

Abstract
HIV anti-retroviral drugs decrease protein synthesis, although the underlying regulatory mechanisms of this process are not fully established. Therefore, we investigated the effects of the HIV protease inhibitor lopinavir (LPV) on protein metabolism. We also characterized the mechanisms that mediate the effects of this drug on elongation factor-2 (eEF2), a key component of the translational machinery. Treatment of C2C12 myocytes with LPV produced a dose-dependent inhibitory effect on protein synthesis. This effect was observed at 15 min and was maintained for at least 4 h. Mechanistically, LPV increased the phosphorylation of eEF2 and thereby decreased the activity of this protein. Increased phosphorylation of eEF2 was associated with increased activity of its upstream regulators AMP-activated protein kinase (AMPK) and eEF2 kinase (eEF2K). Both AMPK and eEF2K directly phosphorylated eEF2 in an in vitro kinase assay suggesting two distinct paths lead to eEF2 phosphorylation. To verify this connection, myocytes were treated with the AMPK inhibitor compound C. Compound C blocked eEF2K and eEF2 phosphorylation, demonstrating that LPV affects eEF2 activity via an AMPK-eEF2K dependent pathway. In contrast, incubation of myocytes with rottlerin suppressed eEF2K, but not eEF2 phosphorylation, suggesting that eEF2 can be regulated independent of eEF2K. Finally, LPV did not affect PP2A activity when either eEF2 or peptide was used as the substrate. Collectively, these results indicate that LPV decreases protein synthesis, at least in part, via inhibition of eEF2. This appears regulated by AMPK which can act directly on eEF2 or indirectly via the action of eEF2K.
Keywords: AMPK, eEF2K, HIV antiretroviral drugs
INTRODUCTION
The regulation of protein synthesis is a complex process involving alterations in the phosphorylation state of many components of the translational machinery. One group of these components consists of the peptide-chain elongation factors (eEFs). Among the various elongation factors, phosphorylation of eEF2 by the eEF2 kinase (eEF2K) is the best characterized mechanism controlling the rate of elongation [Dorovkov et al., 2002; Riis et al., 1990; Ryazanov et al., 1991]. eEF2K, also known as Ca2+/calmodulin kinase III, is a protein kinase which phosphorylates eEF2 on Thr-56 and -58 [Mitsui et al., 1993; Price et al., 1991; Redpath et al., 1993]. The phosphorylation of eEF2 is inversely related to the rate of elongation, thereby contributing to the overall decrease of protein synthesis. Likewise, the activity of eEF2K is regulated through single or multisite phosphorylation when cells are exposed to various stimuli. For example, phosphorylation of eEF2K at the Ser 366 residue following exposure to neurotrophic factor or hormones [e.g., insulin or insulin-like growth factor (IGF)-I] decreases the activity of this kinase, while phosphorylation at other sites in response to stress conditions increases its activity [Browne and Proud, 2002; Inamura et al., 2005]. Hence, the phosphorylation of eEF2K can regulate eEF2 either positively or negatively, depending on the stimuli and the particular residue that is phosphorylated.
A number of signaling pathways are involved in the regulation of eEF2 and eEF2K. For instance, AMPK, mTOR and MEK/ERK signaling mediate increased phosphorylation of eEF2 and eEF2K in response to various stimuli such as low cellular energy level, hypoxia, electrical stimulation [Atherton et al., 2005; McLeod and Proud, 2002; Terai et al., 2005], hormones (e.g. serotonin, phenylephrine) and growth factors (insulin, IGF-I) [Carroll et al., 2004; Proud, 2004; Wang and Proud, 2002]. Interestingly, some data suggest that these pathways are involved in regulating either eEF2 or eEF2K, but not both. For example, incubation of myocytes with alcohol suppresses eEF2K activity, while increasing eEF2 phosphorylation [Hong-Brown et al., 2007]. In contrast, treatment with the mTOR inhibitor rapamycin and the MEK1 inhibitor PD98059 did not affect eEF2K phosphorylation in the presence of cholecystokinin, even though eEF2 phosphorylation was sensitive to these inhibitors [Sans et al., 2004]. The role that AMPK plays in regulating eEF2 is controversial. Published studies report that AMPK is not involved in eEF2 phosphorylation in response to exercise in skeletal muscle cells [Rose et al., 2005]. In contrast, our recent studies suggest that eEF2 can be directly regulated by AMPK in myocytes following alcohol treatment [Hong-Brown et al., 2007]. These latter findings are in agreement with others reporting that stimulation of AMPK by low cellular energy increased eEF2 phosphorylation [McLeod and Proud, 2002].
Lopinavir is a human immunodeficiency virus (HIV)-1 protease inhibitor commonly used in combination with other agents as part of the highly active anti-retroviral therapy (HAART). Anti-retroviral therapy helps control HIV infection by suppressing plasma viral levels and enhancing the immunological status of patients, thereby leading to a decline in morbidity and mortality [Palella et al., 1998; Schwarcz et al., 2000]. Unfortunately, the benefits of this therapy are limited, owing to certain inherent adverse effects that anti-retroviral drugs have on bone, lipid, carbohydrate, and protein metabolism [Ben-Romano et al., 2006; Ergun-Longmire et al., 2006; Jain and Lenhard, 2002]. For example, lopinavir has been reported to contribute to insulin resistance and the development of type 2 diabetes mellitus by inhibiting insulin sensitive glucose transporters in adipocytes and muscle cells [Noor et al., 2006; Yan and Hruz, 2005]. Likewise, lopinavir impairs lipid metabolism causing hyperlipidemia and lipodystrophy [Montes et al., 2005; Prot et al., 2006; Valerio et al., 2005]. At present, the effects of lopinavir on protein metabolism have not been thoroughly investigated. However, there is evidence that HIV-related wasting still occurs in patients treated with these drugs [Mangili et al., 2006].
Previous in vitro and in vivo studies showed that various HIV protease inhibitors impaired protein synthesis, and this response was associated with defects in translation initiation and/or elongation. However, the effect that lopinavir has on these processes has not been reported. The aim of the present study was to determine whether lopinavir influences protein synthesis in C2C12 myocytes. In addition, we studied how proteins synthesis-related signaling events were regulated by this drug. Lopinavir decreased protein synthesis in a dose-and time-dependent manner, and this impairment was associated with an increase in eEF2 phosphorylation. Lopinavir also increased eEF2K phosphorylation and activity. Phosphorylation of eEF2K by lopinavir was mediated via activation of the AMPK pathway. On the other hand, in vitro kinase assays and studies using chemical inhibitors suggested that AMPK can regulate eEF2 independent of eEF2K.
MATERIALS AND METHODS
Lopinavir was provided by the NIH AIDS Research and Reference Reagent Program (Rockville, MD). The majority of the antibodies used in this study were purchased from Cell Signaling Technology (Beverly, MA). These included polyclonal antibodies specific for the phosphorylated (p) form of AMPK-α (Thr 172), p-eEF2 (Thr 56), p-eEF2K (Ser 366), p-acetyl CoA carboxylase (ACC; Ser 79). Antibodies to total AMPK-α, eEF2, eEF2K and ACC were also obtained from the same source. The AMPK and eEF2K inhibitors compound C and rottlerin, respectively, were purchased from CalBiochem (EMD Biosciences, San Diego, CA). 35S-methionine/cysteine (>1000 Ci/mol) was obtained from MP Biomedicals (Aurora, OH). Cell culture media and fetal bovine serum (FBS) were from Gibco, Invitrogen Corporation (Carlsbad, CA).
Cell culture
C2C12 mouse myoblasts were purchased from American Type Culture Collection (Manassas, VA). Cells were cultured in DMEM containing 10% FBS, penicillin (100 U/ml), streptomycin (100 μg/ml) and amphotericin (25 μg/ml). The effect of lopinavir on protein synthesis was determined as previously described [Hong-Brown et al., 2005] with minor modifications. Briefly, for metabolic labeling, cells were incubated in the presence of lopinavir and radioisotope for various periods of time with 10 μCi [35S] in methionine/cysteine- free DMEM media. In preliminary studies, the rate of radiolabel incorporation into protein was linear between 15 min and 24 h (data not shown) indicating there was no significant change in the specific activity of the precursor pool. At the conclusion of experiments, cells were collected and precipitated in 10% TCA, and the incorporation of 35S- methionine/cysteine into TCA-precipitable protein was determined via liquid scintillation counting. The results were then compared with those of the appropriate time-matched control group and data were expressed as a percentage of the control value.
Western immunoblot analysis
C2C12 myocytes were sub-cultured in 6-well plates. Cells were incubated in the presence or absence of lopinavir for 15 min and collected in 2X Laemmli sample buffer (LSB). Equal amounts of protein from cell lysates were electrophoresed on denaturing polyacrylamide gels and transferred to nitrocellulose. The resulting blots were blocked with 5% nonfat dry milk and incubated with the antibodies of interest as described above. Unbound primary antibody was removed by washing with TBS containing 0.05% Tween-20 (ICI Americas, Inc, Wilmington, DE), and blots were incubated with anti-rabbit immunoglobulin conjugated with horseradish peroxidase. Blots were briefly incubated with an enhanced chemiluminescent detection system (Amersham, Bickinghamshire, England) and exposed to Kodak x-ray film (Rochester, NY). The film was scanned (ScanMaker 4, Microtek, Los Angeles, CA) and analyzed with NIH Image 1.6 software.
Assay for eEF2K and AMPK activity
For kinase activity measurements, cells were lysed in 1% NP-40 containing 20 mM Hepes, 150 mM NaCl, and a cocktail of protease and phosphatase inhibitors as described previously [Hong-Brown et al., 2007]. In brief, cell extracts (100-120 μg of protein) were immunoprecipitated overnight with 4-6 μg of specific antibodies against AMPK, eEF2K or eEF2. The antibody-antigen complex was then captured by incubation for 1 h with 40 μl protein A Sepharose (Amersham Biosciences, Piscataway, NJ). Immune complexes were washed with lysis buffer and then incubated with 50 μl reaction buffer A (40 mM Hepes, 0.2 mM AMP, 80 mM NaCl, 0.8 mM DTT, 5 Mm MgCl2, and 0.2 mM [gamma-32P] ATP) or buffer B (50 mM Hepes, 10 mM magnesium-acetate, 100 μM CaCl2, 5 mM DTT, 0.6 μg Ca/calmodulin, and 50 μM [gamma-32P] ATP) for measuring AMPK or eEF2K activity, respectively. The reaction was allowed to proceed for 10-14 min at 30°C, and terminated by addition of 2x LSB with heating for 5 min. Samples were run on SDS-PAGE gels, dried at 80°C and quantitated using a phosphoimager. The results were standardized with total protein, as determined using a BCA protein assay reagent kit (Pierce Biotechnology, Rockford, IL).
Phosphatase assay
Myocytes were incubated with lopinavir as described above and phosphatase activity was measured using a Ser/Thr phosphatase assay kit from Upstate Biotechnology. Cells were lysed with buffer containing 20 mM imidazole-HCl (pH 7.0), 2 mM EDTA, 2 mM EGTA, and a cocktail of protease inhibitors. For detection of PP2A activity, lysates were incubated for 25 min at room temperature with a phosphopeptide (R-K-pT-I-R-R) according to the manufacturer’s protocol. The reaction was then terminated following the addition of malachite green reagent and free phosphatase was quantified by measuring the absorbance at 620 nm. To examine the ability of PP2A to dephosphorylate eEF2, an in vitro phosphatase assay was conducted. For these experiments, eEF2 was immunoprecipitated from untreated cells and used as the substrate, while PP2A was isolated from control and lopinavir treated cells. Substrates and phosphatase were incubated together at room temperature for 25 min, and PP2A activity was quantitated as described above.
Statistical analysis
For experimental protocols with more than two groups, statistical significance was determined using one-way ANOVA followed by the Dunnett’s test to compare all data to the appropriate time-matched control group. For experiments with only two groups, an unpaired Student’s t-test was performed. Data are presented as mean ± SE. Mean values were considered significantly different at P < 0.05.
RESULTS
Effects of lopinavir on basal protein synthesis
Several HIV anti-retroviral drugs have been shown to adversely affect protein synthesis and metabolism. However, little is known regarding the effect that the protease inhibitor lopinavir has on these processes. In our initial experiments, we treated cells with 10 μM lopinavir which is similar to those used previously by others and within the range of concentrations observed in the plasma of patients receiving this drug [Guiard-Schmid et al., 2003; Gutierrez et al., 2003; Hsu et al., 2003]. At this dose, there was no apparent toxic effect, because cell numbers were similar following incubation for 24 h in the presence (50 ±5 ×104) or absence (52 ±7 ×104) of lopinavir.
To determine whether lopinavir altered the basal rate of protein synthesis, myocytes were labeled for various periods of time in media containing 10 μM of the drug. When protein synthesis was assessed, a significant 30% decrease was observed as early as 15 min following treatment, when compared to values from time-matched control cells (Fig. 1A). The inhibitory effect of this drug on protein synthesis was maintained for at least 4 hours. However, the level of inhibition was diminished with longer treatment periods, and no change in the overall rate of protein synthesis was detected when cells were incubated in the presence of lopinavir for 24 h. The lack of an effect at the 24 h time point was most likely due to the relatively short half-life of lopinavir. Furthermore, this result is consistent with our findings examining the reversible nature of the drug effect. For these experiments, cells were incubated in the presence or absence of lopinavir for various periods of time. The drug was then removed and cells were allowed to recover for 1 day prior to labeling. As expected, the inhibitory effect of lopinavir on basal protein synthesis was not sustained when the drug was removed from the media (data not shown).
Fig. 1. Time- and dose-dependent effects of lopinavir (LPV) on protein synthesis.
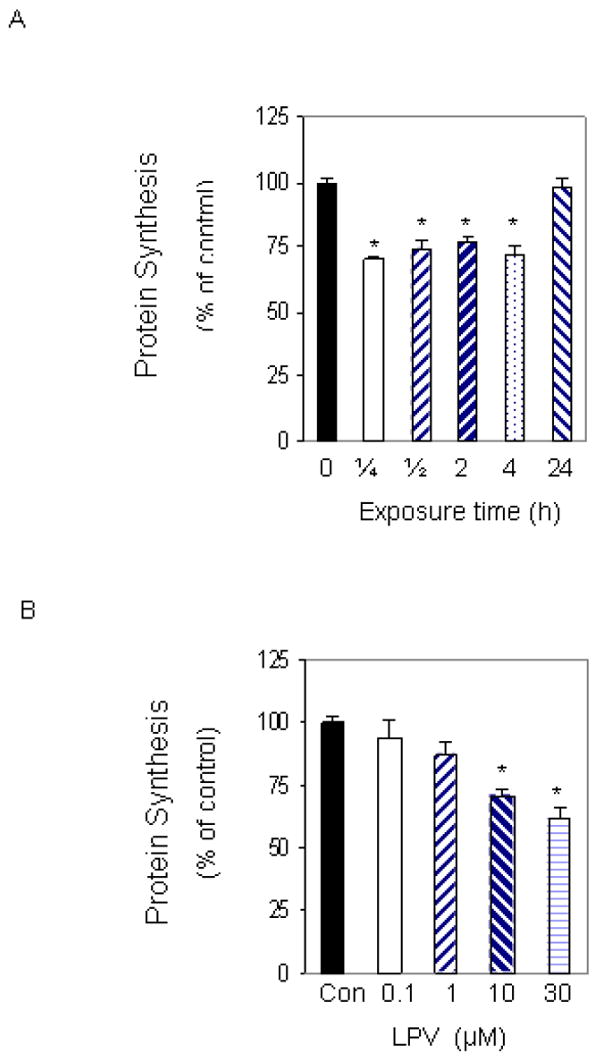
C2C12 myocytes were labeled with [35S] methionine/cysteine in 24 well plates. Cells were labeled in the presence of 10 μM LPV for various periods of time (A). Cells were then collected, and the amount of trichloroacetic acid (TCA) -precipitable radioactivity was determined as described under “Materials and Methods.” C2C12 myocytes were labeled with increasing concentrations of LPV for 15 min and the amount of TCA-precipitable radioactivity was determined (B). Values are compared to time-matched controls that were labeled in the absence of LPV. Each bar represents the mean ± SE of 3-4 independent experiments consisting of 3-6 replicate samples per experiment * P< 0.05 versus matched control value.
To investigate the dose-dependent effect of lopinavir on protein synthesis, C2C12 myocytes were labeled for 15 min in the absence or presence of increasing concentrations of the drug. Treatment of cells with 0.1 or 1 μM lopinavir did not alter the rate of protein synthesis. However, at a concentration of 10 μM, lopinavir significantly decreased protein synthesis by 28%, when compared to untreated control cells (Fig.1B). Incubation of myocytes with higher concentrations of lopinavir did not result in a further decline in protein synthesis. For example, 30 μM lopinavir produced a comparable decrease in protein synthesis when compared to cells treated with 10 μM. Based upon the above results, 10 μM of lopinavir was chosen for use in all subsequent experiments.
Lopinavir affects the phosphorylation state of eEF2
The activity of eEF2 is critical for the elongation step of translation, and this activity is negatively regulated by its phosphorylation. To determine the effect of lopinavir on eEF2 phosphorylation, cells were incubated in the presence or absence of 10 μM lopinavir for 15 min. This time point was chosen because it was sufficient to significantly decrease protein synthesis (see Fig. 1A). As illustrated in Figure 2, lopinavir increased phosphorylation of eEF2 by 2-fold at the 15 min time point relative to the control group. In contrast, this drug had no affect on total eEF2 protein content. Therefore, the observed increase in eEF2 phosphorylation is consistent with the ability of lopinavir to decrease protein synthesis.
Fig. 2. Effects of lopinavir (LPV) on eEF2 phosphorylation.
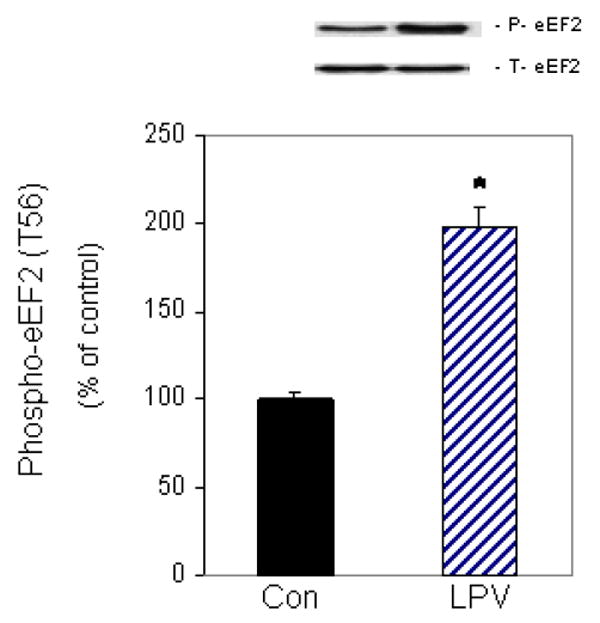
C2C12 myocytes were incubated in the presence or absence of LPV (10 μM) for 15 min. Cell extracts were collected and analyzed via Western blotting using anti-phospho-eEF2 (T56) and total eEF2 antibodies. Results for phosphor-eEF2 are normalized to total eEF2 and are expressed as a percentage of basal control levels. Each bar graph represents mean ± SE of 6 independent experiments consisting of 3-4 replicate samples per experiment. * P< 0.05 versus control values.
Lopinavir-induced phosphorylation of eEF2 is controlled by eEF2K
eEF2 is regulated by the calcium and calmodulin-dependent kinase eEF2K, and the activity of this kinase is mediated by phosphorylation events [Browne et al., 2004]. To examine whether eEF2K plays a role in regulating eEF2 phosphorylation in our system, we first examined the effect of lopinavir on eEF2K phosphorylation. Figure 3A shows that lopinavir increased eEF2K phosphorylation on the Ser 366 residue by 48%, as compared to control values. This increase was in part due to an unexpected, albeit statistically significant, increase in total eEF2K protein content (Fig. 3B).
Fig. 3. Lopinavir (LPV) stimulates eEF2K phosphorylation and activity.
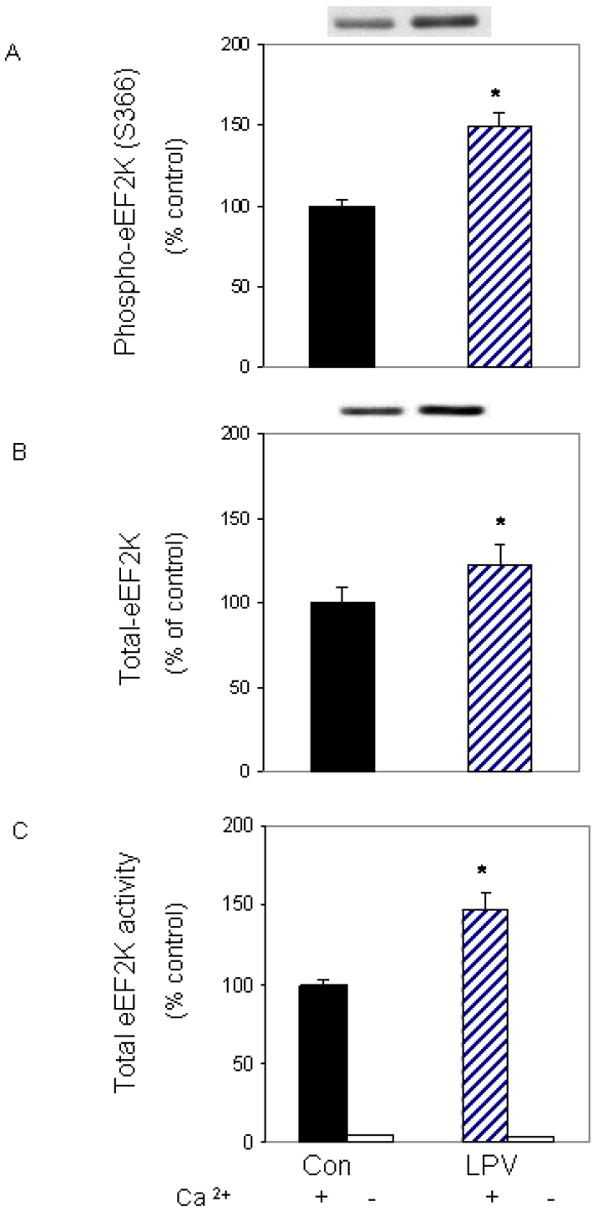
C2C12 myocytes were treated as described in Fig. 2. Cell extracts were analyzed via Western blotting using antibodies that recognize phosphorylated eEF2K at S366 (A) and total eEF2K (B). C: An in vitro eEF2K activity assay was performed in the presence of CaCl2, Ca/calmodulin and ATP, as described in the “Materials and Methods.” Data are mean ± SE of 3-4 independent experiments consisting of 3-6 replicate samples per experiment. * P< 0.05 versus the control value.
The phosphorylation of eEF2K can either inhibit or enhance the activity of its downstream substrate, depending on the site of phosphorylation and the type of stimuli. For example, phosphorylation of Ser 365 following hormone treatment has been shown to decrease the activity of eEF2K [Browne and Proud, 2002; Wang et al., 2001]. In contrast, we observed that lopinavir increased eEF2K phosphorylation of Ser 366 in conjunction with increased eEF2 phosphorylation. Hence, it is possible that other sites on this kinase, such as Ser 398, may also be phosphorylated following lopinavir treatment, thereby activating eEF2 phosphorylation [Browne et al., 2004]. To determine whether lopinavir increased the activity of eEF2K towards eEF2, we performed an in vitro activity assay. For this assay, eEF2 was isolated from control cells and used as a substrate, while eEF2K was immunoprecipitated from cells treated with or without lopinavir. Incubation of myocytes with lopinavir increased eEF2K activity by 50% when compared to control values (Fig. 3C). Hence, our results are consistent with a model in which lopinavir increases eEF2 phosphorylation via the action of eEF2K.
Lopinavir increases eEF2 phosphorylation in an AMPK-dependent manner
The elongation process consumes a considerable amount of cellular energy, thereby linking protein synthesis and signaling pathways that respond to changes in energy levels. For example, the AMPK pathway is activated in response to various stimuli that affect cellular energy levels. Furthermore, AMPK is known to regulate the activity of eEF2K [Browne et al., 2004; Crozier et al., 2005; Horman et al., 2003; Horman et al., 2002]. Thus, AMPK may be an important modulator of the effects of lopinavir on eEF2 phosphorylation. Figure 4A illustrates that lopinavir increased the phosphorylation of AMPK by 2-fold, when compared with control untreated cells. Lopinavir similarly increased the phosphorylation of ACC (Fig. 4B), a known downstream substrate of AMPK. Both of these changes were independent of a change in total AMPK or ACC protein. Thus, these data are indicative of an overall increase in the activity of AMPK in myocytes after exposure to lopinavir.
Fig. 4. Lopinavir (LPV) stimulates phosphorylation of AMPK and its downstream target acetyl-CoA carboxylase (ACC).
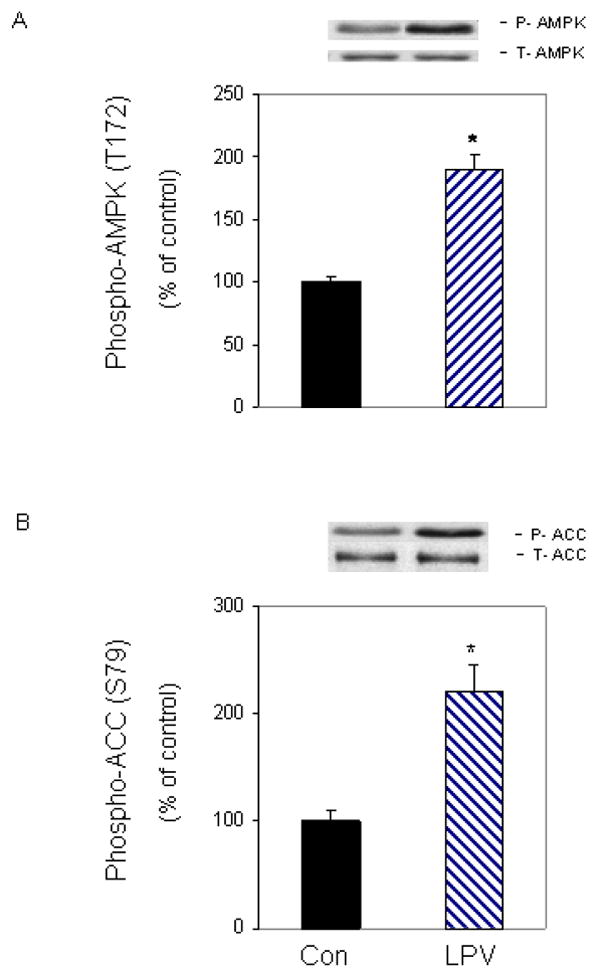
C2C12 myocytes were treated as described in Fig. 2. Western blots were performed with antibodies that recognize phosphorylated AMPK at T172 (A) and phosphorylated ACC at S79 (B). Data are means ± SE of 3-5 independent experiments consisting of 3-4 replicate samples per experiment. * P< 0.05 versus the control value.
To delineate the mode of action of AMPK in regulating eEF2 phosphorylation, we utilized an inhibitor that blocks the activity of this protein. For these experiments, cells were treated with lopinavir in the presence or absence of the inhibitor compound C. As shown in Figure 5, a combined treatment with compound C and lopinavir significantly decreased (60%) the level of phosphorylated eEF2K (panel A), relative to cells treated with lopinavir alone. Likewise, this inhibitor suppressed the increased phosphorylation of eEF2 by 56% when both drugs were present (panel B). Collectively, these data suggest that lopinavir stimulates AMPK to function as an upstream regulator of both eEF2K and eEF2 phosphorylation.
Fig. 5. Lopinavir (LPV) stimulates eEF2 phosphorylation via an AMPK- dependent pathway.
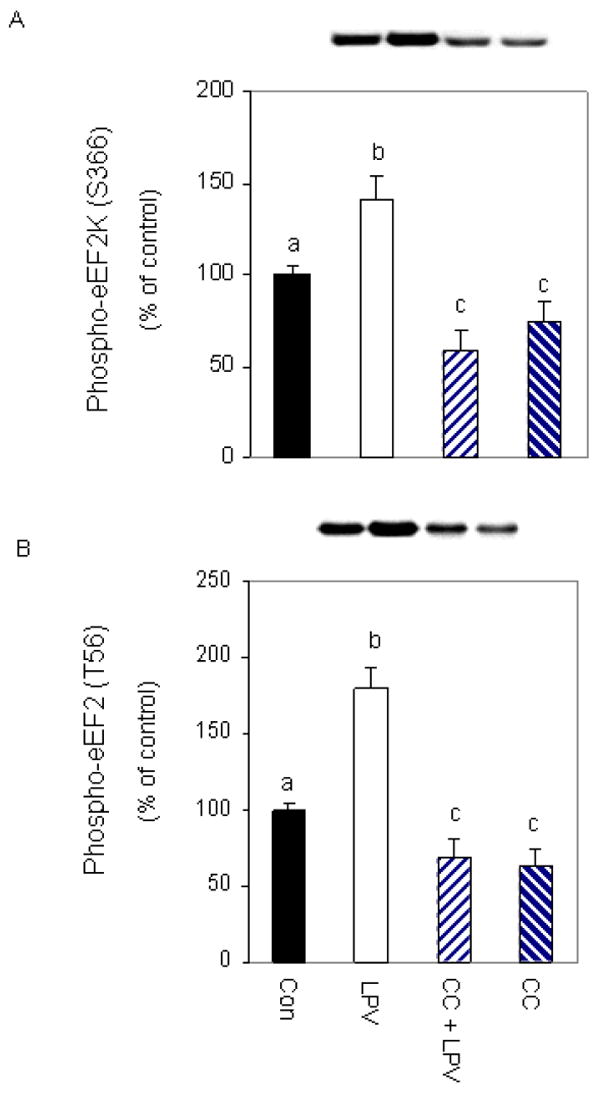
Cells were pre-incubated for 1 h in the presence or absence of the AMPK inhibitor compound C (20 μM) and then treated with 10 μM LPV for 15 min. Cell extracts were analyzed via Western blotting using antiserum that recognizes eEF2K phosphorylated at S366 (A) or eEF2 phosphorylated at T56 (B). Data are mean ± SE of 3-4 independent experiments consisting of 4 replicate samples per experiment. * P< 0.05 versus the control value. Groups with different letters are significantly different from one another (*P < 0.05). Groups with the same letters are not significantly different.
The above data are consistent with a model whereby lopinavir- induced stimulation of AMPK regulates the phosphorylation of eEF2K and eEF2. Note, however, that compound C may have other non-specific effects. To address this issue, we used a complementary approach in which AMPK activity was determined using an in vitro kinase assay in the presence or absence of compound C. In Fig. 6A, we show that lopinavir increases the ability of AMPK to phosphorylate eEF2K. However, if compound C was included in the in vitro reaction mixture, there was a significant effect on the ability of lopinavir to increase AMPK activity. Note that 20 μM compound C had a greater effect on AMPK activity in control versus lopinavir treated samples. However, higher concentrations of compound C reduced this activity further. Thus, these data verify that compound C can directly attenuate the ability of AMPK to phosphorylate its downstream target. As expected, similar results were obtained when ACC was utilized as a substrate in these experiments (Fig. 6B). Lopinavir stimulated the ability of AMPK to activate ACC, and this response was suppressed in the presence of compound C.
Fig. 6. Compound C inhibits AMPK but not eEF2K activity.
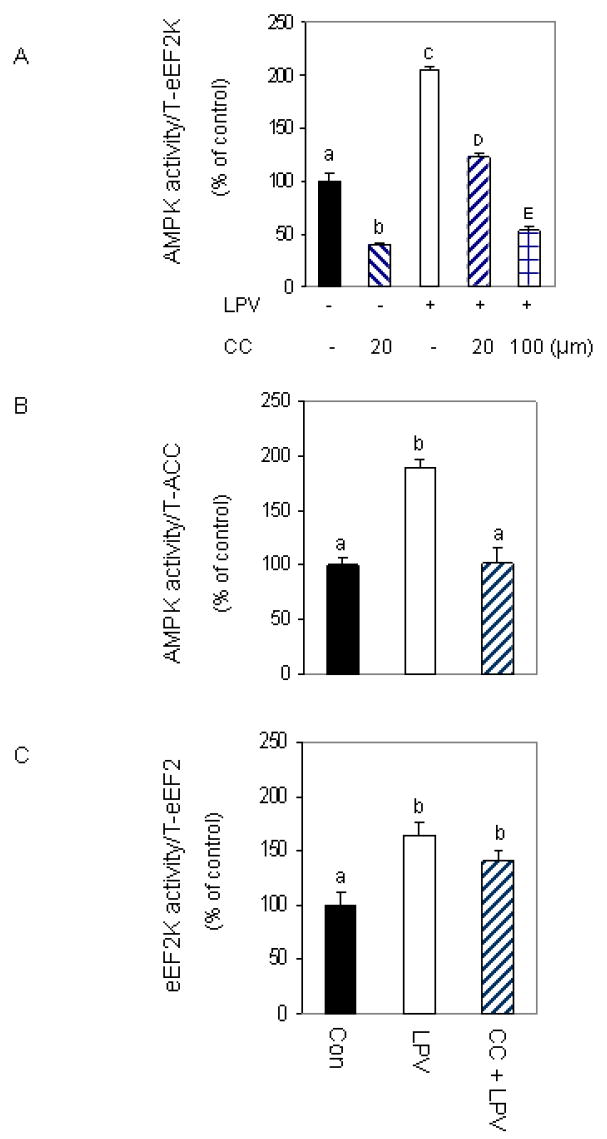
C2C12 myocytes were treated as described in Fig. 2 and the specificity of compound C (CC) to inhibit AMPK was examined using an in vitro AMPK activity assay where eEF2K (panel A) or ACC (panel B) were utilized as substrates. AMPK was immunoprecipitated from 100 μg of lysates and the activity was determined in the presence or absence of 20 μM CC, MgCl2 and AMP as described under “Materials and Methods.” Panel C: an in vitro eEF2K activity was determined using eEF2 as the substrate in the presence of CC, CaCl2, Ca/calmodulin and ATP, as described in the “Materials and Methods.” Data are mean ± SE of 3 independent experiments consisting of 4 replicate samples per experiment. Groups with different letters are significantly different from one another (*P < 0.05). Groups with the same letters are not significantly different.
Because treatment of cells with compound C blocked the effects of lopinavir on both eEF2K and eEF2, this suggests that these two proteins are downstream of AMPK signaling. As such, they can be blocked by the action of compound C on AMPK activity. Alternatively, compound C may directly affect these proteins or other kinases that regulate this pathway. To distinguish between these possibilities, we performed an in vitro eEF2K kinase assay using eEF2 as the substrate. Lopinavir increased the activity of eEF2K to phosphorylate eEF2. However, in contrast to our result on AMPK, the addition of compound C did not block this effect (Fig. 6C). Thus, these data suggest that compound C does not directly inhibit the activity of eEF2K, but instead act independent of this protein.
As reported previously [Hong-Brown et al., 2007], eEF2K is not always required for the transmission of signals to eEF2. Therefore, we next determined whether eEF2K was necessary for the phosphorylation of eEF2 under our experimental conditions. For these experiments, myocytes were treated with lopinavir in the presence or absence of the inhibitor rottlerin, a drug which inhibits eEF2K and PKC delta with equal efficacy [Gschwendt et al., 1994; Parmer et al., 1997; Parmer et al., 1999]. Rottlerin suppressed the stimulatory effect of lopinavir on eEF2K phosphorylation (Fig. 7A). The presence of rottlerin also blocked the ability of lopinavir to increase eEF2K activity. For example, when rottlerin was added to the in vitro reaction mixture, this drug inhibited the stimulatory effect that was otherwise observed following lopinavir treatments (Fig. 7B) Interestingly, rottlerin did not block the effects of lopinavir on eEF2 phosphorylation (Fig. 7C). Thus, these results indicate that lopinavir can stimulate eEF2 phosphorylation independent of the action of eEF2K, perhaps via the action of AMPK. Along these lines, we observed that rottlerin did not block the activity of AMPK under in vitro circumstances. As such, AMPK activity against eEF2K was increased following lopinavir treatment, and this activity remained elevated in the presence of the inhibitor (Fig 7D).
Fig. 7. Lopinavir (LPV) stimulation of eEF2 phosphorylation is eEF2K-independent.

C2C12 myocytes were pre-incubated for 1 h in the presence or absence of 2 μM rottlerin (Rott) and then treated with 10 μM LPV for 15 min. Cell extracts were analyzed via Western blotting using anti-phospho-eEF2K (panel A). Panel B: the specificity of rottlerin to inhibit eEF2K was determined using an in vitro eEF2K activity assay with eEF2 as the substrate. eEF2K was immunoprecipitated from 100 μg of lysates and the activity was determined in the presence or absence of Rott, CaCl2, Ca/calmodulin and ATP as described above. Panel C: C2C12 myocytes were treated as described in panel A. Cell extracts were analyzed via Western blotting using anti- phospho-eEF2 (T56) antibody. Panel D: an in vitro AMPK activity assay was performed using eEF2K as the substrate in the presence or absence of Rott, MgCl2 and AMP as described above. Data are mean ± SE of 4 independent experiments consisting of 3-4 replicate samples per experiment. Groups with different letters are significantly different from one another (*P < 0.05). Groups with the same letters are not significantly different. * P< 0.05 versus the control value.
We previously reported that AMPK can directly phosphorylate eEF2 [Hong-Brown et al., 2007]. Furthermore, the level of activity was shown to increase following treatment with ethanol. Our next experiment examined whether a similar response occurs following treatment of cells with lopinavir. For these studies, an in vitro kinase assay was utilized to measure AMPK activity. eEF2 was isolated from control cells and used as a substrate, while AMPK was immunoprecipitated from cells treated with or without lopinavir. Incubation of myocytes with lopinavir increased AMPK activity towards eEF2 by 2.5-fold, when compared to control values (Fig. 8A). Thus, these data suggest that eEF2 is a direct downstream target of AMPK.
Fig. 8. Lopinavir (LPV) stimulates activity of AMPK.
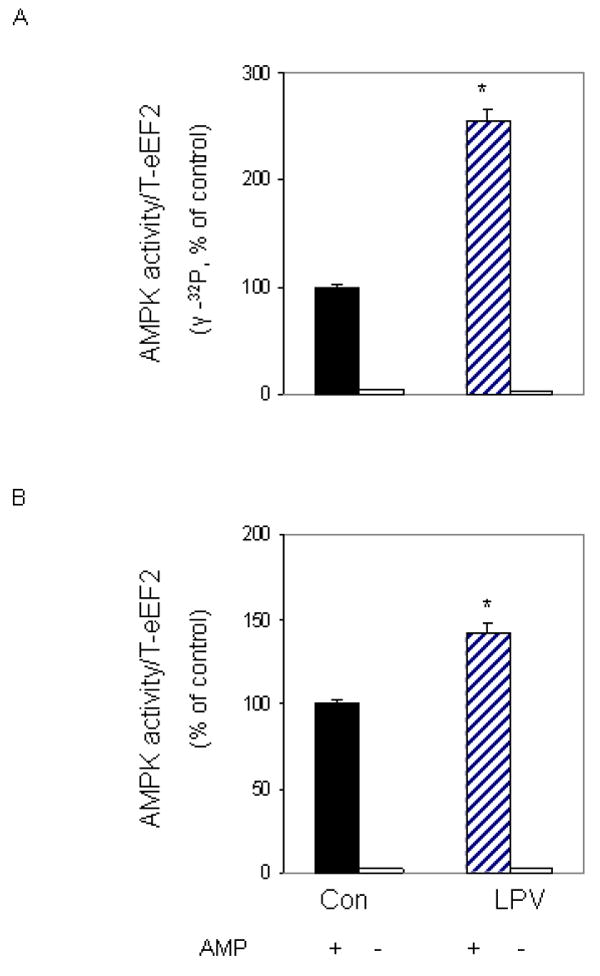
C2C12 myocytes were incubated in the presence or absence of LPV (10 μM) for 15 min. For in vitro kinase activity, AMPK was immunoprecipitated from 100 μg of cell lysates and the activity was assayed using eEF2 as the substrate, while in the presence of MgCl2 and AMP. Reaction mixtures were incubated in the presence (panel A) or absence (panel B) of [γ-32P] ATP as described under “Materials and Methods”. In panel B, reaction mixtures were examined by Western blot, using the anti-phospho eEF2 (T56) antibody. Results are mean ± SE of 3 independent experiments consisting of 4 replicate samples per experiment. * P<0.05 versus control values.
We previously demonstrated the effect of lopinavir on eEF2 phosphorylation using a T56-specific antibody (Fig. 2). Because our in vitro experiments (Fig. 8A) measured the incorporation of [γ32P] ATP label into total eEF2 protein, we could not distinguish whether the eEF2 T56 residue was the site that was phosphorylated following treatment with lopinavir. To address this issue, we performed an in vitro kinase assay as above, albeit using unlabeled ATP. Following completion of the in vitro phosphorylation reaction, the material was subjected to Western blot analysis using the eEF2 antibody that recognized the phosphorylated form of the T56 residue. A significant increase in phosphorylation was observed at the T56 site in cells incubated with lopinavir (Fig. 8B). Note, however, that this increase appeared less than that observed when we examined AMPK activity using [γ32P] ATP incorporation (Fig. 8A), suggesting that multiple sites may be targeted for increased phosphorylation by AMPK following exposure to lopinavir.
Lopinavir-induced increases in eEF2 phosphorylation are not regulated by PP2A phosphatase
The activity of kinases in cells is balanced by the action of general or specific phosphatases. Accordingly, the increased phosphorylation of eEF2 following lopinavir treatment could be due, in part, to a decrease in phosphatase activity. Our final experiments examined whether protein phosphatases play a role in the regulation of eEF2 following incubation with lopinavir. Control and lopinavir-treated cells were harvested and total cell lysates or immunoprecipitated PP2A were examined for phosphatase activity. Data in Table 1 show that lopinavir caused a small, albeit statistically significant, increase in PP2A activity compared to control values. This increase was observed whether the peptide or eEF2 was used as a substrate. Therefore, these results suggest that a decrease in PP2A is not responsible for the increased phosphorylation of eEF2 following lopinavir treatments.
Table 1.
Effects of lopinavir (LPV) on PP2A activity using peptide or total eEF2 as the substrate
Control and LPV treated cell lysates were examined for phosphatase activity as described under “Materials and Methods.” For these experiments, total cell lysates were incubated with a phosphopeptide (R-K-pT-I-R_R) for 20-25 min at room temperature. Alternatively, the ability of PP2A to dephosphorylate eEF2 was assayed using PP2A and eEF2 immunoprecipitates from control and LPV treated cells. Values are mean ± SE of 3-6 experiments consisting of 3-7 replicate samples per experiment.
P<0.05 versus control values (100 %).
DISCUSSION
In this study, we investigated the effects of the anti-retroviral drug lopinavir on protein synthesis and signaling events related to the control of elongation. Our results demonstrate that the basal rate of protein synthesis declined after a relatively acute exposure of myocytes to lopinavir. The rapid effect of this drug was in contrast to previous studies, where cells required a significantly longer treatment (24-48 h) to other HIV-protease inhibitors in order to exert an inhibitory effect [Hong-Brown et al., 2004; Hong-Brown et al., 2005; Janneh et al., 2003]. Our results are also in contrast to reports where levels of proteins such as P-glycoprotein immunoreactive protein were induced following extended exposure to this drug [Vishnuvardhan et al., 2003], indicating that the synthesis of all proteins is not uniformly suppressed. Although lopinavir impaired protein synthesis, it is noteworthy that this drug did not appear affect cell viability. This is in agreement with previously reports in which treatment of kidney cells with lopinavir did not alter cell number, even after several days of drug exposure [Vidal et al., 2006]. Taken together, our findings and published data indicate that protease inhibitors can negatively influence protein metabolism in a variety of cell types.
The mechanisms by which lopinavir alters muscle protein synthesis have not been investigated previously. In general, regulation of protein synthesis involves changes in the phosphorylation state of several key components of the translation machinery including the phosphorylation of the elongation factor eEF2. Although we did not directly determine rates of elongation in the current study, these results are consistent with the observed reduction in protein synthesis in response to lopinavir. The effect of lopinavir on eEF2 is in agreement with previous studies examining various stressors. For example, treatment with the protease inhibitor indinavir decreased the activity of eEF2 in myocytes [Hong-Brown et al., 2004]. Likewise, alcohol or ATP depletion had a similar effect on the phosphorylation state of this elongation factor [Hong-Brown et al., 2007; McLeod and Proud, 2002].
In the present study, we provide evidence that eEF2K is an upstream regulator of eEF2. For example, lopinavir increased eEF2K phosphorylation at Ser 366 and it also increased eEF2K activity. This appears in contrast to others reports where phosphorylation at this same site was correlated with decreased eEF2K activity [Browne and Proud, 2002; Hong-Brown et al., 2007; Wang et al., 2001]. Hence, it is possible that phosphorylation of other eEF2K residues, such as Ser 398, may be responsible for increased kinase activity (Browne et al., 2004). Nevertheless, our in vivo and in vitro data showed that eEF2K activity increased in response to lopinavir, regardless of the sites that were phosphorylated.
Our studies also suggest that eEF2K is not necessarily required for the control of eEF2 phosphorylation. As such, treatment with the inhibitor rottlerin did not prevent the lopinavir-induced increase in eEF2 phosphorylation (Fig. 7C), although it did suppress the stimulatory effect of lopinavir on eEF2K (Fig. 7A). This idea is further supported by our in vitro kinase assay in which we used eEF2 as a substrate to directly measure lopinavir-induced changes in eEF2K activity (Fig. 7B). This activity was blocked when rottlerin was included in the reaction mixture, verifying the ability of this drug to inhibit this step. These data are consistent with previous reports where rottlerin failed to block the stimulatory effect of alcohol on eEF2 phosphorylation, even though it did inhibit the increased activity of eEF2K in response to AICAR, FBS or growth factors [Hong-Brown et al., 2007; Parmer et al., 1997; Parmer et al., 1999]. Thus, these results indicate that there is an alternative mechanism that can control eEF2 activity, without the involvement of eEF2K. This conclusion is in agreement with published studies whereby exercise or treatment with farnesyltransferase induced inactivation of eEF2 in association with inhibition of protein synthesis [Ren et al., 2005; Rose et al., 2005]. These effects were also independent of the activity of eEF2K.
Previously, AMPK was reported to directly stimulate eEF2 phosphorylation following alcohol treatment [Hong-Brown et al., 2007] and this response did not require the action of eEF2K. In the present study, AMPK was observed to activate eEF2K under in vitro conditions, and this activity increased in the presence of lopinavir. In addition, the lopinavir-induced increases in both eEF2K and eEF2 phosphorylation were blocked by the AMPK inhibitor compound C, suggesting that AMPK activates eEF2 via its effects on eEF2K. However, as stated above, data from our rottlerin experiments indicate that eEF2K is not required for this process. Moreover, AMPK was also shown to directly regulate eEF2 following lopinavir treatment (Fig. 8). Collectively, these data are consistent with the hypothesis that AMPK directly acts on eEF2, even when eEF2K and other upstream kinases pathways such as mTOR/S6K1 and ERK1/2 are inhibited (data not shown). Hence, in response to lopinavir, AMPK can regulate eEF2 in a manner that is independent of mTOR/eEF2K and ERK/eEF2K pathways.
Finally, we examined the role that phosphatases may play in regulating the effect of lopinavir on eEF2 phosphorylation. Previously, alcohol has been shown to decrease the PP2A activity against eEF2 [Hong-Brown et al., 2007]. In contrast, lopinavir did not appear to inhibit PP2A activity. Although a role for other phosphatases can not be excluded, the observed changes in eEF2 phosphorylation were most likely due to changes in kinase activity.
We propose a model in which lopinavir increases the phosphorylation and activity of AMPK, thereby leading to an increased phosphorylation and inactivation of eEF2. Based on our in vivo and in vitro inhibitor studies, we suggest that AMPK can act on eEF2 either directly, or indirectly via the action of eEF2K. For example, treatment with the inhibitor compound C blocks the ability of AMPK to phosphorylate either eEF2K or eEF2, although it does not directly inhibit the activity of eEF2K towards eEF2. Likewise, rottlerin treatments block the activity of eEF2K towards eEF2, without affecting AMPK. In summary, these data are in agreement with the decrease in protein synthesis that occurs in myocytes exposed to lopinavir. As such, this should provide insight into the AMPK signaling mechanism regulating this process.
Acknowledgments
Lopinavir was obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH.
This study was supported by NIH DK072909.
LITERATURE CITED
- Atherton PJ, Babraj J, Smith K, Singh J, Rennie MJ, Wackerhage H. Selective activation of AMPK-PGC- 1alpha or PKB-TSC2- mTOR signaling can explain specific adaptive responses to endurance or resistance training-like electrical muscle stimulation. Faseb J. 2005;19:786–8. doi: 10.1096/fj.04-2179fje. [DOI] [PubMed] [Google Scholar]
- Ben-Romano R, Rudich A, Etzion S, Potashnik R, Kagan E, Greenbaum U, Bashan N. Nelfinavir induces adipocyte insulin resistance through the induction of oxidative stress: differential protective effect of antioxidant agents. Antivir Ther. 2006;11:1051–60. [PubMed] [Google Scholar]
- Browne GJ, Finn SG, Proud CG. Stimulation of the AMP-activated protein kinase leads to activation of eukaryotic elongation factor 2 kinase and to its phosphorylation at a novel site, serine 398. J Biol Chem. 2004;279:12220–31. doi: 10.1074/jbc.M309773200. [DOI] [PubMed] [Google Scholar]
- Browne GJ, Proud CG. Regulation of peptide-chain elongation in mammalian cells. Eur J Biochem. 2002;269:5360–8. doi: 10.1046/j.1432-1033.2002.03290.x. [DOI] [PubMed] [Google Scholar]
- Carroll M, Warren O, Fan X, Sossin WS. 5-HT stimulates eEF2 dephosphorylation in a rapamycin-sensitive manner in Aplysia neurites. J Neurochem. 2004;90:1464–76. doi: 10.1111/j.1471-4159.2004.02634.x. [DOI] [PubMed] [Google Scholar]
- Crozier SJ, Vary TC, Kimball SR, Jefferson LS. Cellular energy status modulates translational control mechanisms in ischemic-reperfused rat hearts. Am J Physiol Heart Circ Physiol. 2005;289:H1242–50. doi: 10.1152/ajpheart.00859.2004. [DOI] [PubMed] [Google Scholar]
- Dorovkov MV, Pavur KS, Petrov AN, Ryazanov AG. Regulation of elongation factor-2 kinase by pH. Biochemistry. 2002;41:13444–50. doi: 10.1021/bi026494p. [DOI] [PubMed] [Google Scholar]
- Ergun-Longmire B, Lin-Su K, Dunn AM, Chan L, Ham K, Sison C, Stavola J, Vogiatzi MG. Effects of protease inhibitors on glucose tolerance, lipid metabolism, and body composition in children and adolescents infected with human immunodeficiency virus. Endocr Pract. 2006;12:514–21. doi: 10.4158/EP.12.5.514. [DOI] [PubMed] [Google Scholar]
- Gschwendt M, Muller HJ, Kielbassa K, Zang R, Kittstein W, Rincke G, Marks F. Rottlerin, a novel protein kinase inhibitor. Biochem Biophys Res Commun. 1994;199:93–8. doi: 10.1006/bbrc.1994.1199. [DOI] [PubMed] [Google Scholar]
- Guiard-Schmid JB, Poirier JM, Meynard JL, Bonnard P, Gbadoe AH, Amiel C, Calligaris F, Abraham B, Pialoux G, Girard PM, Jaillon P, Rozenbaum W. High variability of plasma drug concentrations in dual protease inhibitor regimens. Antimicrob Agents Chemother. 2003;47:986–90. doi: 10.1128/AAC.47.3.986-990.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez F, Padilla S, Navarro A, Masia M, Hernandez I, Ramos J, Esteban A, Martin-Hidalgo A. Lopinavir plasma concentrations and changes in lipid levels during salvage therapy with lopinavir/ritonavir-containing regimens. J Acquir Immune Defic Syndr. 2003;33:594–600. doi: 10.1097/00126334-200308150-00007. [DOI] [PubMed] [Google Scholar]
- Hong-Brown LQ, Brown CR, Huber DS, Lang CH. Alcohol regulates eukaryotic elongation factor 2 phosphorylation via an AMP-activated protein kinase-dependent mechanism in C2C12 skeletal myocytes. J Biol Chem. 2007;282:3702–12. doi: 10.1074/jbc.M606593200. [DOI] [PubMed] [Google Scholar]
- Hong-Brown LQ, Brown CR, Lang CH. Indinavir impairs protein synthesis and phosphorylations of MAPKs in mouse C2C12 myocytes. Am J Physiol Cell Physiol. 2004;287:C1482–92. doi: 10.1152/ajpcell.00038.2004. [DOI] [PubMed] [Google Scholar]
- Hong-Brown LQ, Brown CR, Lang CH. HIV antiretroviral agents inhibit protein synthesis and decrease ribosomal protein S6 and 4EBP1 phosphorylation in C2C12 myocytes. AIDS Res Hum Retroviruses. 2005;21:854–62. doi: 10.1089/aid.2005.21.854. [DOI] [PubMed] [Google Scholar]
- Horman S, Beauloye C, Vertommen D, Vanoverschelde JL, Hue L, Rider MH. Myocardial ischemia and increased heart work modulate the phosphorylation state of eukaryotic elongation factor-2. J Biol Chem. 2003;278:41970–6. doi: 10.1074/jbc.M302403200. [DOI] [PubMed] [Google Scholar]
- Horman S, Browne G, Krause U, Patel J, Vertommen D, Bertrand L, Lavoinne A, Hue L, Proud C, Rider M. Activation of AMP-activated protein kinase leads to the phosphorylation of elongation factor 2 and an inhibition of protein synthesis. Curr Biol. 2002;12:1419–23. doi: 10.1016/s0960-9822(02)01077-1. [DOI] [PubMed] [Google Scholar]
- Hsu A, Isaacson J, Brun S, Bernstein B, Lam W, Bertz R, Foit C, Rynkiewicz K, Richards B, King M, Rode R, Kempf DJ, Granneman GR, Sun E. Pharmacokinetic-pharmacodynamic analysis of lopinavir-ritonavir in combination with efavirenz and two nucleoside reverse transcriptase inhibitors in extensively pretreated human immunodeficiency virus-infected patients. Antimicrob Agents Chemother. 2003;47:350–9. doi: 10.1128/AAC.47.1.350-359.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inamura N, Nawa H, Takei N. Enhancement of translation elongation in neurons by brain-derived neurotrophic factor: implications for mammalian target of rapamycin signaling. J Neurochem. 2005;95:1438–45. doi: 10.1111/j.1471-4159.2005.03466.x. [DOI] [PubMed] [Google Scholar]
- Jain RG, Lenhard JM. Select HIV protease inhibitors alter bone and fat metabolism ex vivo. J Biol Chem. 2002;277:19247–50. doi: 10.1074/jbc.C200069200. [DOI] [PubMed] [Google Scholar]
- Janneh O, Hoggard PG, Tjia JF, Jones SP, Khoo SH, Maher B, Back DJ, Pirmohamed M. Intracellular disposition and metabolic effects of zidovudine, stavudine and four protease inhibitors in cultured adipocytes. Antivir Ther. 2003;8:417–26. [PubMed] [Google Scholar]
- Mangili A, Murman DH, Zampini AM, Wanke CA. Nutrition and HIV infection: review of weight loss and wasting in the era of highly active antiretroviral therapy from the nutrition for healthy living cohort. Clin Infect Dis. 2006;42:836–42. doi: 10.1086/500398. [DOI] [PubMed] [Google Scholar]
- McLeod LE, Proud CG. ATP depletion increases phosphorylation of elongation factor eEF2 in adult cardiomyocytes independently of inhibition of mTOR signalling. FEBS Lett. 2002;531:448–52. doi: 10.1016/s0014-5793(02)03582-2. [DOI] [PubMed] [Google Scholar]
- Mitsui K, Brady M, Palfrey HC, Nairn AC. Purification and characterization of calmodulin-dependent protein kinase III from rabbit reticulocytes and rat pancreas. J Biol Chem. 1993;268:13422–33. [PubMed] [Google Scholar]
- Montes ML, Pulido F, Barros C, Condes E, Rubio R, Cepeda C, Dronda F, Antela A, Sanz J, Navas E, Miralles P, Berenguer J, Perez S, Zapata A, Gonzalez-Garcia JJ, Pena JM, Vazquez JJ, Arribas JR. Lipid disorders in antiretroviral-naive patients treated with lopinavir/ritonavir-based HAART: frequency, characterization and risk factors. J Antimicrob Chemother. 2005;55:800–4. doi: 10.1093/jac/dki063. [DOI] [PubMed] [Google Scholar]
- Noor MA, Flint OP, Maa JF, Parker RA. Effects of atazanavir/ritonavir and lopinavir/ritonavir on glucose uptake and insulin sensitivity: demonstrable differences in vitro and clinically. Aids. 2006;20:1813–21. doi: 10.1097/01.aids.0000244200.11006.55. [DOI] [PubMed] [Google Scholar]
- Palella FJ, Jr, Delaney KM, Moorman AC, Loveless MO, Fuhrer J, Satten GA, Aschman DJ, Holmberg SD. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV Outpatient Study Investigators. N Engl J Med. 1998;338:853–60. doi: 10.1056/NEJM199803263381301. [DOI] [PubMed] [Google Scholar]
- Parmer TG, Ward MD, Hait WN. Effects of rottlerin, an inhibitor of calmodulin-dependent protein kinase III, on cellular proliferation, viability, and cell cycle distribution in malignant glioma cells. Cell Growth Differ. 1997;8:327–34. [PubMed] [Google Scholar]
- Parmer TG, Ward MD, Yurkow EJ, Vyas VH, Kearney TJ, Hait WN. Activity and regulation by growth factors of calmodulin-dependent protein kinase III (elongation factor 2-kinase) in human breast cancer. Br J Cancer. 1999;79:59–64. doi: 10.1038/sj.bjc.6690012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price NT, Redpath NT, Severinov KV, Campbell DG, Russell JM, Proud CG. Identification of the phosphorylation sites in elongation factor-2 from rabbit reticulocytes. FEBS Lett. 1991;282:253–8. doi: 10.1016/0014-5793(91)80489-p. [DOI] [PubMed] [Google Scholar]
- Prot M, Heripret L, Cardot-Leccia N, Perrin C, Aouadi M, Lavrut T, Garraffo R, Dellamonica P, Durant J, Le Marchand-Brustel Y, Binetruy B. Long-term treatment with lopinavir-ritonavir induces a reduction in peripheral adipose depots in mice. Antimicrob Agents Chemother. 2006;50:3998–4004. doi: 10.1128/AAC.00625-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proud CG. mTOR-mediated regulation of translation factors by amino acids. Biochem Biophys Res Commun. 2004;313:429–36. doi: 10.1016/j.bbrc.2003.07.015. [DOI] [PubMed] [Google Scholar]
- Redpath NT, Price NT, Severinov KV, Proud CG. Regulation of elongation factor-2 by multisite phosphorylation. Eur J Biochem. 1993;213:689–99. doi: 10.1111/j.1432-1033.1993.tb17809.x. [DOI] [PubMed] [Google Scholar]
- Ren H, Tai SK, Khuri F, Chu Z, Mao L. Farnesyltransferase inhibitor SCH66336 induces rapid phosphorylation of eukaryotic translation elongation factor 2 in head and neck squamous cell carcinoma cells. Cancer Res. 2005;65:5841–7. doi: 10.1158/0008-5472.CAN-04-3141. [DOI] [PubMed] [Google Scholar]
- Riis B, Rattan SI, Clark BF, Merrick WC. Eukaryotic protein elongation factors. Trends Biochem Sci. 1990;15:420–4. doi: 10.1016/0968-0004(90)90279-k. [DOI] [PubMed] [Google Scholar]
- Rose AJ, Broholm C, Kiillerich K, Finn SG, Proud CG, Rider MH, Richter EA, Kiens B. Exercise rapidly increases eukaryotic elongation factor 2 phosphorylation in skeletal muscle of men. J Physiol. 2005;569:223–8. doi: 10.1113/jphysiol.2005.097154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryazanov AG, Rudkin BB, Spirin AS. Regulation of protein synthesis at the elongation stage. New insights into the control of gene expression in eukaryotes. FEBS Lett. 1991;285:170–5. doi: 10.1016/0014-5793(91)80798-8. [DOI] [PubMed] [Google Scholar]
- Sans MD, Xie Q, Williams JA. Regulation of translation elongation and phosphorylation of eEF2 in rat pancreatic acini. Biochem Biophys Res Commun. 2004;319:144–51. doi: 10.1016/j.bbrc.2004.04.164. [DOI] [PubMed] [Google Scholar]
- Schwarcz SK, Hsu LC, Vittinghoff E, Katz MH. Impact of protease inhibitors and other antiretroviral treatments on acquired immunodeficiency syndrome survival in San Francisco, California, 1987-1996. Am J Epidemiol. 2000;152:178–85. doi: 10.1093/aje/152.2.178. [DOI] [PubMed] [Google Scholar]
- Terai K, Hiramoto Y, Masaki M, Sugiyama S, Kuroda T, Hori M, Kawase I, Hirota H. AMP-activated protein kinase protects cardiomyocytes against hypoxic injury through attenuation of endoplasmic reticulum stress. Mol Cell Biol. 2005;25:9554–75. doi: 10.1128/MCB.25.21.9554-9575.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valerio L, Fontas E, Pradier C, Lavrut T, Garraffo R, Dunais B, Cua E, Dellamonica P. Lopinavir/ritonavir combination and total/HDL cholesterol ratio. J Infect. 2005;50:229–35. doi: 10.1016/j.jinf.2004.01.014. [DOI] [PubMed] [Google Scholar]
- Vidal F, Domingo JC, Guallar J, Saumoy M, Cordobilla B, Sanchez de la Rosa R, Giralt M, Alvarez ML, Lopez-Dupla M, Torres F, Villarroya F, Cihlar T, Domingo P. In vitro cytotoxicity and mitochondrial toxicity of tenofovir alone and in combination with other antiretrovirals in human renal proximal tubule cells. Antimicrob Agents Chemother. 2006;50:3824–32. doi: 10.1128/AAC.00437-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishnuvardhan D, Moltke LL, Richert C, Greenblatt DJ. Lopinavir: acute exposure inhibits P-glycoprotein; extended exposure induces P-glycoprotein. Aids. 2003;17:1092–4. doi: 10.1097/01.aids.0000060380.78202.b5. [DOI] [PubMed] [Google Scholar]
- Wang L, Proud CG. Regulation of the phosphorylation of elongation factor 2 by MEK-dependent signalling in adult rat cardiomyocytes. FEBS Lett. 2002;531:285–9. doi: 10.1016/s0014-5793(02)03536-6. [DOI] [PubMed] [Google Scholar]
- Wang X, Li W, Williams M, Terada N, Alessi DR, Proud CG. Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. Embo J. 2001;20:4370–9. doi: 10.1093/emboj/20.16.4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Q, Hruz PW. Direct comparison of the acute in vivo effects of HIV protease inhibitors on peripheral glucose disposal. J Acquir Immune Defic Syndr. 2005;40:398–403. doi: 10.1097/01.qai.0000176654.97392.c7. [DOI] [PMC free article] [PubMed] [Google Scholar]