe DM, Baumgarten CM. Angiotensin II (AT1) receptors and NADPH oxidase regulate Cl- current elicited by beta1 integrin stretch in rabbit ventricular myocytes. J Gen Physiol. 2004;124:273–287. doi: 10.1085/jgp.200409040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- Choi DW. Excitotoxic cell death. J Neurobiol. 1992;23:1261–1276. doi: 10.1002/neu.480230915. [DOI] [PubMed] [Google Scholar]
- Choi HS, Kim JW, Cha YN, Kim C. A quantitative nitroblue tetrazolium assay for determining intracellular superoxide anion production in phagocytic cells. J Immunoassay Immunochem. 2006;27:31–44. doi: 10.1080/15321810500403722. [DOI] [PubMed] [Google Scholar]
- Colton CA, Gilbert DL. Production of superoxide anions by a CNS macrophage, the microglia. FEBS Lett. 1987;223:284–288. doi: 10.1016/0014-5793(87)80305-8. [DOI] [PubMed] [Google Scholar]
- Day BJ, Fridovich I, Crapo JD. Manganic porphyrins possess catalase activity and protect endothelial cells against hydrogen peroxide-mediated injury. Arch Biochem Biophys. 1997;347:256–262. doi: 10.1006/abbi.1997.0341. [DOI] [PubMed] [Google Scholar]
- DeChatelet LR, McPhail LC, Mullikin D, McCall CE. An isotopic assay for NADPH oxidase activity and some characteristics of the enzyme from human polymorphonuclear leukocytes. J Clin Invest. 1975;55:714–721. doi: 10.1172/JCI107981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decher N, Lang HJ, Nilius B, Bruggemann A, Busch AE, Steinmeyer K. DCPIB is a novel selective blocker of I(Cl,swell) and prevents swelling- induced shortening of guinea-pig atrial action potential duration. Br J Pharmacol. 2001;134:1467–1479. doi: 10.1038/sj.bjp.0704413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X, Cadet JL. Methamphetamine-induced apoptosis is attenuated in the striata of copper-zinc superoxide dismutase transgenic mice. Brain Res Mol Brain Res. 2000;83:121–124. doi: 10.1016/s0169-328x(00)00169-8. [DOI] [PubMed] [Google Scholar]
- Ducharme G, Newell EW, Pinto C, Schlichter LC. Small-conductance Cl(-) channels contribute to volume regulation and phagocytosis in microglia. Eur J Neurosci. 2007;26:2119–2130. doi: 10.1111/j.1460-9568.2007.05802.x. [DOI] [PubMed] [Google Scholar]
- Eder C. Regulation of microglial behavior by ion channel activity. J Neurosci Res. 2005;81:314–321. doi: 10.1002/jnr.20476. [DOI] [PubMed] [Google Scholar]
- Eder C, Klee R, Heinemann U. Involvement of stretch-activated Cl- channels in ramification of murine microglia. J Neurosci. 1998;18:7127–7137. doi: 10.1523/JNEUROSCI.18-18-07127.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farber K, Kettenmann H. Physiology of microglial cells. Brain Res Brain Res Rev. 2005;48:133–143. doi: 10.1016/j.brainresrev.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Fogal B, Li J, Lobner D, McCullough LD, Hewett SJ. System x(c)- activity and astrocytes are necessary for interleukin-1 beta-mediated hypoxic neuronal injury. J Neurosci. 2007;27:10094–10105. doi: 10.1523/JNEUROSCI.2459-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friis MB, Vorum KG, Lambert IH. Volume-sensitive NADPH oxidase activity and taurine efflux in NIH3T3 mouse fibroblasts. Am J Physiol Cell Physiol. 2008;294:C1552–C1565. doi: 10.1152/ajpcell.00571.2007. [DOI] [PubMed] [Google Scholar]
- Gschwendt M, Dieterich S, Rennecke J, Kittstein W, Mueller HJ, Johannes FJ. Inhibition of protein kinase C mu by various inhibitors. Differentiation from protein kinase c isoenzymes. FEBS Lett. 1996;392:77–80. doi: 10.1016/0014-5793(96)00785-5. [DOI] [PubMed] [Google Scholar]
- Halliwell B. Oxidative stress and neurodegeneration: where are we now? J Neurochem. 2006;97:1634–1658. doi: 10.1111/j.1471-4159.2006.03907.x. [DOI] [PubMed] [Google Scholar]
- Haskew-Layton RE, Mongin AA, Kimelberg HK. Hydrogen peroxide potentiates volume-sensitive excitatory amino acid release via a mechanism involving Ca2+/calmodulin-dependent protein kinase II. J Biol Chem. 2005;280:3548–3554. doi: 10.1074/jbc.M409803200. [DOI] [PubMed] [Google Scholar]
- Jackson PS, Morrison R, Strange K. The volume-sensitive organic osmolyte-anion channel VSOAC is regulated by nonhydrolytic ATP binding. Am J Physiol. 1994;267:C1203–C1209. doi: 10.1152/ajpcell.1994.267.5.C1203. [DOI] [PubMed] [Google Scholar]
- Kawahara BT, Quinn MT, Lambeth JD. Molecular evolution of the reactive oxygen-generating NADPH oxidase (Nox/Duox) family of enzymes. BMC Evol Biol. 2007;7:109. doi: 10.1186/1471-2148-7-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimelberg HK. Current concepts of brain edema. Review of laboratory investigations. J Neurosurg. 1995;83:1051–1059. doi: 10.3171/jns.1995.83.6.1051. [DOI] [PubMed] [Google Scholar]
- Kimelberg HK, Goderie SK, Higman S, Pang S, Waniewski RA. Swelling-induced release of glutamate, aspartate, and taurine from astrocyte cultures. J Neurosci. 1990;10:1583–1591. doi: 10.1523/JNEUROSCI.10-05-01583.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimelberg HK, Mongin AA. Swelling-activated release of excitatory amino acids in the brain: Relevance for pathophysiology. Contrib Nephrol. 1998;123:240–257. doi: 10.1159/000059916. [DOI] [PubMed] [Google Scholar]
- Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- Kubo M, Okada Y. Volume-regulatory Cl- channel currents in cultured human epithelial cells. J Physiol. 1992;456:351–371. doi: 10.1113/jphysiol.1992.sp019340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert IH. Reactive oxygen species regulate swelling-induced taurine efflux in NIH3T3 mouse fibroblasts. J Membr Biol. 2003;192:19–32. doi: 10.1007/s00232-002-1061-1. [DOI] [PubMed] [Google Scholar]
- Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- Lang F, Ritter M, Gamper N, Huber S, Fillon S, Tanneur V, Lepple-Wienhues A, Szabo I, Bulbins E. Cell volume in the regulation of cell proliferation and apoptotic cell death. Cell Physiol Biochem. 2000;10:417–428. doi: 10.1159/000016367. [DOI] [PubMed] [Google Scholar]
- Lipton P. Ischemic cell death in brain neurons. Physiol Rev. 1999;79:1431–1568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- Liu HT, Tashmukhamedov BA, Inoue H, Okada Y, Sabirov RZ. Roles of two types of anion channels in glutamate release from mouse astrocytes under ischemic or osmotic stress. Glia. 2006;54:343–357. doi: 10.1002/glia.20400. [DOI] [PubMed] [Google Scholar]
- Malarkey EB, Parpura V. Mechanisms of glutamate release from astrocytes. Neurochem Int. 2008;52:142–154. doi: 10.1016/j.neuint.2007.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martyn KD, Frederick LM, von Loehneysen K, Dinauer MC, Knaus UG. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell Signal. 2006;18:69–82. doi: 10.1016/j.cellsig.2005.03.023. [DOI] [PubMed] [Google Scholar]
- McBean GJ. Cerebral cystine uptake: a tale of two transporters. Trends Pharmacol Sci. 2002;23:299–302. doi: 10.1016/s0165-6147(02)02060-6. [DOI] [PubMed] [Google Scholar]
- Mongin AA, Kimelberg HK. ATP potently modulates anion channel-mediated excitatory amino acid release from cultured astrocytes. Am J Physiol Cell Physiol. 2002;283:C569–C578. doi: 10.1152/ajpcell.00438.2001. [DOI] [PubMed] [Google Scholar]
- Mongin AA, Kimelberg HK. Astrocytic swelling in neuropathology. In: Kettenmann H, Ransom BR, editors. Neuroglia. Oxford University Press; Oxford New York: 2005. pp. 550–562. [Google Scholar]
- Nakajima K, Kohsaka S. Microglia: activation and their significance in the central nervous system. J Biochem (Tokyo) 2001;130:169–175. doi: 10.1093/oxfordjournals.jbchem.a002969. [DOI] [PubMed] [Google Scholar]
- Nilius B, Droogmans G. Amazing chloride channels: an overview. Acta Physiol Scand. 2003;177:119–147. doi: 10.1046/j.1365-201X.2003.01060.x. [DOI] [PubMed] [Google Scholar]
- Nilius B, Eggermont J, Voets T, Buyse G, Manolopoulos V, Droogmans G. Properties of volume-regulated anion channels in mammalian cells. Prog Biophys Mol Biol. 1997;68:69–119. doi: 10.1016/s0079-6107(97)00021-7. [DOI] [PubMed] [Google Scholar]
- Okada Y. Volume expansion-sensing outward-rectifier Cl- channel: fresh start to the molecular identity and volume sensor. Am J Physiol Cell Physiol. 1997;273:C755–C789. doi: 10.1152/ajpcell.1997.273.3.C755. [DOI] [PubMed] [Google Scholar]
- Okada Y. Cell volume-sensitive chloride channels: phenotypic properties and molecular identity. Contrib Nephrol. 2006;152:9–24. doi: 10.1159/000096285. [DOI] [PubMed] [Google Scholar]
- Okada Y, Maeno E, Shimizu T, Dezaki K, Wang J, Morishima S. Receptor-mediated control of regulatory volume decrease (RVD) and apoptotic volume decrease (AVD) J Physiol. 2001;532:3–16. doi: 10.1111/j.1469-7793.2001.0003g.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortenblad N, Young JF, Oksbjerg N, Nielsen JH, Lambert IH. Reactive oxygen species are important mediators of taurine release from skeletal muscle cells. Am J Physiol Cell Physiol. 2003;284:C1362–C1373. doi: 10.1152/ajpcell.00287.2002. [DOI] [PubMed] [Google Scholar]
- Park L, Anrather J, Zhou P, Frys K, Pitstick R, Younkin S, Carlson GA, Iadecola C. NADPH-oxidase-derived reactive oxygen species mediate the cerebrovascular dysfunction induced by the amyloid beta peptide. J Neurosci. 2005;25:1769–1777. doi: 10.1523/JNEUROSCI.5207-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piani D, Fontana A. Involvement of the cystine transport system xc- in the macrophage-induced glutamate-dependent cytotoxicity to neurons. J Immunol. 1994;152:3578–3585. [PubMed] [Google Scholar]
- Piani D, Frei K, Do KQ, Cuenod M, Fontana A. Murine brain macrophages induced NMDA receptor mediated neurotoxicity in vitro by secreting glutamate. Neurosci Lett. 1991;133:159–162. doi: 10.1016/0304-3940(91)90559-c. [DOI] [PubMed] [Google Scholar]
- Piani D, Spranger M, Frei K, Schaffner A, Fontana A. Macrophage-induced cytotoxicity of N-methyl-D-aspartate receptor positive neurons involves excitatory amino acids rather than reactive oxygen intermediates and cytokines. Eur J Immunol. 1992;22:2429–2436. doi: 10.1002/eji.1830220936. [DOI] [PubMed] [Google Scholar]
- Qin S, Colin C, Hinners I, Gervais A, Cheret C, Mallat M. System Xc- and apolipoprotein E expressed by microglia have opposite effects on the neurotoxicity of amyloid-beta peptide 1-40. J Neurosci. 2006;26:3345–3356. doi: 10.1523/JNEUROSCI.5186-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Z, Raucci FJ, Jr, Browe DM, Baumgarten CM. Regulation of swelling-activated Cl(-) current by angiotensin II signalling and NADPH oxidase in rabbit ventricle. Cardiovasc Res. 2008;77:73–80. doi: 10.1093/cvr/cvm031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankarapandi S, Zweier JL, Mukherjee G, Quinn MT, Huso DL. Measurement and characterization of superoxide generation in microglial cells: evidence for an NADPH oxidase-dependent pathway. Arch Biochem Biophys. 1998;353:312–321. doi: 10.1006/abbi.1998.0658. [DOI] [PubMed] [Google Scholar]
- Sato H, Kuriyama-Matsumura K, Hashimoto T, Sasaki H, Wang H, Ishii T, Mann GE, Bannai S. Effect of oxygen on induction of the cystine transporter by bacterial lipopolysaccharide in mouse peritoneal macrophages. J Biol Chem. 2001;276:10407–10412. doi: 10.1074/jbc.M007216200. [DOI] [PubMed] [Google Scholar]
- Schlichter LC, Sakellaropoulos G, Ballyk B, Pennefather PS, Phipps DJ. Properties of K+ and Cl- channels and their involvement in proliferation of rat microglial cells. Glia. 1996;17:225–236. doi: 10.1002/(SICI)1098-1136(199607)17:3<225::AID-GLIA5>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Serrander L, Cartier L, Bedard K, Banfi B, Lardy B, Plastre O, Sienkiewicz A, Forro L, Schlegel W, Krause KH. NOX4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem J. 2007;406:105–114. doi: 10.1042/BJ20061903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu T, Numata T, Okada Y. A role of reactive oxygen species in apoptotic activation of volume-sensitive Cl(-) channel. Proc Natl Acad Sci U S A. 2004;101:6770–6773. doi: 10.1073/pnas.0401604101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonsalla PK, Nicklas WJ, Heikkila RE. Role for excitatory amino acids in methamphetamine-induced nigrostriatal dopaminergic toxicity. Science. 1989;243:398–400. doi: 10.1126/science.2563176. [DOI] [PubMed] [Google Scholar]
- Stolk J, Hiltermann TJ, Dijkman JH, Verhoeven AJ. Characteristics of the inhibition of NADPH oxidase activation in neutrophils by apocynin, a methoxy-substituted catechol. Am J Respir Cell Mol Biol. 1994;11:95–102. doi: 10.1165/ajrcmb.11.1.8018341. [DOI] [PubMed] [Google Scholar]
- Strange K, Emma F, Jackson PS. Cellular and molecular physiology of volume-sensitive anion channels. Am J Physiol. 1996;270:C711–C730. doi: 10.1152/ajpcell.1996.270.3.C711. [DOI] [PubMed] [Google Scholar]
- Stutzin A, Hoffmann EK. Swelling-activated ion channels: functional regulation in cell-swelling, proliferation and apoptosis. Acta Physiol (Oxf) 2006;187:27–42. doi: 10.1111/j.1748-1716.2006.01537.x. [DOI] [PubMed] [Google Scholar]
- Takeuchi H, Jin S, Wang J, Zhang G, Kawanokuchi J, Kuno R, Sonobe Y, Mizuno T, Suzumura A. Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J Biol Chem. 2006;281:21362–21368. doi: 10.1074/jbc.M600504200. [DOI] [PubMed] [Google Scholar]
- Varela D, Simon F, Riveros A, Jorgensen F, Stutzin A. NAD(P)H oxidase-derived H2O2 signals chloride channel activation in cell volume regulation and cell proliferation. J Biol Chem. 2004;279:13301–13304. doi: 10.1074/jbc.C400020200. [DOI] [PubMed] [Google Scholar]
- Walder CE, Green SP, Darbonne WC, Mathias J, Rae J, Dinauer MC, Curnutte JT, Thomas GR. Ischemic stroke injury is reduced in mice lacking a functional NADPH oxidase. Stroke. 1997;28:2252–2258. doi: 10.1161/01.str.28.11.2252. [DOI] [PubMed] [Google Scholar]
- Worrell RT, Butt AG, Cliff WH, Frizzell RA. A volume-sensitive chloride conductance in human colonic cell line T84. Am J Physiol. 1989;256:C1111–C1119. doi: 10.1152/ajpcell.1989.256.6.C1111. [DOI] [PubMed] [Google Scholar]
- Yang G, Chan PH, Chen J, Carlson E, Chen SF, Weinstein P, Epstein CJ, Kamii H. Human copper-zinc superoxide dismutase transgenic mice are highly resistant to reperfusion injury after focal cerebral ischemia. Stroke. 1994;25:165–170. doi: 10.1161/01.str.25.1.165. [DOI] [PubMed] [Google Scholar]
- Zeevalk GD, Nicklas WJ, Sonsalla PK. NMDA receptor involvement in two animal models of Parkinson's disease. Neurobiol Aging. 1994;15:269–270. doi: 10.1016/0197-4580(94)90130-9. [DOI] [PubMed] [Google Scholar]
- Zhang W, Wang T, Qin L, Gao HM, Wilson B, Ali SF, Zhang W, Hong JS, Liu B. Neuroprotective effect of dextromethorphan in the MPTP Parkinson's disease model: role of NADPH oxidase. FASEB J. 2004;18:589–591. doi: 10.1096/fj.03-0983fje. [DOI] [PubMed] [Google Scholar]