

Abstract
Spinocerebellar ataxia type 1 is caused by expansion of a translated CAG repeat in ataxin1. The level of the polyglutamine-expanded protein is one of the factors that contribute to disease severity. Here, we show that miR-19, miR-101, and miR-130 co-regulate ataxin1 levels and that their inhibition enhances the cytotoxicity of polyglutamine-expanded ataxin1. This study provides a new candidate mechanism for modulating pathogenesis of neurodegenerative diseases sensitive to protein dosage.
Polyglutamine (polyQ) diseases are dominantly inherited, neurodegenerative disorders caused by expansion of CAG repeats that encode polyglutamine in the disease causing protein. In all these disorders, the polyQ-expanded proteins are toxic, leading to degeneration of specific neurons. Although animal studies have shown that levels of the mutant protein contribute to polyQ disease severity 4,5,6,7, the in vivo mechanisms that regulate protein levels remain to be addressed.
To verify that mutant protein levels contribute to disease severity in the context of SCA1, we evaluated mice that overexpress SCA1 (ATXN1) with 82 CAG repeats under the control of the Purkinje cell-specific Pcp2 promoter (SCA1[82Q]) 4 either in hemizgous or homozygous state. SCA1[82Q]Tg/Tg mice show more severe motor impairment and Purkinje cell pathology compared to SCA1[82Q]Tg/+ mice (Supplementary Fig. 1a,b). These data suggest that increased levels of mutant protein result in more severe disease.
Human ATXN1 contains a long 3´UTR (~7kb), implying that it might contain regulatory elements for posttranscriptional regulation (Fig. 1f and Supplementary Fig. 5a). One mechanism that regulates levels of gene products involves microRNAs (miRNAs), endogenous small non-coding RNAs that bind the 3´UTR of cognate target mRNAs to suppress their expression 8. Interestingly, impairment of miRNA biogenesis in Purkinje cells results in cerebellar degeneration and ataxia in mice 9. Thus, we hypothesized that miRNA-mediated posttranscriptional regulation of ataxin1 (ATXN1) might modulate SCA1 neuropathology by affecting the levels of the protein.
Figure 1.

miR-19, miR-101, and miR-130 regulate ATXN1 levels. (a and b) miR-19a, miR-101 and miR-130a decrease ATXN1 level in HEK293T cells. A representative western blot image (a) and mean relative levels of ATXN1 (negative control =1) and standard deviation (s.d.) (b) are presented. (c and d) 2´-O-methyl inhibitors specific for each miRNA increase ATXN1 levels. (e) Luciferase assays using portions of the ATXN1 3´UTR depicted by nucleotide number within the 3´UTR identify the regions regulated by three different miRNAs. (f) Schematic of human ATXN1 3´UTR shows location of the authentic miR-19, miR-101, and miR-130 target sites conserved in vertebrates and verified by mutagenesis in Supplementary Figures 6 and 7. * P≤0.05 and ** P<0.01.
To test this hypothesis, we searched for evolutionarily conserved miRNA binding sites in the 3´UTR of human ATXN1 using miRNA target prediction databases 10,11. Of the predicted miRNAs, we chose eight different miRNAs as candidates, based on the number of ATXN1 target sites and their neuronal expression. To validate the role of the selected miRNAs in modulating ATXN1 levels, we transfected MCF7 cells, which highly express endogenous ATXN1 (Supplementary Fig. 2a), with each miRNA duplex. miR-19a, miR-101, and miR-130a down-regulated the level of ATXN1 (Supplementary Fig. 2b). Generally, different miRNAs act cooperatively on the same target mRNA to suppress its translation 8. To determine if this is the case for the miRNAs we identified, we transfected different human cell lines (HEK293T, HeLa, and MCF7 cells) either with each individual miRNA (miR-19a, miR-101, and miR-130a) or with all of them combined. We observed a marked decrease in ATXN1 levels upon co-transfection of all three miRNAs; transfection of 120 pmoles of each miRNA individually also decreased ATXN1 levels (Fig. 1a,b and Supplementary Fig. 3a-c). When we used 40 pmoles for each individual miRNA, the reduction in ATXN1 levels was less pronounced in comparison to 40 pmoles of all three combined (Supplementary Fig. 3a). Collectively, these data suggest that miR-19a, miR-101, and miR-130a cooperatively regulate ATXN1 levels. Interestingly, only miR-101 affected both mRNA and protein levels while miR-19a and miR-130a reduced protein level without changing mRNA level in HeLa cells (Supplementary Fig. 3c,d). This suggests that miR-101 induces mRNA degradation as well as translational repression, whereas others act mainly on translation. We also found that mouse ATXN1 is cooperatively regulated by these three miRNAs (Supplementary Fig. 4), suggesting that the mechanism of ATXN1 regulation by miR-19, miR-101, and miR-130 is conserved in mammals. When we used 2´-O-methyl inhibitors against miR-19, miR-101, and miR-130a, we found that ATXN1 levels increased in HEK293T cells (Fig. 1c,d).
To confirm that miR-19, miR-101, and miR-130 directly target the 3´UTR of ATXN1, we constructed firefly luciferase reporter genes linked to partial or full length human ATXN1 3´UTR (Supplementary Fig. 5b) and carried out dual luciferase assays in HeLa cells. In the context of the full length 3´UTR (pGL3-hATXN1 3´UTR 1-7,015), miR-101 dramatically reduced reporter gene expression, whereas miR-19a and miR-130a only marginally did, as compared to the pGL3 control (Fig. 1e). We observed dramatic suppression of two additional reporters containing the partial ATXN1 3´UTR in which miRNA target sites are enriched (pGL3-hATXN1 3´UTR 1-1,600 and 4,200-7,015), with a clear correlation between the number of putative target sites and the degree of down-regulation (Fig. 1e). We then proceeded to identify the authentic miRNA target sites by mutagenizing each putative miRNA target site (except for the first miR-130 target site) conserved in vertebrates as well as one miR-130 target site unique to humans (Supplementary Figs. 6 and 7). Most of the conserved putative miRNA target sites, except for the fourth miR-19 target site and the miR-130 target site unique to humans, are authentic (Fig. 1f and Supplementary Figs. 6 and 7). Together these data show that miR-19, miR-101, and miR-130 directly bind to the ATXN1 3´UTR to suppress the translation of ATXN1.
Cerebellar Purkinje cells are vulnerable in SCA1 patients and animal models where mutant ATXN1 accumulates gradually and causes toxicity 4,12. To probe the possibility that the level of ATXN1 could be regulated by miR-19, miR-101, and miR-130 in Purkinje cells, we investigated the expression pattern and level of these miRNAs in mouse cerebellum. Northern blot analysis demonstrates that they are expressed in the cerebellum (Supplementary Fig. 8) and RNA in situ hybridization revealed that miR-19b, miR-101, and miR-130 are expressed in Purkinje cells (Fig. 2). Of note, however, miR-101 and miR-130 are more abundant than miR-19 in Purkinje cells (Fig. 2), suggesting that miR-101 and miR-130 could dominantly act on ATXN1 in this brain region. Altogether, the expression of these miRNAs in the cells most vulnerable in disease suggests that they could play a role in modulating the toxicity of mutant ATXN1.
Figure 2.
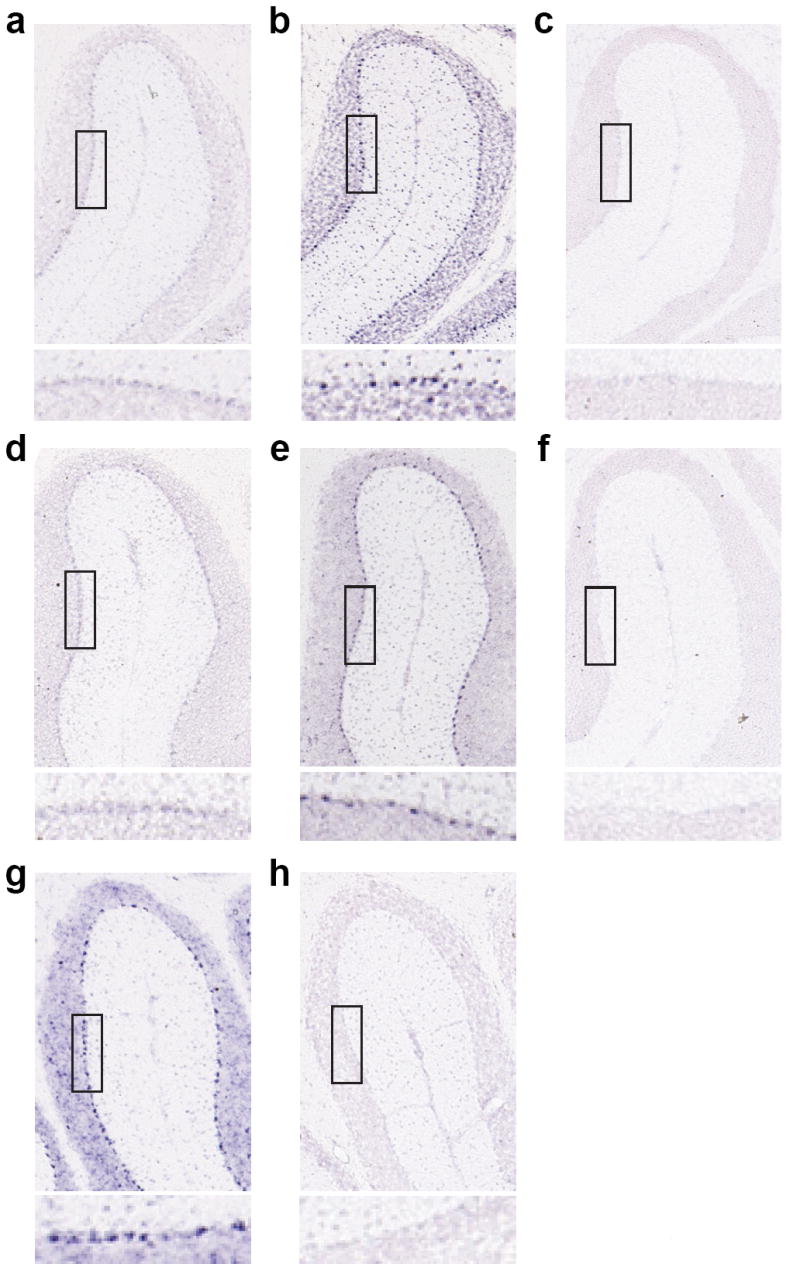
Purkinje cell expression of miR-19, miR-101 and miR-130. In situ hybridization using LNA probes for (a) miR-19b, (b) miR-130a, (c) scrambled (control for miR-19b and miR-130a), (d) miR-101a, (e) miR-101b, (f) scrambled (control for miR-101a and miR-101b), (g) miR-130b, and (e) scrambled (control for miR-130b). Upper panels show miR-19b, miR-101 and miR-130 expression in anterior cerebellar lobules and lower panels are enlarged images of boxed regions to show expression in Purkinje cells.
To investigate whether these miRNAs modulate the cytotoxicity of the polyQ expanded ATXN1, we generated polyQ expanded ATXN1 expression vector containing the wild-type 3´UTR of human ATXN1 (F86Q-3UTR WT) and carried out a cell-based assay of mutant ATXN1 toxicity. We used HEK293T cells because the polyQ expanded ATXN1 induces cell death in this cell line 14 and, more importantly, because molecular mechanisms that contribute to SCA1 pathogenesis in the cerebellum have been reproduced in this cell line in terms of formation of the toxic protein complex containing the polyQ expanded ATXN1 and RBM17 15. We tested whether inhibition of endogenous miR-19, miR-101, and miR-130 enhances cytotoxicity of the polyQ expanded ATXN1 (hATXN1[86Q]) expressed from F86Q-3UTR WT. Inhibition of these three miRNAs increased hATXN1[86Q] levels (Fig. 3b,c). As a consequence, increased hATXN1[86Q] significantly reduced cell viability 48h and 72h after transfection, in comparison to co-transfection with F86Q-3UTR WT and control 2´-O-methyl inhibitor, although only mixture of inhibitors specific for each miRNA (19a+19b+101+130b) also induced slightly more cell death than the control inhibitor (Control) (Fig. 3d). To exclude the possibility that up-regulation of other targets caused by inhibition of these miRNAs could contribute to reduction of cell viability, we generated a hATXN1[86Q] expression vector containing the 3´UTR with mutated miRNA target sites (F86Q-3UTR Mut) ; 3 sites corresponding to miR-19, and 1 site each corresponding to either miR-101 or miR-130 (Fig. 3a). Mutation of these particular miRNA target sites not only increased the level of hATXN1[86Q], but also reduced cell viability (Fig. 3e-g). These data suggest that the inhibition of miRNA-mediated posttranscriptional regulation of mutant ATXN1 enhances its cytotoxicity.
Figure 3.

Inhibition of miRNA-mediated posttranscriptional regulation of polyQ expanded ATXN1 causes more severe cytotoxicity in HEK293T cells. (a) Schematic diagrams of three FLAG-hATXN1[86Q] constructs that either lack the 3´UTR (F86Q), contain a wild type 3´UTR (F86Q-3UTR WT), or contain a 3´UTR harboring mutated miRNA target sites depicted as “x” (F86Q-3UTR Mut). (b and c) A mixture of 2´-O-methyl inhibitors specific for each miRNA (19a+19b+101+130a) increases the level of FLAG-hATXN1[86Q] expressed from F86Q-3UTR WT. A representative western blot image (b) and mean relative levels of FLAG-hATXN1[86Q] (negative control =1) and s.d. (c) are presented. EGFP is a normalization control for transfection efficiency. (d) Lethality is significantly enhanced in cells transfected with the mixture of 2´-O-methyl inhibitors specific for each miRNA and F86Q-3UTR WT compared to cells transfected with 2´-O-methyl inhibitor control and F86Q-3UTR WT. (e and f) Disruption of miRNA target sites in the ATXN1 3´UTR increases FLAG-hATXN1[86Q] level. A representative western blot image (e) and mean relative levels of FLAG-hATXN1[86Q] (F86Q of lane 3 in the western image = 1) and s.d. (f) are presented. (g) Assays of cell viability 48h or 72h after transfection with each vector demonstrate significantly more lethality in cells expressing either F86Q or F86Q-3UTR Mut in comparison to cells expressing F86Q-3UTR WT. * P≤0.05 and ** P<0.01.
In this study, we provide evidence for a novel key regulatory mechanism by discovering miRNAs that regulate the level of a polyQ disease causing protein in mammals and demonstrate the modulatory role of miRNA-mediated mechanisms in ATXN1 toxicity. The identification of this new modulatory pathway of ATXN1 begs the question whether similar regulatory mechanisms exist for other polyQ proteins and for proteins implicated in neurodegenerative diseases that are caused by a gain-of-function mechanism and are sensitive to disease protein levels. Moreover, such findings raise the possibility that mutations in the miRNA binding sites or the miRNA genes themselves might cause neurodegenerative phenotypes owing to accumulation of ATXN1.
Supplementary Material
Acknowledgments
We thank N. Ao, Y. Liu, and A. Liang of the Baylor College of Medicine ISH core for technical assistance; and Dr. V. Narry Kim and members of the Zoghbi laboratory for helpful discussions and comments on the manuscript. This research was supported by NIH grants NS27699 and HD24064 to H.Y.Z. and NS22920 to H.T.O. H.Y.Z. is an investigator with the Howard Hughes Medical Institute.
Footnotes
Author Contributions Y.L. and H.Y.Z. designed the experiments. Y.L. performed the majority of the experiments with the exception of the experiments for comparison between SCA1[82Q]Tg/+ and SCA1[82Q]Tg/Tg mice in Supplementary Fig. 1 (J.R.G. and R.C.S.), and RNA in situ hybridization in Fig. 2 (C.T.). Data analyses and interpretation were conducted by Y.L., H.T.O. and H.Y.Z. Y.L. and H.Y.Z. wrote the paper.
References
- 1.Orr HT, Zoghbi HY. Annu Rev Neurosci. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- 2.Orr HT, et al. Nat Genet. 1993;4:221–6. doi: 10.1038/ng0793-221. [DOI] [PubMed] [Google Scholar]
- 3.Banfi S, et al. Nat Genet. 1994;7:513–20. doi: 10.1038/ng0894-513. [DOI] [PubMed] [Google Scholar]
- 4.Burright EN, et al. Cell. 1995;82:937–48. doi: 10.1016/0092-8674(95)90273-2. [DOI] [PubMed] [Google Scholar]
- 5.Cemal CK, et al. Hum Mol Genet. 2002;11:1075–94. doi: 10.1093/hmg/11.9.1075. [DOI] [PubMed] [Google Scholar]
- 6.Xia H, et al. Nat Med. 2004;10:816–20. doi: 10.1038/nm1076. [DOI] [PubMed] [Google Scholar]
- 7.Huynh DP, Figueroa K, Hoang N, Pulst SM. Nat Genet. 2000;26:44–50. doi: 10.1038/79162. [DOI] [PubMed] [Google Scholar]
- 8.Bartel DP. Cell. 2004;116:281–97. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 9.Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Cell. 2003;115:787–98. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 10.Krek A, et al. Nat Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 11.Schaefer A, et al. J Exp Med. 2007;204:1553–8. doi: 10.1084/jem.20070823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zoghbi HY, Orr HT. Semin Cell Biol. 1995;6:29–35. doi: 10.1016/1043-4682(95)90012-8. [DOI] [PubMed] [Google Scholar]
- 13.Clark HB, et al. J Neurosci. 1997;17:7385–95. doi: 10.1523/JNEUROSCI.17-19-07385.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rich T, Varadaraj A. PLoS ONE. 2007;2:e1014. doi: 10.1371/journal.pone.0001014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lim J, et al. Nature. 2008;452:713–8. doi: 10.1038/nature06731. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.