

Abstract
Pulsatile GnRH (GNRH) differentially regulates LH and FSH subunit genes, with faster frequencies favoring Lhb transcription and slower favoring Fshb. Various intracellular pathways mediate the effects of GNRH, including CaMK II (CAMK2), ERK, and JNK. We examined whether activation of these pathways is regulated by GNRH pulse frequency in vivo. GNRH-deficient rats received GNRH pulses (25 ng i.v. every 30 or 240 min for 8 h, vehicle to controls). Pituitaries were collected 5 min after the last pulse, bisected, and one half processed for RNA (to measure beta subunit primary transcripts [PTs]) and the other for protein. Phosphorylated CAMK2 (phospho-CAMK2), ERK (mitogen-activated protein kinase 1/3 [MAPK1/3], also known as p42 ERK2 and p44 ERK1, respectively), and JNK (MAPK8/9, also known as p46 JNK1 and p54 JNK2, respectively) were determined by Western blotting. The 30-min pulses maximally stimulated Lhb PT (8-fold), whereas 240 min was optimal for Fshb PT (3-fold increase). Both GNRH pulse frequencies increased phospho-CAMK2 4-fold. Activation of MAPK1/3 was stimulated by both 30- and 240-min pulses, but phosphorylation of MAPK3 was significantly greater following slower GNRH pulses (240 min: 4-fold, 30 min: 2-fold). MAPK8/9 activation was unchanged by pulsatile GNRH in this paradigm, but as previous results showed that GNRH-induced activation of MAPK8/9 is delayed, 5 min after GNRH may not be optimal to observe MAPK8/9 activation. These data show that CAMK2 is activated by GNRH, but not in a frequency-dependant manner, whereas MAPK3 is maximally stimulated by slow-frequency GNRH pulses. Thus, the ERK response to slow pulse frequency is part of the mechanisms mediating Fhb transcriptional responses to GNRH..
Keywords: CaMKII, ERK, follicle-stimulating hormone, follistatin, GNRH, gonadotropin-releasing hormone, gonadotropins, JNK, luteinizing hormone, signal transduction
GNRH differentially regulates gonadotropin gene transcription via changes in CaMK II and ERK intracellular signaling and follistatin gene expression.
INTRODUCTION
Luteinizing hormone and FSH are dimeric protein hormones composed of a common glycoprotein α subunit (CGA) and a unique β subunit [1]. The gonadotropin subunit genes are regulated by the hypothalamus in both a coordinate and differential manner, primarily by the decapeptide GNRH that is released in a pulsatile manner. GNRH differentially regulates subunit mRNA synthesis via changes in pulse frequency, with faster-intermediate GNRH pulse frequencies (8- to 60-min pulse intervals) favoring Cga and Lhb and slower-frequency pulses (≥120-min pulse intervals) favoring Fshb [2].
The intracellular mechanisms by which GNRH pulse frequency differentially regulates gonadotropin β subunit transcription are not well understood. The GNRH receptor (GNRHR) is a member of the seven-transmembrane receptor family [3, 4], which activates several members of the G protein family, including Gq and G11 [5, 6]. Activated Gq stimulates phospholipase C, resulting in increased inositol 1,4,5,-triphosphate (IP3 ), elevated diacylglycerol levels, and activation of protein kinase C (PKC) [6–8]. GNRHR binding also stimulates a transient increase in intracellular calcium (Ca2+), derived from IP3-induced release of Ca2+ from intracellular storage pools, and from influx via L-type voltage-gated calcium channels, which subsequently stimulates the activation of several signal transduction pathways [7, 9].
Previous studies reveal that an increase in intracellular calcium is essential for GNRH modulation of gonadotropin subunit gene expression, as Ca2+ channel blockade completely prevents the increases in subunit mRNAs in response to GNRH in rat pituitary cells [10] and rat Lhb promoter activity in LβT2 cells [11]. Our group showed that intermittent changes in intracellular Ca2+ are more effective in stimulating subunit mRNAs than continuous Ca2+ influx [10, 12]. Also similar to GNRH, Ca2+ pulse frequency differentially regulates subunit transcription, with rapid-intermediate frequency selectively stimulating Lhb and slower-frequency pulses being optimal for Fshb [12, 13].
Evidence derived from various cell types reveals that intracellular calcium signals are transduced, in part, via calcium/calmodulin-dependent kinase II (CAMK2). CAMK2 is a multimeric enzyme of 8–12 subunits composed of one or more isoforms (α, β, γ, or δ) [14], with the α (54-kDa) and β (60-kDa) isoforms being predominate in the brain. In its inactive state, the CAMK2 autoinhibitory domain sequesters the catalytic region. When intracellular Ca2+ concentrations increase, Ca2+ binds calmodulin (CALM1), and the Ca2+/CALM1 complex can then bind the autoregulatory region of CAMK2, unmasking the catalytic domain leading to phosphorylation of numerous target substrates. In addition, activated subunits autophosphorylate a threonine residue (T286) on an adjacent subunit within the holoenzyme. This event increases the affinity of the enzyme for Ca2+/CALM1 and prevents the autoinhibitory domain from reassociating with the kinase domain, resulting in Ca2+/CALM1-independent kinase activity and amplification of the original Ca2+ signal. The autophosphorylation nature of CAMK2 is thought to be important in transmitting Ca2+ pulse frequency and amplitude signals, as fast and high-amplitude Ca2+ influxes result in greater and/or sustained Ca2+/CALM1 levels, increasing the magnitude of CAMK2 autophosphorylation and Ca2+/CALM1-independent kinase activity [15].
We have reported that GNRH induces a rapid and transient increase in CAMK2 activation following a single pulse, and treatment with a CaMK inhibitor completely blocks GNRH-induced increases in α subunit transcription or promoter activity, and partially blocks those of Lhb and Fshb [16, 17]. In addition to CAMK2, other investigations showed that members of the mitogen-activated protein kinase (MAPK) pathway are important in transmitting GNRH signals within the gonadotrope. GNRH stimulates activation of MAPK family members ERK (MAPK1/3), JNK (MAPK8/9), and p38 (MAPK11/12/13/14) [2, 4, 8]. GNRH-induced MAPK1/3 activation is mediated via both PKC-dependent and independent mechanisms, resulting in the subsequent activation of several transcription factors (i.e., FOS, JUN, and ELK1) that appear to play important roles in gonadotropin subunit gene transcription [2, 18]. We have previously reported that GNRH pulses are more effective than continuous GNRH in sustaining pituitary MAPK1/3 activation in male rats, that MAPK1/3 phosphorylation is greater following slow-frequency GNRH pulses, and that blocking MAPK1/3 activation suppresses Cga and Fshb mRNA responses to GNRH [19]. The observation that ERK blockade has no effect on GNRH-induced Lhb mRNA or promoter activity [11] suggests that Lhb transcription is driven via a distinct intracellular pathway(s).
GNRH also stimulates the MAPK8/9 pathway via activation of cSrc (SRC) and CDC42 [20, 21]. GNRH-induced MAPK8/9 activation results in increased expression and phosphorylation of JUN [22, 23] and increased ATF3 expression [22]. Furthermore, we have recently reported that blocking JNK activation completely prevented the GNRH-induced increase in Lhb transcription in perifused rat pituitary cells [23]. Of interest, MAPK8/9 also appears to regulate basal Fshb transcription, since blocking activation suppresses basal Fshb primary transcript (PT) levels by half but has no effect on the magnitude of GNRH-stimulated Fshb PT responses [23].
The purpose of the present study was to investigate whether GNRH pulse frequency differentially regulates CAMK2, MAPK8/9, or MAPK1/3 activation in normal rat pituitaries, and whether those changes are correlated with alterations in Lhb and Fshb subunit gene transcription.
MATERIALS AND METHODS
Rat In Vivo Studies
Adult (225–250 g) male Sprague-Dawley rats (Harlan Sprague Dawley Inc., Indianapolis, IN) were used for all experiments. Rats were housed in a light-controlled (lights on 0500–1700 h) and temperature-controlled (25°C) room and allowed access to food and water ad libitum. All surgeries were performed under isoflurane (2.5% isoflurane, balance O2; ISO-THESIA; Vetus Animal Heath; Burns Veterinary Supply Inc., Westbury, NY) anesthesia. At the completion of experiments, rats were euthanized by decapitation under anesthesia. The University of Virginia Animal Care and Use Committee approved the animal experimentation described within this report.
To study the effects of GNRH pulse frequency, we used a GNRH-deficient (castrate + testosterone [TE]-replaced] male rat model. Rats were castrated, and two 20-mm TE-containing silastic implants were inserted s.c. (serum TE levels were 4–5 ng/ml, 24 h postimplantation insertion), as previously described [24]. An indwelling right jugular cannula was also inserted at the time of castration for i.v. GNRH administration. All experiments began 24 h after castration. Rats (n = 6–7 per group) were pulsed with 25-ng GNRH pulses (in 0.25 ml of 0.9% saline/0.1% BSA) either every 30 or 240 min for 8 h (for a total of 17 or 3 GNRH pulses, respectively). Controls were pulsed with vehicle only. Animals were killed 5 min after the last pulse, based on previous data in rats showing that phosphorylated CAMK2 (phospho-CAMK2), phospho-MAPK1/3, and β subunit PT responses to GNRH are maximal 5 min after a pulse [16, 19, 25]. Recent findings also reveal that GNRH simulates a significant (50%) increase in phospho-MAPK8/9 at 5 min after the pulse, although maximal increases are seen 15 min after GNRH [23]. Pituitaries were collected, bisected, and then half processed immediately for protein and the other half snap frozen in liquid nitrogen and stored at −70°C until RNA was extracted.
RNA Preparation and Measurement of Gonadotropin Lhb and Fshb Transcription and Follistatin mRNA
Total pituitary RNA was extracted using the acid guanidinium method [26]. Residual genomic DNA was removed by treatment with 1 unit RNase Free DNase I/μg RNA (Roche Molecular Biochemicals, Indianapolis, IN) at 37°C for 1 h. RNA preparations were confirmed to be DNA free by PCR in the absence of a preceding RT reaction. Lhb and Fshb PTs were measured by quantitative RT-PCR assays as previously described [27]. Follistatin (Fst) mRNA was measured by quantitative real-time PCR using primers previously described [28]. For real-time PCR, 1 μg RNA/sample was reverse transcribed in 20 μl containing: 50 mM Tris HCl, pH 8.3; 75 mM KCl; 3 mM MgCl2; 10 mM dithiothreitol; 0.5 mM each dATP, dCTP, dTTP, and dGTP (dNTPs; Promega, Madison, WI); 150 ng random hexamers (Roche Applied Science, Indianapolis, IN); 200 units Superscript II Reverse Transcriptase (Invitrogen, Carlsbad, CA); and 40 units RNase Inhibitor (Roche Applied Science). Reverse transcription conditions were 10 min at 25°C, 42 min at 42°C, and 15 min at 70°C. Quantitative real-time PCR was conducted on an iCycler instrument (Bio-Rad, Hercules, CA). For Fst mRNA, 25 ng RNA equivalent was used per reaction. Each 30-μl reaction contained: 10 mM Tris HCl, pH 8.3; 50 mM KCl; 3 mM MgCl2, 0.3 μM each forward and reverse primers; 0.3 mM dNTPs; 1 unit TAQ DNA polymerase (Jump Start, Sigma, St. Louis, MO); 6 nM Fluorescein Calibration Dye (Bio-Rad); 0.16× SYBR Green I Dye (Molecular Probes, Invitrogen); and 3% dimethyl sulfoxide (v/v). Polymerase chain reaction conditions were 1 cycle of 95°C for 2 min, followed by 45 cycles of 94°C for 15 sec, 61°C for 30 sec, and 72°C for 30 sec. To confirm amplification of a single product, PCR amplification was followed by melt curve analysis. Each sample was measured against a standard curve of Fst cDNA in pGEM4 [28] at 105 to 10−2 fg plasmid in 1:10 dilutions. All samples and standards were measures in triplicate in the same assay to avoid interassay variablilty. Mean intraassay coefficient of variation was 12.4%.
Pituitary Protein Preparation/Western Immunoblot Assays
For protein isolation, hemipituitaries were homogenized in tissue lysis buffer containing: 50 mM HEPES, 100 mM NaCl, 2 mM EDTA, 1% Nonidet P-40, and protease and phosphatase inhibitor cocktails (P8340 and P5726, respectively, with stocks considered 100×; Sigma). Pituitary protein lysates were resolved by 10% SDS-PAGE electrophoresis and then transferred to nitrocellulose filters. For CAMK2, 100 μg pituitary protein was required per sample. For phospho-CAMK2, the primary antibody (rabbit) was obtained from Promega; this antibody recognizes both the 50-kDa α subunits and the 60-kDa β subunits autophosphorylated on threonine 286 (pT286). The secondary antibody was horseradish peroxidase-conjugated goat anti-rabbit (Millipore, Billerica, MA). Immunoactivity was detected using SuperSignal West Dura chemiluminescent system (Pierce, Rockville, IL), followed by autoradiography. Protein bands were quantified by densitometry using TotalLab Software (Amersham Biosciences, Piscataway, NJ). Following phospho-CAMK2 determination, filters were stripped (0.4 M glycine, 0.2% SDS, 2% Tween-20, pH 2.2) 2× for 30 min and reprobed for total CAMK2 (t-CAMK2). This rabbit anti-CAMK2 antibody (Cell Signaling Technology) detects only the 50-kDa α subunit. Last, as a protein loading control, blots were reprobed for glyceraldehyde 3-phosphate dehydrogenase (GAPDH, 37 kDa; Santa Cruz Biotechnology, Santa Cruz, CA).
Immunoblot assays for ERK and JNK were conducted similarly, but required only 30 μg protein per sample. All primary antibodies for phospho-JNK, t-JNK, phospho-ERK, and t-ERK were obtained from Cell Signaling Technology. The antibodies (phosphorylated and total) for JNK (MAPK8/9) recognize the p46 and p54 isoforms, and the antibodies for ERK (MAPK1/3) recognize both the p42 and p44 isoforms. The antibodies for activated JNK and ERK recognize the dually phosphorylated forms.
Analysis
All data were examined by ANOVA. Significant differences (P < 0.05) were determined posthoc by Duncan Multiple Range test. Prior to analyses, all measurements were transformed to the logarithmic scale to attain equal variation among treatments.
RESULTS
The effects of GNRH pulse frequency on Lhb and Fshb subunit transcription and Fst mRNA are shown in Figure 1. Similar to our previous studies, GNRH pulses every 30 min produced maximal effects on Lhb transcription, increasing Lhb PT 8.6-fold (P < 0.05 vs. controls). GNRH pulses every 240 min also increased Lhb PT, but to a lesser degree (3-fold vs. controls, P < 0.05). Both 30-min and 240-min GNRH pulse intervals increased Fshb PT 2- and 3-fold, respectively (P < 0.05 vs. controls). We previously observed that both fast and slow GNRH pulse frequencies increase Fshb PT initially, but that sustained increases in Fshb transcription (8–24 h) required slow-frequency GNRH pulses, which do not simulate pituitary Fst expression [24]. In this study, we also found that 8 h of slow-frequency GNRH pulses reduced Fst mRNA (30% of controls, P < 0.05), whereas fast-frequency GNRH pulses increased Fst mRNA 14-fold (P < 0.05 vs. controls).
FIG. 1.
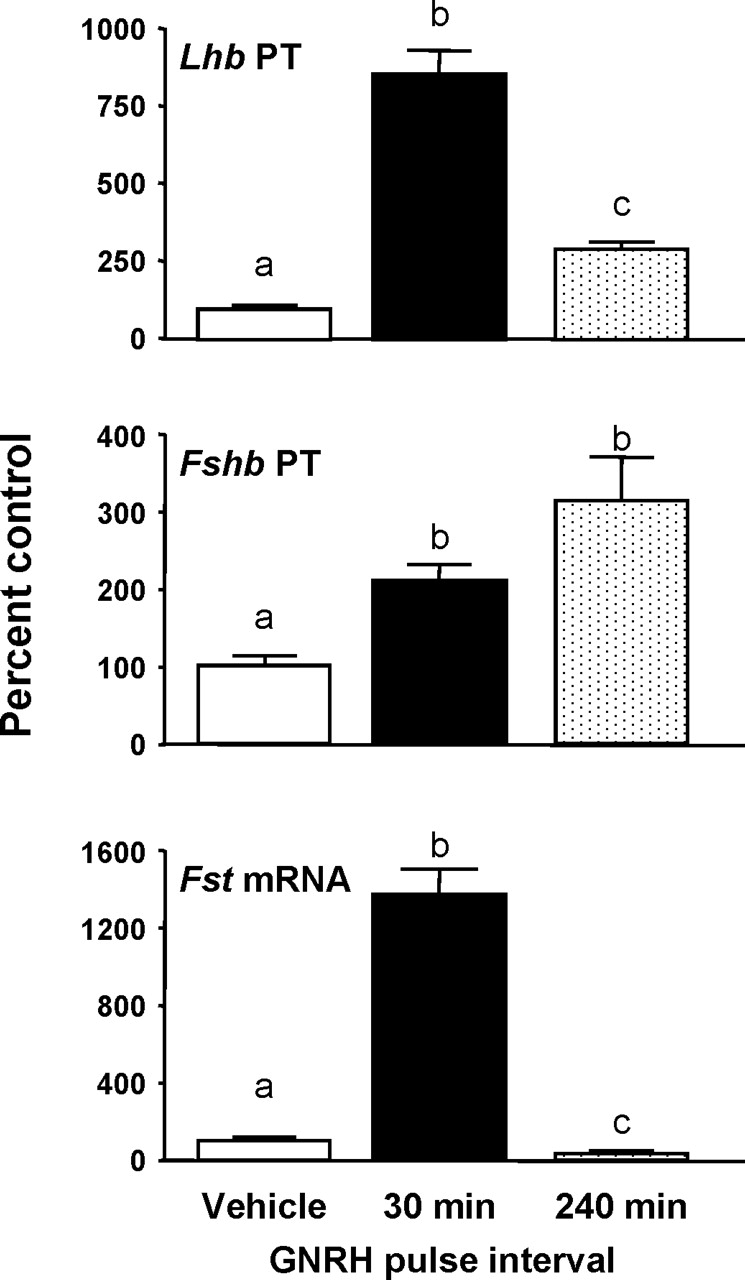
The effect of GNRH pulse frequency on Lhb and Fshb PTs and Fst mRNA. The β subunit PTs and Fst mRNA levels were measured in pituitary RNA from male rats that had been pulsed with GNRH every 30 min or 240 min for 8 h. Rats were euthanized and pituitaries collected 5 min after the last pulse. All data are presented as percentage (±SEM) of controls (n = 6–7 per group). Means with different letters are significantly different (P < 0.05). Control means were: 47.0 ± 7.5 fg competitor RNA/100 ng RNA for Fshb PT, and 96.2 ± 10.5 fg competitor RNA/100 ng RNA for Lhb PT and 12.7 ± 1.0 fg plasmid/100 ng RNA for Fst mRNA.
To determine whether the differential effects of GNRH pulse frequency on Lhb and Fshb transcription are mediated by changes in specific intracellular signaling pathways, we examined the activation of CAMK2, MAPK8/9, and MAPK1/3. Pituitary CAMK2 phosphorylation in control rats was barely detectable (Fig. 2), but GNRH pulses increased phospho-CAMK2 4-fold (P < 0.05), similarly after both GNRH pulse frequencies. Total CAMK2 was not altered by GNRH treatment.
FIG. 2.

The effects of GNRH pulse frequency on autophosphorylated CAMK2 and total CAMK2. Top: Representative Western blots of pituitary protein from male rats pulsed with GNRH every 30 min or 240 min for 8 h (n = 3 from a total of 6–7 per group). Blots were immunostained for autophosphorylated (phospho) CAMK2, total CAMK2, and GAPDH. Protein amounts are 100 μg/lane. Bottom: Changes in CAMK2 phosphorylation and total CAMK2 were quantified by densitometry normalized to GAPDH for protein loading and expressed as percentage of 0 h (±SEM) controls. Points with different letters are significantly different (P < 0.05).
The activation of MAPK8/9 in response to GNRH pulse frequency is presented in Figure 3. GNRH had no effect on MAPK8 phosphorylation. Phosphorylation of MAPK9 tended to increase by 1.5-fold with both GNRH pulse frequencies. In a recent report [23], we also found that phospho-MAPK8/9 increased by 1.5-fold (P < 0.05 vs. controls) 5 min after a GNRH pulse and peaked at 15 min (3-fold increase). The magnitude of the MAPK9 activation in response to GNRH in the present study was similar to those found previously; however, the increase was not significant. Thus, the timing of sampling may not be optimal for observing MAPK8/9 phosphorylation in this study.
FIG. 3.

The effects of GNRH pulse frequency on JNK activation. Top: Representative Western blots of pituitary protein from male rats pulsed with GNRH every 30 min or 240 min for 8 h (n = 3 from a total of 6–7 per group). Blots were immunostained for dually phosphorylated (phospho) MAPK8/9, total MAPK8/9, and GAPDH. Protein amounts are 30 μg/lane. Bottom: Changes in phosphorylated and total MAPK8/9 were quantified by densitometry normalized to GAPDH for protein loading and expressed as percentage of 0 h (±SEM) controls. The MAPK8/9 isoforms (p46 and p54, respectively) were analyzed independently.
As shown in Figure 4, pulsatile GNRH stimulated the activation of both MAPK1 and MAPK3; however, responses to pulse frequency differed. Specifically, MAPK1 increased by 2-fold (P < 0.05 vs. controls) after both GNRH pulse frequencies. In contrast, MAPK3 phosphorylation was maximally stimulated (4-fold) by slower (240-min) pulses of GNRH (P < 0.05 vs. control and 30-min pulse groups). Similar to results noted for CAMK2 and MAPK8/9, GNRH pulse frequency did not alter total MAPK1/3 content.
FIG. 4.

The effects of GNRH pulse frequency on ERK activation. Top: Representative Western blots of pituitary protein from male rats pulsed with GNRH every 30 min or 240 min for 8 h (n = 3 from a total of 6–7 per group). Blots were immunostained for dually phosphorylated (phospho) MAPK1/3, total MAPK1/3, and GAPDH. Protein amounts are 30 μg/lane. Bottom: Changes in phosphorylated and total MAPK1/3 were quantified by densitometry normalized to GAPDH for protein loading and expressed as percentage of 0 h (±SEM) controls. The MAPK1/3 isoforms (p42 and p44, respectively) were analyzed independently.
DISCUSSION
The present study extends earlier work that fast-frequency GNRH pulses favor Lhb transcription and that slow-frequency pulses favor Fshb transcription through mechanisms that involve suppressing pituitary Fst expression and increased ERK activation. This study also demonstrates that pulsatile GNRH increases pituitary CAMK2 autophosphorylation 4-fold, regardless of pulse frequency.
We previously showed that the frequency of Ca2+ signals could mimic the effects of pulsatile GNRH on gonadotropin subunit gene transcription [13]. However, the pathways that encode GNRH/Ca2+ frequency information have not been fully deduced. In rat lactotropes and GH3 cells, CAMK2 mediates TRH-induced prolactin secretion via activation of voltage-gated calcium channels [29, 30]. Also, we reported that inhibition of CAMK2 activation using KN93 partially suppressed GNRH-induced increases in Lhb and Fshb PTs in rat pituitary cells, as well as Cga and Lhb promoter activity in gonadotrope-derived LβT2 cells [16, 17]. Based on those findings, we hypothesized that alterations in GNRH pulse frequency, and hence changes in intracellular Ca2+ pulse frequency, may be encoded via Ca/CAMK2 activity. However, these results showed that CAMK2 autophosphorylation was increased to a similar degree by either rapid (30-min) or slow (240-min) GNRH pulse intervals. This observation does not exclude a potential role for CAMK2 in the transmission of GNRH frequency signals within the gonadotrope, but suggests that CAMK2 is not the only mechanism involved in frequency regulation.
Following a single GNRH pulse, either in vivo in rats or in LβT2 cells, maximal (4- to 5-fold) increases in CAMK2 autophosphorylation occur within 2–5 min [16, 17]. The rapid onset and short half-life of CAMK2 responses to GNRH [16] make this pathway a potential mediator of intracellular frequency signaling. In the present study, we observed a similar (4-fold) increase at the end of 8 h of pulsatile treatment, suggesting that the magnitude of CAMK2 responses to pulsatile GNRH (fast or slow) are sustained for at least 8 h. However, CAMK2 autophosphorylation is an indicator of Ca2+/CALM1 independent activity of the enzyme, not kinase activity. Our previous report [16] suggests that the half-life of pituitary CAMK2 activity following a single GNRH pulse is between 15 and 45 min. Thus gonadotropes receiving GNRH pulses every 30 min may experience elevated CAMK2 kinase activity almost continuously, whereas gonadotropes subjected to slow-frequency GNRH pulses would experience only episodic increases.
CAMK2 is ubiquitous and has many target substrates, including serum response factor (SRF), members of the Ets transcription factor family, cAMP response element-binding protein (CREB), and others [31]. Little is known about the regulation of the substrates of CAMK2 in the gonadotrope, but several important players in gonadotropin β subunit gene regulation, such as EGR1, are potential targets. GNRH-sensitive regions of the Egr1 promoter contain several SRF-binding elements, Ets, and CREB-binding sites. Also, the CAMK2 blocker, KN62, partially suppresses GNRH induction of Egr1 promoter activity in aT3 cells [32]. Further research is needed to determine the downstream target proteins/genes that are stimulated by GNRH pulse activation of CAMK2, as well as how these proteins/genes respond (e.g., activation state and protein and/or mRNA expression) to fast vs. slow intermittent increases in CAMK2 within gonadotrope cells.
Activation of ERK by GNRH is well established, is mediated via both PKC-dependent and independent mechanisms, and results in the phosphorylation and/or activation of a number of transcription factors (i.e., FOS, JUN, the Ets protein ELK1, and EGR1) that are important for β subunit transcription [2, 18]. We have previously reported that a single i.v. GNRH pulse in male rats induces a rapid (within 4 min) and sustained (≥30 min) increase in MAPK1/3 phosphorylation [19]. Furthermore, MAPK1/3 activation responses are prolonged following GNRH pulses vs. continuous GNRH, and are sustained more effectively by slower GNRH pulses (120-min pulse interval) than faster pulse frequencies (30- or 60-min intervals). The present results are consistent with those findings, where we found that a slower GNRH pulse interval (240 min) maximally stimulated ERK activation, specifically MAPK3, to a greater extent than GNRH pulses every 30 min.
The observation that GNRH preferentially activates MAPK3 (also known as ERK1) has been reported previously in αT3 and LβT2 cells [33, 34]. Also, GNRH-induced activation of MAPK3 phosphorylation (relative to MAPK1) is more sensitive to PKC levels, external Ca2+ levels, and SRC inhibition in LβT2 cells [34]. However, the finding that GNRH pulse pattern selectively regulates MAPK3 is a novel observation. Slow GNRH pulse frequencies maximally stimulate Fshb and Gnrhr gene transcription, and we have previously shown that treatment of rat pituitary cultures with the MAP2K1 (also known as MEK1) inhibitor PD098059 (PD) abrogate the GNRH-induced increases in Fshb and Gnrhr mRNAs, but not Lhb mRNA [19]. Similarly, Weck et al. [11] also reported that PD had no effect on GNRH induced Lhb transgene expression in cultured mouse pituitary cells.
Differential effects of GNRH pulse frequency on MAPK1/3 phosphorylation have also been shown in gonadotrope-derived cell lines. Kanasaki et al. [35] found that in perifused LβT2 cells, both fast- and slow-frequency GNRH pulses (30- or 120-min interpulse intervals, respectively) increase MAPK1/3 phosphorylation to the same magnitude (2.5- to 3-fold), but with different time courses. Fast-frequency GNRH pulses increase phospho-MAPK1/3 maximally 10 min after pulse, with a duration of 20–30 min, whereas slow-frequency GNRH pulses increased phospho-MAPK1/3 maximally at 5 min and for a longer duration (40–50 min after pulse). These data dovetail well with the present results, where we found that 5 min after the last GNRH pulse, MAPK1/3 phosphorylation was elevated by both GNRH pulse frequencies, but maximal increases occurred after 240-min pulses. Kanasaki et al. [35] also found that fast-frequency GNRH pulses preferentially stimulated rat Lhb promoter activity, whereas slow GNRH pulses stimulated rat Fshb promoter activity, and GNRH-induced activity of both promoters was blocked by the MAP2K1 inhibitor U0126. The sensitivity of GNRH-induced Fshb transcriptional activity to MAP2K1 blockade in both primary cells or gonadotrope-derived cell lines has been confirmed by others [19, 34, 38], but the regulation of Lhb is mixed, with several reports in cell lines indicating that Lhb promoter activity is MAPK1/3 dependent [20, 34, 37, 39], whereas in primary cells, transgenic mice, and others it is not [11, 19, 36].
GNRH also increases activation of the JNK arm of the MAPK signaling cascade both in vivo and in vitro [20, 23, 34, 37]. In contrast to PKC-dependent MAPK1/3 signaling, MAPKs 8 and 9 are activated via SRC and CDC42 [21, 34, 40]. We previously found that a single GNRH pulse in vivo stimulates MAPK8/9 phosphorylation within 5 min, but is not maximal until 15 min after GNRH [23]. Furthermore, in gonadotrope-derived cell lines, MAPK8/9 activation by GNRH is delayed compared with MAPK1/3 activation, and it does not peak until 30 min after initiation of GNRH treatment [20, 34, 36, 37]. In an effort to optimize experimental design to assess several pathways in this study, we collected pituitaries 5 min after the last GNRH pulse. Although MAPK8 tended to respond to GNRH to a degree similar to that seen in our previous study (1.5-fold), the increase was not significant. This, however, does not mean that MAPK8/9 activation is not important for GNRH-induced β subunit gene transcription. Recently, we reported that blocking MAPK8/9 signaling with SP600125 in perifused rat pituitary cells completely abrogated the GNRH-induced increases in Lhb PT in response to 60-min pulses of GNRH, and suppressed both basal and GNRH-induced Fshb PT by 50% [23]. We also found in LβT2 cells that the GNRH-induced increase in rat Lhb promoter activity was blocked by 80% when cells were cotransfected with dominant negatives for the MAPK8/9-activating kinases, MAP2K4 (also known as MKK4) and/or MAP2K7 (also known as MKK7) [23]. Similarly, in LβT2 cells, GNRH actions on a rat Lhb promoter/reporter were suppressed by cotransfecting with dominant negatives for either JNK or JUN [36]. Still other investigators report that GNRH stimulation of an ovine Fshb promoter/reporter was reduced almost 50% when cotransfected with dominant negatives for CDC42 or JNK [34].
In conclusion, the present study demonstrates that phospho-CAMK2 is upregulated 4-fold by GNRH, but not in a frequency-dependent manner. Also, MAPK3 activation is maximally stimulated by slow-frequency GNRH pulses and is correlated with increased Fshb transcription and suppressed Fst mRNA. Thus, the phospho-MAPK1/3 response coupled with changes in pituitary FST levels, and hence bioavailable activin, likely play significant roles in mediating Fshb transcriptional responses to slower GNRH pulse intervals.
Footnotes
1Supported by U.S. Public Health Service grants HD-33039 and HD-11489 to J.C.M., and the National Institute of Child Health and Human Development/National Institutes of Health through a cooperative agreement (U54-HD28934, Ligand Assay and Analysis Core, Molecular Core) as part of the Specialized Cooperative Centers Program in Reproductive Research to J.C.M. and D.J.H.
REFERENCES
- Gharib SD, Wierman ME, Shupnik MA, Chin WW.Molecular biology of the pituitary gonadotropins. Endocrine Rev 1990; 11: 177–199.. [DOI] [PubMed] [Google Scholar]
- Burger LL, Haisenleder DJ, Dalkin AC, Marshall JC.Regulation of gonadotropin subunit gene transcription. J Mol Endocrinol 2004; 33: 559–584.. [DOI] [PubMed] [Google Scholar]
- Millar RP, Lu ZL, Pawson AJ, Flanagan CA, Morgan K, Maudsley SR.Gonadotropin-releasing hormone receptors. Endocrine Rev 2004; 25: 235–275.. [DOI] [PubMed] [Google Scholar]
- Kaiser UB, Conn PM, Chin WW.Studies of GnRH action using GnRH receptor-expressing pituitary cell lines. Endocrine Rev 1997; 18: 46–70.. [DOI] [PubMed] [Google Scholar]
- Grosse R, Schmid A, Schoneberg T, Herrlich A, Muhn P, Schultz G, Gudermann T.Gonadotropin-releasing hormone receptor initiates multiple signaling pathways by exclusively coupling to G(q/11) proteins. J Biol Chem 2000; 275: 9193–2000.. [DOI] [PubMed] [Google Scholar]
- Liu F, Usui I, Evans LG, Austin DA, Mellon PL, Olefsky JM, Webster NJ.Involvement of both Gq/11 and Gs proteins in gonadotropin-releasing hormone receptor-mediated signaling in LβT2 cells. J Biol Chem 2002; 277: 32099–32108.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojilkovic SS, Catt KJ.Novel aspects of GnRH-induced intracellular signaling and secretion in pituitary gonadotrophs. J Neuroendocrinol 1995; 7: 739–757.. [DOI] [PubMed] [Google Scholar]
- Ando H, Hew CL, Urano A.Signal transduction pathways and transcription factors involved in the gonadotropin-releasing hormone-stimulated gonadotropin subunit gene expression. Comp Biochem Physiol B Biochem Mol Biol 2001; 129: 525–532.. [DOI] [PubMed] [Google Scholar]
- Naor Z.Signal transduction mechanisms of Ca2+ mobilizing hormones: the case of gonadotropin-releasing hormone. Endocrine Rev 1990; 11: 326–353.. [DOI] [PubMed] [Google Scholar]
- Haisenleder DJ, Yasin M, Marshall JC.The regulation of prolactin, thyrotropin and gonadotropin subunit gene expression by pulsatile or continuous calcium signals. Endocrinology 1993; 133: 2055–2061.. [DOI] [PubMed] [Google Scholar]
- Weck J, Fallest PC, Pitt LK, Shupnik MA.Differential gonadotropin-releasing hormone stimulation of rat luteinizing hormone subunit gene transcription by calcium influx and mitogen-activated protein kinase-signaling pathways. Mol Endocrinol 1998; 12: 451–457.. [DOI] [PubMed] [Google Scholar]
- Haisenleder DJ, Yasin M, Marshall JC.Gonadotropin subunit and gonadotropin-releasing hormone receptor gene expression are regulated by alterations in the frequency of calcium pulsatile signals. Endocrinology 1997; 138: 5227–5230.. [DOI] [PubMed] [Google Scholar]
- Haisenleder DJ, Workman LJ, Burger LL, Aylor KW, Dalkin AC, Marshall JC.Gonadotropin subunit transcriptional responses to calcium signals in the rat: evidence for regulation by pulse frequency. Biol Reprod 2001; 65: 1789–1793.. [DOI] [PubMed] [Google Scholar]
- Hudmon A, Schulman H.Structure-function of the multifunctional Ca2+/calmodulin-dependent protein kinase II. Biochem J 2002; 364(pt 3):593–611.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Koninck P, Schulman H.Sensitivity of CaM kinase II to the frequency of calcium oscillations. Science 1998; 279: 227–230.. [DOI] [PubMed] [Google Scholar]
- Haisenleder DJ, Burger LL, Aylor KW, Dalkin AC, Marshall JC.GnRH stimulation of gonadotropin subunit transcription: evidence for the involvement of calcium/calmodulin-dependent kinase II activation in rat pituitaries. Endocrinology 2003; 144: 2768–2774.. [DOI] [PubMed] [Google Scholar]
- Haisenleder DJ, Ferris HA, Shupnik MA.The calcium component of GnRH-stimulated LH subunit gene transcription is mediated by calcium/calmodulin-dependent protein kinase type II. Endocrinology 2003; 144: 2409–2416.. [DOI] [PubMed] [Google Scholar]
- Dobkin-Bekman M, Naidich M, Pawson AJ, Millar RP, Seger R, Naor Z.Activation of mitogen-activated protein kinase (MAPK) by GnRH is cell-context dependent. Mol Cell Endocrinol 2006; 252: 184–190.. [DOI] [PubMed] [Google Scholar]
- Haisenleder DJ, Cox ME, Parsons SJ, Marshall JC.Gonadotropin-releasing hormone pulses are required to maintain activation of mitogen-activated protein kinase: role in stimulation of gonadotrope gene expression. Endocrinology 1998; 139: 3104–3111.. [DOI] [PubMed] [Google Scholar]
- Harris D, Bonfil D, Chuderland D, Kraus S, Seger R, Naor Z.Activation of MAPK cascades by GnRH: ERK and JNK are involved in basal and GnRH-stimulated activity of the glycoprotein hormone LHβ-subunit promoter. Endocrinology 2002; 143: 1018–1025.. [DOI] [PubMed] [Google Scholar]
- Mulvaney JM, Roberson MS.Divergent signaling pathways requiring discrete calcium signals mediate concurrent activation of two mitogen-activated protein kinases by gonadotropin-releasing hormone. J Biol Chem 2000; 275: 14182–14189.. [DOI] [PubMed] [Google Scholar]
- Xie J, Bliss SP, Nett TM, Eersole BJ, Sealfon SC, Roberson MS.Transcript profiling of immediate early genes reveals a unique role of ATF-3 in mediating activation of the glycoprotein hormone alpha-subunit promoter by GnRH. Mol Endocrinol 2005; 19: 2624–2638.. [DOI] [PubMed] [Google Scholar]
- Haisenleder DJ, Burger LL, Walsh HE, Stevens J, Aylor KW, Shupnik MA, Marshall JC.Pulsatile gonadotropin-releasing hormone stimulation of gonadotropin subunit transcription in rat pituitaries: evidence for the involvement of Jun N-terminal kinase but not p38. Endocrinology 2008; 149: 139–145.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burger LL, Dalkin AC, Aylor KW, Haisenleder DJ, Marshall JC.GnRH pulse frequency modulation of gonadotropin subunit gene transcription in normal gonadotropes-assessment by primary transcript assay provides evidence for roles of GnRH and follistatin. Endocrinology 2002; 143: 3243–3249.. [DOI] [PubMed] [Google Scholar]
- Dalkin AC, Burger LL, Aylor KW, Haisenleder DJ, Workman LJ, Cho S, Marshall JC.Regulation of gonadotropin subunit gene transcription by gonadotropin-releasing hormone: measurement of primary transcript ribonucleic acids by quantitative reverse transcription-polymerase chain reaction assays. Endocrinology 2001; 142: 139–146.. [DOI] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N.Single step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 1987; 162: 156–159.. [DOI] [PubMed] [Google Scholar]
- Burger LL, Haisenleder DJ, Wotton GM, Aylor KW, Dalkin AC, Marshall JC.The regulation of FSHβ transcription by gonadal steroids: testosterone and estradiol modulation of the activin intracellular signaling pathway. Am J Physiol Endocrinol Metab 2007; 293: E277–E285.. [DOI] [PubMed] [Google Scholar]
- Kirk SE, Dalkin AC, Yasin M, Haisenleder DJ, Marshall JC.Gonadotropin-releasing hormone pulse frequency regulates expression of pituitary follistatin messenger ribonucleic acid: a mechanism for differential gonadotrope function. Endocrinology 1994; 135: 876–880.. [DOI] [PubMed] [Google Scholar]
- Cui ZJ, Gorelick FS, Dannies PS.Calcium/calmodulin-dependent protein kinase-II activation in rat pituitary cells in the presence of thyrotropin-releasing hormone and dopamine. Endocrinology 1994; 134: 2245–2250.. [DOI] [PubMed] [Google Scholar]
- Cui ZJ, Hidaka H, Dannies PS.KN-62, a calcium/calmodulin-dependent protein kinase II inhibitor, inhibits high potassium-stimulated prolactin secretion and intracellular calcium increases in anterior pituitary cells. Biochim Biophys Acta 1996; 1310: 343–347.. [DOI] [PubMed] [Google Scholar]
- Means AR.Regulatory cascades involving calmodulin-dependent protein kinases. Mol Endocrinol 2000; 14: 4–13.. [DOI] [PubMed] [Google Scholar]
- Duan WR, Ito M, Park Y, Maizels ET, Hunzicker-Dunn M, Jameson JL.GnRH regulates early growth response protein 1 transcription through multiple promoter elements. Mol Endocrinol 2002; 16: 221–233.. [DOI] [PubMed] [Google Scholar]
- Reiss N, Llevi LN, Shacham S, Harris D, Seger R, Naor Z.Mechanism of mitogen-activated protein kinase activation by gonadotropin-releasing hormone in the pituitary of αT3–1 cell line: differential roles of calcium and protein kinase C. Endocrinology 1997; 138: 1673–1682.. [DOI] [PubMed] [Google Scholar]
- Bonfil D, Chuderland D, Kraus S, Shahbazian D, Friedberg I, Seger R, Naor Z.Extracellular signal-regulated kinase, Jun N-terminal kinase, p38, and c-Src are involved in gonadotropin-releasing hormone-stimulated activity of the glycoprotein hormone follicle-stimulating hormone β-subunit promoter. Endocrinology 2004; 145: 2228–2244.. [DOI] [PubMed] [Google Scholar]
- Kanasaki H, Bedecarrats GY, Kam KY, Xu S, Kaiser UB.Gonadotropin-releasing hormone pulse frequency-dependent activation of extracellular signal-regulated kinase pathways in perifused LβT2 cells. Endocrinology 2005; 146: 5503–5513.. [DOI] [PubMed] [Google Scholar]
- Yokoi T, Ohmichi M, Tasaka K, Kimura A, Kanda Y, Hayakawa J, Tahara M, Hisamoto K, Kurachi H, Murata Y.Activation of the luteinizing hormone beta promoter by gonadotropin- releasing hormone requires c-Jun NH2-terminal protein kinase. J Biol Chem 2000; 275: 21639–21647.. [DOI] [PubMed] [Google Scholar]
- Lui F, Austin DA, Mellon PL, Olefsky JM, Webster NJG.GnRH activates ERK 1 and 2 leading to the induction of c-fos and LHβ protein expression in Lβ-T2 cells. Mol Endocrinol 2002; 16: 419–434.. [DOI] [PubMed] [Google Scholar]
- Vasilyev VV, Lawson MA, Dipaolo D, Webster NJ, Mellon PL.Different signaling pathways control acute induction versus long-term repression of LHβ transcription by GnRH. Endocrinology 2002; 143: 3414–3426.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen KA, Santos SJ, Kreidel MK, Diaz AL, Rey R, Lawson MA.Acute regulation of translation initiation by gonadotropin-releasing hormone in the gonadotrope cell line LβT2. Mol Endocrinol 2004; 18: 1301–1312.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson MS, Zhang T, Li HL, Mulvaney JM.Activation of the p38 mitogen-activated protein kinase pathway by gonadotropin-releasing hormone. Endocrinology 1999; 140: 1310–1318.. [DOI] [PubMed] [Google Scholar]