

Abstract
Alzheimer’s Disease (AD) is a neurodegenerative disease resulting in progressive cognitive decline. Amyloid plaque deposits consisting specifically of Aβ peptides that have formed fibrils displaying β-pleated sheet conformation are associated with activated microglia and astrocytes, are colocalized with C1q and other complement activation products, and appear at the time of cognitive decline in AD. APP transgenic mouse models of AD, that lack the ability to activate the classical complement pathway display less neuropathology than do the APPQ+/+ mice, consistent with the hypothesis that complement activation and the resultant inflammation may play a role in the pathogenesis of AD. Further investigation of the presence of complement proteins C3 and C4 in the brain of these mice demonstrate that both C3 and C4 deposition increase with age in APPQ+/+ transgenic mice, as expected with the age-dependent increase in fibrillar Aβ deposition. In addition, while C4 is predominantly localized on the plaques and/or associated with oligodendrocytes in APPQ+/+ mice, little C4 is detected in APPQ−/− brains consistent with a lack of classical complement pathway activation due to the absence of C1q in these mice. In contrast, plaque and cell associated C3 immunoreactivity is seen in both animal models and, surprisingly, is higher in APPQ−/− than in APPQ+/+ mice, providing evidence for alternative pathway activation. The unexpected increase in C3 levels in the APPQ−/− mice coincident with decreased neuropathology provides support for the hypothesis that complement can mediate protective events as well as detrimental events in this disease. Finally, induced expression of C3 in a subset of astrocytes suggests the existence of differential activation states of these cells.
Keywords: complement, C3, C4, transgenic models, Aβ plaques, Alzheimer’s Disease
Introduction
Complement proteins of both the classical and alternative pathways (such as C1q, C4, C3, and Factor B) have been colocalized with fibrillar amyloid plaques and cerebral vascular amyloid in the cerebral cortex and hippocampus of AD patients (Strohmeyer et al. 2000;Eikelenboom and Stam 1984;Stoltzner et al. 2000). The C5b-9 membrane attack complex has been found associated with myelin and membranes in AD brain (Webster et al. 1997), demonstrating that in this disorder the entire complement cascade is activated. In vitro, Aβ fibrils (fAβ) activate both the classical complement pathway by directly binding to C1q (Rogers et al. 1992;Jiang et al. 1994) and the alternative pathway via interactions with C3 (Bradt et al. 1998;Watson et al. 1997). Thus, it was hypothesized that in vivo fAβ activates the complement cascade and contributes to local inflammation, particularly by recruiting glia into the area of the plaque, resulting in neurotoxicity and dementia (Tenner 2001;Cooper et al. 2000;Eikelenboom and Veerhuis 1996). This hypothesis was examined and supported in subsequent in vivo studies using a mouse model of AD with a complete deficiency of the complement protein C1q (APPQ−/−) and thus unable to activate the classical complement pathway. While this APP C1q−/− transgenic mouse demonstrated age-dependent amyloid plaque deposition, there was a 50–60% reduction glial activation (GFAP, MAC-1) surrounding the plaques and a similar significant increase in neuronal markers in the CA3 region of the hippocampus (Fonseca et al. 2004). These data suggest that, at ages when the fibrillar plaque pathology is present, C1q contributes a detrimental effect on neuronal integrity, most likely through the activation of the classical complement cascade and the enhancement of inflammation. However, the source of the residual pathology remains unknown. Potential mechanisms include a complement-independent pathway and/or the activation of the alternative pathway of complement by Aβ, a process which would be unaltered by the deficiency of C1q and which would also lead to the generation of the chemotactic factors, C3a and C5a and recruitment of glia to the plaques.
Interestingly, in another murine model the over-expression of an inhibitor of complement C3, Crry, resulted in increased pathology, suggesting that some complement activation fragments (such as C3b, C3a or C5a) may decrease the neuropathology in mouse models of inflammation including those over expressing mutant APP (Wyss-Coray et al. 2002;Mukherjee and Pasinetti 2000) or limit the detrimental responses to neurodegenerative stimuli in other injury models (Van Beek et al. 2003). In addition, recent in vitro studies from this lab demonstrated a neuroprotective effect of C1q on primary neurons in culture in the absence of any other complement components (Pisalyaput and Tenner 2008). Thus, complement components may also be neuroprotective.
To further investigate complement protein expression in our murine models of AD, immunohistochemistry followed by quantitative image analysis and western blot analysis were used to assess the presence and localization of C3 and C4 in the brains of these mice using antibodies that recognize the murine C4 and that differentiate native C3 and C3 cleaved as a result of activation of the complement cascade.
Material and methods
Animals
Tg (HuAPP605.K670N-M671L)2576 mice from K. Hsiao-Ashe (Hsiao et al. 1996) were crossed with C1q knockout mice (C1qa−/−) (Botto et al. 1998), and mice with APPQ−/−genotype were generated (Fonseca et al. 2004). Non-transgenic littermates or B6/SJL wild type mice were used as age-matched controls. APPPSQ−/− animals were obtained by crossing Tg2576 APP or PS1 (line6.2 on a SW/B6D2F1/J background from University of South Florida) (Holcomb et al. 1998) with C1q−/−. APPQ+/− or APPQ−/− and/or PSQ−/− mice were intercrossed until APPPSQ+/+ and APPPSQ−/− were generated. (All genotypes were confirmed by PCR.)
Tissue collection and immunohistochemistry
Mice at different ages (3, 6, 9, 12, 16 months) were deeply anesthetized with an overdose of pentobarbital (150mg/kg, IP) and then transcardially perfused with cold phosphate-buffered saline (PBS). After dissection, one half of the brain was immediately frozen on dry ice (for Western blots) and the other half fixed overnight with 4% paraformaldehyde in PBS, pH 7.4. Thereafter, fixed tissue was stored in PBS/0.02% Sodium azide (NaN3) at 4°C until use. Fixed brain tissue was sectioned (40 μm) with a vibratome, and coronal sections were collected in PBS (containing 0.02% sodium azide), and stored at 4°C before being stained.
Immunohistochemistry (IHC) was performed on free-floating brain sections. Sections were pretreated with 50% formic acid for 5 min (to enhance subsequent staining of β amyloid) or microwaved (700w) in antigen unmasking solution (Vector Laboratories, Burlingame, CA) (for C3, C4 and CNPase antigens) for 2~5 min. Endogenous peroxidase in tissue was blocked by treating with 3% H2O2 in PBS, 20 min. at room temperature. Nonspecific background staining was blocked by a 2 hour incubation in 2% BSA with 0.3% Triton X-100 (TX). Sections were then incubated with primary antibodies (Table 1) overnight at 4°C, rinsed in PBS with 0.1%TX and incubated with biotinylated secondary antibody (Vector) and streptavidin-horseradish peroxidase (Jackson ImmunoResearch, West Grove, PA) for 1 hour each at room temperature. Finally, the sections were incubated for approximately 2~5 min with diamino-benzidine (DAB) (Vector). Sections were mounted on slides, dehydrated in a series of graded ethanol, cleared with xylene, and then coverslipped with DPX (BHD, Biomedical Specialties, CA). In double-labeling experiments, bound antibodies were detected using Fab′2 FITC-, CY3-, or TRITC-conjugated anti-IgG (Jackson ImmunoResearch, PA). As controls, sections were incubated in parallel without primary antibody or with control IgG of the corresponding species and these sections failed to develop specific staining.
Table 1.
Summary of antibody used in this study
| Antibody | Antigen | Type | Source | Dilution1 | Reference |
|---|---|---|---|---|---|
| C1q | C1q (mouse) | Goat polyclonal | F. Petry | IHC: 16ug/ml | (Fonseca et al. 2004) |
| C1q(1151) | C1q (mouse) | Rabbit polyclonal | A.J.Tenner | IHC: 4ug/ml WB: 5ug/ml | (Huang et al. 1999) |
| C3(11H9) | C3/C3b/iC3b (mouse) | Rat monoclonal | Cell Sciences (HBT) | IHC: 3.3ug/ml | (Kremmer et al. 1990) |
| C3(2/16) | C3/iC3b/C3c (mouse) | Rat monoclonal | J. D. Lambris | IHC: 1:1000 | (Mastellos et al. 2004) |
| C3(2/11) | C3b/iC3b/C3c (mouse) | Rat monoclonal | J. D. Lambris | IHC: 1:1000 | (Mastellos et al. 2004) |
| C3 | C3 (mouse) | Goat polyclonal | Cappel | WB: 1:2000 | |
| C4 | C4/C4b/C4d (mouse) | Rat monoclonal | Cell Sciences (HBT) | IHC: 3.3ug/ml WB: 2ug/ml | (Kremmer et al. 1990) |
| C4 | C4/C4d (mouse) | Rabbit polyclonal | R. Ogata | IHC: 2.4ug/ml WB: 24 ug/ml | (Ogata et al. 1994) |
| MAC-1 | CD11b (mouse) | Rat monoclonal | Serotec | IHC: 10ug/ml | (Anderson et al. 1986) |
| CD45 | CD45 (mouse) | Rat Monoclonal | Serotec | IHC: 1ug/ml | (Brewer et al. 1989) |
| CNPase | CNPase (human) | Mouse monoclonal | Chemicon | IHC: 1ug/ml | (Ehtesham et al. 2002) |
| GFAP | Glial Fibrillary acidic protein (bovine) | Rabbit polyclonal | Dako | IHC: 4ug/ml | (Viale et al. 1991) |
| 6E10 | Aβ1–17 (human) | Mouse monoclonal | Seneteck | IHC: 1ug/ml | (Jung et al. 1999) |
IHC, immunohistochemistry; WB, Western Blot
Image analysis
Immunostaining was observed under a Zeiss Axiovert-200 inverted microscope (Carl Zeiss, Thornwood NY) and images acquired with a Zeiss Axiocam high-resolution digital color camera (1300×1030 pixel) using Axiovision 3.1 software. Digital images were analyzed using KS300 analysis program (Zeiss). Percentage of immunostained area (field area of immunostaining/total image area ×100) was determined for all the markers studied by averaging several images per section that cover all or most of the region of study (cortex and hippocampus). Optical sections (z=1μm) of fluorescently labeled specimens were captured using a FLUOVIEW confocal Microscope (Olympus). All experiments were repeated at least twice, with n= 3–6 animals per group per age per marker. All quantitative comparisons were performed on sections processed at the same time.
Western Blot
Tissue samples were prepared as previously indicated (Fonseca et al. 2004). Briefly, fresh frozen half brain (including only cortex and hippocampus) was homogenized in 10 volumes of Tris-buffered saline (TBS, pH 7.4) containing protease inhibitors (0.5 mM phenylmethylsulfonyl fluoride (PMSF), 20 μg/ml aprotinin, leupeptin and pepstatin and 1mM EDTA; all inhibitors obtained from Sigma). Homogenates were briefly sonicated and centrifuged at 15,000 g for 30 min. Pellets were extracted with 2% SDS in TBS with protein inhibitors and centrifuged at 15,000g for 30 min. Protein concentration in the supernatants was determined with BCA protein assay (Pierce, Rockford, IL). Samples (30 μg protein per lane) were run on 10% SDS polyacrylamide gel under reducing conditions (100 mM DTT). Proteins were transferred to PDVF (Amersham Biosciences, Piscataway, NJ) (300 mA for 2 h). Membranes were blocked with 3% dry milk in 0.1% Tween/TBS overnight, then incubated with primary antibodies for 2 h at RT at the dilutions indicated (Table 1). After washing, blots were incubated with the corresponding HRP-labeled secondary antibodies (1:2000 – 1:5000 dilution) for 1 h. Labeling was detected using ECL system (Amersham Biosciences). Blots were stripped following manufacturer’s instructions (Amersham) and subsequently labeled with actin antibody (1:10,000 Sigma) following same procedures as above to verify equal loading.
Statistical analysis
Single ANOVA statistical analysis was used to assess the significance of the differences in plaque area, and anti C3 and anti C4 reactivity among the animal groups (B6/SJL, APPQ+/+ and APPQ−/−). Standard deviation (S.D.) of the mean is indicated by the error bars on the graphs.
Results
C1q deposition is age-dependent in APP mice brain
As observed in AD brain, C1q immunoreactivity was seen associated with thioflavine-positive Aβ plaques only in the Tg2576 animals (Fonseca et al. 2004) and APP/PS mice models (Matsuoka et al. 2001). To verify and expand these results, brain tissues from APP transgenic mice with (APPQ+/+) and without a functional C1q gene (APPQ−/−) and the nontransgenic control, B6/SJL, were assessed for detectable C1q at 3, 6, 9, 12, and 16 months of age by immunohistochemical analysis. C1q immunoreactivity quantitatively increases with age and amyloid plaque deposition, as shown in Figure 1A for the Tg2576 (APPQ+/+) model. No C1q staining was seen in APPQ−/− animals at any age ((Fonseca et al. 2004) and Figure 1A) nor in the B6/SJL nontransgenic animal (data not shown). Western blot analysis on SDS-extracted brain tissue using a polyclonal anti-mouse C1q confirmed the absence of C1q in the APPQ−/− mice homozygous for the ablated C1q gene (Figure 1B).
Figure 1. C1q deposition is age-dependent in APP Q+/+mice brain, and absence in APPQ−/− mice.

A. Quantification of C1q immunostaining in animals at different ages (3mo–16mo). Data points represent group means ± SD (n=4 animals per age group except n=6 for 16 months, 5 fields/animal including cortex and hippocampus). 12m and 16m APP mice C1q staining was significantly different from 9 month old animals (*, p<0.01). B. Western blot analysis of 16mo APPQ+/+ and APPQ−/− brain extracts using a polyclonal anti-mouse C1q antibody as described in Materials and Methods. The C1q immunoreactive band is only seen in APPQ+/+ mice.
C4 deposition correlates with age in APPQ+/+ transgenic mice but not APPQ−/−
Brain sections from mice at 6, 9, 12 and 16 months of age, were then stained with a monoclonal anti-mouse C4 (HBT) that recognizes various forms of C4 α-chain via an epitope in the C4d region of the molecule. (The C4d region contains the thioester which, upon activation of complement and cleavage of C4 by activated C1, can mediate covalent linkage to the activator.) Distinct C4 immunoreactivity was first detected in the APPQ+/+ mice at 12 months, and this reactivity was increased in 16 month old mice, temporally correlating with the appearance of C1q associated with thioflavine-positive plaques (Figure 1A, 2A, 2C). In contrast, at 12 and 16 months little to no C4 immunoreactivity was seen in APPQ−/− mice. Image analysis of the 16 mo animals results in average % field area of 0, 1.2 and 0.15 for B6/SJL (wild type), APPQ+/+ and APPQ−/− respectively (Figure 2C). Results in 16 mo APPPS1Q+/+ vs APPPS1Q−/− mice also show the lack of immunoreactivity for C4 in APPPSQ−/− confirming the lack of classical pathway activation in the C1q−/− mice (Figure 2B).
Figure 2. APPQ−/− have less C4 reactivity than APPQ+/+ mice.

A. Representative pictures of cortex of APPQ+/+ (left) and APPQ−/− (right) mice at different ages showing immunostaining with anti-mouse C4 (C4d) (HBT). Scale bar: 50μm. B. Sections from cortex of 16 mo old double transgenic APPPS1 Q+/+ and APPPS1Q−/− were stained with anti-mouse C4 as in A. C. Quantification of C4 reactivity by image analysis shows that C4d staining in APPQ+/+ (-□-) mice is significantly higher (* p<0.02) than in APPQ−/− (-▲-) at 16 mo. (n=3 or 4 mice for each genotype at each age). There was no reactivity in the wild type B6/SJL (-○-). Data points represent group means ± SD. D. Western blot analysis of brain extracts from 16m B6SJL, APPQ+/+ and APPQ−/− mice probed with polyclonal anti-mouse C4 (Ogata, Table 1) detect C4 activation fragment, C4d at 42,000 Mr.
Western blot analysis was used as an alternative approach to assess C4 levels in extracts from brain tissue of 16 mo old mice. Using both a polyclonal anti-murine C4 antibody and the same monoclonal antibody against C4d used in the immunohistochemical investigations above, uncleaved C4 was detected in the nontransgenic B6/SJL mice, the APPQ+/+ and the APPQ−/− animals (data not shown). However, the 42,000 Mr activation-induced cleavage fragment of C4, C4d, was clearly present at increased levels in the APPQ+/+ brain extracts relative to the nontransgenic and APPC1q−/− mice (Figure 2D and data not shown). These observations are consistent with the requirement for C1q in C1 to induce cleavage of C4 (and thus allow deposition of C4b/d on plaques).
Some C4d immunoreactivity is seen in extracts of brains of 16 mo nontransgenic mice (Figure 2D), data suggesting an amyloid-independent baseline level of this complement fragment since no amyloid deposits are present in the nontransgenic animals (data not shown). In addition, levels of C4d in the APPQ−/− mice were similar to the nontransgenic mice, levels that may be due to amyloid-independent baseline fragment deposition, similar to the non-transgenic mice. C4d staining is not seen in the brain vasculature in any of the animals studied. However, it is also possible that some C4d in the APPQ−/− (that is clustered as seen in Figure 2A) may be the result of activation of the contact system by β-amyloid as previously seen in in vitro systems (Bergamaschini et al. 1999).
C4 Immunoreactivity is associated with plaques and oligodendrocytes
C4 reactivity was localized with amyloid plaques and cellular structures surrounding the plaque. To identify the C4 labeled structures, sections were double labeled with antibodies specific for astrocytes, microglia and oligodendrocytes as well as thioflavine to assess plaque associated C4. C4 was not associated specifically with astrocytes (Figure 3A) or microglia (Figure 3B) alone, but was found in thioflavine positive plaque areas (data not shown) containing microglia and surrounded by astrocytes (Figure 3A and B). Confocal microscopy following double labeling for C4 and CPNase, an oligodendrocyte specific marker, demonstrated cell-associated C4 reactivity colocalized with oligodendrocytes. The C4 labeled oligodendrocytes were predominantly found in the APPQ+/+ animals (Figure 4, top panel), but some immunopositive cells are present in sections from APPQ−/− animals (Figure 4, bottom panel). There was no C4 detected in association with oligodendrocytes in age matched wild type (B6/SJL) controls.
Figure 3. C4 is not associated with astrocytes or microglia.
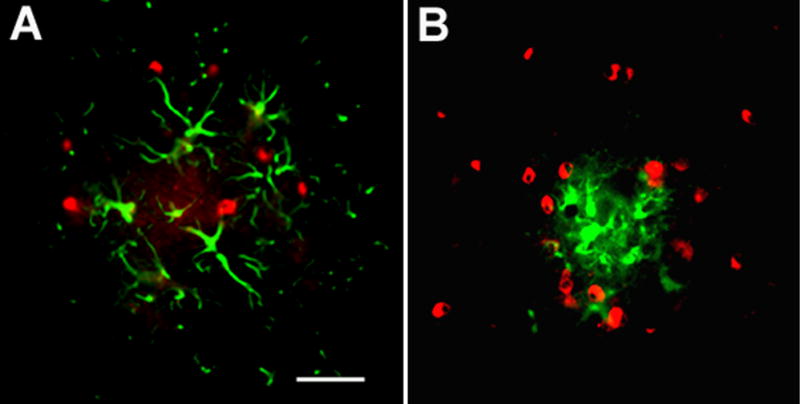
Immunofluorescent double labeling of C4 (red, Ogata anti mouse C4) and either astrocytes (GFAP, green) (A), or microglia (CD45, green) (B) in cortex of 16m APPQ+/+ mice. Scale bar: 25μm.
Figure 4. C4 colocalizes with oligodendrocytes in APP transgenic mouse brain.

Confocal images in brain cortex show colocalization of C4 (Ogata polyclonal anti-mouse C4, red,) with oligodendrocyte cell bodies (anti-CNPase monoclonal, green) in 16mo APPQ+/+ (upper panels). In APPQ−/− (lower panels), C4 immunostaining is lower but also present in oligodendrocyte cell bodies. Right panels are merged confocal images. Scale bar: 25μm.
C3 reactivity is greater in APPQ−/− than in APPQ+/+ mice
We previously reported that while both astrocytic and microglia accumulation and loss of neuronal markers was significantly reduced in APPQ−/− animals, some gliosis and loss of synaptophysin and MAP2 in the CA3 region remained, relative to nontransgenic animals. This observed pathology could be due to either a complement-independent pathway or activation of the alternative pathway of complement by β-amyloid, as reported by Cooper and colleagues in vitro (Bradt et al. 1998). Such alternative pathway activation would be unaltered by the deficiency of C1q. If β-amyloid activation of the alternative pathway was occurring in the APPQ−/− brain, C3 could be covalently linked to the plaques via activation exposed thioester (Bradt et al. 1998). Quantitative image analysis of immunopositive staining with anti-murine C3 showed C3 reactivity in all APP mice starting at 12 mo, but was surprisingly significantly elevated in APPQ−/− relative to APPQ+/+ at 16 mo. Interestingly, this was detected with three distinct anti-C3 monoclonal antibodies (Figure 5A–D), one of which detects full length, native C3 (HBT anti-C3), while anti-C3 monoclonal antibodies 2/16 and 2/11 detects only activated (cleaved) C3 (Mastellos et al. 2004).
Figure 5. C3 immunoreactivity is greater in APPQ−/− than in APPQ+/+.

Anti-C3 staining (HBT) in cortex of APPQ+/+ (left) and APPQ−/− (right) at 12m and 16m shows increased cellular immunoreactivity in APPQ−/− mice. Scale bar: 50 μm. B. Quantification of C3 immunoreactivity in cortex and hippocampus in APPQ+/+ (-□-), APPQ−/− (-▲-) and B6/SJL (-○-) at 6, 9, 12, 16 mo (6m n=3; 9m n=3; 12m n=4; 16m n=4 each genotype) shows significantly higher levels of C3 (anti-C3, HBT) in APPQ−/− than APP at 16 months (* p<0.0 2). Data points are group means ± SD. C. Anti-C3c (Lambris 2/16) staining in 16m APPQ+/+ (left) and APPQ−/− (right) in plaque-like structures. Scale bar: 50 μm D. Image analysis of anti-C3c (2/16) staining. (n=3 animals each genotype; * p < 0.05). Data shown are group means ± SD. E. Western blot of brain extracts from B6/SJL, APPQ+/+ and APPQ−/− at 16 months probed with anti-mouse C3 antibody (Cappel) shows greater reactivity to anti native C3α chain (115,000 Mr) in the APPQ−/− animals.
In APP mice native C3 is upregulated in astrocytes whereas C3 activation cleavage fragment is plaque associated
Morphologically, the staining of these antibodies differed (Figure 5A, 5C) suggesting native and activated C3 associated with different structures. Confocal microscopy imaging of immunofluorescent double labeling demonstrated that native C3 (HBT, Netherlands) was colocalized with the astrocyte marker GFAP (Figure 6A & 6B). In contrast, anti-C3 monoclonal antibodies to activated C3 (2/11 and 2/16) were associated with thioflavine positive plaques (Figure 7 and data not shown). Finally, using a polyclonal anti-C3 (Cappel), Western blot analysis of brain homogenates confirmed the increase in native C3 (C3 α-chain) in APPQ−/− animals relative to the APPQ+/+ (Figure 5E), and further verified the identification of C3 in the brains of these transgenic animals. Thus, the data accordingly suggest both alternative pathway activation by amyloid plaques and an upregulation of C3 synthesis in astrocytes (consistent with reports of others assessing C3 synthesis in cultured astrocytes in response to injury (Levi-Strauss and Mallat 1987;Rus et al. 1992)). This C3 expression appears to be more robust in the classical complement pathway deficient mouse (APPQ−/−) than in the complement sufficient APP mice.
Figure 6. C3 colocalizes with astrocytes in APPQ+/+ and APPQ−/−.
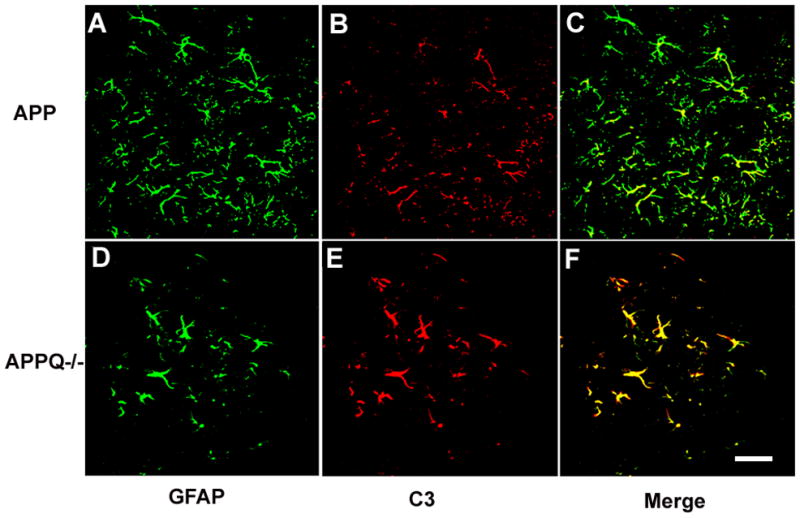
Confocal images of GFAP (green) (A,D) and C3 (red, HBT) (B,E) double labeling in cortex of 16 mo APPQ+/+ (A,B,C) and APPQ−/− (D,E,F) mice. Note fewer astrocytes surrounding plaques in APPQ−/− mice (D) than in APPQ+/+ (A), but the C3 staining appears stronger (E) than in APPQ+/+ (B) Merge of GFAP and C3 (C,F) demonstrates that astrocytes are positive for C3. Scale bar: 25μm
Figure 7. Activated C3 colocalizes with fibrillar amyloid plaques in both APPQ+/+ and APPQ−/− mice.

Representative pictures of 16 mo APPQ+/+ and APPQ−/− sections from cortex that were immunostained with rat anti-mouse C3c (Lambris, 2/11) (red) and with thioflavine (β-sheet structure, green. Scale bar: 25μm.
Discussion
In the C1q-deficient Tg2576 APP transgenic model of amyloid pathology, the decreased level of inflammatory glial cells and the reduced loss of neuronal integrity in CA3 region of the hippocampus (while levels of both total and fibrillar amyloid deposits remain unchanged) suggested a detrimental role for the complement cascade in this neurodegenerative disease (Fonseca et al. 2004). Here we show that while complement activation by the classical pathway was clearly absent in brain of these Tg2576 C1q−/− animals (Figure 1 and 2), fibrillar plaque associated C3b/iC3b, the cleavage product of all three complement activating cascades, was found to be elevated in the classical pathway deficient animals (Figure 5). Since C4 deposition, which could result from either classical or lectin pathway activation, was reduced to baseline levels, this C3 deposition could not be the result of lectin pathway activation. These data are consistent with the activation of the alternative pathway of complement by β-sheet fibrillar amyloid deposits, providing the in vivo correlate of the in vitro Aβ activation of the alternative complement pathway (Bradt et al. 1998;Watson et al. 1997). However, the diminished glial activation and protection of neuronal markers in the C1q-deficent animals indicates that alternative pathway activation does not fully compensate for the lack of classical pathway activation in terms of detrimental consequences. This leads to the question of whether the remaining plaque associated glial activation and/or loss of synaptophysin and MAP2 seen in the C1q−/− Tg2576 mice is due to the activation of the alternative complement pathway and/or to complement-independent events with the activated alternative pathway lacking a detrimental function in this system.
It is important to appreciate that in the absence of C4 deposition (via its thioester), additional “plaque-associated” sites for C3 thioester binding are available and thus the amount of C3 bound to plaques may not quantitatively correlate with the absolute amount of complement activation between these two genotypic models. As a result of this, and given the additional fact that any C3 associated with plaques is long lived due to its covalent nature, deposition of C3 activation fragments may be only loosely correlated with the concentration of C5a, C3a, and/or C5b-9 generated at any given time. Thus, the partial but substantial protection observed in the absence of classical pathway activation (Fonseca et al. 2004) that occurs even when C3 is present on plaques may reflect a difference in the kinetics and/or the extent of complement activation and consequent generation of effector molecules in these models.
While a detrimental role of complement in neurodegeneration has been demonstrated in this and other studies, there is evidence for beneficial effects arising from complement activity which may limit the detrimental responses to neurodegenerative stimuli in certain injury models (Pasinetti et al. 1996;Rus et al. 2005) as reviewed in (Tenner and Pisalyaput 2008). The complement activation products C3a (Van Beek et al. 2001;Boos et al. 2004;Boos et al. 2005) and C5a (Osaka et al. 1999;Mukherjee and Pasinetti 2000;Farkas et al. 2003;van et al. 2003;Sewell et al. 2004;Morgan et al. 2004;Reiman et al. 2005;Woodruff et al. 2006) have been reported to have both protective and detrimental effects in the CNS. Cleavage of C3 in APPQ−/− mice, presumably via alternative pathway activation, and the generation of C3a and C3b may result in neuroprotective activities, thereby contributing to the decreased pathology observed in the APPQ−/− mice in comparison to APP mice. This hypothesis is consistent with the increased pathology observed following inhibition of C3 (Wyss-Coray et al. 2002) by over-expressing Crry, an inhibitor of C3 cleavage and thus also of further downstream complement activation events. These authors suggested a protective role for complement activation, perhaps via the generation of C3b and its opsonizing activity which may contribute to amyloid clearance (Wyss-Coray et al. 2002). If C5a, which is chemotactic for microglia and astrocytes, is generated via the alternative pathway C5 convertase, glia would presumable be recruited into the area of the complement activating fibrillar plaques. However, the influence of C5a in neurodegeneration in AD models is still under debate. Evidence of terminal complement pathway cleavage of C5 and thus generation of C5a and the C5b-9 has not yet been demonstrated in these models. Indeed, the presence of C5a alone in the brain does not necessarily induce or exacerbate inflammation (Reiman et al. 2005). While a direct measure of C5a generation in the brain is problematic (particularly post mortem) as the molecule is short-lived, future investigation of the role of C5a in the progression of disease in these animal models could be approached using C5a receptor antagonists or investigating the effect of a deficiency in either of the C5a receptors (CD88 or the more recently described C5L2).
In another model of neurodegeneration, Fan, et al. demonstrated C1q, C3 and C4 were elevated and localized to areas with fibrillar amyloid deposits in a mouse model of cerebral amyloid angiopathy (Tg-SwDI) (Fan et al. 2007). These results are consistent with the data presented here and with the possibility of a common mechanism of neurotoxicity based on Aβ dependent complement-mediated inflammation and/or neurotoxicity which can result in cognitive loss modeled in each of these systems. This group demonstrated a 2- and 5-fold upregulation of C3 and C1q mRNA, respectively, in the amyloid rich thalamic region of the brains from these transgenics (not in wild type animals). Interestingly, however, in the Fan report, cellular associated C3 immunoreactivity was exclusively localized to microglia (Fan et al. 2007), in contrast to the astrocyte localization in the present study. While it is known that C1q and C3 can be synthesized by neurons, microglia, and astrocytes in the injured brain, the factors involved in the cell and tissue specific regulation of that induced expression are currently an area of active investigation, and ultimately should shed light on the basis for the different apparent cellular sites of synthesis suggested in these studies.
Although C1q plays a central role in the activation of the classical complement cascade, this protein alone has been shown to have multiple effects in disease and homeostasis. For example, it is known that C1q enhances phagocytosis (Botto et al. 1998;Mitchell et al. 1999;Bobak et al. 1987) (and reviewed in (Bohlson et al. 2007)), down regulates the production of proinflammatory molecules in myeloid cells (Fraser et al. 2006;Fraser et al. 2007;Nauta et al. 2004;Yamada et al. 2004), and in vitro has a direct neuroprotective effect on injured neurons (Pisalyaput and Tenner 2008). Neuronal synthesis of C1q has been seen in several injury models (Spielman et al. 2002;Thomas et al. 2000;Fan and Tenner 2005;Rozovsky et al. 1994;Shen et al. 1997) often in the absence of detected induction of other C1 subcomponents C1r and Cls (Veerhuis et al. 1999). These data are consistent with a protective role for C1q in early stages of neuronal injury by mediating the rapid clearance of apoptotic neurons and synaptic debris and/or suppressing the progression of an inflammatory state, hypotheses which are currently under investigation. While clearly more data is needed, if the absence of C1q leads to less modulation (ie higher expression) of proinflammatory gene expression in the brain, the lack of C1q may be the reason that C3 expression in astrocytes is elevated in the APPQ−/− animals.
Upon induction of synthesis of the remaining components of the cascade and the emergence of complement activators (such as fibrillar amyloid plaques in AD (Afagh et al. 1996;Cummings et al. 1996), or amyloid containing drusen in age related macular degeneration (Johnson et al. 2002) or other misfolded or aggregated proteins such as in other dementias (Rostagno et al. 2002)), complement activation could occur and contribute to progression of disease. Indeed, synthesis of most complement factors occurs within the AD brain (Johnson et al. 1992;Shen et al. 1997), and also in “normal” aged brain to a lesser extent (Walker and McGeer 1992). Receptors for complement activation products C5a and C3a are on neurons as well as on microglia and astrocytes [reviewed in (Nataf et al. 1999)], and signature markers (C1q, C4b, C3b/iC3b, C5b-9) indicate that complement activation does occur in the AD brain (Eikelenboom and Stam 1982;Webster et al. 1997;Loeffler et al. 2008) (and data presented here). The consequences of this activation are the generation of C3b, which may facilitate phagocytosis or result in bystander damage due to release of toxic enzymes and oxidative radicals from activated phagocytes [“frustrated phagocytosis”] (Gardiner et al. 1994). C5a is also generated, resulting in recruitment of phagocytic cells to the plaque area and these cells may then be activated by plaque components. Finally, generation of the C5b-9 complex can occur and cause cell lysis if present in high enough concentration or if the expression of the host membrane inhibitor, CD59, is decreased (Yang et al. 2000). However, sublytic C5b-9 has also been shown to be neuroprotective, perhaps via its anti apoptotic effects on oligodendrocytes (Rus et al. 2006). The importance of the balance between activation of complement and the effective regulation of the cascade in neurodegenerative disorders has been most recently evident in the association of specific single nucleotide polymorphisms in complement components C3, C2, Factor B, and Factor H, the latter a complement regulator protein, which together have been shown to contribute >80% of the risk of developing aged related macular degeneration (Hageman et al. 2005;Gold et al. 2006;Maller et al. 2007). Clearly, both the nature of the activators and the local environment also influence development of disease, as these degenerative processes are not seen in all tissues and/or ages.
While the kinetics of all these processes may differ in genetically diverse individuals as well as in the different genetic backgrounds of model mouse strains, the variety of contributing factors may also provide multiple and/or specific targets for regulation of these disease processes both in AD and other neurodegenerative diseases. Given the number of activities as well as regulators of the complement effector system, much more is to be learned of the balance/imbalance that influences health and disease. An recent example of novel functions for these proteins is the interesting data reported by Stevens and colleagues that demonstrates a critical role for C1q and C3 in proper synapse pruning during development but a possible detrimental role in glaucoma (Stevens et al. 2007).
Therapeutic interventions in murine models of Alzheimer’s Disease that target inflammation and oxidative stress have proven successful in reducing amyloid plaque burden/pathology as well as the proinflammatory cytokine IL-1β and indicators of oxidative stress (Yao et al. 2004). If complement activation contributes significantly to the inflammation and subsequent loss of cognitive function, then a specific inhibitor of the C1q interaction site on the activator would be valuable as it would block the pathogenic activation but should not affect the systemic protective functions of the complement system. However, if the early complement activation products (C3b, C3a) have beneficial consequences (such as enhanced clearance and promotion of neurogenesis (Rahpeymai et al. 2006)), an inhibitor of the complement system downstream of these events (such as C5a generation or interaction with cellular receptors) or the upregulation of the beneficial activities, would be optimal (Figure 8). Such therapeutic interventions should be particularly beneficial for individuals entering what has been called the “catastrophic” phase of AD, after cognitive dysfunction becomes clinically apparent, the rate of decline appears to accelerate, and neuritic plaques are observable upon autopsy (Cotman et al. 1996). Although multiple potential targets exist for therapeutic intervention in AD, including immunization to prevent accumulation and/or promote clearance of Aβ (Moore and O’Banion 2002;Zhou et al. 2005;Lemere et al. 2003;Selkoe and Schenk 2003) and/or inhibition of oligomeric amyloid formation, multi point interventions will likely be beneficial at different stages of the disease, and thus a cocktail of therapeutic reagents may be more successful in delaying CNS degeneration and the cognitive impairment characteristic of AD.
Figure 8. Schematic diagram of potential therapeutic drug targeting.
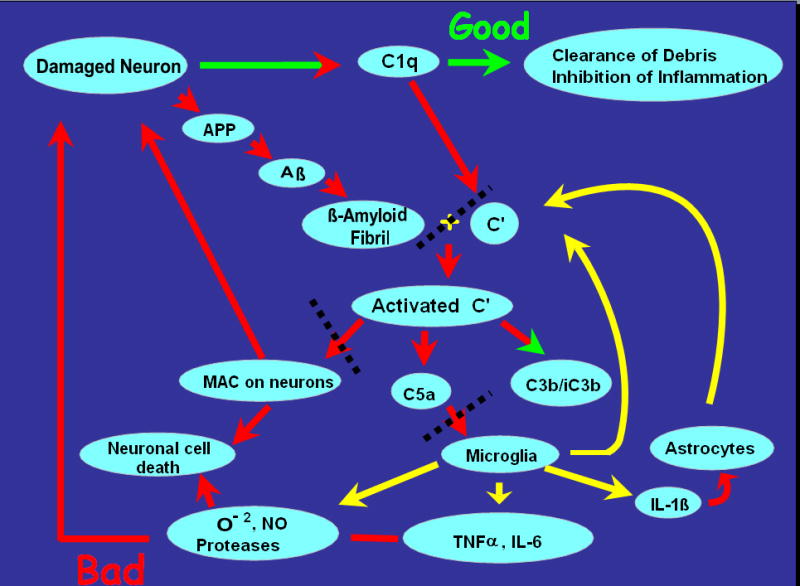
Fibrillar Aβ activates the classical (and alternative) pathway of complement, resulting in C3b/iC3b deposition on plaques, glial recruitment and generation of neurotoxic molecules, and potential lysis by the Membrane attack complex (MAC/C5b-9) of complement. The red and green arrows signify pathways that are probably detrimental and protective respectively. The yellow arrows represent events that are suggested to occur in response to complement activation in vivo but have yet to be definitively proven. The black dotted lines indicate potential targets for intervention/inhibition of detrimental pathways. Modified from (Tenner 2001).
Acknowledgments
This work is supported by NIH NS 35144, AG 00538 and NIH Training Grants AG00096-21 (K.P.). The authors also thank Drs. Karen Hsiao-Ashe (University of Minnesota, Minneapolis), Karen Duff (New York University, NY) and Marina Botto (Imperial College, London) for the Tg2576, APPPS1 and C1q−/− mice, respectively, used to construct the AD models lacking the classical pathway (APP Q−/−), Dr. Franz Petry (Mainz, Germany) for the anti-mouse C1q antibody; Dr. John Lambris (University of Pennsylvania, Philadelphia) for his generous gifts of rat anti-mouse C3 antibodies, and Dr. Ron Ogata (Torrey Pines Medical Institute, La Jolla, CA) for polyclonal anti-mouse C4 antibody. We also thank Irma Hernandez, Jennifer Chen, Xiomara Fernandez and Ozkan Yazan for their assistance with immunohistochemistry staining.
Reference List
- Afagh A, Cummings BJ, Cribbs DH, Cotman CW, Tenner AJ. Localization and cell association of C1q in Alzheimer’s disease brain. Exp Neurol. 1996;138:22–32. doi: 10.1006/exnr.1996.0043. [DOI] [PubMed] [Google Scholar]
- Anderson DC, Miller LJ, Schmalstieg FC, Rothlein R, Springer TA. Contributions of the Mac-1 glycoprotein family to adherence-dependent granulocyte functions: Structure-function assessments employing subunit-specific monoclonal antibodies. J Immunol. 1986;137:15–27. [PubMed] [Google Scholar]
- Bergamaschini L, Canziani S, Bottasso B, Cugno M, Braidotti P, Agostoni A. Alzheimer’s beta-amyloid peptides can activate the early components of complement classical pathway in a C1q-independent manner. Clin exp Immunol. 1999;115:526–533. doi: 10.1046/j.1365-2249.1999.00835.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobak DA, Gaither TG, Frank MM, Tenner AJ. Modulation of FcR Function by Complement: Subcomponent C1q enhances the phagocytosis of IgG-opsonized targets by human monocytes and culture-derived macrophages. J Immunol. 1987;138:1150–1156. [PubMed] [Google Scholar]
- Bohlson SS, Fraser DA, Tenner AJ. Complement proteins C1q and MBL are pattern recognition molecules that signal immediate and long-term protective immune functions. Mol Immunol. 2007;44:33–43. doi: 10.1016/j.molimm.2006.06.021. [DOI] [PubMed] [Google Scholar]
- Boos L, Campbell IL, Ames R, Wetsel RA, Barnum SR. Deletion of the complement anaphylatoxin c3a receptor attenuates, whereas ectopic expression of c3a in the brain exacerbates, experimental autoimmune encephalomyelitis. J Immunol. 2004;173:4708–4714. doi: 10.4049/jimmunol.173.7.4708. [DOI] [PubMed] [Google Scholar]
- Boos L, Szalai AJ, Barnum SR. C3a expressed in the central nervous system protects against LPS-induced shock. Neurosci Lett. 2005;387:68–71. doi: 10.1016/j.neulet.2005.07.015. [DOI] [PubMed] [Google Scholar]
- Botto M, Dell’agnola C, Bygrave AE, Thompson EM, Cook HT, Petry F, Loos M, Pandolfi PP, Walport MJ. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat Genet. 1998;19:56–59. doi: 10.1038/ng0598-56. [DOI] [PubMed] [Google Scholar]
- Bradt BM, Kolb WP, Cooper NR. Complement-dependent proinflammatory properties of the Alzheimer’s disease β-peptide. J Exp Med. 1998;188:431–438. doi: 10.1084/jem.188.3.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer Y, Palmer A, Taube D, Welsh K, Bewick M, Bindon C, Hale G, Waldmann H, Dische F, Parsons V. Effect of graft perfusion with two CD45 monoclonal antibodies on incidence of kidney allograft rejection. Lancet. 1989;2:935–937. doi: 10.1016/s0140-6736(89)90951-3. [DOI] [PubMed] [Google Scholar]
- Cooper NR, Kalaria RN, McGeer PL, Rogers J. Key issues in Alzheimer’s disease inflammation. Neurobiol Aging. 2000;21:451–453. doi: 10.1016/s0197-4580(00)00148-2. [DOI] [PubMed] [Google Scholar]
- Cotman CW, Tenner AJ, Cummings BJ. β-Amyloid converts an acute phase injury response to chronic injury responses. Neurobiol Aging. 1996;17:723–731. doi: 10.1016/0197-4580(96)00117-0. [DOI] [PubMed] [Google Scholar]
- Cummings BJ, Head E, Afagh AJ, Milgram NW, Cotman CW. Beta- amyloid accumulation correlates with cognitive dysfunction in the aged canine. Neurobiology of Learning and Memory. 1996;66:11–23. doi: 10.1006/nlme.1996.0039. [DOI] [PubMed] [Google Scholar]
- Ehtesham M, Kabos P, Kabosova A, Neuman T, Black KL, Yu JS. The use of interleukin 12-secreting neural stem cells for the treatment of intracranial glioma. Cancer Res. 2002;62:5657–5663. [PubMed] [Google Scholar]
- Eikelenboom P, Stam FC. An immunohistochemical study on cerebral vascular and senile plaque amyloid in Alzheimer’s dementia. Virchows Arch B Cell Pathol Incl Mol Pathol. 1984;47:17–25. doi: 10.1007/BF02890185. [DOI] [PubMed] [Google Scholar]
- Eikelenboom P, Stam FC. Immunoglobulins and complement factors in senile plaques. Acta Neuropathol. 1982;57:239–242. doi: 10.1007/BF00685397. [DOI] [PubMed] [Google Scholar]
- Eikelenboom P, Veerhuis R. The role of complement and activated microglia in the pathogenesis of Alzheimer’s disease. Neurobiol Aging. 1996;17:673–680. doi: 10.1016/0197-4580(96)00108-x. [DOI] [PubMed] [Google Scholar]
- Fan R, DeFilippis K, Van Nostrand WE. Induction of complement proteins in a mouse model for cerebral microvascular A beta deposition. J Neuroinflammation. 2007;4:22. doi: 10.1186/1742-2094-4-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan R, Tenner AJ. Differential regulation of Abeta42-induced neuronal C1q synthesis and microglial activation. J Neuroinflammation. 2005;2:1. doi: 10.1186/1742-2094-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkas I, Takahashi M, Fukuda A, Yamamoto N, Akatsu H, Baranyi L, Tateyama H, Yamamoto T, Okada N, Okada H. Complement C5a receptor-mediated signaling may be involved in neurodegeneration in Alzheimer’s disease. J Immunol. 2003;170:5764–5771. doi: 10.4049/jimmunol.170.11.5764. [DOI] [PubMed] [Google Scholar]
- Fonseca MI, Zhou J, Botto M, Tenner AJ. Absence of C1q leads to less neuropathology in transgenic mouse models of Alzheimer’s disease. J Neurosci. 2004;24:6457–6465. doi: 10.1523/JNEUROSCI.0901-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser DA, Arora M, Bohlson SS, Lozano E, Tenner AJ. Generation of inhibitory NF{kappa}B complexes and phosphorylated cAMP response element-binding protein correlates with the activity of complement protein C1q in human monocytes. J Biol Chem. 2007;282:7360–7367. doi: 10.1074/jbc.M605741200. [DOI] [PubMed] [Google Scholar]
- Fraser DA, Bohlson SS, Jasinskiene N, Rawal N, Palmarini G, Ruiz S, Rochford R, Tenner AJ. C1q and MBL, components of the innate immune system, influence monocyte cytokine expression. J Leukoc Biol. 2006;80:107–116. doi: 10.1189/jlb.1105683. [DOI] [PubMed] [Google Scholar]
- Gardiner EE, Mok SS, Sriratana A, Robinson HC, Veitch BJA, Lowther DA, Handley CJ. Polymorphonuclear neutrophils release 35S-labelled proteoglycans into cartilage during frustrated phagocytosis. Eur J Biochem. 1994;221:871–879. doi: 10.1111/j.1432-1033.1994.tb18802.x. [DOI] [PubMed] [Google Scholar]
- Gold B, Merriam JE, Zernant J, Hancox LS, Taiber AJ, Gehrs K, Cramer K, Neel J, Bergeron J, Barile GR, Smith RT, Hageman GS, Dean M, Allikmets R, Chang S, Yannuzzi LA, Merriam JC, Barbazetto I, Lerner LE, Russell S, Hoballah J, Hageman J, Stockman H. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet. 2006;38:458–462. doi: 10.1038/ng1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hageman GS, Anderson DH, Johnson LV, Hancox LS, Taiber AJ, Hardisty LI, Hageman JL, Stockman HA, Borchardt JD, Gehrs KM, Smith RJ, Silvestri G, Russell SR, Klaver CC, Barbazetto I, Chang S, Yannuzzi LA, Barile GR, Merriam JC, Smith RT, Olsh AK, Bergeron J, Zernant J, Merriam JE, Gold B, Dean M, Allikmets R. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U S A. 2005;102:7227–7232. doi: 10.1073/pnas.0501536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, O’Campo K, Hardy J, Prada CM, Eckman C, Younkin S, Hsiao K, Duff K. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med. 1998;4:97–100. doi: 10.1038/nm0198-097. [DOI] [PubMed] [Google Scholar]
- Hsiao KK, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Aβ elevations, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Huang J, Kim LJ, Mealey R, Marsh JrHC, Zhang Y, Tenner AJ, Connolly ES, Pinsky DJ., Jr Neuronal protection in stroke by an sLex-glycosylated complement inhibitory protein. Science. 1999;285:595–599. doi: 10.1126/science.285.5427.595. [DOI] [PubMed] [Google Scholar]
- Jiang H, Burdick D, Glabe CG, Cotman CW, Tenner AJ. β-amyloid activates complement by binding to a specific region of the collagen-like domain of the C1q A chain. J Immunol. 1994;152:5050–5059. [PubMed] [Google Scholar]
- Johnson LV, Leitner WP, Rivest AJ, Staples MK, Radeke MJ, Anderson DH. The Alzheimer’s A{beta}-peptide is deposited at sites of complement activation in pathologic deposits associated with aging and age-related macular degeneration. Proc Natl Acad Sci USA. 2002 doi: 10.1073/pnas.192203399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SA, Lampert-Etchells M, Pasinetti GM, Rozovsky I, Finch C. Complement mRNA in the mammalian brain: Responses to Alzheimer’s disease and experimental brain lesioning. Neurobiol Aging. 1992;13:641–648. doi: 10.1016/0197-4580(92)90086-d. [DOI] [PubMed] [Google Scholar]
- Jung SS, Gauthier S, Cashman NR. Beta-amyloid precursor protein is detectable on monocytes and is increased in Alzheimer’s disease. Neurobiol Aging. 1999;20:249–257. doi: 10.1016/s0197-4580(99)00051-2. [DOI] [PubMed] [Google Scholar]
- Kremmer E, Thierfelder S, Felber E, Hoffmann-Fezer G, Wasiliu M. Monoclonal antibodies to complement components without the need of their prior purification. II Antibodies to mouse C3 and C4. Hybridoma. 1990;9:309–317. doi: 10.1089/hyb.1990.9.309. [DOI] [PubMed] [Google Scholar]
- Lemere CA, Spooner ET, Leverone JF, Mori C, Iglesias M, Bloom JK, Seabrook TJ. Amyloid-beta immunization in Alzheimer’s disease transgenic mouse models and wildtype mice. Neurochem Res. 2003;28:1017–1027. doi: 10.1023/a:1023203122036. [DOI] [PubMed] [Google Scholar]
- Levi-Strauss M, Mallat M. Primary cultures of murine astrocytes produce C3 and Factor B, two components of the alternative pathway of complement activation. J Immunol. 1987;139:2361–2366. [PubMed] [Google Scholar]
- Loeffler DA, Camp DM, Bennett DA. Plaque complement activation and cognitive loss in Alzheimer’s disease. J Neuroinflammation. 2008;5:9. doi: 10.1186/1742-2094-5-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maller JB, Fagerness JA, Reynolds RC, Neale BM, Daly MJ, Seddon JM. Variation in complement factor 3 is associated with risk of age-related macular degeneration. Nat Genet. 2007;39:1200–1201. doi: 10.1038/ng2131. [DOI] [PubMed] [Google Scholar]
- Mastellos D, Prechl J, Laszlo G, Papp K, Olah E, Argyropoulos E, Franchini S, Tudoran R, Markiewski M, Lambris JD, Erdei A. Novel monoclonal antibodies against mouse C3 interfering with complement activation: description of fine specificity and applications to various immunoassays. Mol Immunol. 2004;40:1213–1221. doi: 10.1016/j.molimm.2003.10.019. [DOI] [PubMed] [Google Scholar]
- Matsuoka Y, Picciano M, Malester B, LaFrancois J, Zehr C, Daeschner JM, Olschowka JA, Fonseca MI, O’Banion MK, Tenner AJ, Lemere CA, Duff K. Inflammatory responses to amyloidosis in a transgenic mouse model of Alzheimer’s disease. Am J Pathol. 2001;158:1345–1354. doi: 10.1016/S0002-9440(10)64085-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell DA, Taylor PR, Cook HT, Moss J, Bygrave AE, Walport MJ, Botto M. C1q protects against the development of glomerulonephritis independently of C3 activation. J Immunol. 1999;162:5676–5679. [PubMed] [Google Scholar]
- Moore AH, O’Banion MK. Neuroinflammation and anti-inflammatory therapy for Alzheimer’s disease. Adv Drug Deliv Rev. 2002;54:1627–1656. doi: 10.1016/s0169-409x(02)00162-x. [DOI] [PubMed] [Google Scholar]
- Morgan BP, Griffiths M, Khanom H, Taylor SM, Neal JW. Blockade of the C5a receptor fails to protect against experimental autoimmune encephalomyelitis in rats. Clin Exp Immunol. 2004;138:430–438. doi: 10.1111/j.1365-2249.2004.02646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee P, Pasinetti GM. The role of complement anaphylatoxin C5a in neurodegeneration: implications in Alzheimer’s disease. J Neuroimmunol. 2000;105:124–130. doi: 10.1016/s0165-5728(99)00261-1. [DOI] [PubMed] [Google Scholar]
- Nataf S, Stahel PF, Davoust N, Barnum SR. Complement anaphylatoxin receptors on neurons: new tricks for old receptors? TINS. 1999;22:397–402. doi: 10.1016/s0166-2236(98)01390-3. [DOI] [PubMed] [Google Scholar]
- Nauta AJ, Castellano G, Xu W, Woltman AM, Borrias MC, Daha MR, van Kooten C, Roos A. Opsonization with C1q and mannose-binding lectin targets apoptotic cells to dendritic cells. J Immunol. 2004;173:3044–3050. doi: 10.4049/jimmunol.173.5.3044. [DOI] [PubMed] [Google Scholar]
- Ogata RT, Low PJ, Bradt BM, Cooper NR. Substrate specificities of murine C1s. J Immunol. 1994;152:5890–5895. [PubMed] [Google Scholar]
- Osaka H, Mukherjee P, Aisen PS, Pasinetti GM. Complement-derived anaphylatoxin C5a protects against glutamate- mediated neurotoxicity. J Cell Biochem. 1999;73:303–311. [PubMed] [Google Scholar]
- Pasinetti GM, Tocco G, Sakhi S, Musleh WD, DeSimoni MG, Mascarucci P, Schreiber S, Baudry M, Finch CE. Hereditary deficiencies in complement C5 are associated with intensified neurodegenerative responses that implicate new roles for the C-system in neuronal and astrocytic functions. Neurobiol Dis. 1996;3:197–204. doi: 10.1006/nbdi.1996.0020. [DOI] [PubMed] [Google Scholar]
- Pisalyaput K, Tenner AJ. Complement component C1q inhibits beta-amyloid- and serum amyloid P-induced neurotoxicity via caspase- and calpain-independent mechanisms. J Neurochem. 2008;104:696–707. doi: 10.1111/j.1471-4159.2007.05012.x. [DOI] [PubMed] [Google Scholar]
- Rahpeymai Y, Hietala MA, Wilhelmsson U, Fotheringham A, Davies I, Nilsson AK, Zwirner J, Wetsel RA, Gerard C, Pekny M, Pekna M. Complement: a novel factor in basal and ischemia-induced neurogenesis. Embo J. 2006;25:1364–1374. doi: 10.1038/sj.emboj.7601004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiman R, Torres AC, Martin BK, Ting JP, Campbell IL, Barnum SR. Expression of C5a in the brain does not exacerbate experimental autoimmune encephalomyelitis. Neurosci Lett. 2005;390:134–138. doi: 10.1016/j.neulet.2005.08.022. [DOI] [PubMed] [Google Scholar]
- Rogers J, Cooper NR, Webster S, Schultz J, McGeer PL, Styren SD, Civin WH, Brachova L, Bradt B, Ward P, Lieberburg I. Complement activation by beta-amyloid in Alzheimer disease. Proc Natl Acad Sci. 1992;89:10016–10020. doi: 10.1073/pnas.89.21.10016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostagno A, Revesz T, Lashley T, Tomidokoro Y, Magnotti L, Braendgaard H, Plant G, Bojsen-Moller M, Holton J, Frangione B, Ghiso J. Complement activation in chromosome 13 dementias. Similarities with Alzheimer’s disease. J Biol Chem. 2002;277:49782–49790. doi: 10.1074/jbc.M206448200. [DOI] [PubMed] [Google Scholar]
- Rozovsky I, Morgan TE, Willoughby DA, Dugich-Djordjevich MM, Pasinetti GM, Johnson SA, Finch CE. Selective expression of Clusterin (SGP-2) and complement C1qB and C4 during responses to neurotoxins in vivo and in vitro. Neuroscience. 1994;62:741–758. doi: 10.1016/0306-4522(94)90473-1. [DOI] [PubMed] [Google Scholar]
- Rus H, Cudrici C, Niculescu F. C5b-9 complement complex in autoimmune demyelination and multiple sclerosis: dual role in neuroinflammation and neuroprotection. Ann Med. 2005;37:97–104. doi: 10.1080/07853890510007278. [DOI] [PubMed] [Google Scholar]
- Rus H, Cudrici C, Niculescu F, Shin ML. Complement activation in autoimmune demyelination: dual role in neuroinflammation and neuroprotection. J Neuroimmunol. 2006;180:9–16. doi: 10.1016/j.jneuroim.2006.07.009. [DOI] [PubMed] [Google Scholar]
- Rus HG, Kim LM, Niculescu FI, Shin ML. Induction of C3 expression in astrocytes is regulated by cytokines and Newcastle disease virus. J Immunol. 1992;148:928–933. [PubMed] [Google Scholar]
- Selkoe DJ, Schenk D. Alzheimer’s disease: molecular understanding predicts amyloid-based therapeutics. Annu Rev Pharmacol Toxicol. 2003;43:545–584. doi: 10.1146/annurev.pharmtox.43.100901.140248. [DOI] [PubMed] [Google Scholar]
- Sewell DL, Nacewicz B, Liu F, Macvilay S, Erdei A, Lambris JD, Sandor M, Fabry Z. Complement C3 and C5 play critical roles in traumatic brain cryoinjury: blocking effects on neutrophil extravasation by C5a receptor antagonist. J Neuroimmunol. 2004;155:55–63. doi: 10.1016/j.jneuroim.2004.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Li R, McGeer EG, McGeer PL. Neuronal expression of mRNAs for complement proteins of the classical pathway in Alzheimer brain. Brain Res. 1997;769:391–395. doi: 10.1016/s0006-8993(97)00850-0. [DOI] [PubMed] [Google Scholar]
- Spielman L, Winger D, Ho L, Aisen PS, Shohami E, Pasinetti M. Induction of the complement component C1qB in brain of transgenic mice with neuronal overexpression of human cyclooxygenase-2. Acta Neuropathol (Berl) 2002;103:157–162. doi: 10.1007/s004010100447. [DOI] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- Stoltzner SE, Grenfell TJ, Mori C, Wisniewski KE, Wisniewski TM, Selkoe DJ, Lemere CA. Temporal accrual of complement proteins in amyloid plaques in Down’s syndrome with Alzheimer’s disease. Am J Pathol. 2000;156:489–499. doi: 10.1016/S0002-9440(10)64753-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strohmeyer R, Shen Y, Rogers J. Detection of complement alternative pathway mRNA and proteins in the Alzheimer’s disease brain. Brain Res Mol Brain Res. 2000;81:7–18. doi: 10.1016/s0169-328x(00)00149-2. [DOI] [PubMed] [Google Scholar]
- Tenner AJ. Complement in Alzheimer’s disease: opportunities for modulating protective and pathogenic events. Neurobiol Aging. 2001;22:849–861. doi: 10.1016/s0197-4580(01)00301-3. [DOI] [PubMed] [Google Scholar]
- Tenner AJ, Pisalyaput K. The Complement System in the CNS: Thinking again. In: Lane TE, Carson MJ, Bergmann C, Wyss-Coray T, editors. Central Nervous System Diseases and Inflammation. Springer; New York: 2008. pp. 153–174. [Google Scholar]
- Thomas A, Gasque P, Vaudry D, Gonzalez B, Fontaine M. Expression of a complete and functional complement system by human neuronal cells in vitro. Int Immunol. 2000;12:1015–1023. doi: 10.1093/intimm/12.7.1015. [DOI] [PubMed] [Google Scholar]
- Van Beek J, Elward K, Gasque P. Activation of complement in the central nervous system: roles in neurodegeneration and neuroprotection. Ann N Y Acad Sci. 2003;992:56–71. doi: 10.1111/j.1749-6632.2003.tb03138.x. [DOI] [PubMed] [Google Scholar]
- Van Beek J, Nicole O, Ali C, Ischenko A, MacKenzie ET, Buisson A, Fontaine M. Complement anaphylatoxin C3a is selectively protective against NMDA-induced neuronal cell death. NeuroReport. 2001;12:289–293. doi: 10.1097/00001756-200102120-00022. [DOI] [PubMed] [Google Scholar]
- van BJ, Elward K, Gasque P. Activation of complement in the central nervous system: roles in neurodegeneration and neuroprotection. Ann N Y Acad Sci. 2003;992:56–71. doi: 10.1111/j.1749-6632.2003.tb03138.x. [DOI] [PubMed] [Google Scholar]
- Veerhuis R, Janssen I, De Groot CJ, Van Muiswinkel FL, Hack CE, Eikelenboom P. Cytokines associated with amyloid plaques in Alzheimer’s disease brain stimulate human glial and neuronal cell cultures to secrete early complement proteins, but not C1-inhibitor. Exp Neurol. 1999;160:289–299. doi: 10.1006/exnr.1999.7199. [DOI] [PubMed] [Google Scholar]
- Viale G, Gambacorta M, Coggi G, Dell’Orto P, Milani M, Doglioni C. Glial fibrillary acidic protein immunoreactivity in normal and diseased human breast. Virchows Arch A Pathol Anat Histopathol. 1991;418:339–348. doi: 10.1007/BF01600164. [DOI] [PubMed] [Google Scholar]
- Walker DG, McGeer PL. Complement gene expression in human brain: comparison between normal and Alzheimer disease cases. Brain Res Mol Brain Res. 1992;14:109–116. doi: 10.1016/0169-328x(92)90017-6. [DOI] [PubMed] [Google Scholar]
- Watson MD, Roher AE, Kim KS, Spiegel K, Emmerling MR. Complement interactions with amyloid-β1–42: a nidus for inflammation in AD brains. Amyloid: Int J Exp Clin Invest. 1997;4:147–156. [Google Scholar]
- Webster S, Lue LF, Brachova L, Tenner AJ, McGeer PL, Terai K, Walker DG, Bradt B, Cooper NR, Rogers J. Molecular and cellular characterization of the membrane attack complex, C5b-9, in Alzheimer’s disease. Neurobiol Aging. 1997;18:415–421. doi: 10.1016/s0197-4580(97)00042-0. [DOI] [PubMed] [Google Scholar]
- Woodruff TM, Crane JW, Proctor LM, Buller KM, Shek AB, de VK, Pollitt S, Williams HM, Shiels IA, Monk PN, Taylor SM. Therapeutic activity of C5a receptor antagonists in a rat model of neurodegeneration. FASEB J. 2006;20:1407–1417. doi: 10.1096/fj.05-5814com. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Yan F, Lin AH, Lambris JD, Alexander JJ, Quigg RJ, Masliah E. Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer’s mice. Proc Natl Acad Sci U S A. 2002;99:10837–10842. doi: 10.1073/pnas.162350199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M, Oritani K, Kaisho T, Ishikawa J, Yoshida H, Takahashi I, Kawamoto S, Ishida N, Ujiie H, Masaie H, Botto M, Tomiyama Y, Matsuzawa Y. Complement C1q regulates LPS-induced cytokine production in bone marrow-derived dendritic cells. Eur J Immunol. 2004;34:221–230. doi: 10.1002/eji.200324026. [DOI] [PubMed] [Google Scholar]
- Yang LB, Li R, Meri S, Rogers J, Shen Y. Deficiency of complement defense protein CD59 may contribute to neurodegeneration in Alzheimer’s disease. J Neurosci. 2000;20:7505–7509. doi: 10.1523/JNEUROSCI.20-20-07505.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Chinnici C, Tang H, Trojanowski JQ, Lee VM, Pratico D. Brain inflammation and oxidative stress in a transgenic mouse model of Alzheimer-like brain amyloidosis. J Neuroinflammation. 2004;1:21. doi: 10.1186/1742-2094-1-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Fonseca MI, Kayed R, Hernandez I, Webster SD, Yazan O, Cribbs DH, Glabe CG, Tenner AJ. Novel Abeta peptide immunogens modulate plaque pathology and inflammation in a murine model of Alzheimer’s disease. J Neuroinflammation. 2005;2:28. doi: 10.1186/1742-2094-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]