

Abstract
We have defined some of the mechanisms by which the kinase inhibitor Lapatinib kills HCT116 cells. Lapatinib inhibited radiation-induced activation of ERBB1/2, ERK1/2 and AKT, and radiosensitized HCT116 cells. Prolonged incubation of HCT116 cells with Lapatinib caused cell killing followed by outgrowth of Lapatinib adapted cells. Adapted cells were resistant to serum-starvation –induced cell killing and were cross resistant to multiple therapeutic drugs. Lapatinib was competent to inhibit basal and EGF-stimulated ERBB1 phosphorylation in adapted cells. Co-expression of dominant negative ERBB1 and dominant negative ERBB2 inhibited basal and EGF-stimulated ERBB1 and ERBB2 phosphorylation in parental cells. However in neither parental nor adapted cells did expression of dominant negative ERBB1 and dominant negative ERBB2 recapitulate the cell death promoting effects of Lapatinib. Adapted cells had increased expression of MCL-1, decreased expression of BAX, and decreased activation of BAX and BAK. Over-expression of BCL-XL protected parental cells from Lapatinib toxicity. Knock down of MCL-1 expression enhanced Lapatinib toxicity in adapted cells that was reverted by knock down of BAK expression. Inhibition of caspase function modestly reduced Lapatinib toxicity in parental cells whereas knock down of AIF expression suppressed Lapatinib toxicity. Thus in HCT116 cells Lapatinib adaptation can be mediated by altered expression of pro- and anti-apoptotic proteins that maintain mitochondrial function.
Keywords: Lapatinib, Ras, cell death
Introduction
The ERBB receptor family consists of four members; ERBB1, ERBB2, ERBB3 and ERBB4 (Olayioye et al, 2000; Yarden et al, 2001). These receptors are present on the cell surface as monomers and upon binding ligand, can homo- or hetero-dimerize and autophosphorylate specific tyrosine residues on the cytosolic domain of the receptors which then serve as docking sites for molecules which can mediate downstream intracellular signaling (Hynes et al, 2005; Lin et al, 2004; Nelson et al, 2004). Many cancer types have been shown to have deranged ERBB receptor signaling via various mechanisms such as constitutive receptor activation, impaired receptor down-regulation and increased receptor stimulation via an autocrine loop leading to constitutive downstream pro-survival signaling resulting in aggressive tumors (Hynes et al, 2005; Peschard et al, 2003; Sizeland et al, 1992; Sunpaweravong et al, 2005; Salomon et al, 1995). Hence, studies have been performed to identify mechanisms via which ERBB receptors and their downstream signaling pathways can be inhibited in such tumor cells. Clinically used techniques to inhibit receptor activation include (i) monoclonal antibodies that prevent paracrine ligand binding to the receptors, (ii) quinazoline-derived small molecule tyrosine kinase inhibitors (TKIs) that prevent ATP binding in the kinase domain of the receptors thereby preventing the receptor kinase activity required to activate downstream signaling and (iii) siRNA –mediated knock down of ERBB receptor expression (Imai and Takaoka, 2006; Schmidt-Ullrich et al, 2003).
Lapatinib is a reversible small molecule TKI developed by GlaxoSmithKline that inhibits ERBB1 and ERBB2 activation. The IC50 values for purified ERBB1 and EBB2 inhibition by Lapatinib are 10.2 and 9.8 nM, respectively (Wood et al, 2004; Rusnak et al, 2001; Xia et al, 2006; Xia et al., 2007). Lapatinib showed a >300 fold selectivity for ERBB1 and ERBB2 inhibition when compared to its ability to inhibit a panel of other kinases commonly found in cells (Rusnak et al, 2001). Lapatinib has been studied in Herceptin / Trastuzumab resistant breast cancer lines and induces apoptosis in such cells by inhibiting downstream pro-survival signaling mediated by ERBB1, ERBB2 and also Insulin like growth factor –1 receptor (IGF-1R) (Nahta et al, 2007; Konecny et al, 2006). Combination studies involving Lapatinib and BCL-2 inhibitors in certain cancer cells show synergistic anti-tumor effects (Reed et al, 1996; Witters et al, 2007). Lapatinib (Tykerb) was recently approved by the Food and Drug Administration to be used in combination with capecitabine to treat patients with advanced or metastatic breast cancer that over-express ERBB2 and have been previously treated with other drugs (http://www.cancer.gov/cancertopics/druginfo/fda-lapatinib).
It is generally accepted that cancer patients treated with chemotherapeutic agents usually respond well to the treatment initially resulting in the reduction of tumor size and death of cancer cells. However, months or years later, the cancer can reappear as an aggressive and therapeutically refractory malignancy, that may also be cross-resistant to many other therapeutic drug treatments making such cancers very difficult to manage (Kobayashi et al, 2005). Resistance to Trastuzumab has been suggested to be mediated via the Insulin like growth factor –1 receptor (IGF-1R) signaling that can activate downstream pro-survival pathways (Lu et al, 2001; Camirand et al, 2002). Src and the estrogen receptor have also been implicated in mediating resistance to tyrosine kinase inhibitors, including Lapatinib, by activating and/or re-activating pro-survival signaling pathways (Qin et al, 2006a; Xia et al, 2006). Resistance to ERBB targeted drugs can also occur due to mutations in ERBB receptor kinase domains resulting in the inability of the drug to inhibit the receptor kinase domain or ligand-independent constitutive activation of the receptor (Pao et al, 2005; Sok et al, 2006). Multi-drug resistance pumps, over-expression of anti-apoptotic molecules belonging to the BCL-2 family and constitutive activity of NFκB have also been linked to drug resistance in a wide variety of cancer cell types (Szakacs et al, 2006; Raffo et al, 1995; Baldwin et al, 2001; Sumitomo et al, 1999; Cabannes et al, 1999). Single agent Lapatinib activity has not been noted in the treatment of colon cancer, although various colon cancer cell lines have been reported to express ERBB1 and ERBB2 receptors and Lapatinib can cause apoptosis and inhibition of cell proliferation in vitro and in vivo in colon cancer cell lines (Zhou et al, 2006; Cunningham et al, 2006).
Ionizing radiation is used as a primary treatment for many types of carcinoma. Several groups have discovered that exposure of carcinoma cells to low radiation doses causes activation of growth factor receptors, including ERBB1-4 e.g. (Dent et al, 1999; Dent et al, 2003; Goldkorn et al, 1997). Growth factor signals, via guanine nucleotide exchange factors, activate RAS proteins (Sklar, 1988; Cox and Der, 2003). There are 3 widely recognized isoforms of RAS: Harvey (H), Kirsten (K) and Neuroblastoma (N). GTP-RAS can interact with multiple downstream effector molecules including the Raf-1 protein kinase and the phosphatidyl inositol 3-kinase (PI3K) lipid kinase. Receptor-stimulated guanine nucleotide exchange of ‘RAS’ to the GTP-bound form permits Raf-1 and p110 PI3K to associate with ‘RAS’, resulting in kinase translocation to the plasma membrane environment where activation of these kinases, via a series of complex mechanisms, takes place. RAS contains a GTPase activity that converts bound GTP to GDP resulting in inactivation of the RAS molecule and dissociation of Raf-1 / p110 PI3K back into the cytosol, with their own inactivation.
Mutation of RAS in cancer results in a loss of GTPase activity, generating a constitutively active RAS molecule downstream of growth factor receptors that can lead to elevated activity within intracellular signaling pathways. Thus, expression of a mutated active RAS protein has potential to circumvent the anti-proliferative and tumoricidal impact of inhibiting ERBB receptor function. Approximately one third of human cancers have RAS mutations, primarily the K-RAS isoform, that leads to a radio-protected phenotype (Sklar, 1988; Ellis and Clark, 2000). Of note is that some studies suggest that K-RAS and H-RAS have different but over-lapping signaling specificities to downstream pathways as judged by in vitro cell based studies and in animal knock-out models: thus mutant K-RAS is thought to preferentially activate the Raf-1 / extracellular regulated kinase (ERK1/2) pathway, whereas mutant H-RAS is believed to preferentially activate the PI3K/AKT pathway (Ross et al, 2001; Yan et al, 1998; Liebmann et al, 2001; Chuang et al, 1994; Joneson et al, 1996). It has been argued that ERK1/2 and PI3K signaling downstream of K-RAS and H-RAS, respectively, can in turn control cell growth and cell survival following exposure to multiple growth factors (Dent et al, 1999; Ludde et al, 2001; Morriuchi et al, 2001). Data from our laboratory has argued that K-RAS D13 and H-RAS V12 differentially regulate radiation-induced signaling in HCT116 cells in general agreement with the hypothesis that K-RAS promotes ERK1/2 activation and H-RAS promotes AKT activation (Caron et al, 2005a; Caron et al, 2005b).
HCT116 colon cancer cells express a mutated active K-RAS D13 protein but are also noted to be dependent for their in vitro growth on an ERBB1 – TGFα / epiregulin paracrine loop and totally dependent for their in vivo tumoirgenic potential on both an ERBB1 –epiregulin paracrine loop and K-RAS D13 expression (Baba et al, 2000; Sizemore et al, 1999; Shirasawa et al, 1993). The studies in the present manuscript were initiated to determine to determine the molecular mechanisms by which HCT116 cells survived exposure to Lapatinib.
Materials and Methods
Materials
Dulbecco’s Modified Eagle’s Medium (DMEM), penicillin-streptomycin and 0.25% Trypsin-EDTA were purchased from Invitrogen Life Technologies, Inc. (Carlsbad, CA). HCT116 cells were originally purchased from American Type Culture Collection prior to multiple transfection procedures (Rockville, MD). Fetal bovine serum was purchased from Hyclone, Logan, UT. Trypan blue dye and crystal violet for colony formation assays were purchased from Sigma-Aldrich. For western blot analysis, 8–16% Tris-HCl gels were used (BIORAD, Carlsbad, CA). CMV control virus, ERBB1-CD533 and ERBB2-CD572 were obtained from Dr. Kristoffer Valerie, Virginia Commonwealth University. BCL-XL recombinant adenovirus was obtained from Dr. J. Moltken, University of Cincinnati, Cincinnati, Ohio. Dominant negative (dn) dnIκB (S32A) and dnSTAT3 recombinant adenoviruses purchased from Cell Biolabs (Philadelphia, PA). Control siRNA and siRNA to knock-down AIF (SI02662114, SI02662653), BCL-XL (SI03025141, SI03068352, SI03112018, SI00023191), MCL-1 (SI02781205, SI00131768), BAK (SI00299376, SI02654512) were purchased from Qiagen (Valencia, CA). Lapatinib was obtained from Glaxo Smith Kline (Boston, MA). The IGF-1 receptor inhibitor PPP, the Src family kinase inhibitor PP2, 4-hydroxy Tamoxifen and epidermal growth factor were purchased from Calbiochem (San Diego, CA). Primary antibodies against MCL-1, BCL-XL, BAX, BAK, AIF and cytochrome c were purchased from Cell Signaling (San Diego, CA). ERBB1 (Ab-5) antibody for fluorescence microscopy, primary antibody for active BAK (Ab-1), caspase 8 inhibitor LEHD, caspase 9 inhibitor IETD and pan-caspase inhibitor zVAD were purchased from Calbiochem (San Diego, CA). EGFR (Ab-13 cocktail) and c-ERBB2 (Ab-11 cocktail) to immunoprecipitate ERBB1 and ERBB2 were purchased from NeoMarkers (Freemont, CA). Anti-PhosphoTyr 4G10 antibody was purchased from Upstate (Temecula, CA). Primary antibodies for GAPDH, wild-type p53 (FL-393), mutant p53 (Pab 240), ERK2, active BAX (6A7) and protein A/G Plus agarose beads for immunoprecipitation were purchased from Santa Cruz Biotechnology, (Santa Cruz, CA). Secondary mouse antibody (Alexa Fluor 680 Goat anti-mouse IgG) was purchased from Invitrogen Molecular Probes (Eugene, OR) and secondary rabbit antibody (Anti-Rabbit IgG) was purchased from Rockland (Gilbertsville, PA). UCN-01 was kindly supplied by was provided by the Cancer Treatment and Evaluation Program (CTEP) of the National Cancer Institute. VP-16 was purchased from Sigma (St. Louis, MO). All other Materials and basic Methods of approach were as described in (Caron et al, 2005a; Caron et al, 2005b; Dent et al, 1999)
Methods
Detection of Cell Death by Trypan Blue Assay
After treatment, medium was removed and cells were washed in in 1X PBS. Cells were then harvested by trypsinization with Trypsin/EDTA for ~5 min at 37°C. Because some apoptotic cells detached from the culture substratum into the medium, these cells were also collected by centrifugation of the medium at 1400 RPM for 5 min. The pooled cell pellets were resuspended and mixed with trypan blue dye. Trypan blue stain, in which blue dye-incorporating cells were scored as being dead, was performed by counting of cells using a light microscope and a hemacytometer. The number of dead cells was counted and expressed as a percentage of the total number of cells counted.
Culture of Cells and Drugs Treatments for Colony Formation Assays
Cells were plated (250–1000 cells/well of a 6-well plate). 12 h after plating medium was removed and serum-free medium was added to the cells for 24 or 48 h as indicated. After this, the serum-free media was carefully removed and fresh media (with serum) was added. Colony formation assays were cultured for an additional 8–10 days, after which the media were removed, cells were fixed with methanol, stained with crystal violet, and counted manually.
Immunoprecipitation and Western Blotting
12 hours after plating cells, they were either infected with ERBB1-CD533 and ERBB2-CD572 or control virus for 24h or serum starved and treated with indicated concentrations of Lapatinib or dimethyl sulfoxide (DMSO) for 2h. After either of these treatments, cells were treated with 20ng/ml EGF or vehicle for 10 mins. Cells were then scraped using RIPA buffer (50mM Tris HCl pH 7.4, 150mM NaCl, 1%NP-40, 1% Sodium Deoxycholate, 1mM PMSF, 1mM Sodium orthovanadate, 10mM Sodium Fluoride, 10mM β-glycerophosphate, 0.5mM EGTA, 0.5mM EDTA and protease inhibitor cocktail purchased from (Roche)) and ERBB1 or ERBB2 was immunoprecipitated as indicated, after which samples were boiled for 10 min in whole cell lysis buffer (0.5 M Tris-HCl, pH 6.8, 2% SDS, 10% glycerol, 1% β-mercaptoethanol, 0.02% bromophenol blue). Twelve hours after plating cells, they were also scraped using a non-denaturing lysis buffer (50mM Tris HCl pH 7.4, 150mM NaCl, 1%NP-40, 0.1% Sodium Deoxycholate, protease and phosphatase inhibitor cocktails (Roche)) and mutant p53 was immunoprecipitated after which samples were boiled for 10 min in whole cell lysis buffer.
Cells were also scraped in CHAPS buffer (10mM HEPES, 140mM NaCl, 1% CHAPS) and then active BAK or active BAX was immunoprecipitated. Samples were boiled for 10 min in whole cell lysis buffer. All samples were then loaded on 8%–16% Criterion pre-cast gels (BIORAD) after normalizing total protein and run for about 2 hours. Proteins were then electrophoretically transferred onto 0.22um nitrocellulose membranes and immunoblotted with various primary antibodies as indicated.
Virus Infections
Cells were infected 12h after plating with adenoviruses at an approximate multiplicity of infection of 30 for 4 h with gentle rocking, after which time the media was replaced. Cells were further incubated for 24 h to ensure adequate expression of transduced gene products before drug exposures.
Transfection of Cells with Small Interfering RNA Molecules
RNA interference for down-regulating the expression of AIF, MCL-1, BCL-XL and BAK was performed using validated target sequences designed by Qiagen. For transfection, 20 nM of the annealed siRNA-targeting AIF, MCL-1, BCL-XL or BAK, or the negative control, a "scrambled" (SCR) sequence with no significant homology to any known gene sequences from mouse, rat, or human cell lines, were used. The siRNA molecules were transfected into cells according to the manufacturer's instructions. Cells were cultured for 48h after transfection before any additional experimentation.
Cell Fractionation
12h after plating cells, they were serum starved and treated with 2µM Lapatinib or DMSO for 36h. This experiment was performed on ice at all times. Medium from plates was then aspirated and cells were scraped in buffer (75mM NaCl+8mMNa2HPO4+1mMNa2H2PO4+0.5mMEDTA+ 0.5mMEGTA with freshly added 350ug/ml digitonin, 250mM sucrose, protease and phosphatase inhibitor cocktails (Roche)) and passed through a 25 gauge needle 12 times. After 15 to 30 minutes on ice, cells were spun down at 5000RPM for 1.5 minutes at 4°C to remove cell debris. Pellet was discarded and supernatant was transferred to a new tube and spun down at 13000 RPM for 25 minutes at 4°C. The supernatant obtained is the cytosolic fraction where as the pellet is the mitochondrial fraction. Whole cell lysis buffer was added to the supernatant and the pellet, boiled for 10 minutes and then western blot analysis was performed. This protocol was adapted from Leist et al. “1-Methyl-4-phenylpyridinium induces autocrine excitotoxicity, protease activation, and neuronal apoptosis.” Mol Pharmacol. (1998) 54: 789–801.
Flow Cytometry
Flow cytometric analysis of cells was performed after staining by the the ANNEXIN V-FITC kit (Beckman Coulter) according to the manufacturer’s instructions and read on Beckton Dickinson FACScan.
Data analysis
Comparison of the effects of various treatments was performed following ANOVA using the Student’s t test. Differences with a p-value of < 0.05 were considered statistically significant. Experiments shown are the means of multiple individual points (± SEM).
Results
Lapatinib is a clinically relevant receptor tyrosine kinase inhibitor that binds to the kinase domains of ERBB1 and ERBB2. ERBB1 and ERBB2 have previously been shown to act upstream of RAS proteins in radiation-induced signal transduction pathways and to play a role in protecting tumor cells from the toxic effects of ionizing radiation. Lapatinib blocked radiation-induced tyrosine phosphorylation of ERBB1, ERBB2 and ERBB3 in parental HCT116 cells and in HCT116 cells expressing H-RAS V12 (Figure 1A) (Caron et al, 2005a; Caron et al, 2005b). Inhibition of ERBB family receptor function correlated with Lapatinib inhibiting radiation-induced activation of ERK1/2 and AKT (Figure 1B). Lapatinib radiosensitized parental HCT116 cells expressing K-RAS D13 and HCT116 cells expressing H-RAS V12 (Figure 1C). These findings demonstrate that in the presence of expressed mutated active K-RAS and H-RAS proteins, the pan-ERBB receptor inhibitor Lapatinib can act as a radiosensitizer in HCT116 cells.
Figure 1. Lapatinib blocks radiation-induced activation of ERBB1, ERBB2, ERBB3 and activation of ERK1/2 and AKT in HCT116 cells.


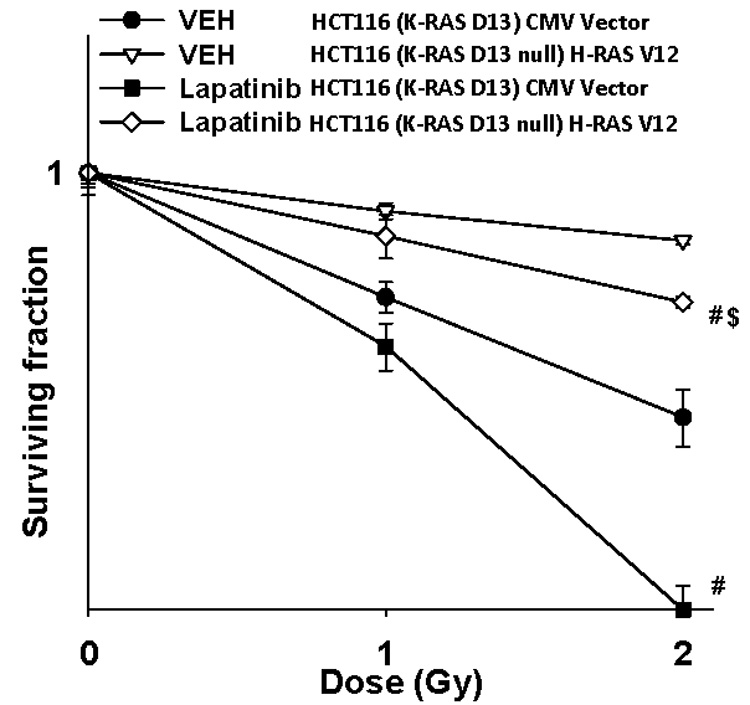
Panel A. Lower graphical sections: Parental HCT116 cells and HCT116 (K-RAS D13 null) cells stably transfected with a plasmid to express H-RAS V12 were plated, and 24h after plating grown in serum depleted medium. Two hours after serum withdrawal, cells were treated with either vehicle (DMSO) or with Lapatinib (2 µM), and 30 min later irradiated (1 Gy). Cells were isolated at the indicated time points and subjected to SDS PAGE to determine the phosphorylation and expression of ERBB1, ERBB2 and ERBB3 as indicated, as previously performed (45, 46). The –Fold alteration in the phosphorylation of ERBB1, ERBB2 and ERBB3 in the presence or absence of Lapatinib after irradiation is presented from 3 separate experiments ± SEM each performed in duplicate for parental HCT116 cells and HCT116 (K-RAS D13 null) cells stably transfected with a plasmid to express H-RAS V12. Upper inset immunoblotting panels: Parental HCT116 cells and HCT116 (K-RAS D13 null) cells stably transfected with a plasmid to express H-RAS V12 were plated, and 24h after plating grown in serum depleted medium. Two hours after serum withdrawal, cells were treated with either vehicle (DMSO) or with Lapatinib (0–2 µM), and 30 min later cells were isolated. The phosphorylation of ERBB1, ERBB2 and ERBB2 and the expression of ERBB1, ERBB2 and ERBB3 were determined by immunoblotting (n = 3). Panel B. Lower graphical sections: Parental HCT116 cells and HCT116 (K-RAS D13 null) cells stably transfected with a plasmid to express H-RAS V12 were plated, and 24h after plating grown in serum depleted medium. Two hours after serum withdrawal, cells were treated with either vehicle (DMSO) or with Lapatinib (2 µM), and 30 min later irradiated (1 Gy). Cells were isolated at the indicated time points and subjected to SDS PAGE and immunoblotting to determine the phosphorylation and expression of ERK1/2 and AKT (S473), as indicated (± SEM, n = 3). Upper inset immunoblotting panels: A representative immunoblotting data set for the lower graphical panels is shown. Panel C. Parental HCT116 and HCT116 (K-RAS D13 null) cells stably transfected with a plasmid to express H-RAS V12 were plated as single cells in sextuplicate and 12h after plating treated with either vehicle (DMSO) or with Lapatinib (2 µM) and 30 min later irradiated (1 Gy, 2 Gy). Lapatinib was removed 96h after irradiation. Colonies were permitted to form over the following 10–14 days after which the media was removed, the colonies fixed, stained with crystal violet and colonies > 50 cells counted. Data are the means of six separate plates from a representative study ± SEM (n = 2). (# p < 0.05 less than corresponding HCT116 cell value without Lapatinib)
The development of resistance to ERBB receptor inhibitors has been observed clinically. In many of these studies, resistance to the ERBB tyrosine kinase inhibitor has been due to mutation of the receptor within its catalytic domain so that the inhibitor no-longer can bind and inhibit receptor tyrosine kinase activity. We initially cultured parental HCT116 cells in 10 µM Lapatinib, a concentration which is below the Cmax for this drug in patients although the average plasma profile of a 1500 mg QD dose peaks at ~2.5 µM; within 72h, many cells (> 95%) became detached and died from this drug exposure (data not shown). Cells were cultured in the presence of Lapatinib for a further ~ 3 months until an essentially homogeneous population of cells grew out from the survivors that were adapted to Lapatinib.
In assays to determine cell survival in the absence of serum with a Lapatinib challenge; Lapatinib adapted cells survived to a significantly greater extent than parental cells (Figures 2A and 2B; Figure S1 in the presence of serum). Lapatinib adapted cells grew more quickly than parental cells in the presence or absence of Lapatinib (Figure S2). In general agreement with these findings, Lapatinib resistant cells had a greater level of survival than parental cells in colony formation assays (Figure 2C). When Lapatinib adapted cells were cultured in the absence of Lapatinib for > 10 flask passages (~2 months), no reversion of the resistant phenotype was observed back to the parental phenotype (Figure 2D). Lapatinib adapted cells were cross resistant to multiple chemotherapeutic agents including VP-16, UCN-01, Taxotere, Oxaliplatin and Doxorubicin (Figures S3–S5). Resistance to Taxotere appeared to be somewhat less than to the other agents. As drug efflux could represent a mechanism of Lapatinib adaptation, particularly as we observed cross-resistance to multiple cytotoxic therapeutic drugs, we performed flow cytometric and immunoblotting analyses to determine the expression of ABC and MDR plasma membrane drug transporters. Little change in the protein levels of any membrane drug transporter was observed, however, comparing wild type and Lapatinib adapted HCT116 cells, arguing that changes in drug efflux was unlikely to be a major component of Lapatinib resistance mechanism under investigation (Fisher and Dent, Unpublished observations).
Figure 2. The generation of Lapatinib resistant HCT116 cells.



Panel A. Parental HCT116 cells (WT) and HCT116 Lapatinib adapted cells (WT-AD) cells were plated, and 24h after plating grown in serum depleted medium in the presence or absence of vehicle (DMSO) or Lapatinib (0–10 µM). Cells were isolated 48h after serum starvation / Lapatinib addition and cell viability determined in triplicate by trypan blue exclusion assay ± SEM. The data shown are a representative study from 3 separate studies. Panel B. Parental HCT116 cells (WT) and HCT116 Lapatinib adapted cells (WT-AD) cells were plated, and 24h after plating grown in serum depleted medium in the presence or absence of vehicle (DMSO) or Lapatinib (0–10 µM). Cells were isolated at the indicated times and cell viability determined by annexin-PI flow cytometric analysis in triplicate ± SEM (n = 2). Panel C. Parental HCT116 cells (WT) and HCT116 Lapatinib adapted cells (WT-AD) cells were plated as single cells in sextuplicate and 12h after plating placed into serum depleted medium for either 24h or 48h in the presence of Lapatinib (2 µM). Media containing serum was re-supplemented back to the cells 24h or 48h after the initiation of serum starvation, as indicated with the removal of Lapatinib from the growth medium. Colonies were permitted to form over the following 10–14 days after which the media was removed, the colonies fixed, stained with crystal violet and colonies > 50 cells counted. Data are the means of four separate plates from a representative study ± SEM (n = 2). (*p < 0.05 less than corresponding HCT116 parental cell value). Panel D. Parental HCT116 cells (WT), HCT116 Lapatinib adapted cells (WT-AD), and HCT116 Lapatinib adapted cells that had been grown for > 2 months in the absence of Lapatinib (WT-AD-REV) were plated, and 24h after plating grown in serum depleted medium in the presence or absence of vehicle (DMSO) or Lapatinib (2 µM). Cells were isolated 48h after serum starvation / Lapatinib addition and cell viability determined in triplicate by trypan blue exclusion assay ± SEM. The data shown are a representative study from 2 separate studies.
Based on the above findings, we examined in molecular detail the role of ERBB receptors in Lapatinib resistance. Co-expression of dominant negative ERBB1 (CD533) and dominant negative ERBB2 (CD572) proteins suppressed basal and EGF-stimulated tyrosine phosphorylation of ERBB1 and ERBB2 in immunoprecipitates from parental HCT116 cells (Figure 3A, upper left inset panel). Co-expression of dominant negative ERBB1 (CD533) and dominant negative ERBB2 (CD572) suppressed basal and EGF-stimulated tyrosine phosphorylation of ERBB1 and ERBB2 in immunoprecipitates from parental HCT116 cells (Figure 3A, upper right inset panel). To our surprise, however, while co-expression of ERBB1 (CD533) and ERBB2 (CD572) acted in a very similar manner as Lapatinib to inhibit ERBB receptor tyrosine phosphorylation, the dominant negative receptors did not recapitulate the toxic effects of Lapatinib in serum-starved parental or adapted cells (Figure 3A, lower graphical panels). Further analyses revealed that although parental and Lapatinib adapted cells expressed similar total cellular amounts of ERBB1 as judged by immunoblotting of whole cell lysate, and that stimulated ERBB1 phosphorylation in response to EGF was inhibited equally well by Lapatinib in both parental and adapted cells, the plasma membrane associated levels of ERBB1 in adapted cells were considerably lower in adapted than those in parental cells. These findings were reflected also in a reduced ability of EGF to stimulate ERK1/2 signaling in adapted cells compared to parental cells (Figures 3B and 3C) Collectively, these findings strongly argue that an ERBB1 receptor mutation has not occurred in Lapatinib adapted HCT116 cells to make these cells resistant to Lapatinib toxicity.
Figure 3. Molecular inhibition of ERBB1 and ERBB2 does not recapitulate the effects of Lapatinib in HCT116 cells: Lapatinib resistant cells express less cell surface ERBB1 than parental cells.



Panel A. Parental HCT116 cells (WT) and HCT116 Lapatinib adapted cells (WT-AD) cells were plated, and 12h after plating infected with either a control empty vector adenovirus, a recombinant adenovirus to express dominant negative ERBB1-CD533, a recombinant adenovirus to express dominant negative ERBB2-CD572 or both viruses to express ERBB1-CD533 and ERBB2-CD572. Twenty four hours after virus infection, cells were grown in serum depleted medium in a similar manner to our prior studies using Lapatinib. Cells were isolated 48h after serum starvation and cell viability determined in triplicate by trypan blue exclusion assay ± SEM. The data shown are a representative study from 3 separate studies. Upper immunoblotting sections. Parental HCT116 cells (WT) were plated, and 12h after plating infected with either a control empty vector adenovirus, or recombinant adenoviruses to express dominant negative ERBB1-CD533 and to express dominant negative ERBB2-CD572. Twenty four hours after virus infection, cells were grown in serum depleted medium for 2h then treated with 20 ng/ml EGF as indicated in the Figure for 10 min. Cells were isolated and subjected to: immunoprecipitation of ERBB1 or ERBB2 followed by SDS PAGE of the immunoprecipitate as indicated followed by anti-phospho-tyrosine blotting to determine the activation of ERBB1/2 proteins. A representative study is shown (n = 2–3). Panel B. Parental HCT116 cells (WT) and HCT116 Lapatinib adapted cells (WT-AD) cells were plated, and 24h after plating grown in serum depleted medium for 2h followed by treatment with vehicle (DMSO) or Lapatinib (0–2 µM), as indicated in the Figure. Thirty minutes after Lapatinib exposure cells were treated, where indicated with 20 ng/ml EGF. Ten minutes after EGF addition, cells were harvested, subjected to lysis, and portions of the lysate either immunoprecipitated to isolate ERBB1 and determine ERBB1 tyrosine phosphorylation or portions of the lysate directly subjected to SDS PAGE followed by SDS PAGE to determine total ERBB1, ERK2 and GAPDH expression and ERK1/2 phosphorylation. A representative study is shown (n = 2–3). Panel C. Parental HCT116 cells (WT) and HCT116 Lapatinib adapted cells (WT-AD) cells were plated, and 24h after plating grown in serum depleted medium for 2h. Cells were fixed but not permeabilized and cell surface levels of ERBB1 determined by immunostaining and examination under fluorescent light microscope. Two representative images are shown from one experiment (n = 2).
We then examined the activities of known signaling pathways whose activities could become altered in the adapted HCT116 cell line. However, almost no difference in basal activities of any pathway, or in the basal activity of any pathway in the presence of Lapatinib, could be observed between parental and adapted cells (data not shown). In HCT116 cells expressing H-RAS V12 and effector mutants of H-RAS V12 that had been characterized to specifically activate: the Raf-MEK-ERK pathway (S35); the RAL-GDS pathway (G37); the PI3K-AKT pathway (C40), only H-RAS V12 expression, but not expression of any H-RAS V12 single point mutant that activated a single signaling pathway, suppressed Lapatinib toxicity (Figure 4A). In contrast to our findings with Lapatinib, for example, expression of H-RAS V12, H-RAS V12 S35 and H-RAS V12 C40, but not H-RAS V12 G37, acted to protect HCT116 cells from the toxic effects of radiation in colony formation assays (Figure 4B). After a 1 Gy radiation exposure, approximating to the shoulder of the survival curve, no statistically significant difference between cell survival for cells expressing H-RAS V12 and H-RAS V12 C40 was observed. Cells expressing H-RAS V12 S35 had a greater level of survival than vector control transfected cells however these cells had significantly less survival than cells expressing H-RAS V12 C40 (After a 1 Gy exposure: vector survival 0.70 ± 0.04; H-RAS V12 S35 survival 0.82 ± 0.04; H-RAS V12 C40 survival 0.90 ± 0.03, p < 0.05 difference between each value). The survival of cells expressing H-RAS V12 S35 was not significantly different from wild type HCT116 cells expressing K-RAS D13.
Figure 4. Lapatinib resistance requires multiple pathways downstream of RAS: lack of involvement of “classical” effectors of resistance.

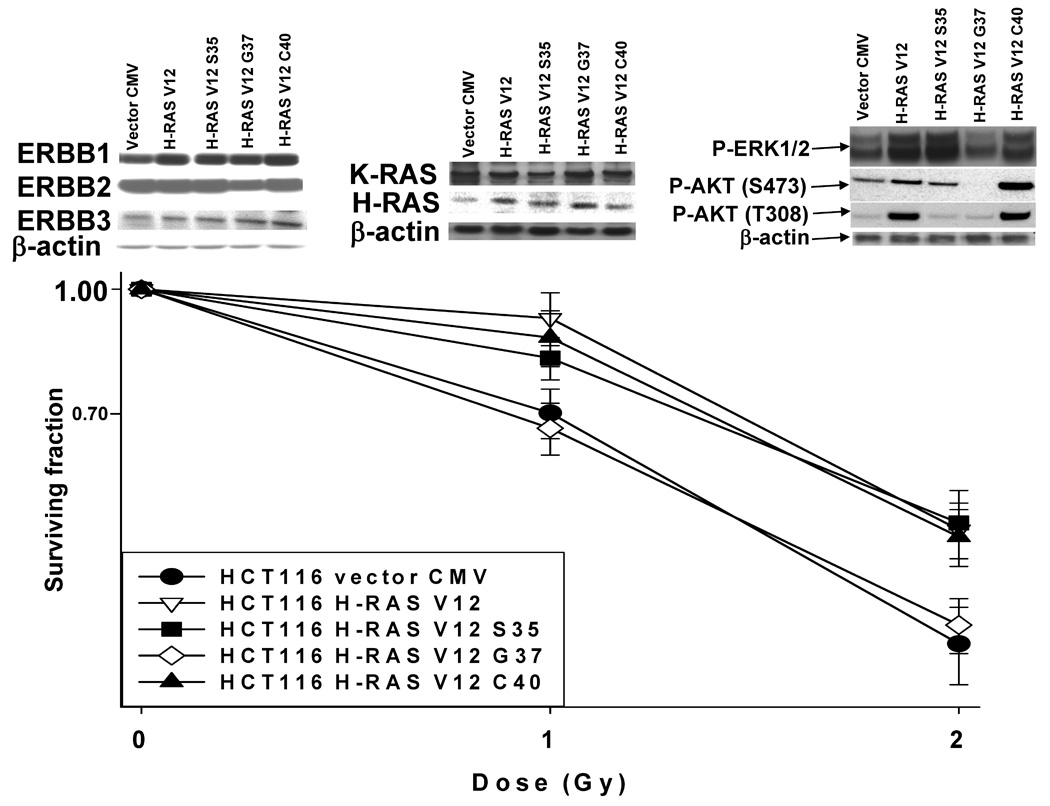


Panel A. Parental HCT116 cells and HCT116 (K-RAS D13 null) cells were stably transfected with the following plasmids: either empty vector control (CMV); H-RAS V12; H-RAS V12 S35; H-RAS V12 G37; H-RAS V12 C40, and selected as described in the Methods (45, 46). Cells were plated, and 24h after plating grown in serum depleted medium in the presence or absence of vehicle (DMSO) or Lapatinib (2 µM). Cells were isolated 48h after drug addition and cell viability determined in triplicate by trypan blue exclusion assay ± SEM. The data shown are a representative study from 2–3 separate studies. (*p < 0.05 less than corresponding value in other HCT116 cell isolates) Panel B. Upper immunoblotting section: HCT116 (K-RAS D13 null) cells stably transfected with plasmids either empty vector control (CMV); H-RAS V12; H-RAS V12 S35; H-RAS V12 G37; H-RAS V12 C40, and selected as described in the Methods. Cells were plated, and 24h after plating grown in serum depleted medium. Two hours after culture in serum depleted medium, cells were harvested for SDS PAGE and immunoblotting analyses. Data are representative of 3–5 separate studies. Blotting panel to the right: Phosphorylation (activity) of protein kinases was determined by immunoblotting using specific antibodies for the phosphorylated forms of ERK1/2, AKT S473, JNK1/2, p38 in the HCT116 cell lines. Total β-actin protein expression was blotted in the same membrane as a loading control; Blotting panel in the center: Expression of K-RAS and H-RAS proteins determined by immunoblotting using RAS isoform specific antibodies. Blotting panel to the left: Expression of ERBB1, ERBB2 and ERBB3 receptor proteins determined by immunoblotting using receptor specific antibodies. Total β-actin protein expression was blotted in the same membrane as a loading control. Lower colony formation graph: Parental HCT116 and HCT116 (K-RAS D13 null) cells stably transfected with plasmids either empty vector control (CMV); H-RAS V12; H-RAS V12 S35; H-RAS V12 G37; H-RAS V12 C40, were plated as single cells in sextuplicate and 12h after plating irradiated (1 Gy, 2 Gy). Colonies were permitted to form over the following 10–14 days after which the media was removed, the colonies fixed, stained with crystal violet and colonies > 50 cells counted. Data are the means of six separate plates from a representative study ± SEM (n = 2). Panel C. Parental HCT116 cells (WT) were plated, and 12h after plating infected with either a control empty vector adenovirus (CMV), a recombinant adenovirus to express constitutively active AKT (caAKT), a recombinant adenovirus to express constitutively active MEK1 (caMEK1) or both viruses to express caAKT and caMEK1. Twenty four hours after virus infection, cells were grown in serum depleted medium in the presence or absence of vehicle (DMSO) or Lapatinib (2 µM). Cells were isolated 48h after drug addition and cell viability determined in triplicate by trypan blue exclusion assay ± SEM. The data shown are a representative study from 3 separate studies. (*p < 0.05 less than corresponding HCT116 CMV infected cell value) Panel D. Left graph: Parental HCT116 cells (WT) and HCT116 Lapatinib adapted cells (WT-AD) cells were plated, and 24h after plating grown in serum depleted, phenol red free medium in the presence or absence of vehicle (DMSO), 4-hydroxytamoxifen (4-OH TAM, 50 nM) or Lapatinib (2 µM); Center graph: HCT116 Lapatinib adapted cells (WT-AD) were plated, and 12h after plating infected with either a control empty vector adenovirus or a recombinant adenovirus to express dominant negative IκB (dnIκB). Twenty four hours after virus infection, cells were grown in serum depleted medium in the presence or absence of vehicle (DMSO) or Lapatinib (0–3 µM). Cells were isolated 48h after drug addition and cell viability determined in triplicate by trypan blue exclusion assay ± SEM. The data shown are a representative study from 2 separate studies; Right graph: HCT116 Lapatinib adapted cells (WT-AD) were plated, and 12h after plating infected with either a control empty vector adenovirus or a recombinant adenovirus to express dominant negative STAT3 (dnSTAT3) Twenty four hours after virus infection, cells were grown in serum depleted medium in the presence or absence of vehicle (DMSO) or Lapatinib (3 µM). Cells were isolated 48h after drug addition and cell viability determined in triplicate by trypan blue exclusion assay ± SEM. The data shown are a representative study from 2 separate studies. Panel E. Parental HCT116 cells (WT) and HCT116 Lapatinib adapted cells (WT-AD) cells were plated, and twenty four hours later grown in serum depleted medium in the presence or absence of vehicle (DMSO), Lapatinib (2 µM), PP2 (10 µM), PPP (250 nM) or the drug combinations indicated in the Figures. Cells were isolated 48h after serum starvation and cell viability determined in triplicate by trypan blue exclusion assay ± SEM. The data shown are a representative study from 2 separate studies.
In general agreement with our short-term cell killing data using Lapatinib exposure and serum starvation, expression of constitutively active MEK1 EE and constitutively active AKT, to a greater extent than the individual activated kinases, suppressed Lapatinib toxicity in parental cells (Figure 4C). In contrast to the use of activated proteins, expression of dominant negative AKT and/or dominant negative MEK1 did not restore Lapatinib sensitivity in adapted cells (Figure S6). As inhibition of ERK1/2 and AKT did not restore Lapatinib sensitivity, we explored whether other mechanisms of Lapatinib resistance were present in HCT116 cells.
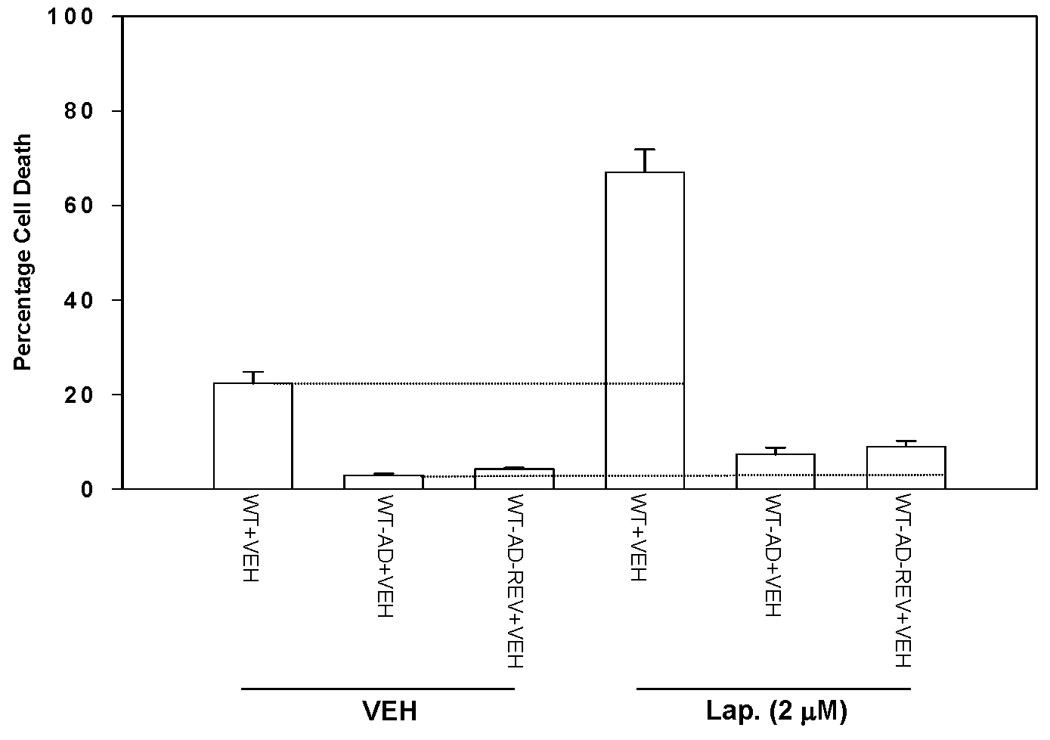
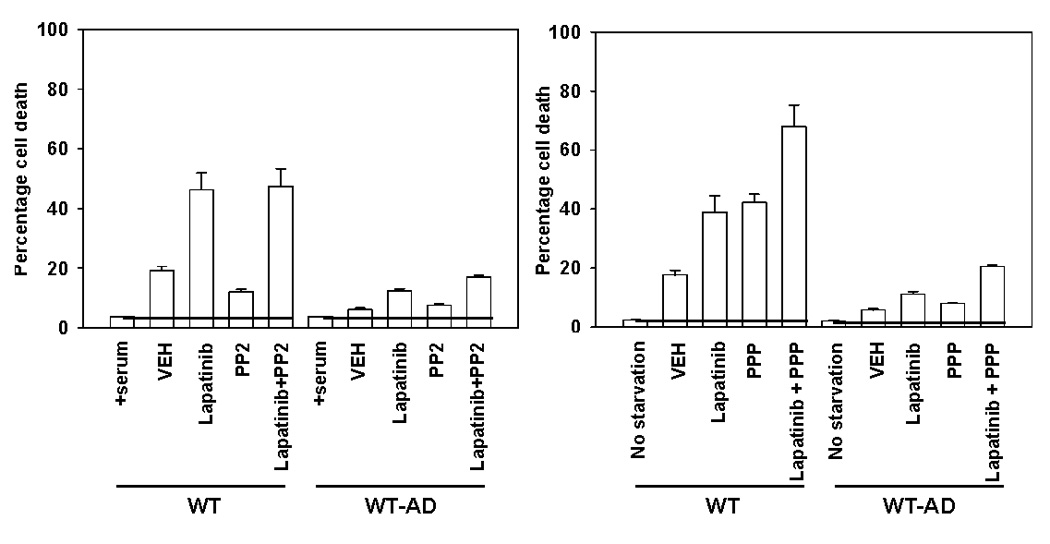
Lapatinib resistance has been linked to re-activation of the estrogen receptor in breast cancer cells and the estrogen receptor is known to be expressed in colon cancer cells (Xia et al, 2006; Cho et al, 2007). However, incubation of adapted cells with the ER inhibitor Tamoxifen did not restore Lapatinib sensitivity (Figure 4C). Similarly, inhibition of NFκB function by over-expression of the IκB super repressor (dominant negative IκB) or inhibition of STAT1 and STAT3 function by expression of a dominant negative STAT3 protein did not restore Lapatinib sensitivity in adapted cells. In some cell types, including colon cancer cells, Src family nonreceptor tyrosine kinases and the insulin like growth factor receptor tyrosine kinase have been linked to the transformed phenotype. However, inhibition of neither Src family kinases using the inhibitor PP2 nor IGF1 receptor function using the inhibitor PPP restored Lapatinib sensitivity (Figure 4D). Of note, inhibition of the IGF1 receptor with PPP caused significant toxicity in parental cells that was abolished in Lapatinib adapted cells arguing that adapted cells were also cross resistant to agents that inhibit the function of other receptor tyrosine kinases that are known to compensate for ERBB1 survival signaling.
Based on our relative lack of success at precisely defining the signaling pathways downstream of ERBB1 and ERBB2 that could be mediating Lapatinib adaptation, we next determined the proximal downstream molecular mechanisms by which serum starved and Lapatinib treated cells die, and the mechanisms by which adaptation was gained. Adapted HCT116 cells expressed higher levels of MCL-1, BCL-XL and p53 than parental cells; these cells expressed lower levels of BAX and BAK than parental cells (Figure 5A, upper blotting section). No obvious changes in the protein expression of CD95, FAS ligand, pro-caspase 8, pro-caspase 9, pro-caspase 3, Apaf-1, A10, Smac/DIABLO, c-FLIP-s, XIAP, BCL-2, BID, BIM, NOXA or PUMA were noted based on immunoblotting analyses (data not shown). Based on the established concept of the so called “apoptotic rheostat,” in which BCL-2 family proteins act in a dynamic balance to suppress the pro-apoptotic signals generated by BH3 domain proteins such as BAX and BAK, our data suggest that adapted cells could be more resistant to Lapatinib than parental cells because they express more of the mitochondrial protective proteins BCL-XL and MCL-1 and that they express less of the mitochondrial toxic proteins BAX and BAK.
Figure 5. Lapatinib resistance is mediated by increased expression of MCL-1, decreased activation of BAK and mutation of p53.
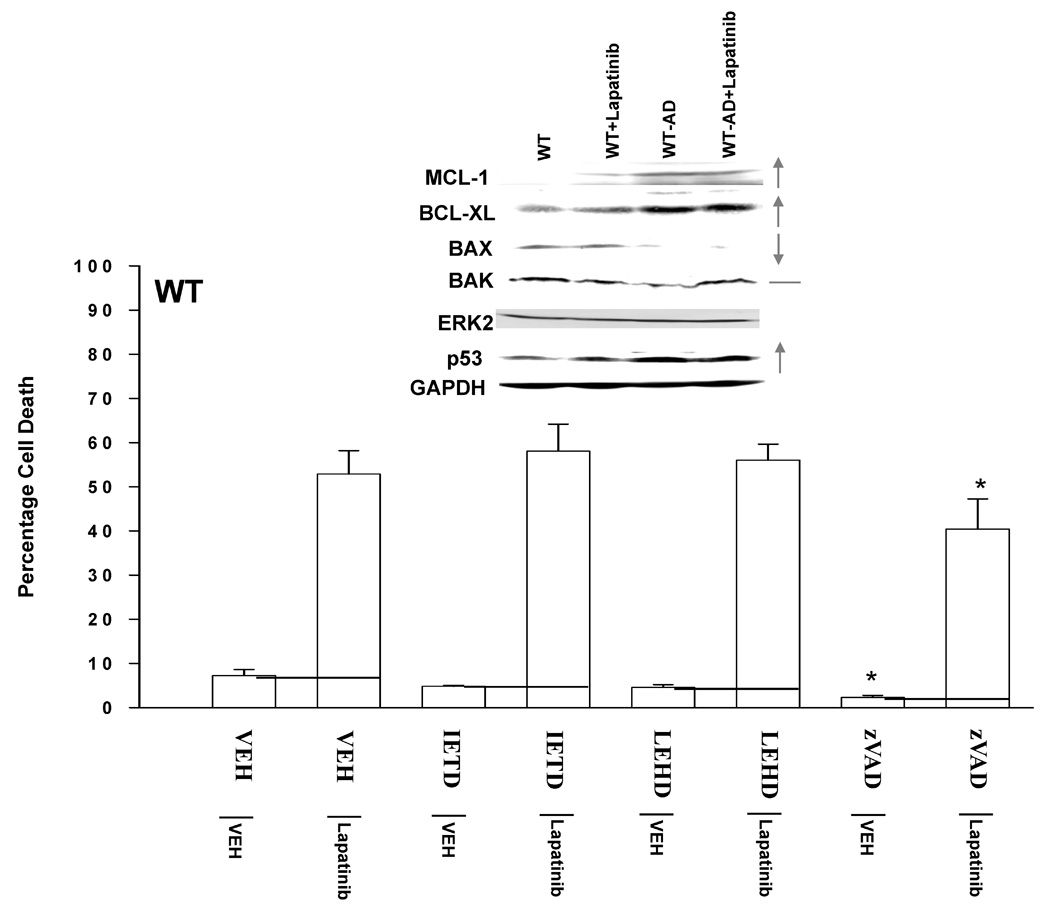


Panel A. Upper immunoblotting section: Parental HCT116 cells (WT) and HCT116 Lapatinib adapted cells (WT-AD) cells were plated, and twenty four hours later grown in serum depleted medium in the presence or absence of vehicle (DMSO), Lapatinib (2 µM). Cells were isolated 48h after Lalatinib addition and subjected to SDS PAGE to determine the expression of multiple proteins, of which are shown in the Figure: BCL-XL; MCL-1; BAX; BAK; p53; ERK2; GAPDH. Lower graphical section: Parental HCT116 cells (WT) were plated, and twenty four hours later grown in serum depleted medium in the presence or absence of vehicle (DMSO) or Lapatinib (2 µM). Cells were, in parallel, grown in the presence or absence of vehicle (DMSO), the caspase 8 inhibitor IETD (50 µM), the caspase 9 inhibitor LEHD (50 µM) or the pan-caspase inhibitor zVAD (50 µM). Cells were isolated 36h after serum starvation and cell viability determined in triplicate by trypan blue exclusion assay ± SEM. The data shown are a representative study from 2 separate studies. (*p < 0.05 less than corresponding HCT116 vehicle control cell values) Panel B. Parental HCT116 cells (WT) were plated, and 12h after plating infected with either a control empty vector adenovirus or a recombinant adenovirus to express BCL-XL. Twenty four hours after virus infection, cells were grown in serum depleted medium in the presence or absence of vehicle (DMSO) or Lapatinib (2 µM). Cells were isolated 48h after drug addition and cell viability determined in triplicate by trypan blue exclusion assay ± SEM. The data shown are a representative study from 2 separate studies. (*p < 0.05 less than corresponding HCT116 cell value). Panel C. As indicated in the two graphs, parental HCT116 cells (WT) and HCT116 Lapatinib adapted cells (WT-AD) cells were plated, and 12h later as indicated transfected with siRNA molecules to reduce the expression of; nothing/control (siSCR); AIF (siAIF); BCL-XL (siBCL-XL); or MCL-1 (siMCL). Forty eight hours after transfection, cells were grown in serum depleted medium in the presence or absence of vehicle (DMSO), Lapatinib (2 µM) or the pan-caspase inhibitor zVAD (50 µM), as indicated. Cells were isolated 36h after serum starvation and cell viability determined in triplicate by trypan blue exclusion assay ± SEM. The data shown are a representative study from 2 separate studies. (#p < 0.05 less than corresponding HCT116 siSCR cell value; ##p < 0.05 less than siAIF + VEH + Lap HCT116 cell value). Panel D. Parental HCT116 cells (WT) and HCT116 Lapatinib adapted cells (WT-AD) cells were plated, and 24h after plating grown in serum depleted medium in the presence or absence of vehicle (DMSO), Lapatinib (2 µM). Thirty six hours after serum depletion / Laptinib addition, cells were isolated for assay of: AIF and cytochrome c release into the cytosol and; by immunoprecipitation to determine the amount of the activated forms of BAX and BAK. A representative from two separate studies is shown.
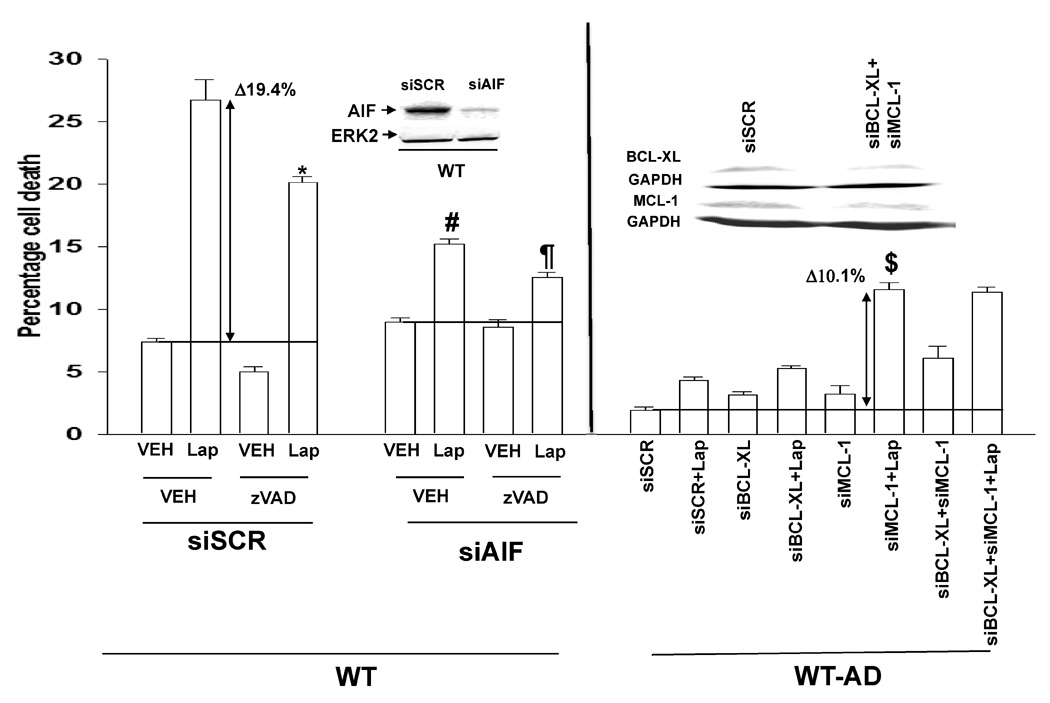
As we observed changes in the expression of proteins who act at the mitochondrion to modulate mitochondrial stability, we next determined whether activation of caspase proteases, and specifically pro-caspase 9, played a role in Lapatinib toxicity. To our surprise, inhibition of caspase function only modestly suppressed Lapatinib toxicity in parental cells treated with Lapatinib (Figure 5A, lower graphical section). In contrast, inhibition of caspases significantly reduced serum-withdrawal –induced cell killing (Figure S7). Inhibition of cathepsin, calpain and serine protease function also caused similar very modest effects on promoting cell survival in Lapatinib treated cells (data not shown). Over-expression of BCL-XL abolished Lapatinib toxicity in parental cells (Figure 5B). Finally, we tested whether apoptosis inducing factor (AIF) played a role in Lapatinib toxicity. Knock down of AIF expression reduced Lapatinib toxicity in parental HCT116 cells, and knock down of AIF expression combined with pan-caspase inhibition almost eliminated Lapatinib toxicity (Figure 5C, left graphical panel).
Knock down of MCL-1 expression, to a greater extent than that of BCL-XL, reverted Lapatinib sensitivity in adapted cells (Figure 5C, right graphical panel). In Figure 5A we noted that the expression levels of pro- and anti-apoptotic proteins were altered comparing parental and adapted HCT116 cells. In parental cells, Lapatinib treatment caused release of AIF into the cytosol whereas in adapted cells, no AIF release was observed (Figure 5D). Thus the induction of cell killing by Lapatinib in parental cells correlated with activation of BAK and BAX. Knock down of BAK activation in adapted cells significantly reduced the reversion of their resistant phenotype by reduced MCL-1 expression (Figure 6A).
Figure 6. Knock down of BAK re-reverts Lapatinib adapted cells after siMCL-1 exposure.


Panel A. HCT116 Lapatinib adapted cells (WT-AD) cells were plated, and 12h later as indicated transfected with siRNA molecules to reduce the expression of; nothing/control (siSCR); MCL-1 (siMCL-1); BAK (siBAK); or MCL-1 and BAK (siMCL+siBAK). Forty eight hours after transfection, cells were grown in serum depleted medium in the presence or absence of vehicle (DMSO) or Lapatinib (2 µM). Cells were isolated 48h after serum starvation and cell viability determined in triplicate by trypan blue exclusion assay ± SEM. The data shown are a representative study from 2 separate studies. (*p < 0.05 less than HCT116 siMCL-1 cell value; **p < 0.05 less than HCT116 siSCR + Lap cell value; ***p < 0.05 less than HCT116 siMCL-1 + Lap cell value; #p < 0.05 greater than HCT116 siSCR cell value; ##p < 0.05 greater than HCT116 siSCR and siMCL-1 cell values; ### p < 0.05 greater than corresponding HCT116 siSCR + Lap cell value). Panel B. Parental HCT116 cells (WT) and HCT116 Lapatinib adapted cells (WT-AD) cells were plated and 24h after plating cells were lysed and prepared for immunoprecipitation and pseudo-immunoprecipitation with beads against mutated inactive p53 followed by SDS PAGE. The SDS PAGE was transferred to nitrocellulose and probed with an anti-p53 antibody and an anti-GAPDH antibody. Data shown are from two representative studies (n = 3).
In Figure 5A, we noted that the expression of p53 was elevated, even though the protein levels of a p53 target protein, BAX, were reduced. In cells that express a mutated p53 protein, the expression of total p53 within a cell is often noted to be elevated. Thus, parental HCT116 cells which express a wild type p53 protein may have in part survived and adapted to Lapatinib exposure by mutating one of their p53 alleles.
Native p53 proteins were immunoprecipitated from parental and Lapatinib resistant HCT116 cells using an antibody that specifically recognizes mutated forms of p53, as judged by recognition of mutant p53 tertiary structure within the DNA binding domain of p53. The p53 proteins were then separated on denaturing SDS PAGE and immunoblotted; Lapatinib resistant cells, but not parental cells, immunoprecipitated a greater amount of “mutant” p53 (Figure 6B). Total poly A mRNA was isolated from adapted HCT116 cells and amplified and sequenced using primers specific for the DNA binding domain of p53. We noted, however, that adapted HCT116 cells did not contain a mutation in p53, suggesting that either our antibody was recognizing an alteration in p53 tertiary conformation in adapted cells unrelated to p53 mutation or that p53 mutation had occurred in a domain unrelated to the DNA binding domain of p53 but that was affecting the tertiary conformation of the DNA binding domain. These findings argue that Lapatinib adaptation in HCT116 cells is mediated by changes in the expression of multiple mitochondrial protective proteins, rather than mutation of ERBB receptors.
Discussion
Previous studies from this group have demonstrated that mutated active forms of K-RAS and H-RAS differentially regulate ERK1/2 and AKT signaling after irradiation. Prior studies from multiple groups have also demonstrated that radiation-induced activation of ERBB1, ERBB2 and ERBB3 is a cytoprotective response. The present studies were proposed to determine the impact of the clinically relevant ERBB1 / ERBB2 inhibitor Lapatinib on tumor cell radiosensitivity and the mechanisms by which HCT116 tumor cells become resistant to the toxic effects of Lapatinib in vitro.
HCT116 cells and the variant cell lines used in this manuscript expressing a mutated active RAS protein were radiosensitized by Lapatinib, even though a priori it would be predicted that activated RAS proteins would tend to overcome the impact of an inhibitor of an upstream receptor tyrosine kinase on radiosensitivity in any cell type. Furthermore, HCT116 cells were sensitive, in the presence or absence of serum, to being killed by doses of Lapatinib that were within the Cmax patient serum concentration of the drug. Multiple studies have argued that tumor cell resistance to therapeutic agents is comprised of the actions of multiple signal transduction pathways, and based on the expression of S35 / G37 / C40 effector domain mutants of H-RAS V12 as well as expression of activated forms of MEK1 and AKT, we concluded that we could prevent Lapatinib –induced cell killing by activating both PI3K-AKT and MEK-ERK1/2 signaling and but not by activating either pathway individually (Dent et al, 2003; Caron et al, 2005a; Caron et al, 2005b). Conversely, our data using point mutants of H-RAS V12 demonstrated that mutant oncogenic RAS is a negative predictor of therapeutic response to Lapatinib exposure.
In the clinic, resistance to the toxic and radio-/chemo-sensitizing effects of ERBB1 receptor inhibitors has been noted primarily with the development of mutations in the tyrosine kinase domain rendering the receptor tyrosine kinase insensitive to the ATP binding site “competitive inhibitor” drug (e.g. Kobayashi et al, 2005; Pao et al 2005). Resistance to Lapatinib in breast cancer cells has been ascribed to reactivation of the estrogen receptor; in a variety of other tumor cell types general resistance to chemotherapeutic drug toxicity has also been linked to hyper-activation of the transcription factor NFκB, the IGF-1 receptor, STAT transcription factors, Src nonreceptor tyrosine kinases, the PI3K-AKT pathway; and to increased levels of drug export pumps. None of these factors appeared to play a primary role in the adaptive resistance of HCT116 cells to Lapatinib.
After many studies we determined that Lapatinib adapted cells expressed higher levels of MCL-1 and BCL-XL and that knock down of MCL-1 expression, but not expression of BCL-XL, significantly reverted the Lapatinib adapted phenotype. Unlike parental cells, Lapatinib adapted cells did not exhibit activation of BAX and BAK following serum starvation and Lapatinib treatment, and knock down of MCL-1 expression will presumably promote modest levels of BAK activation. Knock down of BAK expression restored Lapatinib resistance.
Recent evidence has argued that BAK activation requires simultaneous disruption of its associations with MCL-1 (e.g., by proteins such as NOXA) and BCL-XL (e.g., by proteins such as BAD; e.g. Willis et al, 2005) Moreover, increased production of NOXA can oppose MCL-1 anti-apoptotic functions, leading to simultaneous activation of BAX and BAK (Qin et al, 2006b). In adapted cells we did not observe altered levels of either NOXA or BAD, or altered BAD phosphorylation, arguing against changes in the functions of these proteins in the adaptation process (Unpublished observations). In a recent study we noted that over-expression of BCL-2 or BCL-XL failed to protect leukemia cells from co-treatment with the BCL-2/BCL-XL inhibitor ABT-737 and the CDK inhibitor roscovitine, potentially reflecting an important contribution of MCL-1 down-regulation to the lethality of this drug regimen in leukemic cells (Chen et al, 2007). We found that ectopic expression of MCL-1 diminished the potentiation of ABT-737 lethality by roscovitine, and this likely highlights a central role of MCL-1, and its down-regulation in the synergism of interaction between these agents. This interpretation was further supported by results showing that roscovitine was unable to enhance ABT-737–mediated apoptosis in transformed MCL-1 null mouse embryonic fibroblasts. In these studies, over-expression of MCL-1, but not BCL-2 or BCL-XL, abrogated BAK activation following exposure to ABT-737 and roscovitine, arguing that MCL-1 plays a major role in regulating BAK function. This is consistent with data demonstrating that MCL-1 binds with greater affinity to BAK compared with BCL-XL (IC50 < 10 vs. < 100 nM; Willis et al, 2005).
Whether a strategy combining CDK inhibitors, or other transcriptional repressors capable of down-regulating MCL-1 expression, with BCL-2 / BCL-XL / MCL-1 antagonists such as ABT-737 or Obatoclax will result in enhanced therapeutic efficacy in Lapatinib adapted HCT116 cells will depend upon multiple other factors, including the capacity of such agents to diminish MCL-1 expression in vivo, and whether the therapeutic index is enhanced. In this context, it is noteworthy that ABT-737 and Obatoclax display in vivo anti-tumor selectivity in preclinical studies (Oltersdorf et al, 2005; Trudel et al, 2007). The present findings suggest that in addition to combining BCL-2 / BCL-XL / MCL-1 antagonists with conventional cytotoxic drugs, combination strategies involving targeted agents that down-regulate MCL-1, a protein that can compensate for the loss of BCL-2 / BCL-XL function, could represent a potentially useful alternative approach to subvert Lapatinib resistance.
In our studies to determine the mechanism of Lapatinib resistance we noted that p53 was over-expressed in Lapatinib adapted cells and that the expression of a transcriptional target of p53, BAX, was significantly lower in adapted cells. The expression of p53 is often elevated when mutated. We also noted that on a per molecule basis, the phosphorylation of p53 serine 15 was reduced (Martin and Dent, Unpublished observation). Collectively, this suggested that HCT116 cells, that express a wild type p53 protein, may have undergone a portion of their adaptation process by developing a p53 mutation. In agreement with this hypothesis, using an antibody that specifically immunoprecipitates mutant forms of p53 due to the conformation of the p53 DNA binding domain, we noted that adapted cells but not wild type cells expressed a p53 protein that could be immunoprecipitated by an antibody that recognizes a mutant specific form of p53. However, upon sequencing the coding regions in DNA binding domains of p53, no mutations were noted in the p53 sequences between parental and adapted cells. This suggests that either our antibody was recognizing an alteration in p53 tertiary conformation in adapted cells unrelated to a true p53 mutation but that was in all likelihood still suppressing p53 function, i.e. reduced BAX expression, or that p53 mutation had occurred in a domain unrelated to the DNA binding domain of p53 but that was affecting the tertiary conformation of the DNA binding domain. Further studies beyond the scope of the present manuscript will be required to understand how p53 function, with respect to modulation of all p53 targets and functions, e.g. the extrinsic (CD95) and intrinsic (BAX) apoptosis pathways; senescence; autophagy; metabolism; has been altered in Lapatinib adapted HCT116 cells.
Supplementary Material
Abbreviations
- Lap.
Lapatinib
- ERK
extracellular regulated kinase
- MEK
mitogen activated extracellular regulated kinase
- EGF
epidermal growth factor
- PARP
poly ADP ribosyl polymerase
- PI3K
phosphatidyl inositol 3 kinase
- −/−
null / gene deleted
- ERK
extracellular regulated kinase
- MAPK
mitogen activated protein kinase
- MEK
mitogen activated extracellular regulated kinase
- R
receptor
- JNK
c-Jun NH2-terminal kinase
- dn
dominant negative
- P
phospho-
- ca
constitutively active
- WT
wild type.
Footnotes
This work was funded to P.D. from PHS grants (R01-DK52825, P01-CA104177, R01-CA108520), Department of Defense Awards (DAMD17-03-1-0262); to S.G. from PHS grants (R01-CA63753; R01-CA77141) and a Leukemia Society of America grant 6405-97. A portion of Dr. Yacoub’s funding is from the Department of Radiation Oncology, Virginia Commonwealth University and PD is the holder of the Universal Inc. Professorship in Signal Transduction Research.
References
- Baba I, Shirasawa S, Iwamoto R, et al. Involvement of deregulated epiregulin expression in tumorigenesis in vivo through activated Ki-Ras signaling pathway in human colon cancer cells. Cancer Res. 2000;60:6886–6889. [PubMed] [Google Scholar]
- Baldwin AS. Control of oncogenesis and cancer therapy resistance by the transcription factor NF-κB. J Clin Invest. 2001;107:241–246. doi: 10.1172/JCI11991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabannes E, Khan G, Aillet F, et al. Mutations in the IkBa gene in Hodgkin's disease suggest a tumour suppressor role for Ikappa Balpha. Oncogene. 1999;18:3063–3070. doi: 10.1038/sj.onc.1202893. [DOI] [PubMed] [Google Scholar]
- Camirand A, Lu Y, Pollack M. Co-targeting HER2/ErbB2 and insulin-like growth factor-1 receptors causes synergistic inhibition of growth in HER2-overexpressing breast cancer cells. Med Sci Monit. 2002;8:BR521–BR526. [PubMed] [Google Scholar]
- Caron RW, Yacoub A, Zhu X, et al. H-RAS V12-induced radioresistance in HCT116 colon carcinoma cells is heregulin dependent. Molecular Cancer Therapeutics. 2005a;4:243–255. [PubMed] [Google Scholar]
- Caron RW, Yacoub A, Li M, et al. Activated forms of H-RAS and K-RAS differentially regulate membrane association of PI3K, PDK-1, and AKT and the effect of therapeutic kinase inhibitors on cell survival. Mol Cancer Therapeutics. 2005b;4:257–270. [PubMed] [Google Scholar]
- Chen S, Dai Y, Harada H, Dent P, Grant S. Mcl-1 down-regulation potentiates ABT-737 lethality by cooperatively inducing Bak activation and Bax translocation. Cancer Res. 2007;67:782–791. doi: 10.1158/0008-5472.CAN-06-3964. [DOI] [PubMed] [Google Scholar]
- Cho NL, Javid SH, Carothers AM, Redston M, Bertagnolli MM. Estrogen receptors alpha and beta are inhibitory modifiers of Apc-dependent tumorigenesis in the proximal colon of Min/+ mice. Cancer Res. 2007;67:2366–2372. doi: 10.1158/0008-5472.CAN-06-3026. [DOI] [PubMed] [Google Scholar]
- Chuang E, Barnard D, Hettich L, Zhang XF, Avruch J, Marshall MS. Critical binding and regulatory interactions between Ras and Raf occur through a small, stable N-terminal domain of Raf and specific Ras effector residues. Mol Cell Biol. 1994;14:5318–5325. doi: 10.1128/mcb.14.8.5318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox AD, Der CJ. The dark side of Ras: regulation of apoptosis. Oncogene. 2003;22:8999–9006. doi: 10.1038/sj.onc.1207111. [DOI] [PubMed] [Google Scholar]
- Cunningham MP, Thomas H, Fan Z, Modjtahedi H. Responses of human colorectal tumor cells to treatment with the anti-epidermal growth factor receptor monoclonal antibody ICR62 used alone and in combination with the EGFR tyrosine kinase inhibitor gefitinib. Cancer Res. 2006;66:7708–7715. doi: 10.1158/0008-5472.CAN-06-1000. [DOI] [PubMed] [Google Scholar]
- Dent P, Reardon DB, Park JS, et al. Radiation-induced release of transforming growth factor alpha activates the epidermal growth factor receptor and mitogen-activated protein kinase pathway in carcinoma cells, leading to increased proliferation and protection from radiation-induced cell death. Mol. Biol. Cell. 1999;10:2493–2506. doi: 10.1091/mbc.10.8.2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dent P, Yacoub A, Fisher PB, Hagan MP, Grant S. MAPK pathways in radiation responses. Oncogene. 2003;22:5885–5896. doi: 10.1038/sj.onc.1206701. [DOI] [PubMed] [Google Scholar]
- Ellis CA, Clark G. The importance of being K-RAS. Cell Signaling. 2000;12:425–434. doi: 10.1016/s0898-6568(00)00084-x. [DOI] [PubMed] [Google Scholar]
- Goldkorn T, Balaban N, Shannon M, Matsukuma K. EGF receptor phosphorylation is affected by ionizing radiation. Biochimica Biophysica Acta. 1997;1358:289–299. doi: 10.1016/s0167-4889(97)00063-3. [DOI] [PubMed] [Google Scholar]
- Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5:341–354. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- Imai K, Takaoka A. Comparing antibody and small-molecule therapies for cancer. Nat Rev Cancer. 2006;6:714–727. doi: 10.1038/nrc1913. [DOI] [PubMed] [Google Scholar]
- Joneson T, White MA, Wigler MH, Bar-Sagi D. Stimulation of membrane ruffling and MAP kinase activation by distinct effectors of RAS. Science. 1996;271:810–812. doi: 10.1126/science.271.5250.810. [DOI] [PubMed] [Google Scholar]
- Konecny GE, Pegram MD, Venkatesan N, et al. Activity of the dual kinase inhibitor lapatinib ( GW572016) against HER-2-overexpressing and trastuzumab-treated breast cancer cells. Cancer Res. 2006;66:1630–1639. doi: 10.1158/0008-5472.CAN-05-1182. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- Liebmann C. Regulation of MAP kinase activity by peptide receptor signalling pathway: paradigms of multiplicity. Cell Signaling. 2001;13:777–785. doi: 10.1016/s0898-6568(01)00192-9. [DOI] [PubMed] [Google Scholar]
- Lin NU, Winer EP. Small molecule tyrosine kinase inhibitors. Breast Cancer Res. 2004;6:204–210. doi: 10.1186/bcr919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Zi X, Zhao Y. Insulin-like growth factor-I receptor signaling and resistance to trastuzumab (Herceptin) J Natl Cancer Inst. 2001;93:1852–1857. doi: 10.1093/jnci/93.24.1852. [DOI] [PubMed] [Google Scholar]
- Ludde T, Kubicka S, Plumpe J, Liedtke C, Manns MP, Trautwein C. RAS adenoviruses modulate Cyclin E protein expression and DNA synthesis after partial hepatectomy. Oncogene. 2001;20:5264–5278. doi: 10.1038/sj.onc.1204690. [DOI] [PubMed] [Google Scholar]
- Moriuchi A, Hirono S, Ido A, et al. Additive and inhibitory effects of simultaneous treatment with growth factors on DNA synthesis through MAPK pathway and G1 Cyclins in rat hepatocytes. Biochemical Biophysical Research Communications. 2001;280:368–373. doi: 10.1006/bbrc.2000.4063. [DOI] [PubMed] [Google Scholar]
- Nahta R, Yuan LX, Du Y, Esteva SJ. Lapatinib induces apoptosis in trastuzumab-resistant breast cancer cells: effects on insulin-like growth factor I signaling. Mol Cancer Ther. 2007;6:667–674. doi: 10.1158/1535-7163.MCT-06-0423. [DOI] [PubMed] [Google Scholar]
- Nelson MH, Dolder CR. Lapatinib: a novel dual tyrosine kinase inhibitor with activity in solid tumors. Breast Cancer Res. 2004;6:204–210. [Google Scholar]
- Olayioye MA, Neve RM, Lane HA, Hynes NE. The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J. 2000;19:3159–3167. doi: 10.1093/emboj/19.13.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oltersdorf T, Elmore SW, Shoemaker AR, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- Pao W, Miller VA, Politi KA, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peschard P, Park M. Escape from Cbl-mediated downregulation: a recurrent theme for oncogenic deregulation of receptor tyrosine kinases. Cancer Cell. 2003;3:519–523. doi: 10.1016/s1535-6108(03)00136-3. [DOI] [PubMed] [Google Scholar]
- Qin B, Ariyama H, Baba A, et al. Activated Src and Ras induce gefitinib resistance by activation of signaling pathways downstream of epidermal growth factor receptor in human gallbladder adenocarcinoma cells. Cancer Chemother Pharmacol. 2006a;58:577–584. doi: 10.1007/s00280-006-0219-4. [DOI] [PubMed] [Google Scholar]
- Qin JZ, Xin H, Sitailo LA, Denning MF, Nickoloff BJ. Enhanced killing of melanoma cells by simultaneously targeting Mcl-1 and NOXA. Cancer Res. 2006b;66:9636–9645. doi: 10.1158/0008-5472.CAN-06-0747. [DOI] [PubMed] [Google Scholar]
- Raffo AJ, Perlman H, Chen MW, et al. Overexpression of bcl-2 protects prostate cancer cells fro apoptosis in vitro and confers resistance to androgen depletion in vivo. Caner Res. 1995;55:4438–4445. [PubMed] [Google Scholar]
- Reed JC, Miyashita T, Krajewski S, et al. BCL-2 family proteins: regulators of cell death involved in the pathogenesis of cancer and resistance to therapy. J Cell Biochem. 1996;60:23–32. doi: 10.1002/(SICI)1097-4644(19960101)60:1%3C23::AID-JCB5%3E3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Ross PJ, George M, Cunningham D, DiStefano F, Andreyev HJN, Workman P, Clarke PA. Inhibition of Kirsten RAS expression in human colorectal cancer using rationally selected Kirsten-RAS antisense oligonucleotides. Mol Can Ther. 2001;1:29–41. [PubMed] [Google Scholar]
- Rusnak DW, Lackey K, Affleck K, et al. The effects of the novel, reversible epidermal growth factor receptor/ErbB-2 tyrosine kinase inhibitor, GW2016, on the growth of human normal and tumor-derived cell lines in vitro and in vivo. Mol Cancer Ther. 2001;1:85–94. [PubMed] [Google Scholar]
- Salomon DS, Brandt R, Ciardiello F, Normanno N. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit Rev Oncol Hematol. 1995;19:183–232. doi: 10.1016/1040-8428(94)00144-i. [DOI] [PubMed] [Google Scholar]
- Schmidt-Ullrich RK, Contessa JN, Lammering G, et al. ERBB receptor tyrosine kinases and cellular radiation responses. Oncogene. 2003;22:5855–5865. doi: 10.1038/sj.onc.1206698. [DOI] [PubMed] [Google Scholar]
- Shirasawa S, Furuse M, Yokoyama N, Sasazuki T. Altered growth of human colon cancer cell lines disrupted at activated Ki-ras. Science. 1993;260:85–88. doi: 10.1126/science.8465203. [DOI] [PubMed] [Google Scholar]
- Sizeland AM, Burgess AW. Anti-sense transforming growth factor αoligonucleotides inhibit autocrine stimulated proliferation of a colon carcinoma cell line. Mol Biol Cell. 1992;3:1235–1243. doi: 10.1091/mbc.3.11.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sizemore N, Cox AD, Barnard JA, et al. Pharmacological inhibition of Ras-transformed epithelial cell growth is linked to down-regulation of epidermal growth factor-related peptides. Gastroenterology. 1999;117:567–576. doi: 10.1016/s0016-5085(99)70449-x. [DOI] [PubMed] [Google Scholar]
- Sklar MD. The ras oncogenes increase the intrinsic resistance of NIH 3T3 cells to ionizing radiation. Science. 1988;239:645–647. doi: 10.1126/science.3277276. [DOI] [PubMed] [Google Scholar]
- Sok JC, Coppelli FM, Thomas SM, et al. Mutant epidermal growth factor receptor (EGFRvIII) contributes to head and neck cancer growth and resistance to EGFR targeting. Clin Cancer Res. 2006;12:5064–5073. doi: 10.1158/1078-0432.CCR-06-0913. [DOI] [PubMed] [Google Scholar]
- Sumitomo M, Tachibana M, Nakashima J, et al. An essential role for nuclear factor kappa B in preventing TNF-alpha-induced cell death in prostate cancer cells. J Urol. 1999;161:674–679. [PubMed] [Google Scholar]
- Sunpaweravong P, Sunpaweravong S, Puttawibul P, et al. Epidermal growth factor receptor and cyclin D1 are independently amplified and overexpressed in esophageal squamous cell carcinoma. J Cancer Res Clin Oncol. 2005;131:111–119. doi: 10.1007/s00432-004-0610-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szakacs G, Paterson JK, Ludwig JA, et al. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5:219–234. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- Trudel S, Li ZH, Rauw J, Tiedemann RE, Wen XY, Stewart AK. Preclinical studies of the pan-Bcl inhibitor obatoclax (GX015-070) in multiple myeloma. Blood. 2007;109:5430–5438. doi: 10.1182/blood-2006-10-047951. [DOI] [PubMed] [Google Scholar]
- Willis SN, Chen L, Dewson G, et al. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005;19:1294–1305. doi: 10.1101/gad.1304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witters LM, Witkoski A, Planas-Silva MD, et al. Synergistic inhibition of breast cancer cell lines with a dual inhibitor of EGFR-HER-2/neu and a Bcl-2 inhibitor. Oncol Rep. 2007;17:465–469. [PubMed] [Google Scholar]
- Wood ER, Truesdale AT, McDonald OB, et al. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004;64:6652–6659. doi: 10.1158/0008-5472.CAN-04-1168. [DOI] [PubMed] [Google Scholar]
- Xia W, Bacus S, Hegde P, et al. A model of acquired autoresistance to a potent ErbB2 tyrosine kinase inhibitor and a therapeutic strategy to prevent its onset in breast cancer. Proc Natl Acad Sci USA. 2006;103:7795–7800. doi: 10.1073/pnas.0602468103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia W, Husain I, Liu L, et al. Lapatinib antitumor activity is not dependent upon phosphatase and tensin homologue deleted on chromosome 10 in ErbB2-overexpressing breast cancers. Cancer Res. 2007;67:1170–1175. doi: 10.1158/0008-5472.CAN-06-2101. [DOI] [PubMed] [Google Scholar]
- Yan J, Roy S, Apolloni A, Lane A, Hancock JF. RAS isoforms vary in their ability to activate Raf-1 and phosphoinositide 3-kinase. J Biol Chem. 1998;273:24052–24056. doi: 10.1074/jbc.273.37.24052. [DOI] [PubMed] [Google Scholar]
- Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nature Rev Mol Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- Zambetti GP. The p53 mutation "gradient effect" and its clinical implications. J Cell Physiol. 2007;213:370–373. doi: 10.1002/jcp.21217. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Li S, Hu YP, et al. Blockade of EGFR and ErbB2 by the novel dual EGFR and ErbB2 tyrosine kinase inhibitor GW572016 sensitizes human colon carcinoma GEO cells to apoptosis. Cancer Res. 2006;66:404–411. doi: 10.1158/0008-5472.CAN-05-2506. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.