

Abstract
Lacticin 481 is a lanthionine-containing bacteriocin (lantibiotic) produced by Lactococcus lactis subsp. lactis. The final steps of lacticin 481 biosynthesis are proteolytic removal of an N-terminal leader sequence from the prepeptide LctA and export of the mature lantibiotic. Both proteolysis and secretion are performed by the dedicated ATP-binding cassette (ABC) transporter LctT. LctT belongs to the family of AMS (ABC transporter maturation and secretion) proteins whose prepeptide substrates share a conserved double-glycine type cleavage site. The in vitro activity of a lantibiotic protease has not yet been characterized. This study reports the purification and in vitro activity of the N-terminal protease domain of LctT (LctT150), and its use for the in vitro production of lacticin 481. The G(-2)A(-1) cleavage site and several other conserved amino acid residues in the leader peptide were targeted by site-directed mutagenesis to probe the substrate specificity of LctT as well as shed light upon the role of these conserved residues in lantibiotic biosynthesis. His10-LctT150 did not process most variants of the double glycine motif and processed mutants of Glu-8 only very slowly. Furthermore, incorporation of helix-breaking residues in the leader peptide resulted in greatly decreased proteolytic activity by His10-LctT150. On the other hand, His10-LctT150 accepted all peptides containing mutations in the propeptide or at non-conserved positions of LctA. In addition, the protease domain of LctT was investigated by site-directed mutagenesis of the conserved residues Cys12, His90, and Asp106. The proteolytic activities of the resulting mutant proteins are consistent with a cysteine protease.
Keywords: protease, lantibiotic, bacteriocin, ABC transporter
Lantibiotics are a class of ribosomally synthesized antimicrobial peptides that undergo extensive post-translational modification (1-3). These modifications include enzyme-catalyzed dehydration of serine and threonine residues followed by cyclization of the resulting dehydro amino acids through reaction with cysteine residues to form thio-ether linkages (Figure 1). Following dehydration and cyclization, a leader peptide that is not modified is removed by a protease to give rise to the biologically active species. The modified peptide is secreted from the cell just prior to or immediately following the cleavage event by a transport protein. This sequence of biosynthetic events is shown in Figure 1 for lacticin 481 (1, 4). Serine and threonine residues of the lacticin 481 precursor peptide, LctA, are first dehydrated and then cyclized by the bifunctional protein LctM (5). The unmodified leader sequence is removed and mature lacticin 481 is exported by the LanT protein LctT (6). Because LctM and LctT are bifunctional enzymes, lacticin 481 is a member of the class II lantibiotics (7). In class I lantibiotics distinct proteins perform the dehydration (LanB), cyclization (LanC), cleavage (LanP), and export functions (LanT).
Figure 1.

Biosynthesis of lacticin 481.
The role(s) of the leader peptide in lantibiotic biosynthesis has been the subject of much speculation (8, 9). It may act in a protective role for the producing organism, as its removal is required for bioactivity (10-15). The leader peptide could also serve as a recognition motif for the transport machinery because non-lantibiotic peptides attached to leader peptides are secreted (16, 17), or as a scaffold for the lantibiotic synthetases (11, 18-20). Recently, it was shown that the leader sequence of LctA, while important for processing by LctM, is not absolutely required for activity (21). When just the propeptide was provided to the enzyme, dehydration still proceeded but with much decreased efficiency. These results demonstrated the importance of the leader sequence for efficient dehydration prompting an investigation of the conserved residues in the leader peptide by site-directed mutagenesis. The accompanying paper describes the results of these studies that indicate none of the conserved leader residues are absolutely required for dehydration activity, but that the secondary structure of the peptide as well as Leu-7 are important for efficient processing by LctM (22). In this work, we investigated the importance of the conserved residues in the leader peptide for proteolytic processing.
In the biosynthesis of most class I lantibiotics, removal of the leader peptide is performed by a dedicated LanP serine-type protease (23). However, lanP genes are absent from the biosynthetic gene clusters of class II lantibiotics, and the lanT genes instead encode an N-terminal peptidase domain in the transporter proteins that is absent in the class I lantibiotic ABC transporters. This domain is proposed to remove the leader sequence from the modified precursor peptides concomitant with export of the mature species. Håvarstein and co-workers recognized a correlation between bacteriocin transporters that contain this N-terminal proteolytic domain and the presence of a double glycine motif in the leader sequences of their cognate prepeptide substrates (24). They showed that the N-terminal 150 amino acids of the bacteriocin transporter LagD were sufficient for lactococcin G precursor cleavage. This investigation was the first of three studies on the in vitro activity of non-lantibiotic bacteriocin protease domains, with more recent studies describing in vitro hydrolysis of ComC by the N-terminal domain of ComA (25), and cleavage of the colicin V precursor by the protease domain of CvaB (26). With the exception of a very recent report on ComC (27), these previous studies focused mainly on characterization of the protease with investigation of substrate recognition receiving little attention. The study of lantibiotic proteases has been restricted to limited in vivo mutagenesis of the leader peptides (10, 13, 15, 28, 29).
Herein, we report the first purification and in vitro activity of a lantibiotic protease, the N-terminal domain of LctT responsible for the maturation of lacticin 481. The N-terminal 150 amino acids were overexpressed and purified as a soluble His10-fusion protein designated His10-LctT150. The importance of conserved leader peptide residues and the substrate scope of proteolysis by His10-LctT150 were examined by subjecting various His6-LctA mutant peptides to the protease followed by mass spectrometric analysis. His10-LctT150 did not process most variants of the double glycine motif and processed mutants of Glu-8 only very slowly. Furthermore, incorporation of helix-breaking residues in the leader peptide resulted in greatly decreased protease activity. On the other hand, His10-LctT150 accepted all mutant peptides containing mutations in the propeptide or at non-conserved positions of LctA. In the course of this study, the first complete in vitro synthesis of lacticin 481 was achieved by treating His6-LctA consecutively with His6-LctM and His10-LctT150.
Materials and Methods
General Methods
Molecular biology techniques were carried out using standard protocols. Mass spectrometry was performed by the Mass Spectrometry Laboratory at the University of Illinois using a Voyager matrix-assisted laser desorption ionization time of flight (MALDI-TOF) spectrometer. Unless otherwise noted, all substrates including chimera were cloned, overexpressed, and purified according to (22).
Construction of Plasmids pGSTLctT150 and pHis10-LctT150
L. lactis CNRZ 481 was grown in M17 broth supplemented with 0.5% glucose (GM17). Genomic DNA was isolated using a DNAzol kit from Molecular Research Center, Inc., and was used as the template for generating the 450 bp fragment corresponding to the N-terminal 150 amino acids of LctT. This PCR product was obtained using a forward primer containing a BamHI restriction site (primer 1, Table S1) and a reverse primer containing an XhoI site (primer 2, Table S1). The PCR product and pGEX-6p-1 (GE Healthcare Life Sciences) were digested with BamHI-XhoI and ligated to produce the vector pGSTLctT150 encoding the N-terminal 150 amino acids of LctT fused at its N-terminus to a glutathione S-transferase tag and a PreScission™Protease cleavage site (LEVLFQGP). pGSTLctT150, was used as template for amplification of a 450 bp lctT150 gene fragment containing a 5′-NdeI site and a 3′-BamHI site (primers 3 and 4, Table S1). The PCR product and pET16b (Novagen) were digested with NdeI-BamHI and ligated to produce the vector pHis10LctT150 encoding the N-terminal 150 amino acids of LctT fused at its N-terminus to a His10-tag (30) containing a Factor Xa site (MGHHHHHHHHHHSSGHIEGRH). The correct sequence of the lctT inserts were confirmed by sequencing at the Biotechnology Center at the University of Illinois at Urbana-Champaign.
Construction of plasmids pGSTLctT150(C12A) and pGSTLctT150(C12S)
The lctT150 gene was amplified from pGSTLctT150 by PCR employing forward primers encoding the C12A or C12S mutation and containing a BamHI restriction site (Table S1, primers 5 and 6, respectively.) and the wild-type reverse primer with XhoI site (Table S1, primer 2). The PCR products and vector pGEX-6p-1 (GE Healthcare Life Sciences) were digested with BamHI-XhoI, and ligated to produce the plasmids pGSTLctT150(C12A) and pGSTLctT150(C12S).
Construction of plasmids pGSTLctT150(H90A), pGSTLctT150(H90N), and pGSTLctT150(D106A)
The entire pGSTLctT150 plasmid was amplified by QuikChange Site-Directed Mutagenesis (Stratagene) using the appropriate mutant forward primers (Table S1, primers 8, 10, and 12) and reverse primers (Table S1, primers 7, 9, and 11).
Construction of plasmids for mutants of His10-LctT150: C12A, C12S, H90A, H90N, and D106A
The lctT150 mutant genes were amplified by PCR from the corresponding pGSTLctT150 mutant plasmids using the wild-type NdeI forward primer (Table S1, primer 3) and wild-type BamHI reverse primer (Table S1, primer 4). The mutant genes and pET16b were digested with NdeI-BamHI and ligated producing plasmids encoding the N-terminal 150 amino acids of LctT with the appropriate single mutation fused at its N-terminus to a His10-tag.
Construction of plasmids for mutants of His6-LctA: E-13A, L-7A, A-1G, and K1A
A 5′ gene fragment was amplified by PCR from the pHis6-LctA plasmid (11) using the T7 forward primer and an internal mutant reverse primer (Table S1, primers 11, 13, 15, and 17). An overlapping 3′ gene fragment was also generated by PCR with the T7 reverse primer and an internal mutant forward primer (Table S1, primers 12, 14, 16, and 18). Finally, a nested PCR with the overlapping gene fragments and the T7 forward and reverse primers was used to generate full-length LctA mutants. After digestion of the corresponding genes and pET15b with NdeI and BamHI, ligation produced plasmids encoding LctA point-mutants fused at their N-termini to a hexa-histidine tag (MGSSHHHHHHSSGLVPRGSH).
Overexpression of wild-type and mutant His10-LctT150
The E. coli strain BL21(DE3) containing pHis10-LctT150 (wild-type or mutant) was grown overnight in 5 mL of Luria-Bertani (LB) medium containing 100 μg/mL ampicillin. The cells were harvested at 4750g and the supernatant decanted. The cell pellet was resuspended in 1 mL of fresh LB, and the suspension used to inoculate 500 mL of LB medium containing 100 μg/mL ampicillin. The culture was incubated at 37 °C with aeration until OD600 = 0.7. Isopropyl β-d-1-thiogalactopyranoside (IPTG) was added to a final concentration of 0.5 mM. The culture was then grown at 25 °C for 5 h and the cells harvested by centrifugation at 11,900g for 25 min at 4 °C. The cell pellet was frozen in liquid nitrogen and stored at -80 °C.
Purification of wild-type and mutant His10-LctT150
His10-LctT150 was isolated by affinity chromatography using a 5 mL HiTrap chelating column (GE Healthcare Life Sciences) charged with 0.1 M CoCl2 and equilibrated with phosphate start buffer (50 mM Na2HPO4, 500 mM NaCl, pH 7.5 at 4 °C). The cell pellet was thawed and resuspended in 10 mL of phosphate start buffer. The cells were lysed by sonication with a Sonics & Materials Inc. Vibra Cell™ and the cell lysate was cleared by centrifugation at 17,400g for 30 min at 4 °C. The supernatant was filtered through a 0.45 μm syringe-tip filter (Millipore) and applied to the HiTrap column. The column was washed with two column volumes of phosphate start buffer and the protein eluted using a step gradient of imidazole. Three column volumes each of phosphate start buffer containing 0 mM, 50 mM, 100 mM, 250 mM, 500 mM, and 1 M imidazole were used. The fractions containing protein, as visualized by SDS-PAGE analysis, were concentrated by Amicon filtration with a YM-1 ultrafiltration membrane (Millipore). Glycerol was added to the concentrated protein to generate a 10% (v/v) solution, which was frozen in liquid N2 and stored at -80 °C.
Cleavage of His6-LctA substrates by His10-LctT150
Assays were performed with 2 mg/mL stock solutions of each of the substrates shown in Tables 1-3. Wild-type and mutant His6-LctA peptides and His10-LctT150 were incubated in the presence of 50 mM Na2HPO4 pH 7.5, 50 mM Na2SO4, and 1 mM DTT (buffer A) at 25 °C for 2-4 h. The presence of reductant was required for His10-LctT150 activity. The final concentration of each peptide was 0.4 mg/mL (∼ 50 μM) and that of His10-LctT150 was 0.2 mg/mL (10 μM). Reactions were quenched by the addition of 1% trifluoroacetic acid (TFA) to a final concentration of 0.1% and samples for mass analysis were prepared by C18 Zip-tip (Millipore). Reaction progress was monitored by MALDI-TOF mass spectrometry.
Table 1.
His10-LctT150 cleavage of wild-type and G(-2)A(-1) mutant His6-LctA peptides.a
| Peptide | Full length (calc.) | Leader (calc.) | Propeptide (calc.) | Resultb |
|---|---|---|---|---|
| LctA wt | 7709 (7709) | 4754 (4754) | 2973 (2973) | Ala-1 / Lys1 |
| G-2V | 7752 (7751) | 4797 (4796) | 2973 (2973) | Ala-1 / Lys1* |
| G-2E | 7781 (7781) | — (4826) | — (2973) | No cleavage |
| G-2K | 7781 (7780) | — (4825) | — (2973) | No cleavage |
| A-1I | 7752 (7751) | — (4796) | — (2973) | No cleavage |
| A-1D | 7754 (7753) | — (4798) | — (2973) | No cleavage |
| A-1K | 7765 (7766) | — (4811) | — (2973) | No cleavage |
| A-1G | 7695 (7695) | 4738 (4740) | 2971 (2973) | Gly-1 / Lys1 |
Masses for peaks observed by MALDI mass spectrometry are shown, with calculated values in parentheses, (—) indicates no cleavage products were observed, and (*) indicates reduced enzyme activity based on the intensity of the product ion relative to the substrate ion.
Site of observed cleavage, indicating N-terminal amino acid / C-terminal amino acid with respect to cleavage site.
Table 3.
His10-LctT150 cleavage of wild-type and propeptide mutant His6-LctA peptides and chimeric substrates.a
| Peptide | Full Length (calc.) | Leader (calc.) | Propeptide (calc.) | Resultb |
|---|---|---|---|---|
| LctA wt | 7709 (7709) | 4754 (4754) | 2973 (2973) | Ala-1 / Lys1 |
| K1A | 7652 (7652) | 4753 (4754) | 2914 (2916) | Ala-1 / Ala1 |
| G3I/G5H | 7850 (7845) | 4755 (4754) | 3107 (3109) | Ala-1 / Lys1 |
| K1-G2insAAAc | 7925 (7922) | 4683 (4683) | — (3257) | Gly-2 / Ala-1 |
| 4755 (4754) | 3187 (3186) | Ala-1 / Lys1 | ||
| 5153 (5152) | — (2788) | Gly2 / Gly3 | ||
| 5210 (5209) | — (2731) | Gly3 / Val4 | ||
| LctMutA | 8427 (8423) | 5127 (5126) | 3317 (3317) | Ala-1 / Asn1 |
| LctRumA | 7858 (7853) | 5131 (5126) | 2748 (2747) | Ala-1 / Gly1 |
| LctNukA | 7769 (7767) | 4752 (4752) | 3029 (3031) | Ala-1 / Lys1* |
| LctSpaS | 8573 (8568) | 5126 (5126) | 3462 (3465) | Ala-1 / Trp1* |
| LctNisA | 8605 (8601) | — (5126) | — (3498) | No cleavage |
Masses for peaks observed by MALDI mass spectrometry are shown, with calculated values in parentheses, and (*) indicates reduced enzyme activity based on the intensity of the product ion relative to the substrate ion. Based on the relative intensities of the leader peptide and the propeptide in all mass spectra, the ionization efficiency for the leader peptide is likely much higher, potentially because of the His-tag. In some cases, the propeptide was not detected at all (—) in the expected mass range, presumably because of poor ionization efficiency and ion suppression.
Site of observed cleavage, indicating N-terminal amino acid / C-terminal amino acid with respect to cleavage site.
For full linear amino acid sequence, see Chatterjee et. al (35).
Cleavage of His6-LctA substrates by His10-LctT150 at low substrate concentrations
His6-LctA (wild-type, V-12A, L-7E, D-6P) and His6-MutLctA were incubated with His10-LctT150 in 1 mL of diluted cleavage buffer (10 mM Na2HPO4 pH 7.5, 10 mM Na2SO4, and 0.2 mM DTT) at 25 °C for 4 h. The final concentration of each peptide was 0.2 μM and that of His10-LctT150 was 0.5 μM. The reactions were quenched by addition of 1% TFA and then concentrated 10-fold by a Centrivap concentrator (Labconco). Samples for mass analysis were prepared by C18 Zip-tip (Millipore) and reaction progress was monitored by MALDI-TOF mass spectrometry.
In vitro Synthesis of Lacticin 481
His6-LctA and His6-LctM were incubated in the presence of 50 mM MOPS pH 7.2, 2.5 mM ATP, 2.5 mM TCEP, and 10 mM MgCl2 at 25 °C for 2-4 h. The dehydrated material was applied to a C18 Zip-tip and eluted in 10 μL of 50% acetonitrile, 0.3% TFA. The solvent was removed by a Centrivap concentrator, and the residue dissolved in 10 μL of buffer A. This product mixture was incubated with 10 μL of 0.5 mg/mL His10-LctT150 at 25 °C for 12 h. Aliquots for MS analysis were quenched with 1% TFA, partially purified by Zip-tip and analyzed by MALDI-TOF mass spectrometry.
Synthesis and Activity of Short Peptide Substrates
The 10-mer peptide DLILGAKGGS corresponding to the P1-P6 and P1′-P4′ residues of LctA was synthesized using standard Fmoc-based solid phase peptide synthesis and subsequent HPLC purification using previously described protocols (31). In addition, the peptide Ac-LILGA-pNP (with pNP = C-terminal para-nitrophenyl ester) was prepared by solid phase peptide synthesis of AcLILGA and subsequent esterification. Both peptides were incubated with His10-LctT150 or with GST-LctT150 but no activity was observed by either LC-MS or UV-vis spectroscopy.
Results
Cloning and isolation of LctT150
Part of the lctT gene corresponding to the N-terminal 150 amino acids was amplified by PCR from genomic DNA of L. lactis CNRZ 481. This specific truncation of the LctT protein was chosen for study as sequence alignment with known bacteriocin AMS proteins followed by application of a previously reported algorithm (32) identified a possible interdomain linker region between residues 125-175. The gene fragment was inserted into pGEX-6p-1 to generate an N-terminal glutathione S-transferase (GST) fusion of the LctT protease domain (LctT150). This fusion protein was heterologously overexpressed in E. coli and purified by glutathione affinity chromatography. Incubation of the protein with His6-LctA resulted in very low protease activity and surprisingly cleavage between Gly-2 and Ala-1 (Figure S1). Given the low activity and the incorrect position of proteolysis, this fusion protein was not further investigated. The 450 bp lctT150 gene fragment was instead inserted into pET16b to produce an N-terminal ten histidine (His10) fusion of LctT150. His10-LctT150 was heterologously expressed in E. coli as a soluble fusion-protein and purified by Co2+-NTA affinity chromatography, resulting in 2 mg of fusion protein per liter of cell culture. It should be noted that attempts to purify this fusion protein by Ni2+ affinity chromatography were unsuccessful.
His10-LctT150 activity with wild-type His6-LctA
Small-scale assays were performed with wild-type His6-LctA in order to assess enzyme activity. All assays were performed at 25 °C for 2-4 h at pH 7.5 and reaction progress was monitored by MALDI-TOF mass spectrometry. Figure 2A shows the mass spectrum for His10-LctT150-catalyzed cleavage of His6-LctA. A small peak for starting material was observed at 7709 Da, along with two proteolysis products. The peak at 4754 Da corresponds to residues -25 to - 1 of His6-LctA, which is the N-terminal fragment resulting from amide bond hydrolysis at the predicted site between Ala-1 and Lys1 (Figure 1). The corresponding C-terminal product, LctA1-27 was observed at 2973 Da. The amide hydrolysis reaction did not require any metals or cofactors and was not inhibited by EDTA. The presence of a thiol reducing agent (DTT or TCEP) was required for activity during assay but not during purification or for storage of the enzyme. Quantitative analysis to determine kinetic parameters was hampered by the previously documented poor solubility of the substrate peptide (33, 34). Attempts to use a synthetic truncated substrate DLILGA-KGGS corresponding to the P1-P6 and P1′-P4′ residues of LctA were unsuccessful, as this peptide was not processed by the enzyme. Similarly, a para-nitrophenyl ester of a truncated substrate was not hydrolyzed by His10-LctT150.
Figure 2.
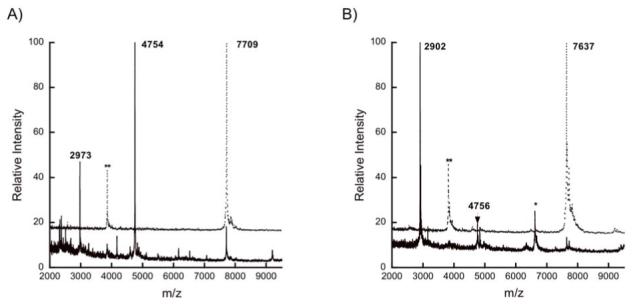
In vitro reconstitution of LctT proteolytic activity. MALDI mass spectra for (A) linear His6-LctA and (B) His6-LctM-processed His6-LctA after incubation with (solid lines) or without (dotted lines) His10-LctT150. The double asterisk (**) indicates a peak corresponding to doubly charged starting material, and the single asterisk (*) to a triply charged His10-LctT150 ion.
The same cleavage pattern was observed when His6-LctA was dehydrated and cyclized using His6-LctM (Figure 1) and then subjected to cleavage by His10-LctT150 (Figure 2B). The His6-LctM-processed His6-LctA was detected at 7637 Da after the loss of four water molecules. After His10-LctT150 treatment, the leader sequence was observed at 4756 Da and lacticin 481 was detected at 2902 Da, indicating that the loss of four water molecules occurred in the propeptide. Comparing Figures 2a and 2b, it appears that the ionization efficiency of the C-terminal fragment is enhanced upon dehydration and cyclization, but this observation was not further investigated. These results are the first in vitro biosynthesis of authentic lacticin 481 as previous in vitro studies before the availability of LctT150 utilized the commercial protease Lys-C, which produces Δ1-lacticin 481 (11, 34, 35). As no difference was observed in LctT activity between the linear and cyclized substrates, further studies were continued with linear peptides for simplicity.
Proteolytic activity of His10-LctT150 mutant proteins
Three conserved residues of LctT proposed to be involved in peptide bond cleavage (Cys12, His90, and Asp106) (24) were mutated to investigate their importance in amide bond hydrolysis. The involvement of these residues in catalysis has been suggested, but never experimentally tested for lantibiotic proteases. Mutation of Cys12 to either Ser or Ala resulted in loss of all proteolytic activity (Figure 3A and 3B, respectively) consistent with the LctT protease domain being a cysteine protease. Surprisingly, mutation of His90 to Ala resulted in incorrect cleavage between Gly-2 and Ala-1 (Figure 3C). Mutation of Asp106 to Ala had no effect on peptidase activity (Figure 3D) indicating this residue is not essential for catalysis.
Figure 3.
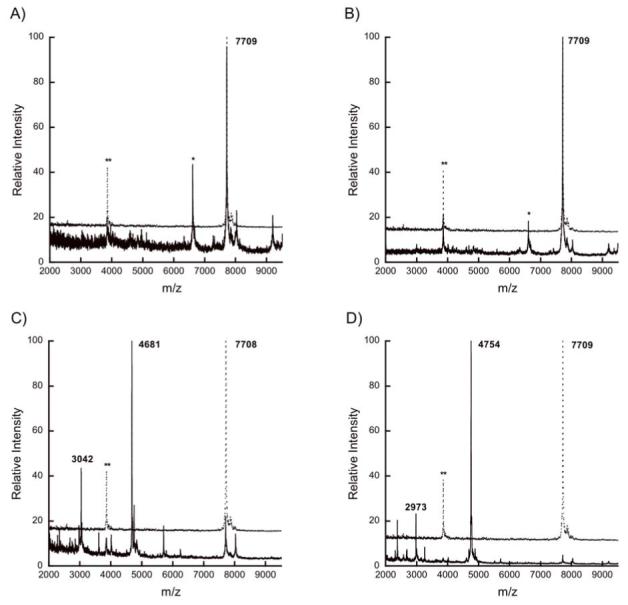
MALDI mass spectra for His6-LctA wild-type after incubation with (solid lines) or without (dotted lines) (A) His10-LctT150 C12S, (B) His10-LctT150 C12A, (C) His10-LctT150 H90A, or (D) His10-LctT150 D106A. The double asterisk (**) indicates a peak corresponding to doubly charged starting material, and the single asterisk (*) to a triply charged His10-LctT150 ion.
His10-LctT150 activity with His6-LctA double glycine mutant peptides
Enzymatic assays were performed with a number of His6-LctA mutant peptides. Because LctT was predicted to recognize the double glycine motif at the cleavage site, seven peptides with mutations at residues -2 and -1 were generated. The results of the assays with these mutants are summarized in Table 1. Mutation of Gly-2 or Ala-1 to a charged residue (Glu, Asp, or Lys) completely abolished cleavage by His10-LctT150 (Figure 4A and Figure S2a-c). Exchange of Ala-1 for Ile also resulted in complete inhibition of proteolysis (Figure S2d). Out of seven double glycine motif mutant peptides, only His6-LctA(G-2V) (Figure S2e) and A-1G (Figure 4B) were accepted as substrates by His10-LctT150. However, the G-2V peptide was not a good substrate and was processed to a much lesser extent than the wild type sequence under identical conditions (Figure S2e).
Figure 4.
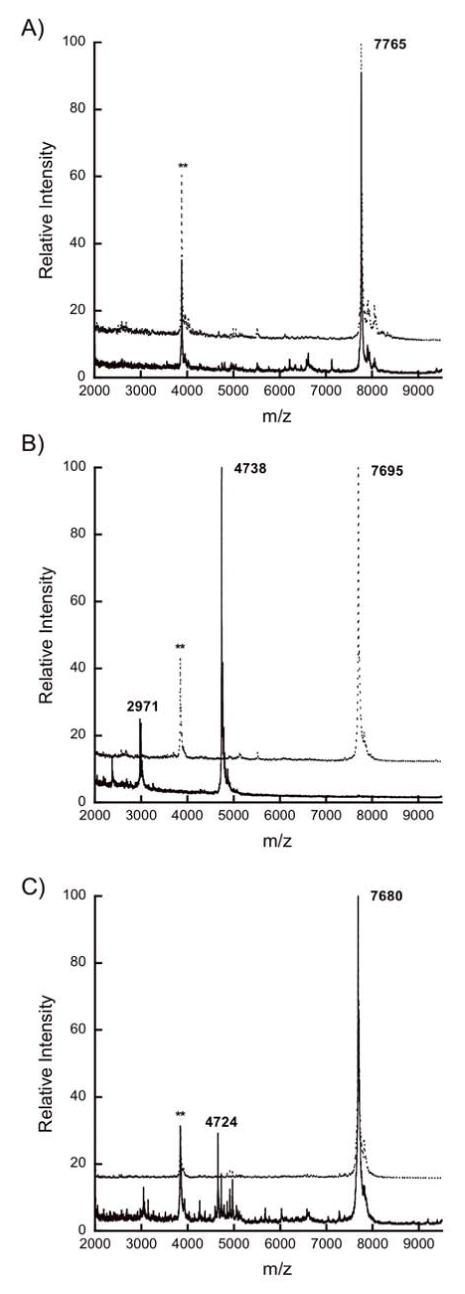
MALDI mass spectra for (A) His6-LctA A-1K, (B) His6-LctA A-1G, and (C) His6-LctA E-8P after incubation with (solid lines) or without (dotted lines) His10-LctT150. The double asterisk (**) indicates a peak corresponding to doubly charged starting material ion.
His10-LctT150 catalyzed proteolysis of His6-LctA leader sequence mutant peptides
In order to evaluate the importance of other conserved residues in the leader sequence besides Gly-2 and Ala-1, the mutants E-13A, V-12A, E-8A, L-7A, L-7E, L-7K, and L-5Q were investigated. Assay results for these peptides are summarized in Table 2 and show that these mutations did not prohibit hydrolysis by His10-LctT150 (Figure S3), except in the case of E-8A for which the cleavage efficiency was reduced (Figure S3d). As discussed above, we have not been able to obtain quantitative kinetic parameters and efforts to use competition assays to determine relative catalytic efficiencies with respect to the wild type sequence provided unreliable results. Qualitatively, however, with the exception of E-8A all mutant substrates were processed with very similar efficiencies as wild type LctA based on relative intensities of the substrate and product ions observed by MALDI-TOF MS under identical assay conditions.
Table 2.
His10-LctT150 cleavage of wild-type and leader sequence mutant His6-LctA peptides.a
| Peptide | Full length (calc.) | Leader (calc.) | Propeptide (calc.) | Resultb |
|---|---|---|---|---|
| LctA wt | 7709 (7709) | 4754 (4754) | 2973 (2973) | Ala-1 / Lys1 |
| E-13A | 7653 (7651) | 4697 (4696) | 2972 (2973) | Ala-1 / Lys1 |
| V-12A | 7677 (7681) | 4727 (4726) | 2975 (2973) | Ala-1 / Lys1 |
| V-12P | 7708 (7707) | 4753 (4752) | — (2973) | Ala-1 / Lys1* |
| E-8A | 7652 (7651) | 4696 (4696) | — (2973) | Ala-1 / Lys1* |
| E-8P | 7680 (7677) | 4724 (4722) | — (2973) | Ala-1 / Lys1* |
| 4653 (4651) | — (3044) | Gly-2 / Ala-1* | ||
| L-7A | 7668 (7667) | 4712 (4712) | 2972 (2973) | Ala-1 / Lys1 |
| L-7E | 7727 (7725) | 4772 (4770) | 2975 (2973) | Ala-1 / Lys1 |
| L-7K | 7728 (7724) | 4773 (4769) | — (2973) | Ala-1 / Lys1 |
| D-6P | 7695 (7691) | 4739 (4736) | 2975 (2973) | Ala-1 / Lys1 |
| L-5Q | 7725 (7724) | 4769 (4769) | 2973 (2973) | Ala-1 / Lys1 |
| L-5P | 7695 (7693) | 4738 (4738) | — (2973) | Ala-1 / Lys1* |
| I-4P | 7695 (7693) | 4742 (4738) | — (2973) | Ala-1 / Lys1* |
| MutLctA | 8085 (8081) | 5127 (5126) | 2973 (2973) | Gly-1 / Lys1 |
Masses for peaks observed by MALDI mass spectrometry are shown, with calculated values in parentheses, and (*) indicates reduced enzyme activity based on the intensity of the product ion relative to the substrate ion. Based on the relative intensities of the leader peptide and the propeptide in all mass spectra, the ionization efficiency for the leader peptide is likely much higher, potentially because of the His-tag. In some cases, the propeptide was not detected at all (—) in the expected mass range, presumably because of poor ionization efficiency and ion suppression.
Site of observed cleavage, indicating N-terminal amino acid / C-terminal amino acid with respect to cleavage site.
Five additional mutant peptides (V-12P, E-8P, D-6P, L-5P, and I-4P) were generated to disrupt potential secondary structure in the leader peptide that could influence the activity of LctT150, as structure prediction programs suggest the leader sequence may be helical (22). Interestingly, all of these mutants except D-6P resulted in reduced efficiency of cleavage by His10-LctT150 (Figure S3). As a representative example, Figure 4C shows the level of cleavage of His6-LctA E-8P by His10-LctT150 under conditions where His6-LctA would be completely processed. The effects of these mutations are not entirely due to disruption of secondary structure, however. For example, for Glu-8, mutation to Ala had a similar effect on proteolysis as mutation to Pro (Figure S3d), suggesting the glutamate side chain provides an important recognition point. On the other hand, mutation of Val-12 to Ala and Leu-5 to the isosteric Gln resulted in substrates that were processed with similar efficiencies as the wild type LctA peptide (Figures S3b and S3i, respectively), suggesting that the observed decreased proteolysis with V-12P and L-5P (Figure S3j) is related to disruption of the structure of the peptide.
In addition to these point mutants, the possible importance of the non-conserved residues in the leader sequence was investigated via a chimera (MutLctA) joining the leader sequence of the mutacin II prepeptide, MutA, to the propeptide of LctA (22). The leader peptides from MutA and LctA share significant sequence homology, with 11 out of 24 residues identical in the two peptides (Figure 5). Therefore, the chimeric peptide amounted to a mutant in which about 55% of the leader peptide residues were mutated simultaneously, with the mutations concentrated in non-conserved positions. We do note that the leader peptides of these two peptides differ in the double glycine motif residues with the MutA leader ending in G(-2)G(-1) as opposed to G(-2)A(-1) for LctA. However, because the LctA A-1G mutant was a good substrate for His10-LctT150 as shown above, this difference was not considered a problem. As shown in Figure 6A, the hybrid peptide was a substrate for His10-LctT150, displaying about the same proteolytic efficiency as the His6-LctA and His6-LctA A-1G substrates.
Figure 5.

Sequence alignment of lantibiotic and non-lantibiotic bacteriocin prepeptides. The residues in red indicate the positions that are fully conserved within that class, and those in blue are highly conserved. For the non-lantibiotic bacteriocins, only the leader sequences are shown. The site of proteolysis is indicated by the arrow.
Figure 6.
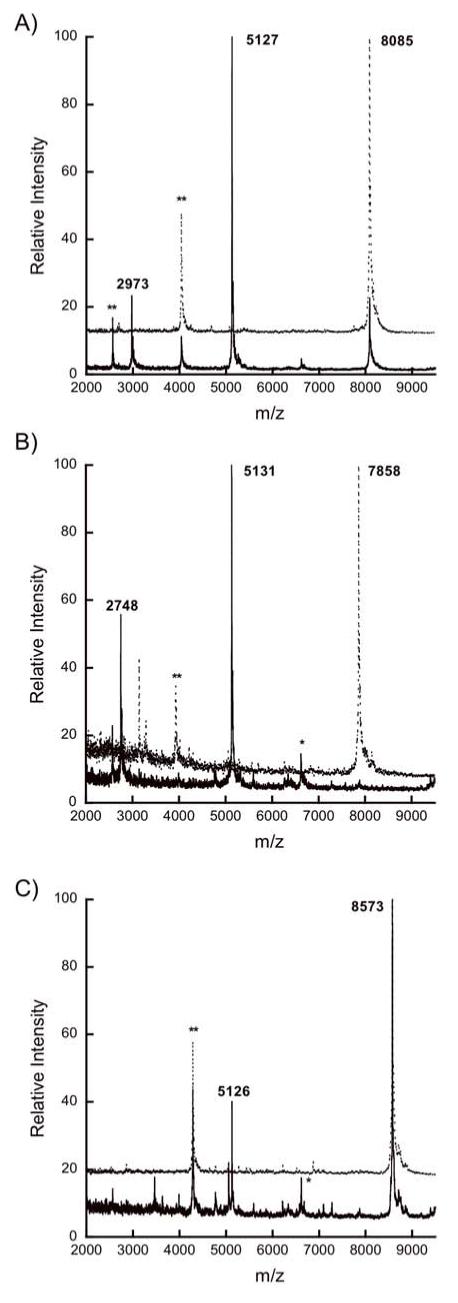
MALDI mass spectra for (A) His6-MutLctA, (B) His6-LctRumA, and (C) His6-LctSpaS after incubation with (solid lines) or without (dotted lines) His10-LctT150. The double asterisk (**) indicates a peak corresponding to doubly charged starting material, and the single asterisk (*) to a triply charged His10-LctT150 ion.
As has been reported previously (34), the LctA peptide has very poor solubility (∼1 μM) in aqueous buffer at pH 7, which has precluded quantitative assessment of the concentration dependence of enzyme activity. All data discussed so far were obtained at the solubility limit. One possible explanation for the apparent lack of effect of several of the mutations in LctA discussed above is that at the concentrations used the observed activities report on kcat and that this parameter was unaffected by the mutations. If so, the possibility still existed that these mutations could affect Km. To test potential binding effects, several of the His6-LctA substrates (wild-type, E-13A, V-12A, L-7E, L-7A, L-5Q, D-6P, A-1G, K1A, and His6-MutLctA) were incubated with His10-LctT150 at lower substrate and enzyme concentrations (0.2 μM) and in a larger reaction volume to obtain sufficient material for MS analysis. Comparison of the data so obtained showed that V-12A, L-7A, L-5Q, A-1G, and MutLctA were processed to a similar extent as wild type His6-LctA, but E-13A, L-7E, and D-6P were poorer substrates under these conditions (Figure S4).
His10-LctT150 catalyzed proteolysis of His6-LctA propeptide mutant peptides
Three His6-LctA peptides containing mutations in the P1′ - P5′ sites were generated to determine if the propeptide residues C-terminal to the cleavage site are important for proteolysis by His10-LctT150. The results for these peptides are summarized in Table 3. Mutation of Lys1 in the P1′ position to Ala did not abolish proteolysis (Figure S5a). Similarly, simultaneous mutation of Gly3 in the P3′ position to Ile and Gly5 in the P5′ position to His resulted in a peptide that was accepted by His10-LctT150 (Figure S5b). Analysis of a previously generated mutant in which three Ala residues were inserted between Lys1 and Gly2 (35) showed that this peptide was hydrolyzed in the correct location after Ala-1 but also after Gly-2, Gly5 and Gly6 (Figure S5c).
His10-LctT150 Cleavage of His6-LctA Chimeras
In addition to the MutLctA chimeric substrate discussed above, five other chimeric peptides were generated in order to investigate the role of the propeptide as a whole in cleavage by LctT. These peptides contain the LctA leader sequence fused at its C-terminus to the propeptide of other lantibiotics including MutA (mutacin II), RumA (ruminococcin), SpaS (subtilin), NukA (nukacin ISK-1), and NisA (nisin) (for sequences, see Figure 5). Both the LctRumA and LctMutA peptides were cleaved efficiently by His10-LctT150 (Figures 6B and S6a, respectively). The LctNukA chimera was cleaved correctly (Figure S6b), albeit to a lesser degree than the other class II chimeras, in agreement with a previous report on the in vivo processing of preNukA by LctT (36). Interestingly, fusion of the LctA leader to the class I lantibiotic subtilin propeptide (LctSpaS) resulted in a peptide that was a substrate for His10-LctT150 although under identical assay conditions, it was not cleaved to the same degree as the class II chimeric substrates (Figure 6C). LctNisA was not a substrate for His10-LctT150 (Figure S6c).
Discussion
The biosynthesis of lantibiotics has been extensively studied for several decades, but relatively little is known about the function of the leader peptides. Several roles have been proposed, including acting as a scaffold for the modification enzymes, allowing the prepeptide to be recognized for export, or protecting the producing organism (9). The leader sequences within each lantibiotic class share several conserved residues that could contribute to these functions (Figure 5). Thus far, studies on the importance of these residues have been conducted via in vivo mutagenesis in which the leader sequence was changed in a lantibiotic producing bacterial strain. For class I lantibiotics, such studies on nisin biosynthesis demonstrated that the highly conserved leader residues in the -18 to -15 region (Figure 5) are essential for lanthionine incorporation into the precursor peptide, while Ala-4 and Arg-1 are essential for processing by NisP (10). This same region of the Pep5 precursor peptide, however, appears unimportant for correct modification to the mature species, but is important for biosynthetic efficiency (29). Similarly, five mutations of conserved residues in the leader peptide of the class II lantibiotic mutacin II (E-13K, V-12L, E-8K, L-7K, and I-4D) resulted in decreased production of mutacin II as compared to a wild-type Streptococcus mutans strain. Two MutA mutant peptides, G-1A and G-2A, prevented the cleavage and/or transport event for production of mutacin II, as dehydrated premutacin accumulation was observed in the cell membranes of these mutant strains (15). The G-2A mutation in the LctA peptide also dramatically reduced in vivo cleavage by His10-LctT150 (13). Although these studies have provided valuable insights into the importance of certain leader peptide residues for lantibiotic production, to date a comprehensive analysis of their significance for lanthionine formation and proteolytic processing is not available. In this work, we dissected the importance of leader peptide residues for these two processes, reporting on the effects of alteration of the substrate sequence with respect to dehydration and cyclization in the accompanying paper, and disclosing in this study the first in vitro analysis of the importance of the leader peptide for proteolytic processing.
Because the bifunctional LctT transporter/protease is an integral membrane protein of 691 amino acids (6), we developed a heterologous expression system for only the protease domain. Analogous to similar studies on the protease domain of bifunctional transporters for non-lantibiotic bacteriocins (24-26), expression in E. coli of the first 150 residues of LctT resulted in an active protease that correctly hydrolyzed the amide bond between Ala-1 and Lys1 in LctA. These findings indicate that the transmembrane transporter domain of LctT is not required for substrate recognition and proteolysis. Furthermore, His10-LctT150 processed LctM-treated LctA containing the three lanthionine rings about equally well as the linear LctA peptide, demonstrating that the rings are not responsible for substrate recognition. This finding is in contrast to the serine proteases NisP and PepP involved in removal of the leader peptides of the class I lantibiotics nisin and Pep5, for which linear and dehydrated NisA and PepA were not substrates in vivo (17, 37). Håvarstein and coworkers proposed that transport and proteolysis of pre-bacteriocins by ABC transporter maturation and secretion (AMS) proteins may be intimately connected and that substrate recognition for transport may reside in the protease domain (24). Our observation that proteolysis by His10-LctT150 does not depend on the presence of lanthionine rings suggests that AMS proteins can be used to secrete non-lantibiotic peptides attached to the leader sequence of their cognate bacteriocin, similar to previous reports demonstrating such activity for the NisT transporter involved in nisin biosynthesis (17, 19, 38).
Based on sequence homology with papain-like cysteine proteases in the MEROPS database (39), the class II lantibiotic LanT proteins have been proposed to utilize a conserved Cys-His pair in the active site of their protease domains (Figure 7). Although no structural information is currently available on protease domains of bifunctional ABC-type transporters, mutational analysis of the protease domains of the non-lantibiotic transporters LagD, ComA, and CvaB support the importance of these two residues (24, 25). Our results provide additional experimental support for this model for the protease domains of the lantibiotic subgroup of AMS proteins as C12A and C12S mutants of His10-LctT150 were devoid of proteolytic activity. Interestingly, mutation of the putative catalytic histidine to alanine did not prevent cleavage but caused incorrect proteolysis after Gly-2 instead of Ala-1. This surprising result indicates that His90 is not essential for catalysis under the assay conditions employed, and may instead play a role in the regiospecificity of amide bond hydrolysis. This observation is in contrast to previous studies on cleavage of the colicin V precursor by the protease domain of CvaB and hydrolysis of ComC by ComA in which mutation of the corresponding His to Ala abolished protease activity (25, 26). Similar to mutation of His90, replacement of the conserved Asp residue with Ala in His10-LctT150 did not affect the cleavage reaction, suggesting Asp106 is not essential for catalysis. This residue has not been investigated in LagD, ComA, or CvaB. One possible explanation for the apparent unimportance of His90 and Asp106 involves the protonation state of the protein. The assays in this study were conducted at pH 7.5, but lacticin 481 production has been reported to be induced at pH 5 in the lactic acid bacterium Lactococus lactis (40). If the active site of LctT is in an environment of low pH, Cys may be protonated in the absence of His90 and/or Asp106, resulting in an inactive protease. Unfortunately, we were unable to test this hypothesis, as His10-LctT150 was unstable at pH 5. It has been reported that the activity of the protease domain of CvaB is Ca2+ dependent, but like ComA (25) no metal requirement was observed for LctT150.
Figure 7.

Sequence alignment of the protease domain of LctT with papain and the non-lantibiotic bacteriocin peptidase domains of CvaB, ComA, and LcnC. The first and last numbers indicate amino acid positions and the numbers in parentheses denote the number of excluded residues. The conserved active site residues Cys-His-Asp/Asn and the oxyanion hole Gln are highlighted in blue and marked with asterisks.
The cleavage site for removal of leader peptides of certain members of both lantibiotic and non-lantibiotic bacteriocins is governed by GG or GA, historically called the double Gly motif (41-44). As mentioned above, the importance of this motif for lantibiotic biosynthesis has been investigated through a limited set of mutations. In non-lantibiotic systems, the motif has been probed in one in vitro study that showed that mutation of either of the two Gly in the ComC substrate decreased the catalytic efficiency of proteolysis by the protease domain of ComA by 500 to 1,000-fold (25). In this investigation, an extensive analysis of the double Gly motif showed that mutation of either Gly-2 or Ala-1 to a charged residue completely abolished His10-LctT150 protease activity. Exchange of Ala-1 for Ile also resulted in loss of peptidase activity. Interestingly, mutation of Gly-2 to Val did not prevent cleavage but did reduce the efficiency, as only small amounts of cleavage products were observed under conditions that lead to full conversion of His6-LctA. Only the LctA A-1G mutation allowed His10-LctT150 to retain good proteolytic activity.
With the exception of mutations of Glu-8, replacement of the other conserved residues did not dramatically alter proteolytic processing by His10-LctT150. Glu-8 is also implicated as an important residue for proteolysis and/or transport by its conservation in the double Gly type leader peptides of non-lantibiotic bacteriocins (Figure 5). Since these peptides do not undergo any post-translational modifications other than proteolysis (43), the conservation of Glu-8 is likely to be related to proteolysis and secretion. With respect to the observation that mutation of the other conserved residues did not prevent proteolysis, it is important to emphasize that we have been unable to obtain kinetic parameters because of the insolubility of LctA, despite various attempts with truncated synthetic peptides. As a result, we have no information regarding kcat and Km values and some of the mutants might still be poor substrates under physiological conditions of low substrate concentration as shown for instance for L-7E, E-13A, and D-6P.
As discussed in the accompanying paper, secondary structure predicting programs anticipate α-helical character for the LctA leader peptide between Asn-17 and Leu-3 (22). A helical structure of lantibiotic leader peptides has been suggested previously on the basis of circular dichroism experiments performed with synthetic leader peptides of class I lantibiotics nisin, subtilin, gallidermin, epidermin, and Pep5 (45, 46). These studies were carried out in the presence of trifluoroethanol, which induces helicity, but the biosynthetic proteins may similarly promote or recognize a helical structure, as has recently been demonstrated for binding of the prepeptide ComA to the ComA peptidase domain (27). Circular dichroism and NMR studies also demonstrated α-helical character in the -15 to -5 region of the leader sequence for the non-lantibiotic bacteriocin carnobacteriocin B2 (47). This helix was proposed to be a key recognition element for export and processing and possibly also for keeping the bacteriocin inactive until removal of the leader peptide. In the current work, four of the five Pro mutants in the predicted helical region of His6-LctA, V-12P, E-8P, L-5P, and I-4P were poor substrates for His10-LctT150 indicating putative secondary structure of the leader may indeed be important for substrate recognition. Surprisingly, D-6P was a relatively good substrate being processed about 60% at the highest concentrations allowed by its solubility limit. Under these conditions, wild type LctA is almost completely proteolyzed. At low substrate concentration, however, D-6P also was a poor substrate, indicating its reduced ability to bind to the enzyme. Hence, all Pro mutants appear to reduce the efficiency of processing by His10-LctT150.
The recognition of a helical secondary structure by LctT may also explain the additional incorrect processing observed when three Ala residues were inserted between K1 and G2 (LctA-K1G2insAAA). Secondary structure predicting programs indicate an extension of the α-helical region of the leader peptide in this mutant to the second inserted Ala residue. If the active site of LctT is positioned such that it cleaves at a sequence of two consecutive small amino acids (Gly or Ala) immediately C-terminal to a helical region, the observed cleavage between Gly5 and Gly6 and between Gly6 and Ser7 in this mutant can be rationalized. According to this hypothesis, the lack of cleavage between these residues in wild type His6-LctA (in wt LctA these residues are Gly2, Gly3, and Ser4) is because they are not located immediately C-terminal to a helical region. If this hypothesis is correct, LctT150 and other such domains of AMS proteins may have interesting potential for application in biotechnology to remove expression tags similar to the use of Factor Xa, PreScission protease, thrombin, and enterokinase. The demonstrated high promiscuity of His10-LctT150 with respect to the residues C-terminal to the cleavage site as well as the high fidelity with which it cleaved those peptides further highlight its potential, although at low substrate concentrations several of the mutants in which the propeptide was mutated were processed slower than wild type LctA (Figure S4). Because, LctT150 appears to recognize only the residues N-terminal to the cleavage site, tag removal is traceless.
A different strategy was employed to probe the importance of non-conserved residues for proteolytic processing to test the possibility that the determinants for substrate specificity for a particular lantibiotic are imparted by the non-conserved residues that differ from compound to compound. The chimeric substrate His6-MutLctA consisting of the mutacin II leader peptide and lacticin 481 propeptide contained 12 simultaneous mutations of mostly non-conserved residues (Figure 5). Nevertheless, it was processed by His10-LctT150 with about the same efficiency as the LctA prepeptide indicating that these residues are not important for substrate recognition and catalysis. A similar conclusion was reached for the dehydration and cyclization events in the accompanying paper (22).
As previously discussed, formation of the lanthionine rings was also not required for proteolysis by LctT150, suggesting that the propeptide is not important for substrate recognition. However, the N-terminus of lacticin 481 has a linear sequence of eight residues (Figure 1), which might be important for leader sequence removal. Non-conservative mutations of residues in the P1′-P5′ positions (K1A, G3I, G5H), however, did not affect proteolysis by His10-LctT150, and several chimeric peptides containing multiple simultaneous mutations in these positions (eg P1′: Asn, Trp, Gly; P2′: Lys, Arg, Asn; P3′: Ser, Trp, Gly, Lys (Table 3 and Figure 5)) were likewise processed. Thus, the residues C-terminal to the cleavage site are not important, which may also explain why some two-component lantibiotic biosynthetic gene clusters contain only a single LanT protein despite dramatic differences between the two propeptides of their substrates (12, 48). A possible exception to the high promiscuity with respect to the residues C-terminal to the cleavage site may be a branched residue at the P1′ position because the nisin chimera with an Ile at this position was not accepted. An alternative explanation for the lack of activity with LctNisA is that secondary structure prediction programs anticipate an α-helix for the leader sequence of LctNisA in addition to β-strand character C-terminal to the cleavage site. The presence of this additional secondary structure may be responsible for inhibition of protease activity.
In summary, the double Gly motif is critical for proteolysis with only an Ala-1Gly mutation in LctA tolerated by the protease. Collectively, the results with mutations of conserved residues to Pro residues support the hypothesis that the region immediately N-terminal to the cleavage site is recognized as a helix by His10-LctT150. Interestingly, Glu-8 appears important for substrate recognition by LctT but mutations other than Pro are tolerated at this position by LctM. Thus, it appears that different residues of the leader peptide are important for specific events in the overall post-translational modification process of lacticin 481 biosynthesis.
Supplementary Material
Acknowledgements
The authors thank Gregory C. Patton and Lisa E. Cooper for the preparation of His6-LctA mutant peptides, and Moushumi Paul for generation of the chimeric peptides. All mass spectra were recorded on a Voyager mass spectrometer funded in part by the National Institutes of Health (RR 11966).
Abbreviations
- AMS
ABC transporter maturation and secretion
- Dha
2,3-didehydroalanine
- Dhb
2,3-didehydrobutyrine
- DTT
dithiothreitol
- GST
glutathione S-transferase
- IPTG
isopropyl β-d-1-thiogalactopyranoside
- LB
Luria-Bertani
- LanB
class I lantibiotic dehydratase enzymes
- LanC
class I lantibiotic cyclase enzymes
- LanP
class I lantibiotic protease
- LanM
class II lantibiotic synthetase enzymes
- LanT
class I lantibiotic transporter or class II lantibiotic ABC transporter/protease
- LctT
lacticin 481 transporter/protease
- LctM
lacticin 481 synthetase
- MALDI-TOF MS
matrix assisted laser desorption ionization - time of flight mass spectrometry
- RP-HPLC
reverse phase high performance liquid chromatography
- TCEP
triscarboxyethylphosphine
- TFA
trifluoroacetic acid
Footnotes
Supported by a research grant from the National Institutes of Health (GM58822 to WAV) and a Chinoree T. and Kimiyo Enta pre-doctoral fellowship to LAFI.
References
- 1.Chatterjee C, Paul M, Xie L, van der Donk WA. Biosynthesis and Mode of Action of Lantibiotics. Chem. Rev. 2005;105:633–684. doi: 10.1021/cr030105v. [DOI] [PubMed] [Google Scholar]
- 2.Cotter PD, Hill C, Ross RP. Bacterial lantibiotics: strategies to improve therapeutic potential. Curr. Protein Pept. Sci. 2005;6:61–75. doi: 10.2174/1389203053027584. [DOI] [PubMed] [Google Scholar]
- 3.Willey JM, van der Donk WA. Lantibiotics: Peptides of Diverse Structure and Function. Annu. Rev. Microbiol. 2007;61:477–501. doi: 10.1146/annurev.micro.61.080706.093501. [DOI] [PubMed] [Google Scholar]
- 4.Dufour A, Hindré T, Haras D, Le Pennec JP. The biology of the lantibiotics of the lacticin 481 subgroup is coming of age. FEMS Microbiol. Rev. 2007;31:134–167. doi: 10.1111/j.1574-6976.2006.00045.x. [DOI] [PubMed] [Google Scholar]
- 5.Xie L, van der Donk WA. Post-Translational Modifications during Lantibiotic Biosynthesis. Curr. Opin. Chem. Biol. 2004;8:498–507. doi: 10.1016/j.cbpa.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 6.Rincé A, Dufour A, Le Pogam S, Thuault D, Bourgeois CM, Le Pennec JP. Cloning, expression, and nucleotide sequence of genes involved in production of lactococcin DR, a bacteriocin from Lactococcus lactis subsp. lactis. Appl. Environ. Microbiol. 1994;60:1652–1657. doi: 10.1128/aem.60.5.1652-1657.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pag U, Sahl HG. Multiple activities in lantibiotics--models for the design of novel antibiotics? Curr. Pharm. Des. 2002;8:815–833. doi: 10.2174/1381612023395439. [DOI] [PubMed] [Google Scholar]
- 8.Jung G. Lantibiotics-Ribosomally synthesized biologically active polypeptides containing sulfide bridges and α,β-dehydroamino acids. Angew. Chem. Intl. Ed. Engl. 1991;30:1051–1068. [Google Scholar]
- 9.Sahl H-G, Jack RW, Bierbaum G. Biosynthesis and Biological Activities of Lantibiotics with Unique Post-Translational Modifications. Eur. J. Biochem. 1995;230:827–853. doi: 10.1111/j.1432-1033.1995.tb20627.x. [DOI] [PubMed] [Google Scholar]
- 10.van der Meer JR, Rollema HS, Siezen RJ, Beerthuyzen MM, Kuipers OP, de Vos WM. Influence of amino acid substitutions in the nisin leader peptide on biosynthesis and secretion of nisin by Lactococcus lactis. J. Biol. Chem. 1994;269:3555–3562. [PubMed] [Google Scholar]
- 11.Xie L, Miller LM, Chatterjee C, Averin O, Kelleher NL, van der Donk WA. Lacticin 481: in vitro reconstitution of lantibiotic synthetase activity. Science. 2004;303:679–681. doi: 10.1126/science.1092600. [DOI] [PubMed] [Google Scholar]
- 12.McClerren AL, Cooper LE, Quan C, Thomas PM, Kelleher NL, van der Donk WA. Discovery and in vitro biosynthesis of haloduracin, a new two-component lantibiotic. Proc. Natl. Acad. Sci. USA. 2006;103:17243–17248. doi: 10.1073/pnas.0606088103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Uguen P, Hindré T, Didelot S, Marty C, Haras D, Le Pennec JP, Vallee-Rehel K, Dufour A. Maturation by LctT Is Required for Biosynthesis of Full-Length Lantibiotic Lacticin 481. Appl. Environ. Microbiol. 2005;71:562–565. doi: 10.1128/AEM.71.1.562-565.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Corvey C, Stein T, Dusterhus S, Karas M, Entian KD. Activation of subtilin precursors by Bacillus subtilis extracellular serine proteases subtilisin (AprE), WprA, and Vpr. Biochem. Biophys. Res. Commun. 2003;304:48–54. doi: 10.1016/s0006-291x(03)00529-1. [DOI] [PubMed] [Google Scholar]
- 15.Chen P, Qi FX, Novak J, Krull RE, Caufield PW. Effect of amino acid substitutions in conserved residues in the leader peptide on biosynthesis of the lantibiotic mutacin II. FEMS Microbiol. Lett. 2001;195:139–144. doi: 10.1111/j.1574-6968.2001.tb10511.x. [DOI] [PubMed] [Google Scholar]
- 16.Izaguirre G, Hansen JN. Use of alkaline phosphatase as a reporter polypeptide to study the role of the subtilin leader segment and the SpaT transporter in the posttranslational modifications and secretion of subtilin in Bacillus subtilis 168. Appl. Environ. Microbiol. 1997;63:3965–3971. doi: 10.1128/aem.63.10.3965-3971.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuipers A, De Boef E, Rink R, Fekken S, Kluskens LD, Driessen AJ, Leenhouts K, Kuipers OP, Moll GN. NisT, the transporter of the lantibiotic nisin, can transport fully modified, dehydrated and unmodified prenisin and fusions of the leader peptide with non-lantibiotic peptides. J. Biol. Chem. 2004;279:22176–22182. doi: 10.1074/jbc.M312789200. [DOI] [PubMed] [Google Scholar]
- 18.Li B, Yu J-PJ, Brunzelle JS, Moll GN, van der Donk WA, Nair SK. Structure and Mechanism of the Lantibiotic Cyclase Involved in Nisin Biosynthesis. Science. 2006;311:1464–1467. doi: 10.1126/science.1121422. [DOI] [PubMed] [Google Scholar]
- 19.Rink R, Wierenga J, Kuipers A, Kluskens LD, Driessen AJM, Kuipers OP, Moll GN. Production of dehydroamino acid-containing peptides by Lactococcus lactis. Appl. Environ. Microbiol. 2007;73:1792–1796. doi: 10.1128/AEM.02350-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kluskens LD, Kuipers A, Rink R, de Boef E, Fekken S, Driessen AJ, Kuipers OP, Moll GN. Post-translational Modification of Therapeutic Peptides By NisB, the Dehydratase of the Lantibiotic Nisin. Biochemistry. 2005;44:12827–12834. doi: 10.1021/bi050805p. [DOI] [PubMed] [Google Scholar]
- 21.Levengood MR, Patton GC, van der Donk WA. The Leader Peptide is not Required for Post-translational Modification by Lacticin 481 Synthetase. J. Am. Chem. Soc. 2007;129:10314–10315. doi: 10.1021/ja072967+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Patton GC, Paul M, Cooper LE, Chatterjee C, van der Donk WA. The Importance of the Leader Sequence for Directing Lanthionine Formation in Lacticin 481. Biochemistry. 2008 doi: 10.1021/bi800277d. accompanying paper. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Siezen RJ, Kuipers OP, de Vos WM. Comparison of lantibiotic gene clusters and encoded proteins. Antonie van Leeuwenhoek. 1996;69:171–184. doi: 10.1007/BF00399422. [DOI] [PubMed] [Google Scholar]
- 24.Håvarstein LS, Diep DB, Nes IF. A family of bacteriocin ABC transporters carry out proteolytic processing of their substrates concomitant with export. Mol. Microbiol. 1995;16:229–240. doi: 10.1111/j.1365-2958.1995.tb02295.x. [DOI] [PubMed] [Google Scholar]
- 25.Ishii S, Yano T, Hayashi H. Expression and characterization of the peptidase domain of Streptococcus pneumoniae ComA, a bifunctional ATP-binding cassette transporter involved in quorum sensing pathway. J. Biol. Chem. 2006;281:4726–4731. doi: 10.1074/jbc.M512516200. [DOI] [PubMed] [Google Scholar]
- 26.Wu KH, Tai PC. Cys32 and His105 are the critical residues for the calcium-dependent cysteine proteolytic activity of CvaB, an ATP-binding cassette transporter. J. Biol. Chem. 2004;279:901–909. doi: 10.1074/jbc.M308296200. [DOI] [PubMed] [Google Scholar]
- 27.Kotake Y, Ishii S, Yano T, Katsuoka Y, Hayashi H. Substrate recognition mechanism of the peptidase domain of the quorum-sensing-signal-producing ABC transporter ComA from Streptococcus. Biochemistry. 2008;47:2531–2538. doi: 10.1021/bi702253n. [DOI] [PubMed] [Google Scholar]
- 28.Uguen M, Uguen P. The LcnC homologue cannot replace LctT in lacticin 481 export. FEMS Microbiol. Lett. 2002;208:99–103. doi: 10.1111/j.1574-6968.2002.tb11067.x. [DOI] [PubMed] [Google Scholar]
- 29.Neis S, Bierbaum G, Josten M, Pag U, Kempter C, Jung G, Sahl HG. Effect of leader peptide mutations on biosynthesis of the lantibiotic Pep5. FEMS Microbiol. Lett. 1997;149:249–255. doi: 10.1111/j.1574-6968.1997.tb10337.x. [DOI] [PubMed] [Google Scholar]
- 30.Hochuli E, Dobeli H, Schacher A. New metal chelate adsorbent selective for proteins and peptides containing neighbouring histidine residues. J. Chromatogr. 1987;411:177–184. doi: 10.1016/s0021-9673(00)93969-4. [DOI] [PubMed] [Google Scholar]
- 31.Chatterjee C, Miller LM, Leung YL, Xie L, Yi M, Kelleher NL, van der Donk WA. Lacticin 481 Synthetase Phosphorylates its Substrate during Lantibiotic Production. J. Am. Chem. Soc. 2005;127:15332–15333. doi: 10.1021/ja0543043. [DOI] [PubMed] [Google Scholar]
- 32.Udwary DW, Merski M, Townsend CA. A method for prediction of the locations of linker regions within large multifunctional proteins, and application to a type I polyketide synthase. J. Mol. Biol. 2002;323:585–598. doi: 10.1016/s0022-2836(02)00972-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.You YO, van der Donk WA. Mechanistic investigations of the dehydration reaction of lacticin 481 synthetase using site-directed mutagenesis. Biochemistry. 2007;46:5991–6000. doi: 10.1021/bi602663x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paul M, Patton GC, van der Donk WA. Mutants of the Zinc Ligands of Lacticin 481 Synthetase Retain Dehydration Activity but Have Impaired Cyclization Activity. Biochemistry. 2007;46:6268–6276. doi: 10.1021/bi7000104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chatterjee C, Patton GC, Cooper L, Paul M, van der Donk WA. Engineering Dehydro Amino Acids and Thioethers into Peptides using Lacticin 481 Synthetase. Chem. Biol. 2006;13:1109–1117. doi: 10.1016/j.chembiol.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 36.Nagao J, Aso Y, Shioya K, Nakayama J, Sonomoto K. Lantibiotic engineering: molecular characterization and exploitation of lantibiotic-synthesizing enzymes for peptide engineering. J. Mol. Microbiol. Biotechnol. 2007;13:235–242. doi: 10.1159/000104749. [DOI] [PubMed] [Google Scholar]
- 37.Meyer C, Bierbaum G, Heidrich C, Reis M, Süling J, Iglesias-Wind MI, Kempter C, Molitor E, Sahl H-G. Nucleotide Sequence of the Lantibiotic Pep5 Biosynthetic Gene Cluster and Functional Analysis of PepP and PepC. Eur. J. Biochem. 1995;232:478–489. doi: 10.1111/j.1432-1033.1995.tb20834.x. [DOI] [PubMed] [Google Scholar]
- 38.Rink R, Kluskens LD, Kuipers A, Driessen AJ, Kuipers OP, Moll GN. NisC, the Cyclase of the Lantibiotic Nisin, Can Catalyze Cyclization of Designed Nonlantibiotic Peptides. Biochemistry. 2007;46:13179–13189. doi: 10.1021/bi700106z. [DOI] [PubMed] [Google Scholar]
- 39.Rawlings ND, Morton FR, Barrett AJ. MEROPS: the peptidase database. Nucleic Acids Res. 2006;34:D270–272. doi: 10.1093/nar/gkj089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hindré T, Le Pennec JP, Haras D, Dufour A. Regulation of lantibiotic lacticin 481 production at the transcriptional level by acid pH. FEMS Microbiol. Lett. 2004;231:291–298. doi: 10.1016/S0378-1097(04)00010-2. [DOI] [PubMed] [Google Scholar]
- 41.Håvarstein LS, Holo H, Nes IF. The leader peptide of colicin V shares consensus sequences with leader peptides that are common among peptide bacteriocins produced by gram-positive bacteria. Microbiology. 1994;140(Pt 9):2383–2389. doi: 10.1099/13500872-140-9-2383. [DOI] [PubMed] [Google Scholar]
- 42.Piard JC, Kuipers OP, Rollema HS, Desmazeaud MJ, de Vos WM. Structure, organization, and expression of the lct gene for lacticin 481, a novel lantibiotic produced by Lactococcus lactis. J. Biol. Chem. 1993;268:16361–16368. [PubMed] [Google Scholar]
- 43.Klaenhammer TR. Genetics of bacteriocins produced by lactic acid bacteria. FEMS Microbiol. Rev. 1993;12:39–85. doi: 10.1111/j.1574-6976.1993.tb00012.x. [DOI] [PubMed] [Google Scholar]
- 44.Dirix G, Monsieurs P, Dombrecht B, Daniels R, Marchal K, Vanderleyden J, Michiels J. Peptide signal molecules and bacteriocins in Gram-negative bacteria: a genome-wide in silico screening for peptides containing a double-glycine leader sequence and their cognate transporters. Peptides. 2004;25:1425–1440. doi: 10.1016/j.peptides.2003.10.028. [DOI] [PubMed] [Google Scholar]
- 45.Beck-Sickinger AG, Jung G. In: Nisin and Novel Lantibiotics. Jung G, Sahl H-G, editors. ESCOM; Leiden: 1991. pp. 218–230. [Google Scholar]
- 46.Weil HP, Beck-Sickinger AG, Metzger J, Stevanovic S, Jung G, Josten M, Sahl HG. Biosynthesis of the lantibiotic Pep5. Isolation and characterization of a prepeptide containing dehydroamino acids. Eur. J. Biochem. 1990;194:217–223. doi: 10.1111/j.1432-1033.1990.tb19446.x. [DOI] [PubMed] [Google Scholar]
- 47.Sprules T, Kawulka KE, Gibbs AC, Wishart DS, Vederas JC. NMR solution structure of the precursor for carnobacteriocin B2, an antimicrobial peptide from Carnobacterium piscicola. Eur. J. Biochem. 2004;271:1748–1756. doi: 10.1111/j.1432-1033.2004.04085.x. [DOI] [PubMed] [Google Scholar]
- 48.Dougherty BA, Hill C, Weidman JF, Richardson DR, Venter JC, Ross RP. Sequence and analysis of the 60 kb conjugative, bacteriocin-producing plasmid pMRC01 from Lactococcus lactis DPC3147. Mol. Microbiol. 1998;29:1029–1038. doi: 10.1046/j.1365-2958.1998.00988.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.