

Abstract
Iron–sulfur (Fe–S) clusters are essential for numerous biological processes, including mitochondrial respiratory chain activity and various other enzymatic and regulatory functions. Human Fe–S cluster assembly proteins are frequently encoded by single genes, and inherited defects in some of these genes cause disease. Recently, the spectrum of diseases attributable to abnormal Fe–S cluster biogenesis has extended beyond Friedreich ataxia to include a sideroblastic anemia with deficiency of glutaredoxin 5 and a myopathy associated with a deficiency of a Fe–S cluster assembly scaffold protein, ISCU. Mutations within other mammalian Fe–S cluster assembly genes could be causative for human diseases that manifest distinctive combinations of tissue-specific impairments. Thus, defects in the iron–sulfur cluster biogenesis pathway could underlie many human diseases.
Iron–sulfur proteins: biological roles and relevance
Iron–sulfur (Fe–S) clusters are ancient biological prosthetic groups that are essential for many fundamental processes including photosynthesis and respiration (reviewed in Refs. [1,2]). The most common Fe–S clusters in eukaryotes are the [2Fe–2S] and [4Fe–4S] clusters, which are formed by tetrahedrally coordinated iron atoms with bridging sulfides and are most often ligated to the protein through cysteine residues. The chemical reactivities of iron and sulfur, together with variations in the composition, redox potential, oxidation state, physical accessibility of the cluster and effects of the local protein environment, enable these versatile cofactors to accept or donate single electrons, catalyze enzymatic reactions or function as regulatory proteins. For instance, Fe–S clusters are essential components of respiratory electron transfer complexes as well as the tricarboxylic acid cycle (TCA cycle) enzymes, aconitase and succinate dehydrogenase (Figure 1a). In addition, the Fe–S clusters within DNA repair enzymes Fanconi anemia group J protein (FancJ) and Xeroderma pigmentosum group D protein (XPD) facilitate DNA damage recognition and repair [3]. Moreover, Fe–S cluster assembly and disassembly in mammalian iron regulatory protein-1 (IRP1) alters the active site conformation and accessibility and determines whether IRP1 binds its mRNA targets in response to oxidative stress and intracellular iron levels (reviewed in Refs. [4–6]).
Figure 1.

Disruption of iron–sulfur (Fe–S) proteins can have a myriad of deleterious cellular consequences. Because Fe–S clusters are essential electron carriers and enzyme cofactors in many proteins, defects in Fe–S clusters biogenesis can disrupt many cellular process. (a) Fe–S clusters are essential cofactors in energy metabolism. Fe–S proteins are among the most important electron carriers in nature and are particularly important in the mitochondrial respiratory chain, in which up to 12 different Fe–S clusters shuttle electrons through complexes I–III. Electrons from the oxidation of NADH and succinate are transferred through a chain of redox centers consist of flavins, Fe–S clusters, ubiquinone (Q and QH2), hemes and copper centers (CuA and CuB) to reduce O2 to H2O. The free energy of electron transport is coupled to ATP synthesis. In addition, the ability of Fe–S clusters to coordinate ligands and stabilize protein structures also allows them to facilitate various enzymatic functions. For instance, mitochondrial aconitase is an integral part of the tricarboxylic acid cycle (TCA cycle), and its [4 Fe–4S] cluster is essential for substrate binding and activation. (b) Because Fe–S proteins play a critical role in a wide range of cellular activities, mutations or pathological conditions that disrupt Fe–S cluster stability or biogenesis/repair are associated with several human diseases. For instance, germline mutations of the gene encoding succinate dehydrogenase subunit B (SDHB), a Fe–S protein in respiratory complex II, are a major cause of cancer of the kidney, adrenal gland and thyroid gland [63]. Mutations that destabilize the Fe–S clusters in DNA repair enzymes XPD and FancJ are associated with the phenotypes in patients with trichothiodystrophy and Fanconi anemia respectively [3]. Severe defects in Fe–S cluster biogenesis/repair can profoundly decrease the activities of respiratory complexes, the TCA cycle and the heme biosynthesis pathway [7–9], resulting in decreased energy production and increased oxidative stress. In addition, disruption of Fe–S cluster biogenesis can lead to mitochondrial iron overload and cytosolic iron depletion. In the cytosol, defects in Fe–S cluster biogenesis might affect cytosolic aconitase (c-aconitase) activity and therefore citrate metabolism, which can disrupt the balance between glycolysis and fatty acid biosynthesis. Defects in cytosolic Fe–S cluster biogenesis/repair might also impair ribosome biogenesis, and purine catabolism pathways.
Because Fe–S clusters play a critical role in a wide range of cellular activities, significant disruptions in Fe–S cluster biogenesis and repair are expected to also affect numerous basic cellular processes (Figure 1b). Indeed, defects in this important biological pathway are now recognized as the cause of several human diseases. Over the last decade, studies of Friedriech ataxia revealed that deficiency of the protein frataxin (FXN) results in the loss of Fe–S protein activities, mitochondrial iron overload, oxidative damage and ultimately mitochondrial failure [7]. In the last 2 years, defects in Fe–S cluster assembly genes have been shown to be responsible for two more human diseases: a splicing defect in glutaredoxin 5 (GLRX5) was implicated as a cause of sideroblastic anemia [8], and recently, a splicing defect in the iron–sulfur cluster scaffold ISCU in skeletal muscle was shown to cause myopathy with severe exercise intolerance and deficiencies in succinate dehydrogenase and aconitase activity [9,10]. Here, we summarize the mechanisms of Fe–S cluster biogenesis and highlight recent discoveries of human diseases caused by mutations in Fe–S cluster assembly genes.
Iron–sulfur cluster biogenesis
Among the various theories about the origin of life, one possibility is that life originated in an ‘iron–sulfur world’. Conditions in the primordial atmosphere were likely favorable for spontaneous Fe–S cluster assembly, and early life forms might have used Fe–S clusters to capture and release electrons in primitive metabolic reactions [11]. For instance, a central metabolic pathway of intermediary metabolism, the TCA cycle, might be derived from an ancient reductive citric acid cycle that used two Fe–S enzymes: aconitase and succinate dehydrogenase. Because ferrous iron bioavailability is limited in the oxidative environment of modern earth and because Fe–S proteins are required for basic intermediary metabolism, it is not surprising that nearly all organisms use highly conserved proteins and enzymes to facilitate Fe–S cluster synthesis. The basic components of this process were originally identified in bacterial operons [1], and homologs of these proteins have been identified in eukaryotes, including the Saccharomyces cerevisiae (reviewed in Ref. [2]) and human genomes (reviewed in Ref. [12]).
Studies in yeast, zebrafish and plants have shown that up to 20 different proteins are involved in eukaryotic Fe–S cluster biogenesis (Table 1). A cysteine desulfurase (IscS or SufS in bacteria, NFS1 or ISCS in human) is required to abstract the sulfur atom from cysteine and to donate the sulfur to a scaffold protein, such as human ISCU, on which nascent clusters are assembled (Figure 1). In many organisms, iron delivery might be facilitated by Frataxin [13]. In vitro studies have shown that scaffold proteins can assemble either [2Fe–2S] or [4Fe–4S] clusters and donate these clusters to recipient proteins [14,15]. Redox proteins such as ferredoxin 1 (FDX1) and ferredoxin reductase (FDXR) are thought to facilitate cluster assembly on scaffold proteins [2], whereas heat shock 70-kDa protein 9 (HSCPA9) and a co-chaperone HSCB are believed to use energy derived from ATP to drive conformational changes in scaffold proteins that facilitate cluster transfer to intermediate or final recipient proteins [1]. In addition to the basic components discussed above, LYR motif containing 4 protein (LYRM4, also called ISD11) seems to be important for appropriate folding and function of the cysteine desulfurase [16–18], and GLRX5 homologs have been implicated in Fe–S cluster biogenesis in yeast [19], zebrafish [20] and humans [8]. Other proteins including NUBP1, NUBP2, NARF and CIAO1 (Table 1) are thought to be important for cytosolic Fe–S protein maturation [2,21].
Table 1.
List of human iron-sulfur cluster assembly genes and their homologs
| Symbol | Full name | Chromosomal localization | S. cerevisiae homologues | Proposed function | Lethal mutations in model system | Refs |
|---|---|---|---|---|---|---|
| NFS1 or ISCS | NFS1 nitrogen fixation 1 homolog | 20q11.22 | NFS1 | Cysteine desulfurase; provides sulfur for Fe–S cluster assembly | Deletion: lethal in S. cerevisiae | [48,65] |
| LYRM4 | LYR motif containing 4 | 6p25.1 | ISD11 | ISCS folding | Deletion: lethal in S. cerevisiae | [16,17,66] |
| ISCU | Iron-sulfur cluster scaffold homolog (E. coli) | 12q24.1 | ISU1, ISU2 | Scaffold in mammalian mitchondria and cytosol | Double deletion of ISU1 and ISU2 is lethal | [67,68] |
| NFU1 | NFU1 iron-sulfur cluster scaffold homolog (S. cerevisiae) | 2p15-p13 | NFU1 | Potential alternative scaffold in mitchondria and cytosol | Deletion is synthetic lethal with ssq1 in S. cerevisiae | [68] |
| FXN | Frataxin | 9q13 | YFH1 | Iron chaperone | Deletion: lethal in mouse Synthetic lethal with Succinate dehydrogenase subunit b (Sdhb) in C.elegans | [13,36,69] |
| GLRX5 | Glutaredoxin 5 | 14q32.13 | GRX5 | Deglutathionylates cysteine residues of target proteins or acts as an intermediate Fe–S cluster carrier protein | Deletion is synthetic lethal with grx2 in S. cerevisiae Deletion: lethal in zebrafish | [20,42] |
| FDX 1 | Ferredoxin 1 | 11q22 | YAH1 | Provides reducing equivalents for initial cluster formation | Deletion: lethal in S. cerevisiae | [66,70] |
| FDXR | Ferredoxin reductase | 17q24-q25 | ARH1 | Provides reducing equivalents for initial cluster formation | Deletion: lethal in S. cerevisiae | [66,70] |
| HSPA9 | Heat shock 70 kDa protein 9 (mortalin) | 5q31.1 | SSQ1 | Facilitates cluster transfer from scaffold to recipient proteins by promoting conformational changes | ND | [70] |
| HSCB | HscB iron-sulfur cluster co-chaperone homolog (E. coli) | 22q12.1 | JAC1 | Cochaperone that might facilitate cluster transfer | Deletion: lethal in S. cerevisiae | [66,71] |
| ISCA1 | Iron-sulfur cluster assembly 1 homolog (S. cerevisiae) | 9q21.33 | ISA1 | Likely either a scaffold or regulatory protein | ND | [72,73] |
| ISCA1L | Iron-sulfur cluster assembly 1 homolog (S. cerevisiae)-like | 5q12.1 | ISA1 | ISCA1 homolog | ND | |
| ISCA2 | Iron-sulfur cluster assembly 2 homolog (S. cerevisiae) | 14q24.3 | ISA2 | ISCA1 homolog | ND | |
| NUBP2 | Nucleotide binding protein2 (MinD homolog, E. coli) | 16p13.3 | CFD1 | Scaffold proteins for cytosolic Fe–S cluster maturation | Deletion: lethal in S. cerevisiae | [74,75] |
| GFER | growth factor, augmenter of liver regeneration (ERV1 homolog, S. cerevisiae) | 16p13.3-p13.12 | ERV1 | Fe–S cluster biogenesis in mitochondrial intermembrane space | Deletion: lethal in S. cerevisiae | [66,76] |
| NUBP1 | Nucleotide binding protein1 (MinD homolog, E. coli) | 16p13.13 | NBP35 | Scaffold proteins for cytosolic Fe–S cluster maturation | Deletion: lethal in S. cerevisiae | [74,77] |
| CIAO1 | Cytosolic iron-sulfur protein assembly 1 homolog (S. cerevisiae) | 2q11.2 | CIA1 | Cytosolic Fe–S cluster maturation, transcriptional regulation | Deletion: lethal in S. cerevisiae | [66,78,79] |
| NARF | Nuclear prelamin A recognition factor | 17q25.3 | Nar1 | Cytosolic Fe–S cluster maturation | Deletion: lethal in S. cerevisiae | [66,80] |
| C1orf69 | Chromosome 1 open reading frame 69 | 1q42.13 | Iba57 | Maturation of subset of mitochondrial Fe–S proteins | ND | [81] |
Abbreviations: Fe–S, iron–sulfur; ND, not determined.
Another important finding from genetic and biochemical studies is that Fe–S cluster biogenesis is required for the regulation of mitochondrial iron homeostasis. Many yeast strains that were depleted of Fe–S cluster biogenesis proteins (e.g. nfs1, yfh1, isu1, isu2, isa1, isa2, nfu1, ssq1, jac1, yah1) showed marked iron accumulation in their mitochondria [2]. As discussed in more detail below, mitochondrial iron overload is also a prominent feature of human Fe–S cluster assembly disorders.
Iron–sulfur cluster biogenesis genes implicated in human diseases
Human disease mutations have now been reported for Frataxin, GLRX5 and ISCU (Figure 2). Notably, none of these human mutations is completely inactivating, which is not surprising because work in various model systems has revealed that severe defects in Fe–S cluster assembly can be lethal. In S. cerevisiae, deletions of genes involved in Fe–S cluster assembly, including NFS1, ISD11, JAC1, YAH1, ARH1, CFD1, ERV1, NBP35, NAR1 and CIA1 are lethal (Table 1). In addition, synthetic lethality was observed in pairwise combinations of several other Fe–S cluster assembly genes including ISU1, ISU2, NFU1, SSQ1 and GRX5. Notably, deletions of the GLRX5 homolog in zebrafish and of Frataxin in mouse are lethal during embryogenesis, and exhaustive gene disruption experiments in human colon cancers cells do not yield FDXR null clones, strongly suggesting that FDXR is essential for viability [22]. Furthermore, siRNA studies demonstrate that depletion of mammalian Fe–S cluster assembly factors, such as ISCU, ISCS and NARF, can inactivate various metabolic enzymes including aconitase and xanthine oxidase, misregulate proteins involved in iron metabolism and decrease cell viability [21,23,24]. It is therefore probable that complete inactivation of the human Fe–S assembly proteins would also be profoundly deleterious.
Figure 2.

Iron–sulfur (Fe-S) cluster biogenesis in eukaryotes. Fe–S cluster biogenesis is a complex pathway involving many enzymes and proteins. (a) In the mitochondria and cytosol of mammalian cells, cysteine desulfurases (m-ISCS and c-ISCS) remove sulfur from free cysteine and donate the sulfur to scaffold proteins (e.g. m-ISCU and c-ISCU), whereas frataxin (FXN) is proposed to be an iron chaperone that delivers iron in an accessible form. Scaffold proteins can assemble and donate either [2Fe–2S] (not shown) or [4Fe–4S] clusters to recipient proteins. In the mitochondria, redox proteins such as FDX and FDXR likely deliver reducing equivalents needed for cluster assembly, and protein chaperones HSPA9/HSCB and glutaredoxin GRX5 facilitate the transfer of the cluster from scaffolds to recipient proteins. In the cytosol, additional factors including NUBP1, NUBP2, NARF and CIAO1 might facilitate cluster transfer. ISCS and ISCU are shown in ribbon structures. Other mitochondrial cluster biogenesis factors are shown in light blue ovals and cytoplasmic factors in dark blue ovals. (b) In yeast mitochondria, cysteine desulfurase Nfs1p, Isd11p, frataxin homolog Yfh1p and redox proteins Yah1p and Arh1p facilitate cluster assembly in scaffold proteins, whereas chaperone proteins Ssq1p and Jac1p and glutaredoxin Grx5p facilitate cluster transfer from scaffold to recipient proteins. The scaffold proteins Isu1p, Isu2p and Yfh1p have thus far only been detected in the mitochondria matrix, leading to the proposal that cluster assembly occurs exclusively in the mitochondria matrix, and preformed clusters are exported through Atm1p and transferred to cytosolic recipients through Cfd1p, Nar1p, Cia1p and Nbp35p. Nfs1p and Isu1p are shown in ribbon structures. Other mitochondrial cluster biogenesis factors are shown in light blue ovals and cytoplasmic factors in dark blue ovals.
Friedreich ataxia: the role of Frataxin in iron–sulfur cluster assembly
Friedreich ataxia (FRDA) is an autosomal recessive neurodegenerative disorder caused by deficiency of the nuclear-encoded protein, Frataxin (Table 2). Frataxin deficiency leads to progressive spinocerebellar neurodegeneration associated with gait and limb ataxia, dysarthria (a neuromuscular speech disorder) and muscle weakness, as well as cardiomyopathy and diabetes [25]. The neurologic manifestations result from degeneration of the dorsal root ganglia, the posterior columns, spinocerebellar tracts, corticospinal tracts of the spinal cord and large myelinated fibers in the peripheral nerves. Ataxia usually develops during childhood or the early teens, and patients generally die from cardiomyopathy in early adulthood.
Table 2.
Clinical, metabolic, biochemical and molecular findings in patients presenting iron–sulfur cluster biogenesis defects
| Disorder (occurrence) | Etiology | Clinical features | Biochemical features | Refs |
|---|---|---|---|---|
| Friedreich ataxia (1–2 per 50 000 live births) | GAA expansion in the Fxn gene results in reduced mRNA | progressive limb and gait ataxia, dysarthria, absent tendon reflexes, hypertrophic cardiomyopathy, death occurs in the 30s | Deficiency of aconitase and complex I-III activities in endomyocardial biopsy; mitochondrial DNA depletion in heart and skeletal muscle; increased mitochondrial iron in heart, liver, and spleen | [7,26,29] |
| Sideroblastic anemia with GLRX5 deficiency (one case) | Homozygous point mutation in GLRX5 results in splicing defect and reduced mRNA | Microcytic anemia, hepatosplenomegaly | Ringed sideroblasts, increased iron staining in erythroblasts and macrophages, decreased aconitase activity in patient peripheral blood mononuclear cells | [8] |
| Myopathy with ISCU deficiency (19 cases in northern Sweden) | Homozygous intronic mutation in ISCU results in splicing defect and reduced mRNA | Severe exercise intolerance, shortness of breath, cardiac palpitations, lactic acidosis, muscle swelling and pain, myoglobinuria | Deficiency in skeletal muscle succinate dehydrogenase and aconitase; iron-rich mitochondrial inclusion in skeletal muscle biopsy | [9,10,54] |
Expansion of the unstable GAA triplet repeat in FXN is the most common causal mutation of FRDA [26] (Figure 2). Interestingly, the GAA expansion occurs in the intronic sequence, thus distinguishing this disease from Huntington’s and other trinucleotide repeat diseases. Normal alleles have 7–38 repeats, whereas FRDA alleles have 66–1700 repeats, resulting in significantly decreased Frataxin expression. Longer repeats cause a more profound Frataxin deficiency and are associated with earlier onset and increased severity of the disease. Furthermore, recent studies suggest that the progressive pathology in the dorsal root ganglia results from an age-dependent, tissue-specific, increased expansion of the GAA triplet-repeat sequence [27]. It has been proposed that the long GAA repeats interfere with transcription by forming aberrant DNA structures or by inducing the formation of repressive heterochromatin [28].
Analysis of patient tissue samples revealed that activities of two Fe–S proteins, mitochondrial aconitase and succinate dehydrogenase, were markedly decreased, suggesting that the synthesis or stability of Fe–S clusters is defective in FRDA patients [29,30]. Although structural, complementation and biochemical studies on the function of Frataxin have yielded complex and somewhat contradictory results [31–33], many findings indicate that Frataxin’s main function is to serve as an iron-binding chaperone protein for Fe–S cluster assembly [13]. Frataxin is a small protein that contains an acidic surface that can bind several ferrous iron atoms, whereas other surfaces enable Frataxin to bind the scaffold protein, ISCU, or the heme biosynthetic enzyme, Ferrochelatase, thus potentially allowing Frataxin to readily donate ferrous iron for incorporation into nascent Fe–S clusters as they are assembled on ISCU [13,34].
Interestingly, YFH1 (the yeast FXN homolog)-deficient yeast strains develop profound mitochondrial iron overload; similarly, mitochondrial iron overload is also observed in the hearts and brains of FRDA patients [35] (Box 1) and heart-specific knockout mouse models [36]. These results suggest that eukaryotic mitochondrial iron homeostasis is tightly regulated and that a Fe–S protein might be involved in signaling information about the status of mitochondrial iron stores to the nucleus. In the absence of an appropriate signal, mitochondrial iron uptake might be enhanced, efflux might be diminished, and iron might accumulate within insoluble precipitates in the mitochondrial matrix or within the iron sequestration protein, mitochondrial Ferritin [32]. The resulting mitochondrial iron overload might cause mitochondrial damage via iron-catalyzed oxidation reactions [37]. Furthermore, an interference with mitochondrial iron metabolism can cause disruptions in other subcellular compartments. In yeast, YFH1 deletion increases the expression of >70 nuclear genes, including many that are involved in cellular iron uptake [38]. mRNA levels of numerous transcripts, including ISCU, decrease in Frataxin-deficient mouse heart tissue [39]. In human cells, disruption of Fe–S cluster biogenesis and mitochondrial iron homeostasis can activate iron regulatory proteins (IRPs) [30,23,40], thereby triggering increased cellular iron uptake. The dysregulation of mitochondrial iron homeostasis and activation of IRPs has the potential to engage the cell in a vicious cycle in which increased cellular iron uptake further exacerbates mitochondrial damage.
Box 1
Detection of mitochondrial iron overload might provide a valuable diagnostic tool for diseases associated with defects in iron–sulfur (Fe–S) cluster biogenesis. Mitochondrial iron overload has been found in Friedreich ataxia (FRDA; FXN deficiency), a myopathy with ISCU deficiency as well as sideroblastic anemias (Figure I). Perls’ Prussian blue staining is the most common histochemical test for nonheme ferric iron and involves the treatment of biopsy samples with acid solutions of ferrocyanides. Any nonheme ferric ion present in the tissue reacts with the ferrocyanide and forms a bright blue insoluble pigment called Prussian blue, or ferric ferrocyanide. The sensitivity of Prussian blue staining can be further enhanced using 3,3′-diaminobenzidine (DAB). Ferric ferrocyanide can oxidize DAB in the presence of hydrogen peroxide to produce an intense brown black stain. Detection of the distinctive punctate iron staining pattern might be valuable for diagnosing other diseases associated with defects in Fe–S cluster biogenesis, because many yeast strains that were depleted of other Fe–S cluster biogenesis factors also showed marked mitochondrial iron accumulation.
Figure I.
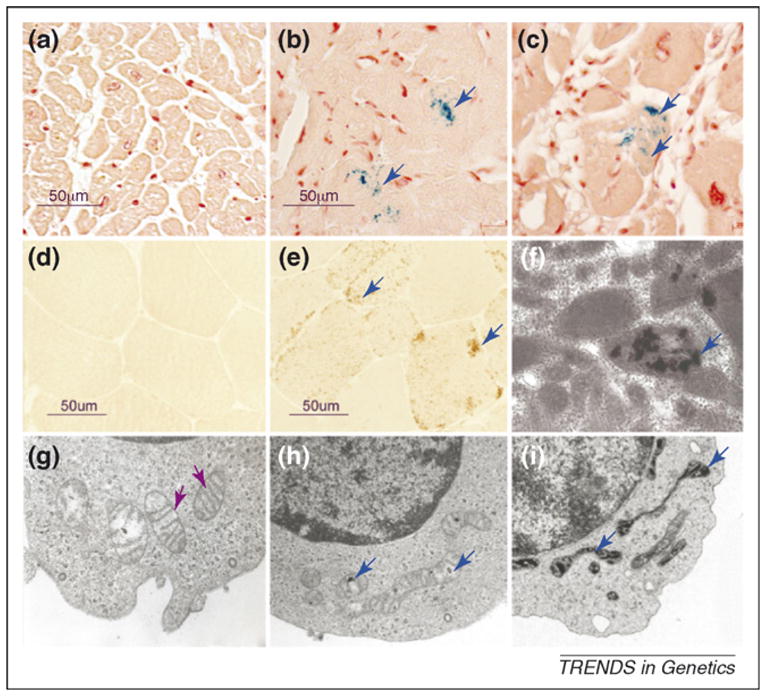
Mitochondrial iron overload in Friedreich ataxia, a myopathy with ISCU deficiency, and an acquired siderblastic anemia. Mitochondrial overload was detected in patient samples using histochemical stain and electron microscopy. Blue arrows indicate mitochondrial iron deposits and purple arrows indicate well preserved cristae and a relative absence of iron deposits. Prussian blue staining revealed that excess ferric iron was present in the hearts of a 9-year-old patient (b) and a 26-year-old patient (c) with FRDA compared with the heart of a unaffected 35-year-old individual (a). Biochemical and electron microscopic analyses have previously indicated that iron accumulates mainly in the mitochondria. In normal skeletal muscle biopsies, absence of brown DAB-enhanced Prussian blue staining is indicative of normal levels of ferric iron (d), whereas brown punctate deposits reveal mitochondrial iron overload throughout the individual muscle fibers of patients with myopathy and ISCU deficiency (e), a finding that has been confirmed by electron microscopy (f) [54]. In human sideroblastic anemias, dark deposits within distorted mitochondria are caused by iron overload [64]. Electron microscopy analysis of normoblasts (normal red cell precursors) of a patient from an early stage in development of acquired sideroblastic anemia showed that most of the mitochondria were still well preserved (as evidenced by well preserved cristae) (g), whereas in the sideroblasts at later stages, the mitochondria became distorted and increasingly loaded with iron, which appears as black deposits within the matrix (h and i).
A glutaredoxin 5 mutation causes inherited human sideroblastic anemia
Sideroblastic anemia describes a distinct group of anemias in which red cell precursors contain iron-overloaded mitochondria that congregate in a ring around the nucleus [41]. Recently, a mutation that causes GLRX5 splicing defects was identified as the cause of a sideroblastic-like microcytic anemia in a human patient (Figure 2). The patient had severe anemia, an enlarged liver and spleen and iron overload with bronze diabetes and cirrhosis [8] (Table 2). Interestingly, the anemia was worsened by blood transfusions but partially reversed by iron chelation therapy. DNA sequencing and RT-PCR experiments showed that a single mutation in the penultimate nucleotide of exon 1 interfered with splicing, resulting in only ~10% of wild-type mRNA expression.
Early experiments in yeast [19] demonstrated that GRX5 might be involved in facilitating the transfer of Fe–S clusters from the scaffold to target proteins [2], although the exact function of GRX5 is still unclear. One proposal suggested that the deglutathionylation activity of glutaredoxins was important for repairing mixed disulfides between glutathione and Fe–S cluster assembly factors in the oxidizing environment of the mitochondrial matrix [42]. Other studies in plants indicated that glutaredoxins might function as scaffold proteins for the assembly and delivery of [2Fe–2S] clusters [43].
In zebrafish, Glutaredoxin 5 mutations cause anemia; decreased heme synthesis was found to be caused by the IRP1-mediated translational repression of erythroid aminolevulinate synthase (eALAS), which catalyzes the first step in heme biosynthesis. [20]. IRP1 and IRP2 are mammalian RNA-binding proteins that play important roles in regulating intracellular iron metabolism (reviewed in Refs. [4,5]). IRP2 is regulated by iron and oxygen-dependent protein degradation, whereas IRP1 regulation largely depends on the presence or absence of its Fe–S cluster. When IRP1 contains an intact Fe–S cluster, it functions as a cytosolic aconitase [44], but when it lacks the Fe–S cluster, it binds RNA stem-loop structures known as iron-responsive elements (IREs) [45]. IREs are found in the transcripts of major iron metabolism transcripts including ferritin H and L subunits, transferrin receptor 1 (TfR1), one isoform of the divalent metal transporter 1, and ferroportin. Binding of IRP1 or IRP2 to an IRE can repress translation of transcripts such as ferritin or can increase TfR1 transcript levels by stabilizing the mRNA [4,5].
Both human and zebrafish genomes contain two separate ALAS genes; the ALAS2 (zebrafish eALAS) transcript contains an IRE in its 5′UTR and is highly expressed in erythroid cells, whereas the ALAS1 transcript is expressed in other tissues and lacks an IRE. Thus, eALAS expression is repressed by apo-IRP1–IRE binding, and zebrafish heme biosynthesis accordingly decreases when IRE–IRP1 binding activity increases [20]. Conversely, in mouse models, eALAS expression increases when IRPs are knocked out [46,47], indicating that the IRP–IRE regulatory system is important in regulating erythroid heme synthesis.
In the GLRX5-deficient patient and in the zebrafish model, IRP1 becomes an active IRE binding protein that blocks heme biosynthesis by inhibiting eALAS translation. How does GLRX5 deficiency result in the loss of the IRP1 Fe–S cluster? The prevailing model, largely based on studies in yeast, proposes that Fe–S clusters are assembled exclusively in mitochondria and that assembled Fe–S clusters are exported through the transporter Atm1p (ABCB7 in humans) to supply Fe–S clusters to recipient proteins in the cytosol and nucleus ([2]). According to this model, if GLRX5 deficiency interfered with mitochondrial Fe–S cluster biogenesis, IRP1 would fail to acquire a Fe–S cluster.
However, with the discoveries of functional cytosolic NFS1, ISCU, NFU1, and Frataxin in human cells [23,48–51], and the recent discoveries of the ISCU and Frataxin homologs in the cytosol of microsporidia Trachipleistophora hominis [52], it is evident that the initial steps in Fe–S cluster biogenesis are not confined to the mitochondrial matrix in all eukaryotes. More importantly, cytosolic iron depletion can directly impair de novo biogenesis of cytosolic Fe–S proteins, and several lines of evidence indicate that disruption of Fe–S cluster biogenesis causes cytosolic iron depletion associated with mitochondrial iron overload. In yeast, radioactive iron labeling experiments indicated that disruption of Fe–S cluster biogenesis triggered the accumulation of iron in the mitochondria at the expense of cytosolic iron [53]. In human cells, siRNA-mediated silencing of ISCU resulted in increased IRE-binding activity of IRP1 and IRP2 and increased IRP2 protein levels [23]. Similarly, IRP2 levels are elevated in FRDA patients’ lymphoblasts [40]. Because IRP2 is regulated through iron-dependent changes in protein turnover and is not known to rely on a Fe–S cluster for its regulation, these results imply that cytosolic iron depletion develops. Thus, it seems that cells that are unable to synthesize Fe–S clusters lose the ability to regulate mitochondrial iron homeostasis. Therefore, another possible explanation for the activation of IRP1 IRE-binding activity is that the loss of mitochondrial GLRX5 disrupts the regulation of mitochondrial iron homeostasis, resulting in mitochondrial iron overload and concomitant cytosolic iron depletion, thereby preventing de novo cytosolic cluster assembly.
Although GLRX5 is expressed in many human tissues, ALAS2 is almost exclusively expressed in erythroid cells. Thus, the tissue specificity of the human sideroblastic anemia could result from the fact that activation of IRP1–IRE-binding activity represses heme synthesis in erythroid cells but not in other tissues.
A splice mutation in ISCU causes myopathy in patients of Swedish descent
Recently, a mutation in the Fe–S cluster assembly scaffold factor, ISCU, was found to be the cause of an inherited skeletal muscle disease found among patients from northern Sweden [9,10]. Patients develop muscle weakness, along with exercise-induced acidosis and myoglobinuria (reddish urine caused by muscle degeneration and excretion of myoglobin) [54,55]. Aconitase and succinate dehydrogenase activities are markedly reduced and mitochondrial iron overload develops in patient muscle samples [54] (Box 1). Studies of distantly related affected individuals identified a region of human chromosome 12 that contained the gene responsible for disease, and ISCU was among the candidates [9,10]. Biochemical studies showed that ISCU expression was indeed markedly diminished in patient skeletal muscle samples, and DNA sequencing revealed that the myopathy allele contains a single G to C mutation near the middle of the final ISCU intron. Analysis of patient transcripts revealed that the G to C mutation promoted the incorporation of a cryptic exon from within the intron into the ISCU transcript, leading to insertion of a premature stop codon (Figure 2). The loss of mitochondrial and cytosolic aconitase activities in the muscle samples of patients with ISCU deficiency is consistent with previous studies of cells depleted of ISCU by siRNA-mediated silencing [23]. Furthermore, the patients’ skeletal muscle mitochondria were markedly overloaded with ferric iron [54] [9] (Box 1), and the punctate ferric-iron staining was reminiscent of the iron staining pattern observed in cells depleted of ISCU by siRNA-mediated silencing [23]. Thus, ISCU loss results in a muscle disease in which Fe–S cluster assembly is impaired, and mitochondrial iron overload develops in conjunction with functional impairment of mitochondrial Fe–S enzymes. Careful comparisons of primary muscle cultures, fibroblasts and lymphoblasts will be needed to determine whether and why the cryptic exon is preferentially expressed in muscle cells.
Potential roles of ABCB7 in X-linked hereditary sideroblastic anaemia
The disease gene for X-linked sideroblastic anemia with ataxia encodes a mitochondrial transporter known as ABCB7 (homolog of Atm1p in yeast). ATM1 deletion leads to mitochondrial iron accumulation, suggesting that it exports an as yet uncharacterized molecule that is important for mitochondrial iron homeostasis [56,57]. Although the activities of two cytosolic Fe–S enzymes (xanthine oxidase and cytosolic aconitase) were impaired in ABCB7 knockdown experiments, succinate dehydrogenase activity was not impaired [57–59], suggesting that ABCB7 is not involved in the basic process of mitochondrial Fe–S cluster biogenesis. Whereas investigators originally postulated that this transporter exported Fe–S clusters for incorporation into cytosolic proteins [2], proteins involved in the early steps of mammalian Fe–S cluster biogenesis are also found in the cytosol [23,48–51] (Figure 3); therefore, the sole purpose of ABCB7 might not be export of Fe–S clusters for use in the cytosol of mammalian cells. Notably, siRNA studies and knockout mouse model studies also show evidence of cytosolic iron deficiency in ABCB7-depleted cells, as indicated by increased IRP1 and IRP2 IRE-binding activities, increased IRP2 levels [59] and decreased expression of ferritin, an iron-storage protein [58]. Thus, it is probable that the ABCB7 substrate might have an important role in the regulation of overall mitochondrial iron homeostasis.
Figure 3.

Mutations in Friedreich ataxia, sideroblastic anemia with GLRX5 deficiency and myopathy with ISCU deficiency. In >98% of Friedreich ataxia (FRDA) cases, the defect is a GAA repeat expansion in the first intron of the Frataxin (FXN) gene. A small subset of patients have one expanded allele and a second allele harboring a premature stop codon or point mutation. In the patient with GLRX5 deficiency, a homologous silent mutation in exon 1 interferes with splicing and drastically reduces levels of fully processed mature GLRX5 mRNA. In myopathy with ISCU deficiency, a single G to C mutation in the fourth intron of ISCU activates a weak splice acceptor site and reduces the production of normal ISCU protein.
Concluding remarks and future perspectives
Iron–sulfur (Fe–S) clusters are essential cofactors for proteins involved in many biological processes, and several human diseases have been linked to defects in Fe–S proteins and their biogenesis factors. Disease can appear in the central nervous system and heart (FRDA mutations), in developing red blood cells (GLRX5 deficiency) or in skeletal muscles (myopathy with deficiencies in ISCU). The fact that defects in a basic essential metabolic pathway common to all tissues can cause such diverse disease presentations raises the possibility that mutations in other conserved Fe–S cluster biogenesis genes might underlie diseases for which the cause is presently unknown (Table 1). An analogy might be found in the spectrum of diseases attributable to pathogenic mtDNA mutations, which also have strikingly distinctive and unexplained tissue-specific presentations [60]. Because deletions of homologs of many human Fe–S genes are lethal in model systems, it is possible that partially disabling mutations of Fe–S assembly genes near disease-linked chromosomal locations could be the cause of diseases, particularly those in which mitochondrial function is compromised. By evaluating diseases that have been mapped to the chromosomal locations of human Fe–S cluster assembly proteins, it seems possible that NFU1 (a putative scaffold protein) might be a good candidate for infant multiple mitochondrial malfunctions syndrome [61]; likewise, HSCB (a chaperone partner) is an interesting candidate for an inherited optic atrophy syndrome [62]. Disease gene discovery can be considerably streamlined by using antibodies to measure the levels of the candidate FeS cluster biogenesis protein in affected tissue [9].
Because both mitochondrial iron overload and mitochondrial failure are common features in model systems and in patients with deficiency in Fe–S cluster biogenesis factors, we also suggest that diseases that are characterized by mitochondrial failure should be analyzed for the presence of mitochondrial iron overload using Prussian blue or 3, 3′-diaminobenzidine (DAB)-enhanced Prussian blue staining [23]. If mitochondrial iron overload is a prominent feature of the disease, the activities of Fe–S proteins, such as mitochondrial aconitase and succinate dehydrogenase, should be measured to evaluate whether Fe–S cluster biogenesis is impaired. The causes of many human sideroblastic anemias, which share the feature of mitochondrial iron overload, remain unknown [41]. The discovery of a sideroblastic anemia patient with a GLRX5 mutation suggests that components of the Fe–S cluster assembly machinery should be assessed in the abnormal erythroid cells from patients with sideroblastic anemias of unknown cause, because mutations of genes in the Fe–S pathway could arise in bone marrow stem cells in mature individuals whose germline cells would be unaffected.
The basic genetic mutations that underlie FRDA, anemia with GLRX5 deficiency and myopathy with ISCU deficiency have the potential to affect all tissues. Therefore, we wonder if some tissues are better protected than others by unrecognized redundancy or low levels of residual activity. Are tissue-specific vulnerabilities, similar to the example of erythroid-specific ALAS and its unique vulnerability to translational repression by activated apo-IRP, at play? Furthermore, are splicing abnormalities favored in specific tissues because of tissue-specific ancillary factors, as might be the case for myopathy with ISCU deficiency? Finally, might there be a common way to treat all of these diseases, based on learning how to circumvent problems with Fe–S cluster biogenesis and mitochondrial iron overload? The fact that at least three highly distinct human diseases result from a compromise in the basic process of Fe–S cluster assembly holds promise that greater understanding of Fe–S cluster biogenesis will yield more effective diagnostics and treatments for a related group of human diseases.
Acknowledgments
The authors thank Arnulf Koeppen for generously providing the FRDA heart pictures, Dr. Djaldetti for the EMs of a sideroblastic anemia patient, and Karen Ayyad and Ron Haller for providing the Perls’ DAB stain and EM of skeletal muscle from patients with ISCU deficiency. This work was supported by the intramural program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, Bethesda, MD.
References
- 1.Johnson DC, et al. Structure, function, and formation of biological iron-sulfur clusters. Annu Rev Biochem. 2005;74:247–281. doi: 10.1146/annurev.biochem.74.082803.133518. [DOI] [PubMed] [Google Scholar]
- 2.Lill R, Muhlenhoff U. Maturation of iron-sulfur proteins in eukaryotes: mechanisms, connected processes, and diseases. Annu Rev Biochem. 2008;77:669–700. doi: 10.1146/annurev.biochem.76.052705.162653. [DOI] [PubMed] [Google Scholar]
- 3.Rudolf J, et al. The DNA repair helicases XPD and FancJ have essential iron-sulfur domains. Mol Cell. 2006;23:801–808. doi: 10.1016/j.molcel.2006.07.019. [DOI] [PubMed] [Google Scholar]
- 4.Wallander ML, et al. Molecular control of vertebrate iron homeostasis by iron regulatory proteins. Biochim Biophys Acta. 2006;1763:668–689. doi: 10.1016/j.bbamcr.2006.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rouault TA. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat Chem Biol. 2006;2:406–414. doi: 10.1038/nchembio807. [DOI] [PubMed] [Google Scholar]
- 6.Cairo G, Recalcati S. Iron-regulatory proteins: molecular biology and pathophysiological implications. Expert Rev Mol Med. 2007;9:1–13. doi: 10.1017/S1462399407000531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Babady NE, et al. Advancements in the pathophysiology of Friedreich’s Ataxia and new prospects for treatments. Mol Genet Metab. 2007;92:23–35. doi: 10.1016/j.ymgme.2007.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Camaschella C, et al. The human counterpart of zebrafish shiraz shows sideroblastic-like microcytic anemia and iron overload. Blood. 2007;110:1353–1358. doi: 10.1182/blood-2007-02-072520. [DOI] [PubMed] [Google Scholar]
- 9.Mochel F, et al. Splice mutation in the iron-sulfur cluster scaffold protein ISCU causes myopathy with exercise intolerance. Am J Hum Genet. 2008;82:652–660. doi: 10.1016/j.ajhg.2007.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Olsson A, et al. Myopathy with lactic acidosis is linked to chromosome 12q23.3-24.11 and caused by an intron mutation in the ISCU gene resulting in a splicing defect. Hum Mol Genet. 2008;17:1666–1672. doi: 10.1093/hmg/ddn057. [DOI] [PubMed] [Google Scholar]
- 11.Wachtershauser G. On the chemistry and evolution of the pioneer organism. Chem Biodivers. 2007;4:584–602. doi: 10.1002/cbdv.200790052. [DOI] [PubMed] [Google Scholar]
- 12.Rouault TA, Tong WH. Opinion: Iron-sulphur cluster biogenesis and mitochondrial iron homeostasis. Nat Rev Mol Cell Biol. 2005;6:345–351. doi: 10.1038/nrm1620. [DOI] [PubMed] [Google Scholar]
- 13.Bencze KZ, et al. The structure and function of frataxin. Crit Rev Biochem Mol Biol. 2006;41:269–291. doi: 10.1080/10409230600846058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yabe T, et al. The Arabidopsis chloroplastic NifU-like protein CnfU, which can act as an iron-sulfur cluster scaffold protein, is required for biogenesis of ferredoxin and photosystem I. Plant Cell. 2004;16:993–1007. doi: 10.1105/tpc.020511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chandramouli K, et al. Formation and properties of [4Fe-4S] clusters on the IscU scaffold protein. Biochemistry. 2007;46:6804–6811. doi: 10.1021/bi6026659. [DOI] [PubMed] [Google Scholar]
- 16.Adam AC, et al. The Nfs1 interacting protein Isd11 has an essential role in Fe/S cluster biogenesis in mitochondria. EMBO J. 2006;25:174–183. doi: 10.1038/sj.emboj.7600905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wiedemann N, et al. Essential role of Isd11 in mitochondrial iron-sulfur cluster synthesis on Isu scaffold proteins. EMBO J. 2006;25:184–195. doi: 10.1038/sj.emboj.7600906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shan Y, et al. Mitochondrial frataxin interacts with ISD11 of the NFS1/ISCU complex and multiple mitochondrial chaperones. Hum Mol Genet. 2007;16:929–941. doi: 10.1093/hmg/ddm038. [DOI] [PubMed] [Google Scholar]
- 19.Rodriguez-Manzaneque MT, et al. Grx5 is a mitochondrial glutaredoxin required for the activity of iron/sulfur enzymes. Mol Biol Cell. 2002;13:1109–1121. doi: 10.1091/mbc.01-10-0517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wingert RA, et al. Deficiency of glutaredoxin 5 reveals Fe-S clusters are required for vertebrate haem synthesis. Nature. 2005;436:1035–1039. doi: 10.1038/nature03887. [DOI] [PubMed] [Google Scholar]
- 21.Song D, Lee FS. A Role for IOP1 in Mammalian Cytosolic Iron-Sulfur Protein Biogenesis. J Biol Chem. 2008;283:9231–9238. doi: 10.1074/jbc.M708077200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hwang PM, et al. Ferredoxin reductase affects p53-dependent, 5-fluorouracil-induced apoptosis in colorectal cancer cells. Nat Med. 2001;7:1111–1117. doi: 10.1038/nm1001-1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tong WH, Rouault TA. Functions of mitochondrial ISCU and cytosolic ISCU in mammalian iron-sulfur cluster biogenesis and iron homeostasis. Cell Metab. 2006;3:199–210. doi: 10.1016/j.cmet.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 24.Fosset C, et al. RNA silencing of mitochondrial m-Nfs1 reduces Fe-S enzyme activity both in mitochondria and cytosol of mammalian cells. J Biol Chem. 2006;281:25398–25406. doi: 10.1074/jbc.M602979200. [DOI] [PubMed] [Google Scholar]
- 25.Pandolfo M. Friedreich’s ataxia: clinical aspects and pathogenesis. Semin Neurol. 1999;19:311–321. doi: 10.1055/s-2008-1040847. [DOI] [PubMed] [Google Scholar]
- 26.Campuzano V, et al. Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271:1423–1427. doi: 10.1126/science.271.5254.1423. [DOI] [PubMed] [Google Scholar]
- 27.De Biase I, et al. Progressive GAA expansions in dorsal root ganglia of Friedreich’s ataxia patients. Ann Neurol. 2007;61:55–60. doi: 10.1002/ana.21052. [DOI] [PubMed] [Google Scholar]
- 28.Herman D, et al. Histone deacetylase inhibitors reverse gene silencing in Friedreich’s ataxia. Nat Chem Biol. 2006;2:551–558. doi: 10.1038/nchembio815. [DOI] [PubMed] [Google Scholar]
- 29.Rotig A, et al. Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia. Nat Genet. 1997;17:215–217. doi: 10.1038/ng1097-215. [DOI] [PubMed] [Google Scholar]
- 30.Seznec H, et al. Friedreich ataxia: the oxidative stress paradox. Hum Mol Genet. 2005;14:463–474. doi: 10.1093/hmg/ddi042. [DOI] [PubMed] [Google Scholar]
- 31.Pastore C, et al. Understanding the binding properties of an unusual metal-binding protein–a study of bacterial frataxin. FEBS J. 2007;274:4199–4210. doi: 10.1111/j.1742-4658.2007.05946.x. [DOI] [PubMed] [Google Scholar]
- 32.Zanella I, et al. The effects of frataxin silencing in HeLa cells are rescued by the expression of human mitochondrial ferritin. Biochim Biophys Acta. 2008;1782:90–98. doi: 10.1016/j.bbadis.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 33.Gakh O, et al. Mitochondrial iron detoxification is a primary function of frataxin that limits oxidative damage and preserves cell longevity. Hum Mol Genet. 2006;15:467–479. doi: 10.1093/hmg/ddi461. [DOI] [PubMed] [Google Scholar]
- 34.Wang T, Craig EA. Binding of yeast frataxin to the scaffold for Fe-S cluster biogenesis, isu. J Biol Chem. 2008;283:12674–12679. doi: 10.1074/jbc.M800399200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Michael S, et al. Iron and iron-responsive proteins in the cardiomyopathy of Friedreich’s ataxia. Cerebellum. 2006;5:257–267. doi: 10.1080/14734220600913246. [DOI] [PubMed] [Google Scholar]
- 36.Puccio H, et al. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat Genet. 2001;27:181–186. doi: 10.1038/84818. [DOI] [PubMed] [Google Scholar]
- 37.Anderson PR, et al. Hydrogen peroxide scavenging rescues frataxin deficiency in a Drosophila model of Friedreich’s ataxia. Proc Natl Acad Sci U S A. 2008;105:611–616. doi: 10.1073/pnas.0709691105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Foury F, Talibi D. Mitochondrial control of iron homeostasis. A genome wide analysis of gene expression in a yeast frataxin-deficient strain. J Biol Chem. 2001;276:7762–7768. doi: 10.1074/jbc.M005804200. [DOI] [PubMed] [Google Scholar]
- 39.Schoenfeld RA, et al. Frataxin deficiency alters heme pathway transcripts and decreases mitochondrial heme metabolites in mammalian cells. Hum Mol Genet. 2005;14:3787–3799. doi: 10.1093/hmg/ddi393. [DOI] [PubMed] [Google Scholar]
- 40.Li K, et al. Iron-dependent regulation of frataxin expression: implications for treatment of Friedreich ataxia. Hum Mol Genet. 2008 doi: 10.1093/hmg/ddn127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bottomley SS. Congenital sideroblastic anemias. Curr Hematol Rep. 2006;5:41–49. [PubMed] [Google Scholar]
- 42.Herrero E, de la Torre-Ruiz MA. Monothiol glutaredoxins: a common domain for multiple functions. Cell Mol Life Sci. 2007;64:1518–1530. doi: 10.1007/s00018-007-6554-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bandyopadhyay S, et al. Chloroplast monothiol glutaredoxins as scaffold proteins for the assembly and delivery of [2Fe-2S] clusters. EMBO J. 2008;27:1122–1133. doi: 10.1038/emboj.2008.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dupuy J, et al. Crystal structure of human iron regulatory protein 1 as cytosolic aconitase. Structure. 2006;14:129–139. doi: 10.1016/j.str.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 45.Walden WE, et al. Structure of dual function iron regulatory protein 1 complexed with ferritin IRE-RNA. Science. 2006;314:1903–1908. doi: 10.1126/science.1133116. [DOI] [PubMed] [Google Scholar]
- 46.Cooperman SS, et al. Microcytic anemia, erythropoietic protoporphyria, and neurodegeneration in mice with targeted deletion of iron-regulatory protein 2. Blood. 2005;106:1084–1091. doi: 10.1182/blood-2004-12-4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Galy B, et al. Altered body iron distribution and microcytosis in mice deficient in iron regulatory protein 2 (IRP2) Blood. 2005;106:2580–2589. doi: 10.1182/blood-2005-04-1365. [DOI] [PubMed] [Google Scholar]
- 48.Land T, Rouault TA. Targeting of a human iron-sulfur cluster assembly enzyme, nifs, to different subcellular compartments is regulated through alternative AUG utilization. Mol Cell. 1998;2:807–815. doi: 10.1016/s1097-2765(00)80295-6. [DOI] [PubMed] [Google Scholar]
- 49.Tong WH, et al. Subcellular compartmentalization of human Nfu, an iron-sulfur cluster scaffold protein, and its ability to assemble a [4Fe-4S] cluster. Proc Natl Acad Sci U S A. 2003;100:9762–9767. doi: 10.1073/pnas.1732541100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Acquaviva F, et al. Extra-mitochondrial localisation of frataxin and its association with IscU1 during enterocyte-like differentiation of the human colon adenocarcinoma cell line Caco-2. J Cell Sci. 2005;118:3917–3924. doi: 10.1242/jcs.02516. [DOI] [PubMed] [Google Scholar]
- 51.Condo I, et al. A pool of extramitochondrial frataxin that promotes cell survival. J Biol Chem. 2006;281:16750–16756. doi: 10.1074/jbc.M511960200. [DOI] [PubMed] [Google Scholar]
- 52.Goldberg AV, et al. Localization and functionality of microsporidian iron-sulphur cluster assembly proteins. Nature. 2008;452:624–628. doi: 10.1038/nature06606. [DOI] [PubMed] [Google Scholar]
- 53.Knight SA, et al. Mt-Hsp70 homolog, Ssc2p, required for maturation of yeast frataxin and mitochondrial iron homeostasis. J Biol Chem. 1998;273:18389–18393. doi: 10.1074/jbc.273.29.18389. [DOI] [PubMed] [Google Scholar]
- 54.Haller RG, et al. Deficiency of skeletal muscle succinate dehydrogenase and aconitase. Pathophysiology of exercise in a novel human muscle oxidative defect. J Clin Invest. 1991;88:1197–1206. doi: 10.1172/JCI115422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Larsson LE, et al. Hereditary metabolic myopathy with paroxysmal myoglobinuria due to abnormal glycolysis. J Neurol Neurosurg Psychiatry. 1964;27:361–380. doi: 10.1136/jnnp.27.5.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chloupkova M, et al. MDL1 is a high copy suppressor of ATM1: evidence for a role in resistance to oxidative stress. J Mol Biol. 2003;331:155–165. doi: 10.1016/s0022-2836(03)00666-1. [DOI] [PubMed] [Google Scholar]
- 57.Kispal G, et al. The mitochondrial proteins Atm1p and Nfs1p are essential for biogenesis of cytosolic Fe/S proteins. EMBO J. 1999;18:3981–3989. doi: 10.1093/emboj/18.14.3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cavadini P, et al. RNA silencing of the mitochondrial ABCB7 transporter in HeLa cells causes an iron-deficient phenotype with mitochondrial iron overload. Blood. 2007;109:3552–3559. doi: 10.1182/blood-2006-08-041632. [DOI] [PubMed] [Google Scholar]
- 59.Pondarre C, et al. The mitochondrial ATP-binding cassette transporter Abcb7 is essential in mice and participates in cytosolic iron-sulfur cluster biogenesis. Hum Mol Genet. 2006;15:953–964. doi: 10.1093/hmg/ddl012. [DOI] [PubMed] [Google Scholar]
- 60.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Seyda A, et al. A novel syndrome affecting multiple mitochondrial functions, located by microcell-mediated transfer to chromosome 2p14-2p13. Am J Hum Genet. 2001;68:386–396. doi: 10.1086/318196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Barbet F, et al. A third locus for dominant optic atrophy on chromosome 22q. J Med Genet. 2005;42:e1. doi: 10.1136/jmg.2004.025502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.King A, et al. Succinate dehydrogenase and fumarate hydratase: linking mitochondrial dysfunction and cancer. Oncogene. 2006;25:4675–4682. doi: 10.1038/sj.onc.1209594. [DOI] [PubMed] [Google Scholar]
- 64.Djaldetti M, et al. Ultrastructural features of bone marrow cells from patients with acquired sideroblastic anemia. Microsc Res Tech. 2004;63:155–158. doi: 10.1002/jemt.20024. [DOI] [PubMed] [Google Scholar]
- 65.Li J, et al. Yeast mitochondrial protein, Nfs1p, coordinately regulates iron-sulfur cluster proteins, cellular iron uptake, and iron distribution. J Biol Chem. 1999;274:33025–33034. doi: 10.1074/jbc.274.46.33025. [DOI] [PubMed] [Google Scholar]
- 66.Giaever G, et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 2002;418:387–391. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- 67.Tong WH, Rouault T. Distinct iron-sulfur cluster assembly complexes exist in the cytosol and mitochondria of human cells. EMBO J. 2000;19:5692–5700. doi: 10.1093/emboj/19.21.5692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schilke B, et al. Evidence for a conserved system for iron metabolism in the mitochondria of Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 1999;96:10206–10211. doi: 10.1073/pnas.96.18.10206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vazquez-Manrique RP, et al. Reduction of Caenorhabditis elegans frataxin increases sensitivity to oxidative stress, reduces lifespan, and causes lethality in a mitochondrial complex II mutant. FASEB J. 2006;20:172–174. doi: 10.1096/fj.05-4212fje. [DOI] [PubMed] [Google Scholar]
- 70.Muhlenhoff U, et al. Components involved in assembly and dislocation of iron-sulfur clusters on the scaffold protein Isu1p. EMBO J. 2003;22:4815–4825. doi: 10.1093/emboj/cdg446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sun G, et al. Identification of a novel candidate gene in the iron-sulfur pathway implicated in ataxia-susceptibility: human gene encoding HscB, a J-type co-chaperone. J Hum Genet. 2003;48:415–419. doi: 10.1007/s10038-003-0048-9. [DOI] [PubMed] [Google Scholar]
- 72.Fontecave M, Ollagnierde-Choudens S. Iron-sulfur cluster biosynthesis in bacteria: mechanisms of cluster assembly and transfer. Arch Biochem Biophys. 2007 doi: 10.1016/j.abb.2007.12.014. [DOI] [PubMed] [Google Scholar]
- 73.Balasubramanian R, et al. Regulatory roles for IscA and SufA in iron homeostasis and redox stress responses in the cyanobacterium Synechococcus sp. strain PCC 7002. J Bacteriol. 2006;188:3182–3191. doi: 10.1128/JB.188.9.3182-3191.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Netz DJ, et al. The Cfd1-Nbp35 complex acts as a scaffold for iron-sulfur protein assembly in the yeast cytosol. Nat Chem Biol. 2007;3:278–286. doi: 10.1038/nchembio872. [DOI] [PubMed] [Google Scholar]
- 75.Roy A, et al. A novel eukaryotic factor for cytosolic Fe-S cluster assembly. EMBO J. 2003;22:4826–4835. doi: 10.1093/emboj/cdg455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lange H, et al. An essential function of the mitochondrial sulfhydryl oxidase Erv1p/ALR in the maturation of cytosolic Fe/S proteins. EMBO Rep. 2001;2:715–720. doi: 10.1093/embo-reports/kve161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vitale G, et al. NBP35 encodes an essential and evolutionary conserved protein in Saccharomyces cerevisiae with homology to a superfamily of bacterial ATPases. Gene. 1996;178:97–106. doi: 10.1016/0378-1119(96)00341-1. [DOI] [PubMed] [Google Scholar]
- 78.Balk J, et al. The essential WD40 protein Cia1 is involved in a late step of cytosolic and nuclear iron-sulfur protein assembly. Mol Cell Biol. 2005;25:10833–10841. doi: 10.1128/MCB.25.24.10833-10841.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Srinivasan V, et al. Structure of the yeast WD40 domain protein Cia1, a component acting late in iron-sulfur protein biogenesis. Structure. 2007;15:1246–1257. doi: 10.1016/j.str.2007.08.009. [DOI] [PubMed] [Google Scholar]
- 80.Balk J, et al. The hydrogenase-like Nar1p is essential for maturation of cytosolic and nuclear iron-sulphur proteins. EMBO J. 2004;13:773–780. doi: 10.1038/sj.emboj.7600216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gelling C, et al. Mitochondrial Iba57p is required for Fe/S cluster formation on aconitase and activation of radical SAM enzymes. Mol Cell Biol. 2008;28:1851–1861. doi: 10.1128/MCB.01963-07. [DOI] [PMC free article] [PubMed] [Google Scholar]