

Abstract
Recent studies in mice suggest that stress-activated c-Jun N-terminal protein kinase 2 (JNK2) plays a pathologic role in acetaminophen (APAP)-induced liver injury (AILI), a major cause of acute liver failure (ALF). In contrast, we present evidence that JNK2 can have a protective role against AILI. When male C57BL/6J wild type (WT) and JNK2−/− mice were treated with 300mg APAP/kg, 90% of JNK2−/− mice died of ALF compared to 20% of WT mice within 48 h. The high susceptibility of JNK2−/− mice to AILI appears to be due in part to deficiencies in hepatocyte proliferation and repair. Therefore, our findings are consistent with JNK2 signaling playing a protective role in AILI and further suggest that the use of JNK inhibitors as a potential treatment for AILI, as has been recommended by other investigators, should be reconsidered.
Keywords: Acetaminophen, Cyclin D1, Hepatoprotection, c-Jun N-terminal kinase 2, JNK2, Liver injury, Proliferating cell nuclear antigen, Repair
Overdose of APAP, a popular antipyretic and analgesic, is a leading cause of ALF resulting in approximately 1,800 deaths per year in the United States [1]. Although the initiating events in AILI have been attributed to reactive metabolite and protein adduct formation as well as glutathione depletion [2–4], the downstream signaling pathways controlling or promoting the severity of AILI have become of a great interest to many researchers [5–14]. One of these pathways involves JNK, which when activated is known to influence important cellular events, including alterations in gene expression [15], cell death [16], cellular proliferation and survival [17–20].
Recent studies involving AILI in JNK2−/− mice have led to conflicting results where JNK2−/− mice were found to be either less susceptible [21] or equally susceptible [22] to AILI when compared to WT mice. In contrast, we present evidence here suggesting that JNK2 instead can have a protective role in AILI.
Materials and methods
Chemicals
Chemicals were purchased from the following commercial sources: APAP (Sigma, St. Louis, MO); alanine aminotransferase (ALT) kit (Teco Diagnostics, Anaheim, CA); complete protease inhibitor cocktail tablets (Roche Molecular Biochemicals, Indianapolis, IN); sodium orthovanadate (Calbiochem, La Jolla, CA); hematoxylin (Sigma, St. Louis, MO); reduced glutathione (GSH) (Sigma, St. Louis, MO); primary antibodies, SAPK/JNK, phospho-SAPK/JNK (Thr183/Tyr185) (Cell Signaling Technology, Danvers, MA); phospho-c-Jun (Ser63) (SantaCruz Biotech, Santa Cruz, CA); proliferating cell nuclear antigen (PCNA) and diaminobenzidine (DAB) (Dako North America, Carpinteria, CA); cyclin D1 antibody (SantaCruz Biotech, Santa Cruz, CA); secondary antibodies, horseradish peroxidase (HRP)-conjugated anti-rabbit and anti-mouse IgG, and immobilon Western HRP chemiluminescent substrate (Millipore, Billerica, MA). Anti-APAP-protein adduct serum was kindly provided by Drs. Jack Hinson and Neil Pumford (University of Arkansas, Little Rock, AR) and anti-rabbit cytochrome P450 2E1 was a generous gift from Dr. Dennis R. Koop (Department of Pharmacology, Oregon Health Science University, Portland, OR).
Animals and APAP treatment
Seven to 9 week-old male WT, JNK1−/− and JNK2−/− mice on a C57BL/6J background were purchased from Jackson Laboratories (Bar Harbor, ME). Mice were acclimated for at least 1 week to a 12-h light/dark cycle in a humidity and temperature-controlled, specific-pathogen-free environment in microisolator autoclaved cages. Mice were allowed autoclaved food and water ad libitum until experimental use. Before each study, mice were fasted overnight (14–16h; free access to water) to uniformly deplete hepatic GSH stores [23]. Food supplies were restored after intraperitoneal administration of APAP (300mg/kg in warm saline; 20ml/kg) or saline vehicle (20ml/kg). All maintenance of animals conformed to the guidelines for humane treatment set by the Association for Assessment and Accreditation for Laboratory Animal Care International’s Guide for the Care and Use of Laboratory Animals and by the National Institutes of Health.
Sera and tissue collection
Blood samples were collected and allowed to clot in microtainer serum separator tubes (Becton Dickinson and Co., Franklin Lakes, NJ) for approximately 2h at room temperature and then centrifuged. Serum was separated and used for ALT measurements. A portion of the left and right lateral liver lobes from euthanized mice were fixed in 10% buffered formalin (Fischer Scientific, Fair Lawn, NJ) with the remainder snap–frozen and stored at −80 °C for further study. Fixed tissue was embedded in paraffin, processed by standard histological techniques, and stained with hematoxylin and eosin (H&E; American Histolabs, Gaithersburg, MD).
Assessment of liver injury
Liver injury was assessed by measuring serum ALT activity using a microtiter plate adaptation of a commercially available kit and through histopathological examination of H&E-stained liver sections under light microscopy. Quantitative evaluation of liver injury in H&E-stained sections was analyzed morphometrically by light microscopy using Axiovision software (v4.5; Carl Zeiss Microimaging, Inc., Thornwood, NY) and a 64-point grid [24]. Approximately 10% of the total area of each liver section was examined. All slides were coded and randomized before evaluation.
Assessment of liver regeneration
PCNA was measured to estimate the extent of proliferating hepatocytes and liver repair [25]. Briefly, paraffin sections were mounted on poly (L-lysine)-treated slides (American Histolabs), deparaffinized and incubated in 10 mM citrate buffer (pH 6.0) for antigen retrieval. Slides were then treated with 3% hydrogen peroxide, blocked with 5% bovine serum albumin, and incubated with PCNA antibody (1:100) overnight at 4°C. HRP-linked anti-mouse IgG secondary antibody was applied for 1 h at room temperature followed by the addition of DAB. The slides were counterstained with 0.1% hematoxylin and PCNA stained cells were counted at an original magnification of 20×. Greater than 95% of the liver area was analyzed for dark brown PCNA positive S-phase cells in both WT and JNK2−/− mice.
Preparation of liver homogenate and immunoblot analysis
Liver homogenates were prepared and SDS–PAGE immunoblot analysis was performed as previously described [26]. Protein changes were visualized with Immobilon Western HRP Chemiluminescent substrate via a Kodak Station 2000 RT Imager and software (Eastman Kodak, Rochester, NY), as previously reported [26].
Statistical analyses
Results are expressed as means ±SEM for all the experiments. Data expressed as percentages were subject to arcsine square root transformations prior to analysis. Single comparisons were analyzed by Student’s t-test where appropriate. Multiple comparisons of homogeneous data were analyzed by one-way or two-way ANOVA, as appropriate, and group means were compared using Student–Newman–Keuls posthoc test. The criterion for statistical differences was p ≤ 0.05 for all comparisons.
Results
Enhanced susceptibility ofJNK2−/− Mice to AILI
When WT and JNK2−/− mice were treated with APAP (300 mg/ kg), JNK2−/− mice were found to be significantly more susceptible to liver injury at 12, 24, and 32 h after treatment than were WT mice, as determined by elevated serum ALT activities (Fig. 1A), a biomarker for liver injury. We repeated these experiments separately four additional times and consistently found the same results (data not shown). In contrast, serum ALT activities of JNK1−/− mice did not differ from those of WT mice following APAP treatment (data not shown). Differences in liver injury were confirmed histopathologically (Fig. 2) and by morphometric analysis of the size of necrotic regions in the liver 24 h post-APAP treatment (percent lesion area from 4–6 animals per group): WT mice, 43.5 ± 1.4; JNK1−/− mice, 47.6 ± 2.5; JNK2−/− mice, 59.0 ± 2.6 (significantly different from APAP-treated WT and JNK1−/− mice). In a separate survival study, 90% of JNK2−/− mice were dead 48 h after APAP treatment compared to only 20% of WT mice (Fig. 1B).
Fig. 1.

Time course for APAP-induced liver injury and survival in WT and JNK2−/− mice. (A) Serum ALT activities for APAP-treated WT and JNK2−/− mice. (B) Percent survival for APAP-treated WT and JNK2−/− mice. Results represent the mean±SEM of 6 animals/group for (A) and 10 WT and 8 JNK2−/− animals for (B). * Significantly different from APAP-treated WT mice at the same time.
Fig. 2.

Representative photomicrographs of H&E-stained liver sections from WT, JNK1−/− and JNK2−/− mice 24 h after treatment with saline vehicle or APAP. (A) Liver from a saline-treated WT mouse exhibited normal liver architecture with no histopathological changes as did livers from saline-treated JNK1−/− and JNK2−/− mice (photomicrographs not shown). (B) Liver from an APAP-treated WT mouse. Moderate perivenous coagulative necrosis was observed along with marked ballooning hydropathy at lesion periphery, minimal hemorrhage and minimal to definite inflammatory infiltrate within the lesion area. (C) Liver from APAP-treated JNK1−/− mouse. Histopathological changes were similar to that observed in APAP-treated WT mice. (D) Liver from an APAP-treated JNK2−/− mouse. Marked to severe perivenous coagulative necrosis, often bridging, was observed along with minimal ballooning hydropathy at the lesion periphery and marked disruption to the sinusoidal architecture, minimal hemorrhage and moderate inflammatory infiltrate within the lesion area. Original magnification: 10×.
Similar levels of APAP–protein adducts in livers of WT and JNK2−/− mice
APAP–protein adducts were measured in liver homogenates by immunoblot analysis [6] to evaluate whether exacerbated AILI in JNK2−/− mice was the result of enhanced APAP bioactivation. Although some minor alterations were observed in the band patterns and intensities of APAP–protein adducts in liver homogenates between WT and JNK2−/− mice 1.5 h after APAP treatment, the overall quantitative nature of the APAP–protein adducts did not differ significantly between these two groups of mice (data not shown). Moreover, the constitutive levels of hepatic cyp2e1 and GSH levels were similar in WT and JNK2−/− mice before APAP administration (data not shown).
Impaired liver regeneration inJNK2−/− mice
PCNA, a nuclear protein that is upregulated in cycling cells and plays a key role in cell growth, was measured immunohistochemically in the livers of WT and JNK2−/− mice to estimate the extent of proliferating hepatocytes and liver repair [25]. Twenty-four hours after WT and JNK2−/− mice were treated with saline, very few nuclei of hepatocytes stained positively for this marker (Fig. 3A and B, respectively). In contrast, the number of hepatocyte nuclei staining positively for PCNA in WT mice increased markedly 24 h after APAP treatment and were located in most cases at the periphery of perivenous necrotic regions (Fig. 3C). Although an increase in PCNA staining was also seen in the livers of APAP-treated JNK2−/− mice (Fig. 3D) as compared to saline-treated control mice (Fig. 3B), the number of positively stained nuclei was significantly less in JNK2−/− mice when compared to WT mice following APAP treatment. WT mice: 299.7 ± 40.7 (n = 6); JNK2−/− mice, 145 ± 21.4 (n = 5; significantly different from APAP-treated WT mice).
Fig. 3.
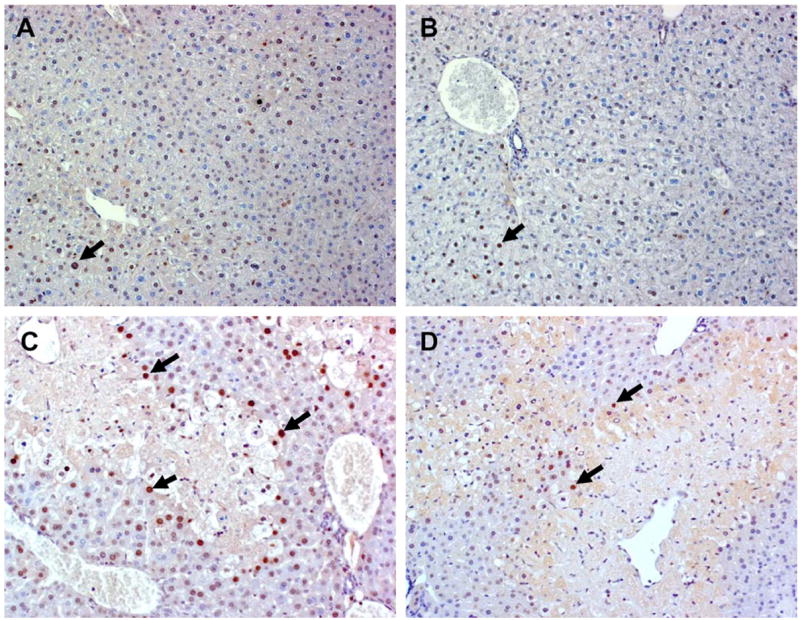
Representative photomicrographs of PCNA immunostaining in hepatocyte nuclei of WT and JNK2−/− mice 24 h after treatment with saline vehicle or APAP. (A) Liver from a saline-treated WT mouse exhibited minimal staining and as did the liver from a saline-treated JNK2−/− mouse (B). (C) Liver from an APAP-treated WT mouse showed a increased number of PCNA positive stained S-phase cells at the periphery of a necrotic lesion, while much less staining was seen in cells surrounding the necrotic lesion in the liver of a JNK2−/− mouse (D). Solid arrows point to PCNA positive S-phase cells, which are characterized by intense brown nuclear staining. Original magnification: 20×.
JNK–cyclin D1 signaling
The expression of JNK2 protein in liver homogenates of WT mice remained unchanged 1.5 h after APAP-treatment (Fig. 4A). Hepatic JNK1 protein levels were also unchanged both in WT and JNK2−/− mice following APAP treatment, although the levels of JNK1 in the JNK2−/− mice were lower than those in the WT mice. In contrast, phosphorylated forms of JNK2, JNK1, and c-Jun (a substrate for JNKs) [27], were very low in WT mice before APAP treatment, but increased considerably 1.5 h post-treatment (Fig. 4A). A similar change in the phosphorylated JNK1 and c-Jun was seen in livers of JNK2−/− mice following APAP administration, but not to the same extent as was found in the livers of WT mice.
Fig. 4.
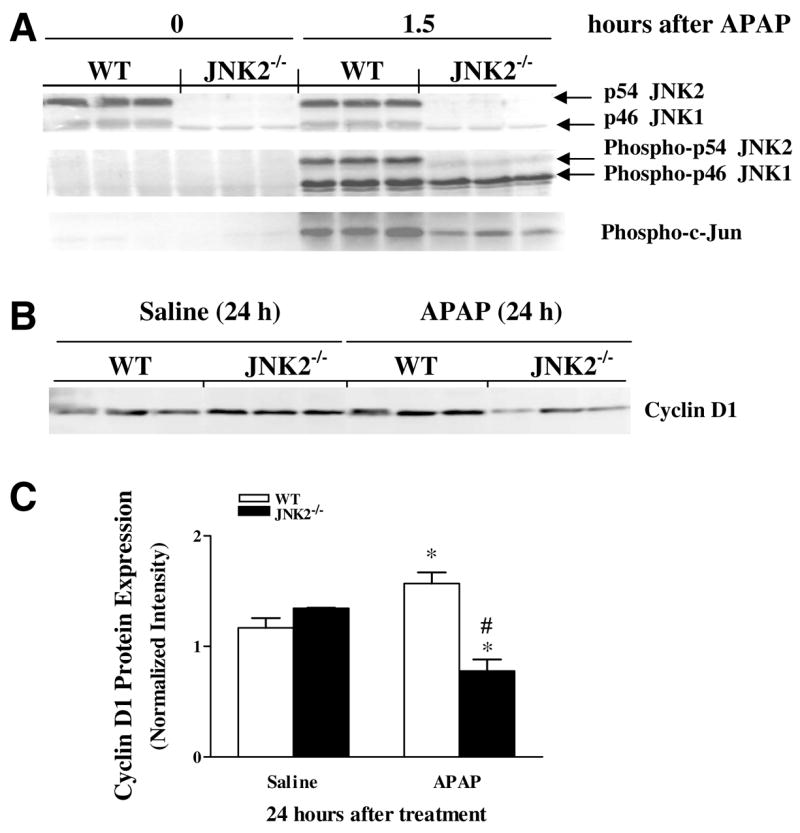
Representative immunoblots of JNK, phospho-JNK, phospho-c-Jun and cyclin D1 in livers from APAP-treated WT and JNK2−/− mice. (A) p54 JNK2, p46 JNK1, phospho-p54 JNK2, phospho-p46 JNK1, and phospho-c-Jun expression in livers from WT and JNK2−/− mice at 0 (before APAP) and 1.5 h after APAP. (B) Hepatic cyclin D1 protein expression in WT and JNK2−/− mice 24h post-administration of APAP or saline vehicle. (C) Scanned intensity for cyclin D1 protein quantification normalized to total protein (Ponceau red). Results represent the mean±SEM of 3 animals per group. *Statistically significant from respective saline-treated group. #Statistically significant from APAP-treated WT group.
Protein expression of cyclin D1, another important regulator of cell cycling and proliferation [28,29] increased significantly in WT mice at 24 h after APAP as compared to saline-treated controls (Fig. 4B). However, hepatic cyclin D1 expression in JNK2−/− mice fell significantly 24 h after APAP when compared to respective saline controls and APAP-treated WT mice at the same time point (Fig. 4C). Overall, this suggests that JNK2 signaling pathway may be a major regulator of cyclin D1 expression following AILI.
Discussion
In this study, we provide evidence for JNK2 playing a protective role in AILI by showing that JNK2−/− mice were more susceptible to both AILI and subsequent death than were WT mice (Figs. 1 and 2). In contrast, JNK1−/− and WT mice did not differ in susceptibility to AILI (Fig. 2), confirming reports by other investigators [21,22]. Since APAP-treated JNK2−/− and WT mice showed the same overall quantitative nature of APAP–protein adducts with only minor differences in protein band pattern in immunoblots of liver homogenates (data not shown), it is unlikely that JNK2 regulates the severity of AILI by modulating APAP bioactivation to its reactive and toxic metabolite, N-acetyl-p-benzoquinone imine [2–4].
Several studies have established liver repair and regeneration as a critical determinant in the final outcome of toxicant-induced liver injury (see review, [30]). Accordingly, since JNK signaling has been reported to activate genes involved in cell cycling and liver cell proliferation and regeneration by phosphorylating c-Jun, a major contributor to the functioning of the transcription factor activating protein-1 (AP-1) [17,27,31], we determined whether JNK2−/− mice exhibited a deficiency in this signaling pathway. Indeed, JNK2−/− mice were observed to have significantly lower hepatic protein levels not only of phospho-c-Jun (Fig. 4A), but also of the downstream cell cycling proteins PCNA (Fig. 3D) and cyclin D1 (Fig. 4B and C) compared to WT mice following APAP treatment. Lower levels of phospho-JNK1 were also found in APAP-treated JNK2−/− mice compared to WT mice (Fig. 4A), possibly contributing to PCNA and cyclin D1 deficiencies observed in the APAP-treated JNK2−/− mice. Taken together, these findings suggest deficiencies in the signaling pathway for liver regeneration and repair are, in part, responsible for the decreased survival observed in APAP-treated JNK2−/− mice (Fig. 1B) and that JNK2 plays a protective role in AILI. In agreement with our findings, others also suggest that JNK-c-Jun-AP-1 signaling pathway might also protect against AILI by inducing the synthesis of heme oxygenase- 1 (HO-1) [12,13], which is protective in several models of hepatic injury including AILI [32].
In contrast to our findings, other investigators studying AILI in JNK deficient mice have obtained variable results, suggesting APAP-treated JNK2−/− mice were either less susceptible [21 ] or did not differ in AILI susceptibility [22] compared to APAP-treated WT mice. In the later case [22], however, a representative histological figure in the manuscript actually shows more liver damage in a section of liver from a JNK2−/− mouse compared to that of a WT mouse treated with APAP.
While the basis for the contradictory results in JNK2−/− mice susceptibility to AILI is not clear, different doses of APAP were used in each study, which might affect the results, and in one case, APAP was dissolved in DMSO [21], which is known to have a profound effect on APAP metabolism [33] and innate-immune response after APAP [34]. Nevertheless, it remains to be determined whether these factors contribute to the conflicting findings.
The role of JNKs in AILI has also been studied in WT mice in vivo with the use of pharmacological inhibitors [14,21,22,35] and anti-sense oligonucleotides [21 ]. Treatment of mice with pharmacological inhibitors that blocked the activity of both JNK1 and 2 reduced AILI and mortality, while administration of JNK2 but not JNK1 anti-sense oligonucleotides partially inhibited AILI when administered individually. It was proposed that JNK had a pathological role in AILI in part by contributing to mitochondrial injury [14,21,35]. Potential limitations of these studies, as acknowledged by the investigators [21,22] is that JNK inhibitors may not be specific and as reported can affect other signaling events that could lead to the misinterpretation of results [36–39]. Further, some of the JNK inhibitors were dissolved in vehicles containing DMSO [14,21] and polyethylene glycol [22,35], which may affect various parameters at the cellular and molecular level [34,40–43].
In conclusion, our findings suggest that JNK2 plays a protective role against AILI in mice at least, in part, by modulating hepatocellular regeneration and repair. Although other research in mice provides evidence for JNK having a pathologic role in AILI, we feel that it is prudent to reconsider the use of JNK inhibitors as a therapeutic intervention in AILI [21,22,35,44].
Acknowledgments
The authors thank John W. George (NHLBI, NIH, Bethesda, MD) for expert animal care and technical assistance. This work was supported by the Intramural Research Program of the NIH and the NHLBI.
References
- 1.Kaplowitz N. Idiosyncratic drug hepatotoxicity. Nat Rev Drug Discov. 2005;4:489–499. doi: 10.1038/nrd1750. [DOI] [PubMed] [Google Scholar]
- 2.James LP, Mayeux PR, Hinson JA. Acetaminophen-induced hepatotoxicity. Drug Metab Dispos. 2003;31:1499–1506. doi: 10.1124/dmd.31.12.1499. [DOI] [PubMed] [Google Scholar]
- 3.Jaeschke H, Bajt ML. Intracellular signaling mechanisms of acetaminophen-induced liver cell death. Toxicol Sci. 2006;89:31–41. doi: 10.1093/toxsci/kfi336. [DOI] [PubMed] [Google Scholar]
- 4.Kaplowitz N. Acetaminophen hepatoxicity: what do we know, what don't we know, and what do we do next? Hepatology. 2004;40:23–26. doi: 10.1002/hep.20312. [DOI] [PubMed] [Google Scholar]
- 5.Jaeschke H, Knight TR, Bajt ML. The role of oxidant stress and reactive nitrogen species in acetaminophen hepatotoxicity. Toxicol Lett. 2003;144:279–288. doi: 10.1016/s0378-4274(03)00239-x. [DOI] [PubMed] [Google Scholar]
- 6.Bourdi M, Eiras DP, Holt MP, Webster MR, Reilly TP, Welch KD, Pohl LR. Role of IL-6 in an IL-10 and IL-4 double knockout mouse model uniquely susceptible to acetaminophen-induced liver injury. Chem Res Toxicol. 2007;20:208–216. doi: 10.1021/tx060228l. [DOI] [PubMed] [Google Scholar]
- 7.Bourdi M, Reilly TP, Elkahloun AG, George JW, Pohl LR. Macrophage migration inhibitory factor in drug-induced liver injury: a role in susceptibility and stress responsiveness. Biochem Biophys Res Commun. 2002;294:225–230. doi: 10.1016/S0006-291X(02)00466-7. [DOI] [PubMed] [Google Scholar]
- 8.Ishida Y, Kondo T, Ohshima T, Fujiwara H, Iwakura Y, Mukaida N. A pivotal involvement of IFN-gamma in the pathogenesis of acetaminophen-induced acute liver injury. FASEB J. 2002;16:1227–1236. doi: 10.1096/fj.02-0046com. [DOI] [PubMed] [Google Scholar]
- 9.Goldring CE, Kitteringham NR, Elsby R, Randle LE, Clement YN, Williams DP, McMahon M, Hayes JD, Itoh K, Yamamoto M, Park BK. Activation of hepatic Nrf2 in vivo by acetaminophen in CD-1 mice. Hepatology. 2004;39:1267–1276. doi: 10.1002/hep.20183. [DOI] [PubMed] [Google Scholar]
- 10.Chan K, Han XD, Kan YW. An important function of Nrf2 in combating oxidative stress: detoxification of acetaminophen. Proc Natl Acad Sci USA. 2001;98:4611–4616. doi: 10.1073/pnas.081082098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chiu H, Brittingham JA, Laskin DL. Differential induction of heme oxygenase-1 in macrophages and hepatocytes during acetaminophen-induced hepatotoxicity in the rat: effects of hemin and biliverdin. Toxicol Appl Pharmacol. 2002;181:106–115. doi: 10.1006/taap.2002.9409. [DOI] [PubMed] [Google Scholar]
- 12.Chiu H, Gardner CR, Dambach DM, Brittingham JA, Durham SK, Laskin JD, Laskin DL. Role of p55 tumor necrosis factor receptor 1 in acetaminophen-induced antioxidant defense. Am J Physiol Gastrointest Liver Physiol. 2003;285:G959–G966. doi: 10.1152/ajpgi.00219.2003. [DOI] [PubMed] [Google Scholar]
- 13.Elsby R, Kitteringham NR, Goldring CE, Lovatt CA, Chamberlain M, Henderson CJ, Wolf CR, Park BK. Increased constitutive c-Jun N-terminal kinase signaling in mice lacking glutathione S-transferase Pi. J Biol Chem. 2003;278:22243–22249. doi: 10.1074/jbc.M301211200. [DOI] [PubMed] [Google Scholar]
- 14.Hanawa N, Shinohara M, Saberi B, Gaarde WA, Han D, Kaplowitz N. Role of JNK translocation to mitochondria leading to inhibition of mitochondria bio-energetics in acetaminophen-induced liver injury. J Biol Chem. 2008;283:13565–13577. doi: 10.1074/jbc.M708916200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bogoyevitch MA, Kobe B. Uses for JNK: the many and varied substrates of the c-Jun N-terminal kinases. Microbiol Mol Biol Rev. 2006;70:1061–1095. doi: 10.1128/MMBR.00025-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Czaja MJ. The future of GI and liver research: editorial perspectives. III. JNK/AP-1 regulation of hepatocyte death. Am J Physiol Gastrointest Liver Physiol. 2003;284:G875–G879. doi: 10.1152/ajpgi.00549.2002. [DOI] [PubMed] [Google Scholar]
- 17.Schwabe RF, Bradham CA, Uehara T, Hatano E, Bennett BL, Schoonhoven R, Brenner DA. c-Jun-N-terminal kinase drives cyclin D1 expression and proliferation during liver regeneration. Hepatology. 2003;37:824–832. doi: 10.1053/jhep.2003.50135. [DOI] [PubMed] [Google Scholar]
- 18.Westwick JK, Weitzel C, Leffert HL, Brenner DA. Activation of Jun kinase is an early event in hepatic regeneration. J Clin Invest. 1995;95:803–810. doi: 10.1172/JCI117730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wisdom R, Johnson RS, Moore C. c-Jun regulates cell cycle progression and apoptosis by distinct mechanisms. EMBO J. 1999;18:188–197. doi: 10.1093/emboj/18.1.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhong Z, Schwabe RF, Kai Y, He L, Yang L, Bunzendahl H, Brenner DA, Lemasters JJ. Liver regeneration is suppressed in small-for-size liver grafts after transplantation: involvement of c-Jun N-terminal kinase, cyclin D1, and defective energy supply. Transplantation. 2006;82:241–250. doi: 10.1097/01.tp.0000228867.98158.d2. [DOI] [PubMed] [Google Scholar]
- 21.Gunawan BK, Liu ZX, Han D, Hanawa N, Gaarde WA, Kaplowitz N. c-Jun N-terminal kinase plays a major role in murine acetaminophen hepatotoxicity. Gastroenterology. 2006;131:165–178. doi: 10.1053/j.gastro.2006.03.045. [DOI] [PubMed] [Google Scholar]
- 22.Henderson NC, Pollock KJ, Frew J, Mackinnon AC, Flavell RA, Davis RJ, Sethi T, Simpson KJ. Critical role of c-jun (NH2) terminal kinase in paracetamol- induced acute liver failure. Gut. 2007;56:982–990. doi: 10.1136/gut.2006.104372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bartolone JB, Sparks K, Cohen SD, Khairallah EA. Immunochemical detection of acetaminophen-bound liver proteins. Biochem Pharmacol. 1987;36:1193–1196. doi: 10.1016/0006-2952(87)90069-4. [DOI] [PubMed] [Google Scholar]
- 24.Yee SB, Bourdi M, Masson MJ, Pohl LR. Hepatoprotective role of endogenous interleukin-13 in a murine model of acetaminophen-induced liver disease. Chem Res Toxicol. 2007;20:734–744. doi: 10.1021/tx600349f. [DOI] [PubMed] [Google Scholar]
- 25.Greenwell A, Foley JF, Maronpot RR. An enhancement method for immuno-histochemical staining of proliferating cell nuclear antigen in archival rodent tissues. Cancer Lett. 1991;59:251–256. doi: 10.1016/0304-3835(91)90149-c. [DOI] [PubMed] [Google Scholar]
- 26.Bourdi M, Masubuchi Y, Reilly TP, Amouzadeh HR, Martin JL, George JW, Shah AG, Pohl LR. Protection against acetaminophen-induced liver injury and lethality by interleukin 10: role of inducible nitric oxide synthase. Hepatology. 2002;35:289–298. doi: 10.1053/jhep.2002.30956. [DOI] [PubMed] [Google Scholar]
- 27.Jaeschke A, Karasarides M, Ventura JJ, Ehrhardt A, Zhang C, Flavell RA, Shokat KM, Davis RJ. JNK2 is a positive regulator of the cjun transcription factor. Mol Cell. 2006;23:899–911. doi: 10.1016/j.molcel.2006.07.028. [DOI] [PubMed] [Google Scholar]
- 28.Fu M, Wang C, Li Z, Sakamaki T, Pestell RG. Minireview: Cyclin D1: normal and abnormal functions. Endocrinology. 2004;145:5439–5447. doi: 10.1210/en.2004-0959. [DOI] [PubMed] [Google Scholar]
- 29.Wang C, Li Z, Fu M, Bouras T, Pestell RG. Signal transduction mediated by cyclin D1: from mitogens to cell proliferation: a molecular target with therapeutic potential. Cancer Treat Res. 2004;119:217–237. doi: 10.1007/1-4020-7847-1_11. [DOI] [PubMed] [Google Scholar]
- 30.Mehendale HM. Tissue repair: an important determinant of final outcome of toxicant-induced injury. Toxicol Pathol. 2005;33:41–51. doi: 10.1080/01926230590881808. [DOI] [PubMed] [Google Scholar]
- 31.Michalopoulos GK. Liver regeneration. J Cell Physiol. 2007;213:286–300. doi: 10.1002/jcp.21172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Farombi EO, Surh YJ. Heme oxygenase-1 as a potential therapeutic target for hepatoprotection. J Biochem Mol Biol. 2006;39:479–491. doi: 10.5483/bmbrep.2006.39.5.479. [DOI] [PubMed] [Google Scholar]
- 33.Jaeschke H, Cover C, Bajt ML. Role of caspases in acetaminophen-induced liver injury. Life Sci. 2006;78:1670–1676. doi: 10.1016/j.lfs.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 34.Masson MJ, Carpenter LD, Graf ML, Pohl LR. Pathogenic role of NKT and NK cells in acetaminophen-induced liver injury is dependent on the presence of DMSO. Hepatology. 2008 doi: 10.1002/hep.22400. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Latchoumycandane C, Goh CW, Ong MM, Boelsterli UA. Mitochondrial protection by the JNK inhibitor leflunomide rescues mice from acetaminophen-induced liver injury. Hepatology. 2007;45:412–421. doi: 10.1002/hep.21475. [DOI] [PubMed] [Google Scholar]
- 36.Bogoyevitch MA, Boehm I, Oakley A, Ketterman AJ, Barr RK. Targeting the JNK MAPK cascade for inhibition: basic science and therapeutic potential. Biochim Biophys Acta. 2004;1697:89–101. doi: 10.1016/j.bbapap.2003.11.016. [DOI] [PubMed] [Google Scholar]
- 37.Bain J, McLauchlan H, Elliott M, Cohen P. The specificities of protein kinase inhibitors: an update. Biochem J. 2003;371:199–204. doi: 10.1042/BJ20021535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee KH, Kim SE, Lee YS. SP600125, a selective JNK inhibitor, aggravates hepatic ischemia-reperfusion injury. Exp Mol Med. 2006;38:408–416. doi: 10.1038/emm.2006.48. [DOI] [PubMed] [Google Scholar]
- 39.Hamilton LC, Vojnovic I, Warner TD. A771726, the active metabolite of leflunomide, directly inhibits the activity of cyclo-oxygenase-2 in vitro and in vivo in a substrate-sensitive manner. Br J Pharmacol. 1999;127:1589–1596. doi: 10.1038/sj.bjp.0702708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kishioka T, lida C, Fujii K, Nagae R, Onishi Y, Ichi I, Kojo S. Effect of dimethyl sulphoxide on oxidative stress, activation of mitogen activated protein kinase and necrosis caused by thioacetamide in the rat liver. Eur J Pharmacol. 2007;564:190–195. doi: 10.1016/j.ejphar.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 41.lida C, Fujii K, Koga E, Washino Y, Ichi I, Kojo S. Inhibitory effect of dimethyl sulfoxide (DMSO) on necrosis and oxidative stress caused by D-galactosamine in the rat liver. J Nutr Sci Vitaminol (Tokyo) 2007;53:160–165. doi: 10.3177/jnsv.53.160. [DOI] [PubMed] [Google Scholar]
- 42.Bertuglia S, Veronese FM, Pasut G. Polyethylene glycol and a novel developed polyethylene glycol-nitric oxide normalize arteriolar response and oxidative stress in ischemia-reperfusion. Am J Physiol Heart Circ Physiol. 2006;291:H1536–H1544. doi: 10.1152/ajpheart.01114.2005. [DOI] [PubMed] [Google Scholar]
- 43.Hauet T, Mothes D, Goujon JM, Carretier M, Eugene M. Protective effect of polyethylene glycol against prolonged cold ischemia and reperfusion injury: study in the isolated perfused rat kidney. J Pharmacol Exp Ther. 2001;297:946–952. [PubMed] [Google Scholar]
- 44.Schwabe RF. Cell death in the liver-all roads lead to JNK. Gastroenterology. 2006;131:314–316. doi: 10.1053/j.gastro.2006.05.029. [DOI] [PubMed] [Google Scholar]