

Abstract
The direct binding of bacteria to platelets is a central interaction in the pathogenesis of infective endocarditis. GspB is a serine-rich, cell wall glycoprotein of Streptococcus gordonii that mediates the binding of this organism to human platelets in vitro. To assess the contribution of this adhesin to the pathogenesis of endocarditis, we compared the virulence of S. gordonii M99 (which expresses GspB) with an isogenic, gspB mutant (PS846) in two rat models of endovascular infection. In the first group of experiments, animals were infected intravenously with M99 or PS846, and sacrificed 72 h later, to assess levels of bacteria within cardiac vegetations, kidneys, and spleens. When inoculated with 105 CFU, rats infected with PS846 had significantly lower densities of organisms within vegetations (mean: 3.84 log10 CFU/g) as compared with M99-infected rats (6.67 log10 CFU/g; P < 0.001). Marked differences were also seen in rats co-infected with M99 and PS846, at a 1:1 ratio. While M99 was found at high levels within vegetations, kidneys and spleens (mean log10 CFU/g: 6.62, 5.07 and 4.18, respectively) PS846 was not detected within these tissues. Thus, platelet binding by GspB appears to be a major interaction in the pathogenesis of endocarditis due to S. gordonii.
Keywords: Endocarditis, Platelets, Streptococci, Virulence, Adhesins, Bacterial
1. Introduction
The binding of bacteria to platelets is a major interaction in the pathogenesis of infective endocarditis. This process may be important both for the initial adherence of bacteria in the bloodstream to the surface of cardiac valves, and for the subsequent formation of macroscopic vegetations. Platelet binding by bacteria in vitro has been shown to occur both directly (i.e., via a ligand-receptor interaction), and through bridging molecules, such as fibrinogen and fibronectin [1]. For at least several Gram-positive species, binding to human platelets is mediated by a family of large, serine-rich glycoprotein adhesins, of which gordonii surface protein B (GspB) is among the best-characterized [2]. GspB is a large (286 kDa), cell wall-anchored protein of Streptococcus gordonii strain M99 that binds human platelets through its interaction with a platelet membrane receptor, glycoprotein (GP) Ibα [3,4]. This high affinity binding (kd = 2.38 × 10−8 M) occurs through the interaction of the N-terminal basic region of GspB with trisaccharide NeuAcα(2–3)Galβ(1–3)GalNAc (sialyl T-antigen) groups on GPIbα. Loss of GspB expression results in approximately a 70% reduction in platelet binding by M99, as compared with the parent strain [2,5].
Although platelet binding by GspB in vitro has been examined in detail, the importance of this interaction in vivo is unknown. Studies of two homologs of GspB (SraP of Staphylococcus aureus and Hsa of S. gordonii Challis) have shown that these serine-rich adhesins contribute significantly to virulence, as measured by animal models of infective endocarditis [6,7]. However, GspB differs considerably from Hsa and SraP, both in terms of the amino acid sequence of their putative ligand binding domains, and in their binding properties. Hsa binds not only sialyl T-antigen, but also α(2–3) sialyllactosamine [4,8]. While SraP expression is associated with increased platelet binding by S. aureus, its platelet receptor has not been identified. Thus, the role of GspB in the pathogenesis of endocarditis cannot be inferred from studies of these other serine-rich adhesins. Moreover, previous studies of SraP and Hsa did not address the impact of these adhesins on the extracardiac complications of this disease, such as splenic or renal infection due to septic emboli. For these reasons, we examined the effect of GspB expression on the course of endocardial infection.
2. Results
2.1. Binding of bacteria to rat platelets in vitro
Loss of GspB expression results in a significant reduction in binding to human platelets by strain M99 [2,5]. To assess whether GspB had a similar impact on the interaction of M99 with rat platelets, we compared M99 and PS846 (an isogenic variant of M99 containing a nonpolar disruption of gspB [9]) for their adherence to these cells in vitro. Binding of PS846 to rat platelets was found to be significantly lower than that of M99 (mean percent inoculum bound: 0.342 ± 0.045 versus 0.942 ± 0.110, respectively; P = 0.002). This represented a 64% reduction in binding, which is comparable to values observed for human platelets, and demonstrates that GspB also mediates binding to rat platelets.
2.2. Effect of GspB expression on streptococcal endocarditis
To examine the role of GspB in the pathogenesis of infective endocarditis, we compared strains M99 and PS846 for their relative ability to produce infective endocarditis, as measured in two well-characterized rat models of aortic valve infection. In the first group of experiments, catheterized animals were infected intravenously with 105 or 106 CFU of M99 or PS846. This inoculum range encompasses the 95% infectious dose of strain M99, and is comparable to that used with other streptococcal strains in this model [7,10]. Animals were sacrificed 72 h later to assess levels of bacteria within tissues. When challenged with an inoculum of 105 CFU, rats infected with PS846 had significantly lower densities of organisms within vegetations (mean = 3.84 ± 1.61 log10 CFU/g; N = 7) as compared with M99-infected rats (mean = 6.67 ± 1.97 log10 CFU/g; P < 0.001; N = 7). In addition, the mean weight of vegetations in the PS846 group was only 58% of that seen in the M99 group (mean ± SD: 13.0 ± 6.5 mg versus 22.2 ± 6.4 mg, respectively; P = 0.026). Loss of GspB expression was also associated with significantly reduced levels of organisms within kidneys (Fig. 1). When a higher inoculum (106 CFU) was used to infect rats, no differences were seen between the two strains, as measured by CFU per gram of tissue within vegetations, kidneys, or spleens (data not shown). At this higher inoculum, animals infected with PS846 did have smaller vegetations, as compared with rats infected with M99, though this difference only bordered on statistical significance (mean vegetation weight ± SD: 28.7 ± 12.6 mg versus 54.6 ± 31.2 mg, respectively; P = 0.056).
Fig. 1.
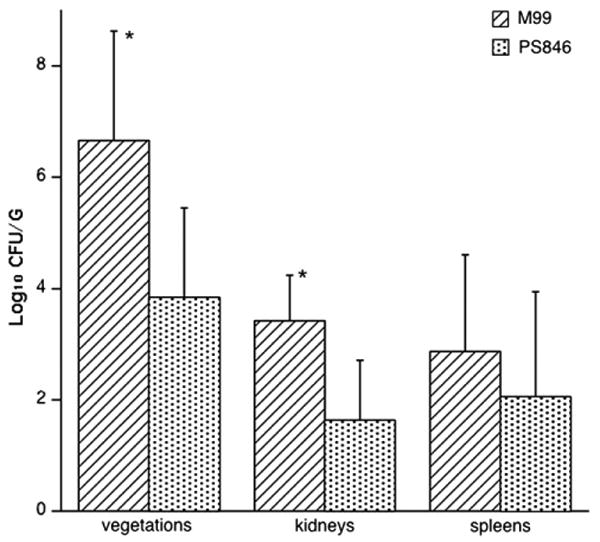
Effect of GspB expression on virulence. Levels of bacteria (mean ± SD) in vegetations, kidneys, and spleens recovered from rats infected with either M99 or PS846. *P≤0.05.
An even more pronounced difference in virulence was seen when the strains were compared in a co-infection model, in which animals were simultaneously infected with 1 × 105 CFU of each strain (Fig. 2). While M99 grew to a mean density of 6.62 ± 1.24 log10 CFU/g of vegetation, PS846 could not be detected within vegetations of co-infected animals (N = 8; P < 0.0001; lower limit of detection: < 2.1 log10 CFU/g). Similarly, although M99 reached high levels within the kidneys (mean ± SD = 5.07 ± 0.64 log10 CFU/g) and spleens (4.18 ± 0.93 log10 CFU/g), PS846 was not detected in either of these organs (lower limits of detection: <0.9 log10 CFU/g and 0.7 CFU/g for kidneys and spleens, respectively). We then re-analyzed these findings by comparing the ratio of M99 to PS846 within tissues, with the CFU of each strain normalized for the inoculum (competition index). When assessed by this approach, the ratio of M99 to the mutant was significantly different in all tissues examined (mean ± SD log10 normalized ratio in vegetations = 4.59 ±1.16; kidneys = 4.27 ± 0.51; spleens = 3.58 ± 0.94; P < 0.00001 for all tissues).
Fig. 2.
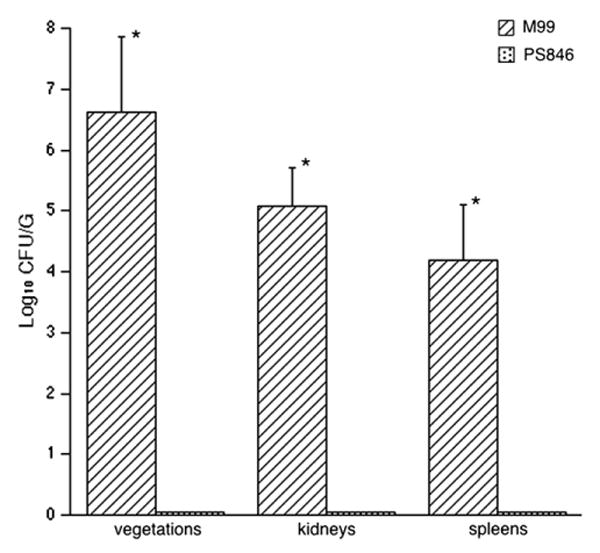
Effect of GspB expression on rats co-infected with M99 and PS846. PS846 could not be detected in any tissues harvested. See text for lower limits of detection. *P < 0.0001.
To confirm these findings, we also compared M99 with another isogenic mutant (PS436), in which gspB is disrupted by insertion of the vector pVA891 [2]. Catheterized animals (N = 6) were co-infected with M99 (6.0 × 105 CFU) and PS436 (6.6 × 105 CFU), as described above, and sacrificed 72 h later to assess the course of infection. As compared with M99, levels of PS436 were significantly lower in vegetations (log10 CFU/g 6.89 ± 1.07 versus 4.95 ± 1.29 for M99 and PS436, respectively; P = 0.02), kidneys (4.44 ± 0.80 versus 3.24 ± 3.24 ± 0.88; P = 0.03), and spleens (4.40 ± 0.76 versus 3.30 ± 0.66; P = 0.02). When these results were re-analyzed as competition indices, PS436 again proved less virulent, with a mean ± SD log10 normalized ratio (M99:PS436) in vegetations = 1.97 ± 1.24, P = 0.01; kidneys = 1.24 ± 0.99, P = 0.02; spleens = 1.15 ± 0.87; P = 0.02. Thus, loss of GspB expression was again associated with reduced virulence.
2.3. Effect of GspB expression on early bacteremia and the initial adherence of bacteria to the endovascular surface
We also examined whether GspB expression had an impact on the levels of bacteremia or the initial attachment of bacteria to the endocardium. Catheterized rats were injected IV with 107 or 109 CFU of M99 and PS846 at a 1:1 ratio, and blood samples were obtained 1 min later for quantitative cultures. At 30 min post inoculation, additional blood samples were obtained for culture, followed by the sacrificing of animals and the harvesting of the aortic valves. At both inocula, M99 achieved higher levels of bacteremia at 1 or 30 min post injection, as compared with PS846 (Table 1). Similarly, M99 had greater binding to the endocardium at 30 min post-infection, versus PS846. However, these differences did not reach statistical significance.
Table 1.
Clearance of bacteremia and valvular adherence following simultaneous IV challenge with S. gordonii M99 and PS846 at a 1:1 ratio
| Strain | Inoculum (# of animals) | Bacteremia (mean ± SD log10 CFU/ml) | Adherence at 30 min (mean ± SD log10 CFU/g of vegetation) | |
|---|---|---|---|---|
| 1 min | 30 min | |||
| M99 | 107 CFU (6) | 2.84 ± 1.65 | 1.50 ± 0.86 | 2.66 ± 0.25 |
| PS846 | 2.44 ± 1.40 | 1.33 ± 0.68 | 2.24 ± 0.57 | |
| M99 | 109 CFU (5) | 5.59 ± 0.23 | 4.10 ± 0.73 | 4.67 ± 0.35 |
| PS846 | 5.52 ± 0.15 | 3.84 ± 0.55 | 4.37 ± 0.27 | |
3. Discussion
GspB and its homologs comprise an expanding family of highly conserved, cell wall-anchored surface proteins of Gram-positive bacteria [11–16]. These proteins share a number of distinct properties, including large size, extensive serine-rich domains, and carbohydrate modification. GspB is encoded within a locus that also encodes proteins for its glycosylation, and an accessory Sec system that is dedicated to the export of this glycoprotein substrate [2,3,5,9,17–20]. The homologs of GspB are all encoded within similar loci, and where examined, these proteins have also been found to be modified with carbohydrate moieties [6,12,13,21]. Moreover, their transport to the cell surface is also likely to be dependent on an accessory Sec locus [14,22,23]. In view of the overall conserved organization and size of these loci, and in some cases, the presence of genes encoding transposases, it has been proposed that these regions may be novel pathogenicity islands [19].
Expression of these serine-rich proteins by bacteria has been linked to virulence in several models of infection. As noted above, both SraP and Hsa have been shown to contribute to pathogenicity, as measured by animal models of infective endocarditis [6,7]. The role of this family of glycoproteins in virulence appears to extend beyond the vascular system. The glycoprotein Srr-2 of Streptococcus agalactiae has been associated with enhanced virulence in a mouse model of neonatal sepsis [14]. Similarly, the serine-rich glycoprotein PsrP of Streptococcus pneumoniae has been shown to be important for virulence in a murine model of pneumonia and bacteremia [23]. Thus, these proteins appear to be a highly conserved group of virulence factors of Gram-positive bacteria that may contribute to pathogenesis in a broad range of in vivo settings.
Although the serine-rich glycoproteins share some features, they can differ considerably in primary amino acid sequence and function. The N-terminal region of GspB, which contains the platelet glycoprotein Ibα-binding domain of the protein [4], has limited similarity to other members of this family. This probably explains why only some homologs of GspB (Hsa, and SrpA of Streptococcus sanguinis) can bind this receptor, while other homologs (e.g., SraP) appear to interact with platelets via a binding site other than GPIbα. In addition, some homologs, such as Srr-1 of Streptococcus agalactiae, do not appear to bind platelets, but rather, have affinity for other receptors (e.g., keratin) and cells [15], which may determine the type of infection associated with its expression.
Our results indicate that GspB is an important virulence determinant in the pathogenesis of infective endocarditis due to S. gordonii, as measured by two models of endovascular infection. In the first set of experiments, rats infected with 105 CFU of the GspB mutant strain (PS846), had significantly fewer bacteria (CFU/g) within vegetations, as compared with animals infected with the parent strain (M99). Moreover, the concentrations of bacteria within kidneys were also significantly reduced in PS846-infected rats. This may reflect the significantly smaller vegetations found in these animals. In some clinical studies of patients with infective endocarditis, vegetation size has been correlated directly with risk of systemic embolization [24]. Thus, the smaller vegetations observed in PS846-infected rats may have been less likely to produce septic emboli to peripheral organs. At a higher inoculum (106CFU) these differences between M99 and PS846 were not observed. This finding is consistent with a previous study of S. gordonii DL1, where loss of Hsa expression was associated with both a decreased ability to establish infection, and a lower density of organisms within vegetations, but only when tested at a low inoculum [7]. Similar results have been described in studies of S. aureus and the role of ClfA in endocarditis, where differences in virulence between a wild type strain and an isogenic mutant could only be detected at lower inocula [25–27]. Binding of bacteria to the endovascular surface appears to be multi-factorial [1,28,29], so it is possible that at higher inocula, secondary mechanisms of attachment may be sufficient for adherence and the initiation of infection.
Even more dramatic differences were observed with our second model of endocarditis, in which rats were co-infected with M99 and PS846 at a 1:1 ratio. Under these conditions, the parent strain achieved high titers within vegetations (4.2 × 106 CFU/g), kidneys (1.2 × 105 CFU/g), and spleens (1.5 × 104 CFU/g). Of note, these levels were comparable to those seen in animals infected with M99 alone in the first set of experiments, indicating that the presence of PS846 in the inoculum had no effect on the ability of M99 to establish infection. In marked contrast, PS846 could not be detected in either the vegetations or organs recovered from any of the co-infected animals. To confirm the results of these co-infection studies, we also compared M99 with a second, independently-derived gspB mutant, strain PS436. When tested in the co-infection model, loss of GspB expression was again associated with significantly reduced levels of bacteria within tissues. These results further indicate that loss of GspB expression results in a profound reduction in virulence.
The reduced virulence seen with loss of GspB expression does not appear to be due to differences in the rate of clearance of organisms from the blood, since co-infected animals had comparable levels of bacteremia at 1 and 30 min after inoculation. Moreover, M99 and PS846 had similar levels of initial binding to the endocardium, as measured by CFU/g at the 30 min time-point. By 72 h post-infection, however, marked disparities in virulence became apparent, indicating that this adhesin may be more important for events subsequent to the initial colonization of the valve, such as the ability of bacteria to proliferate on the valve surface, the deposition of platelets and formation of macroscopic vegetations, and the re-seeding of the valve by bacteria from metastatic foci. The differences observed in numbers of bacteria within vegetations, as well as in vegetation weights and degree of extracardiac involvement, are all consistent with this hypothesis.
As for the exact mechanism by which GspB enhances virulence, our in vitro studies demonstrate that, as was seen with human platelets, the binding of M99 to rat platelets in vitro is also largely mediated by GspB. Although the rat platelet receptor for GspB is unknown, these cells are also extensively glycosylated [30], suggesting that sialyl T-antigen or a similar carbohydrate moiety may also serve as the binding site for this adhesin.
4. Conclusion
Our combined results indicate that GspB-mediated binding is likely to occur in vivo, and this interaction is important for virulence. Moreover, platelet-bacterial binding by GspB may be critical for the persistence and proliferation of bacteria on the valve surface, as well as for the eventual development of mature vegetations. These findings are concordant with earlier work of other platelet adhesins (Hsa, SraP, ClfA) and indicate that the binding of bacteria with platelets may be most important for these later stages of pathogenesis.
5. Materials and methods
5.1. Bacterial strains and media
S. gordonii M99 is a previously-described strain of S. gordonii that was recovered from the blood of a patient with infective endocarditis [31]. PS846 is an isogenic variant of M99 that contains a nonpolar disruption of gspB (M99 ΔgspB∷ pEVP3; CmR) and is significantly reduced in its ability to bind human platelets in vitro [9]. Insertion of pEVP3 into this locus is stable in vitro, even in the absence of antibiotic selection. PS436 is a previously-described [2] variant of M99 that contains a stable pVA891 insertion within gspB (M99∷ pVA891; Ermr). This strain also has reduced platelet binding compared with M99. Todd Hewitt broth (THB), or Todd Hewitt agar (THA) containing 8% (v/v) sheep blood were used as culture media. Chloramphenicol (5 μg/ml) was added to solid media when appropriate. Growth rates of M99 and PS846 in THB and THA were comparable.
5.2. Platelet binding assay
The binding of M99 and PS846 to rat platelets in vitro was assessed as described previously for human platelets [32]. In brief, washed bacteria (approximately 1.5 × 106 CFU) were added to monolayers of rat platelets that were immobilized in microtiter plate wells, and then treated with Blocking Reagent (Roche). After 2 h of incubation at room temperature, unbound bacteria were removed by repeated washing, and the bound bacteria were then recovered by treatment with trypsin (1 mg/ml in Dulbecco's phosphate buffered saline [Sigma]) for 30 min. The bound bacteria were enumerated by plating serial dilutions onto solid media, and binding was calculated as a percentage of the input inoculum.
5.3. Rat models of infective endocarditis
Infective endocarditis was produced in Sprague-Dawley female rats (250–300 g) as described previously [33,34]. In brief, the animals were first anesthetized with ketamine (35 mg/kg) and xylazine (10 mg/kg). A sterile polyethylene catheter was surgically placed across the aortic valve of each animal, such that the tip was positioned in the left ventricle. Catheters were left in place throughout the study. Seven days post-catheterization, the animals were infected intravenously with strain M99 or PS846 (inoculum: 105, or 106 CFU suspended in sterile 0.9% NaCl). At 72 h post-infection, the rats were euthanized with thiopental (100 mg IP). Animals were included in the final analysis only if the catheters were correctly positioned across the aortic valve at the time of sacrifice, and if macroscopic vegetations were seen. All cardiac vegetations, as well as samples of the kidneys and spleens, were harvested, weighed, homogenized in saline, serially diluted, and plated onto THA for quantitative culture. After 48 h of incubation at 37 °C, bacterial colonies were counted. The number of bacteria within tissues were expressed as the log10 CFU per gram of tissue. Differences between means were compared for statistical significance by the unpaired t-test, using P ≤ 0.05 as the threshold for significance.
The relative virulence of M99 and PS846 was also compared in a competition model of infective endocarditis, as described previously [6,35]. In brief, rats were catheterized as detailed above, and then infected IV with an inoculum containing 1 × 105 CFU of both strains (i.e., a 1:1 mixture of M99 and PS846). At 72 h post-infection, animals were sacrificed, and tissues were harvested and processed as detailed above. Homogenates of the tissues were plated onto THA and THA containing 5 μg/ml of chloramphenicol, to determine the number of CFU/g of M99 and PS846 within tissues. Differences in means were compared for statistical significance as described above. The data were also analyzed by calculating a “competition index,” which was defined as the ratio of M99 and PS846 within tissues for each animal, normalized by the ratio of organisms in the inoculum. The mean of the log10 normalized ratios was tested against the hypothesized ‘no effect’ mean value of 0, as described previously, using a paired t-test [35]. Studies comparing the relative virulence of M99 and PS436 were performed and analyzed as detailed above.
We also examined the initial or early in vivo adherence of M99 and PS846 to the endocardium. Catheterized animals were co-infected intravenously with M99 and PS846 at a 1:1 ratio (total inoculum: 107 or 109 CFU). Blood samples for quantitative cultures were obtained 1 and 30 min after infection, at which time the rats were sacrificed. Bacterial densities in blood were expressed as the mean ± SD log10 CFU/ml. Endocardial vegetations were recovered, washed with 1 ml PBS to remove any surface blood and non-adherent bacterial cells, and then processed as described above. The in vivo adherence was expressed as the mean ± SD log10 CFU/g vegetation. Differences between means were compared for statistical significance by the unpaired t-test.
Acknowledgments
This work was supported by the Department of Veterans Affairs and the VA Merit Review program, grants RO1AI41513 (PMS), RO1AI057433 (PMS), and AI039108-09(ASB) from the National Institutes of Health, and American Heart Association National Scientist Development grant 0630219N (YQX).
References
- 1.Fitzgerald JR, Foster TJ, Cox D. The interaction of bacterial pathogens with platelets. Nat Rev Microbiol. 2006;4(6):445–57. doi: 10.1038/nrmicro1425. [DOI] [PubMed] [Google Scholar]
- 2.Bensing BA, Sullam PM. An accessory sec locus of Streptococcus gordonii is required for export of the surface protein GspB and for normal levels of binding to human platelets. Mol Microbiol. 2002;44(4):1081–94. doi: 10.1046/j.1365-2958.2002.02949.x. [DOI] [PubMed] [Google Scholar]
- 3.Bensing BA, Lopez JA, Sullam PM. The Streptococcus gordonii surface proteins GspB and Hsa mediate binding to sialylated carbohydrate epitopes on the platelet membrane glycoprotein Ibalpha. Infect Immun. 2004;72(11):6528–37. doi: 10.1128/IAI.72.11.6528-6537.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takamatsu D, Bensing BA, Cheng H, Jarvis GA, Siboo IR, Lopez JA, et al. Binding of the Streptococcus gordonii surface glycoproteins GspB and Hsa to specific carbohydrate structures on platelet membrane glycoprotein Ibα. Mol Microbiol. 2005;58(2):380–92. doi: 10.1111/j.1365-2958.2005.04830.x. [DOI] [PubMed] [Google Scholar]
- 5.Takamatsu D, Bensing BA, Sullam PM. Two additional components of the accessory sec system mediating export of the Streptococcus gordonii platelet-binding protein GspB. J Bacteriol. 2005;187(11):3878–83. doi: 10.1128/JB.187.11.3878-3883.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Siboo IR, Chambers HF, Sullam PM. Role of SraP, a serine-rich surface protein of Staphylococcus aureus, in binding to human platelets. Infect Immun. 2005;73(4):2273–80. doi: 10.1128/IAI.73.4.2273-2280.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takahashi Y, Takashima E, Shimazu K, Yagishita H, Aoba T, Konishi K. Contribution of sialic acid-binding adhesin to pathogenesis of experimental endocarditis caused by Streptococcus gordonii DL1. Infect Immun. 2006;74(1):740–3. doi: 10.1128/IAI.74.1.740-743.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takamatsu D, Bensing BA, Prakobphol A, Fisher SJ, Sullam PM. Binding of the streptococcal surface glycoproteins GspB and Hsa to human salivary proteins. Infect Immun. 2006;74(3):1933–40. doi: 10.1128/IAI.74.3.1933-1940.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bensing BA, Takamatsu D, Sullam PM. Determinants of the streptococcal surface glycoprotein GspB that facilitate export by the accessory Sec system. Mol Microbiol. 2005;58(5):1468–81. doi: 10.1111/j.1365-2958.2005.04919.x. [DOI] [PubMed] [Google Scholar]
- 10.Sullam PM, Frank U, Yeaman MR, Tauber MG, Bayer AS, Chambers HF. Effect of thrombocytopenia on the early course of streptococcal endocarditis. J Infect Dis. 1993;168(4):910–4. doi: 10.1093/infdis/168.4.910. [DOI] [PubMed] [Google Scholar]
- 11.Handley PS, Correia FF, Russell K, Rosan B, DiRienzo JM. Association of a novel high molecular weight, serine-rich protein (SrpA) with fibril-mediated adhesion of the oral biofilm bacterium Streptococcus cristatus. Oral Microbiol Immunol. 2005;20(3):131–40. doi: 10.1111/j.1399-302X.2004.00190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu H, Mintz KP, Ladha M, Fives-Taylor PM. Isolation and characterization of Fap1, a fimbriae-associated adhesin of Streptococcus parasanguis FW213. Mol Microbiol. 1998;28(3):487–500. doi: 10.1046/j.1365-2958.1998.00805.x. [DOI] [PubMed] [Google Scholar]
- 13.Takahashi Y, Konishi K, Cisar JO, Yoshikawa M. Identification and characterization of hsa, the gene encoding the sialic acid-binding adhesin of Streptococcus gordonii DL1. Infect Immun. 2002;70(3):1209–18. doi: 10.1128/IAI.70.3.1209-1218.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seifert KN, Adderson EE, Whiting AA, Bohnsack JF, Crowley PJ, Brady LJ. A unique serine-rich repeat protein (Srr-2) and novel surface antigen (epsilon) associated with a virulent lineage of serotype III Streptococcus agalactiae. Microbiology. 2006;152(Pt 4):1029–40. doi: 10.1099/mic.0.28516-0. [DOI] [PubMed] [Google Scholar]
- 15.Samen U, Eikmanns BJ, Reinscheid DJ, Borges F. The surface protein Srr-1 of Streptococcus agalactiae binds human keratin 4 and promotes adherence to epithelial HEp-2 cells. Infect Immun. 2007 doi: 10.1128/IAI.00717-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Plummer C, Wu H, Kerrigan SW, Meade G, Cox D, Ian Douglas CW. A serine-rich glycoprotein of Streptococcus sanguis mediates adhesion to platelets via GPIb. Br J Haematol. 2005;129(1):101–9. doi: 10.1111/j.1365-2141.2005.05421.x. [DOI] [PubMed] [Google Scholar]
- 17.Bensing BA, Gibson BW, Sullam PM. The Streptococcus gordonii platelet binding protein GspB undergoes glycosylation independently of export. J Bacteriol. 2004;186(3):638–45. doi: 10.1128/JB.186.3.638-645.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takamatsu D, Bensing BA, Sullam PM. Four proteins encoded in the gspB-secY2A2 operon of Streptococcus gordonii mediate the intracellular glycosylation of the platelet-binding protein GspB. J Bacteriol. 2004;186(21):7100–11. doi: 10.1128/JB.186.21.7100-7111.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takamatsu D, Bensing BA, Sullam PM. Genes in the accessory sec locus of Streptococcus gordonii have three functionally distinct effects on the expression of the platelet-binding protein GspB. Mol Microbiol. 2004;52(1):189–203. doi: 10.1111/j.1365-2958.2004.03978.x. [DOI] [PubMed] [Google Scholar]
- 20.Bensing BA, Siboo IR, Sullam PM. Glycine residues in the hydrophobic core of the GspB signal sequence route export toward the accessory Sec pathway. J Bacteriol. 2007;189(10):3846–54. doi: 10.1128/JB.00027-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stephenson AE, Wu H, Novak J, Tomana M, Mintz K, Fives-Taylor P. The Fap1 fimbrial adhesin is a glycoprotein: antibodies specific for the glycan moiety block the adhesion of Streptococcus parasanguis in an in vitro tooth model. Mol Microbiol. 2002;43(1):147–57. doi: 10.1046/j.1365-2958.2002.02725.x. [DOI] [PubMed] [Google Scholar]
- 22.Chen Q, Wu H, Fives-Taylor PM. Investigating the role of secA2 in secretion and glycosylation of a fimbrial adhesin in Streptococcus parasanguis FW213. Mol Microbiol. 2004;53(3):843–56. doi: 10.1111/j.1365-2958.2004.04116.x. [DOI] [PubMed] [Google Scholar]
- 23.Obert C, Sublett J, Kaushal D, Hinojosa E, Barton T, Tuomanen EI, et al. Identification of a candidate Streptococcus pneumoniae core genome and regions of diversity correlated with invasive pneumococcal disease. Infect Immun. 2006;74(8):4766–77. doi: 10.1128/IAI.00316-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tischler MD, Vaitkus PT. The ability of vegetation size on echocardiography to predict clinical complications: a meta-analysis. J Am Soc Echocardiogr. 1997;10(5):562–8. doi: 10.1016/s0894-7317(97)70011-7. [DOI] [PubMed] [Google Scholar]
- 25.Sullam PM, Bayer AS, Foss WM, Cheung AL. Diminished platelet binding in vitro by Staphylococcus aureus is associated with reduced virulence in a rabbit model of infective endocarditis. Infect Immun. 1996;64(12):4915–21. doi: 10.1128/iai.64.12.4915-4921.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moreillon P, Entenza JM, Francioli P, McDevitt D, Foster TJ, Francois P, et al. Role of Staphylococcus aureus coagulase and clumping factor in pathogenesis of experimental endocarditis. Infect Immun. 1995;63(12):4738–43. doi: 10.1128/iai.63.12.4738-4743.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Siboo IR, Cheung AL, Bayer AS, Sullam PM. Clumping factor A mediates binding of Staphylococcus aureus to human platelets. Infect Immun. 2001;69(5):3120–7. doi: 10.1128/IAI.69.5.3120-3127.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thiene G, Basso C. Pathology and pathogenesis of infective endocarditis in native heart valves. Cardiovasc Pathol. 2006;15(5):256–63. doi: 10.1016/j.carpath.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 29.Moreillon P, Que YA. Infective endocarditis. Lancet. 2004;363(9403):139–49. doi: 10.1016/S0140-6736(03)15266-X. [DOI] [PubMed] [Google Scholar]
- 30.Toor B, McGregor JL, McGregor L, Clemetson KJ. Comparison of the major membrane glycoproteins and proteins of human, rabbit and rat blood platelets. Thromb Res. 1982;26(5):317–28. doi: 10.1016/0049-3848(82)90250-x. [DOI] [PubMed] [Google Scholar]
- 31.Sullam PM, Valone FH, Mills J. Mechanisms of platelet aggregation by viridans group streptococci. Infect Immun. 1987;55(8):1743–50. doi: 10.1128/iai.55.8.1743-1750.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bensing BA, Rubens CE, Sullam PM. Genetic loci of Streptococcus mitis that mediate binding to human platelets. Infect Immun. 2001;69(3):1373–80. doi: 10.1128/IAI.69.3.1373-1380.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vernachio JH, Bayer AS, Ames B, Bryant D, Prater BD, Syribeys PJ, et al. Human immunoglobulin G recognizing fibrinogen-binding surface proteins is protective against both Staphylococcus aureus and Staphylococcus epidermidis infections in vivo. Antimicrob Agents Chemother. 2006;50(2):511–8. doi: 10.1128/AAC.50.2.511-518.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peerschke EI, Bayer AS, Ghebrehiwet B, Xiong YQ. gC1qR/p33 blockade reduces Staphylococcus aureus colonization of target tissues in an animal model of infective endocarditis. Infect Immun. 2006;74(8):4418–23. doi: 10.1128/IAI.01794-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mitchell J, Siboo IR, Takamatsu D, Chambers HF, Sullam PM. Mechanism of cell surface expression of the Streptococcus mitis platelet binding proteins PblA and PblB. Mol Microbiol. 2007;64(3):844–57. doi: 10.1111/j.1365-2958.2007.05703.x. [DOI] [PubMed] [Google Scholar]