

Summary
Integrins are transmembrane receptors that bind to extracellular matrix proteins and convey anchorage-dependent signals regulating normal cell proliferation. Integrin signals within the tumor micro-environment also impact cancer cell survival and invasion during tumor progression. These integrin-associated signaling events are transduced in part through the activation of non-receptor protein-tyrosine kinases. Focal adhesion kinase (FAK) is activated by β-subunit integrins in both normal and transformed cells. As genetic inactivation of β1 integrin or FAK yield early embryonic lethal phenotypes associated with decreased cell proliferation, and dominant-negative inhibition of FAK can cause increased cell apoptosis, there is a concern that FAK inhibition may have cytotoxic effects on cell growth or survival. However, FAK-specific small molecule inhibitors do not directly impact cell growth in culture, but yet show potent anti-tumor growth effects in vivo. Additionally, recent studies have shed new insight into the FAK kinase-independent regulation of cell proliferation and survival mediated by the FAK N-terminal FERM (band 4.1, ezrin, radixin, moesin homology) domain. Herein, we review the role of the FAK FERM domain in both the intrinsic regulation of FAK kinase activity and how FERM-mediated nuclear localization of FAK promotes enhanced cell survival through the inhibition of tumor suppressor p53 activation during development and under conditions of cellular stress. As we find that FAK FERM-mediated regulation of p53 occurs in human carcinoma cells, elevated FAK expression in tumors may promote both kinase-dependent and –independent survival mechanisms. We discuss how the pharmacological inhibition of FAK kinase activity may impact tumor progression through combined effects of blocking both tumor- and stromal-associated signaling regulating neovascularization.
Keywords: Integrin, FAK, Pyk2, FERM, cell survival, p53, tumor growth, angiogenesis
Introduction
Integrins are transmembrane receptors that bind to extracellular matrix proteins (ECM) such as laminin, collagen, vitronectin, and fibronectin1. These ECM proteins are major constituents of the tumor stroma and also function as important signaling initiators within the tumor micro-environment2, 3. In normal cells, integrins convey anchorage-dependent signals regulating cell proliferation and survival4. Much research has been accomplished in understanding how various cell surface receptors (G-protein-linked, growth factor, and cytokine receptors) function to activate intracellular signaling cascades leading to changes in gene expression events, cell survival, cell proliferation, and cell motility5. What is not readily appreciated is that many of these responses do not occur when integrin-mediated cell adhesion is disrupted in normal cells6, 7. In addition, integrin signals impact tumor cell survival and can greatly affect tumor progression or metastasis (Fig. 1A)8-10. Moreover, as β1 integrin (CD29) is one of several biomarkers used to identify putative cancer stem cells11, there is a need to understand how integrins generate intracellular signals to affect cell responses.
Figure 1.
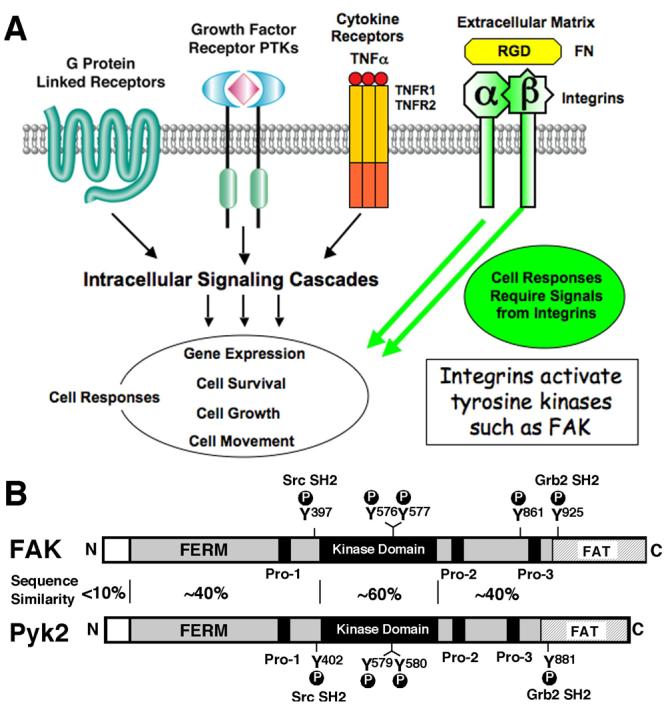
Signaling by integrins through FAK. (A) G-protein-linked, growth factor receptor, and cytokine stimuli activate intracellular signaling cascades and lead to diverse cell responses that require signals from integrin binding to extracellular matrix proteins such as fibronectin (FN). As integrins do not possess intrinsic catalytic activities, the association with and activation of protein-tyrosine kinases (PTKs) such as FAK can link integrins to modulation of cell responses. (B) FAK and Pyk2 contain a central kinase domain flanked by large N- and C-terminal domains with regions of sequence similarity surrounding phosphorylation sites (P) and proline-rich regions (Pro-1 to Pro-3) that are binding sites for Src-homology 3 (SH3) domain-containing proteins such as the adaptor protein, p130Cas. FAK contains 5 major sites of tyrosine phosphorylation (Y397, Y576, Y577, Y861, and Y925) where phosphorylation of either Y397 or Y925 facilitate the binding of Src-homology 2 (SH2) domain containing proteins such as Src kinase and the Grb2 adaptor protein, respectively. The corresponding and conserved phosphorylated tyrosines in Pyk2 are also indicated. The regional amino acid sequence identities between FAK and Pyk2 are indicated with the highest conservation (60%) in the kinase domains. Both proteins contain a FERM (band 4.1, ezrin, radixin, moesin homology) domain in the N-terminal region as well as a FAT (focal adhesion targeting) domain in the C-terminal region that indirectly links FAK to integrins. The FERM and FAT domains of FAK and Pyk2 may have conserved as well as divergent functions33, 54.
Integrins exist in α and β-subunit heterodimers at the cell surface with short cytoplasmic domains that functionally link changes in ECM composition to alterations in the intracellular actin cytoskeleton network12-14. These points of connection are termed focal adhesions that comprise a complex multi-protein scaffolding and signaling unit that mediates cell adhesion and tensile forces important for cell motility15. In tumor cells, focal adhesion structures can become enriched with proteases and these structures are termed invadopodia16, 17, sites that degrade the surrounding ECM and facilitate tumor invasion into the surrounding stroma. As integrin cytoplasmic domains do not possess intrinsic catalytic activity, signaling events must be mediated by associated proteins18. Protein-tyrosine kinases (PTKs) play important roles in mediating signals generated by integrin clustering and cytoplasmic PTK focal adhesion kinase (FAK) indirectly associates with integrins at focal adhesions and is activated by integrin clustering19-21. As FAK activation can also be enhanced by G-protein-linked22, growth factor23, and cytokine stimuli such as tumor necrosis factor α (TNFα)24, research efforts have been directed toward understanding how FAK-associated signaling may control responses such as cell survival and migration25, 26. As FAK expression and tyrosine phosphorylation are up-regulated in many late-stage cancers27, 28, there is a need to determine the role(s) of FAK during tumor progression and what effects small molecule inhibitors targeting FAK activity may have on cell function29, 30. Here, we review recent findings that shed new insights into the regulation of FAK activation and how the role of FAK in promoting normal cell survival-proliferation may be distinct from the role of FAK catalytic activity in tumor progression.
FAK Domain Structure and Signal Connections
FAK and Pyk2 are cytoplasmic tyrosine kinases that comprise a distinct branch of the kinome tree31. FAK is composed of N-terminal FERM (band 4.1, ezrin, radixin, moesin homology), kinase, and C-terminal focal adhesion targeting (FAT) domains (Fig. 1B). The FAT domain indirectly links FAK to integrins and focal adhesions through the binding of proteins such as paxillin. The FERM domain links FAK to growth factor receptors at the plasma membrane and can also facilitate nuclear translocation of FAK under conditions of cellular stress (discussed in detail below). Pyk2 shares a similar domain structure with FAK having ∼40% amino acid sequence identities in the FERM and C-terminal FAT domain regions and ∼60% conservation within the central kinase domain. Although the FAT domain of Pyk2 can bind focal adhesion-associated proteins such as paxillin and can become activated by cell binding to ECM proteins32, it does not strongly localize to focal adhesions in fibroblasts33, suggesting that target protein binding differences exist between the FAT domains of FAK and Pyk2.
This differential linkage to integrins may underlie the distinct expression patterns of FAK and Pyk2. FAK is ubiquitously expressed whereas Pyk2 is expressed primarily in hematopoietic and neuronal cell types34, 35. Very few cells express high levels of both FAK and Pyk2 and no analyses to date have revealed cells lacking both kinases. Interestingly, upon knockdown of FAK expression in primary fibroblasts or endothelial cells, a compensatory up-regulation of Pyk2 expression occurs through an undefined mechanism32, 36, 37. Although this does not occur in all cell types, it is hypothesized that Pyk2 up-regulation may reflect an important cellular function shared by both FAK and Pyk2. This likely involves the regulation of a common signaling target as both FAK and Pyk2 contain conserved proline-rich sites (Pro-1 to Pro-3) that bind Src-homology 3 (SH3) containing proteins such as p130Cas and conserved tyrosine phosphorylation sites, some of which serve as SH2 binding sites for Src-family PTKs or the Grb2 adaptor protein (Fig. 1B). Upon integrin activation, FAK autophosphorylates on Y397 creating an SH2 binding site for Src-family PTKs and forms a signaling complex that phosphorylates p130Cas, paxillin, and other focal adhesion-associated proteins19, 38. Maximal FAK catalytic activation occurs after Src-mediated phosphorylation of FAK within the kinase domain at Y576/57720 and maximal Src activation occurs after FAK phosphorylation of Src within the kinase domain at Y41839. Src-mediated phosphorylation of FAK at Y925 promotes Grb2 adaptor binding and is linked to the activation of the Ras-ERK2 mitogen-activated protein kinase cascade. The FAK-Src complex can phosphorylate various targets leading to the activation of several downstream signaling pathways as well as Rho-family GTPase modulation21.
FERM Regulation of FAK Kinase Activity
Despite the low level of sequence conservation between the FAK FERM and other FERM domain-containing proteins40, 41, the FAK FERM crystal structure revealed that it forms a predicted 3-lobed (F1 to F3) structure42. Early studies found that FERM-domain truncations of FAK yield proteins with increased tyrosine phosphorylation and associated activity43, 44, thus supporting a negative regulatory role in FAK activation. As the FAK FERM can bind the FAK kinase domain and can inhibit FAK activity in trans45, a direct auto-inhibitory mechanism for FAK regulation was proposed. The crystal structure of FAK residues 31-686 encompassing the FERM and kinase domains supported this model46. There are two major points of regulatory contact: binding of the F1 FERM lobe to a linker segment containing Y397 and a hydrophobic pocket within the F2 FERM lobe binding to F596 within the FAK kinase domain. FAK FERM-mediated inhibition of FAK kinase activity therefore results from steric inhibition of target protein access to the catalytic cleft and to the Y397 autophosphorylation site. It is speculated that release of FAK FERM binding to the catalytic domain will allow for FAK autophosphorylation at Y397, recruitment of Src-family PTKs by SH2 binding to the Y397 site, and full FAK activation following Src phosphorylation of FAK in the kinase activation loop46.
Although mutations within the binding interface of the FAK FERM F2 region or kinase domain can generate activated FAK constructs46, the binding of proteins47 or phospholipids48, 49 to the FAK FERM domain can lead to increase FAK Y397 phosphorylation, and fluorescence resonance energy transfer biosensors can discriminate between auto-inhibited and active conformations of FAK49, other studies suggest that integrin-mediated FAK activation involves additional steps. Notably, FERM-truncated FAK displays high catalytic activity, but retains responsiveness to integrin-mediated regulation50. As FAK autophosphorylation occurs primarily through an intermolecular mechanism51, clustering of FAK via binding of paxillin52 or p130Cas53 at focal adhesions is also an important step in FAK activation. Interestingly, the Pyk2 FERM domain may not act in an auto-inhibitory fashion to regulate Pyk2 kinase activity54. Instead, as Pyk2 has been shown to be activated in response to increases in calcium34, the calcium-dependent binding of calmodulin to the Pyk2 FERM domain and the formation of multimeric Pyk2-calmodulin complexes may lead to increased Pyk2 activation55. Clearly, as the FAK and Pyk2 FERM domains may selectively bind target proteins56-58, future studies using mutagenesis based upon known surface residues of the FAK FERM domain will likely yield novel insights into the distinct binding modules present with the FAK and Pyk2 FERM domains.
Inhibition of Integrin Signaling or FAK and Effects on Cell Survival
Evidence in support of a signaling linkage between ECM proteins such as fibronectin (FN) and FAK come from gene targeting studies in mice where FN or FAK loss yields similar early embryonic lethal phenotypes59-61. Embryos lacking FN or FAK contain differentiated endothelial cells (ECs) or their precursors, but fail to develop functioning blood vessels62. These cardiovascular abnormalities contribute to the FAK-null embryo lethality, as the transport of blood cells from their site of synthesis to the embryonic vasculature is impaired63. Isolation of primary FAK-null ECs from embryos was problematic as they did not proliferate in cell culture and showed high levels of apoptosis62. FAK-null EC cell growth was facilitated by inactivation of the p53 tumor suppressor, but FAK−/−p53−/− ECs exhibited defects in cell motility and were unable to form tubular structures in vitro. Conditional deletion of endogenous floxed FAK by crossing with mice expression and EC-specific transgene for the Cre recombinase also results in an embryonic lethal vascular defect phenotype as performed by two independent groups 64, 65. However, although both studies found that loss of EC FAK expression was associated with increased cell apoptosis in vitro and in vivo, there was disagreement with regard to the role of FAK in EC motility. These results may stem from differences in either mouse background or inefficient Cre-mediated FAK excision during development. In addition, as Pyk2 expression is elevated in a compensatory fashion upon Cre-mediated deletion of FAK in adult ECs36, phenotypes and growth regulation differences may be masked by the presence of Pyk2 within FAK-null ECs. In FAK-null fibroblasts, Pyk2 expression is important for enhancing cell growth, but Pyk2 was also causing cellular morphology alterations above and beyond what was due to the loss of FAK37.
Together, these studies support an essential function of FAK in the regulation of both cell survival and motility. One of the key questions is whether the role of FAK in promoting cell proliferation-survival and motility are separable or related. For ECs, dominant-negative over-expression of the FAK C-terminal domain termed FRNK potently blocks growth factor-stimulated cell motility66. FRNK acts as a competitive inhibitor of integrin-FAK linkage and blocks both motility and EC capillary tube formation34, 67. Whereas a number of early studies suggested that FRNK over-expression promoted cell apoptosis68, this is not a general phenomena69-71. We postulate that loss of FAK expression and the ensuing problems in cell proliferation-survival are not equivalent to dominant-negative experiments blocking FAK activation by FRNK over-expression. Moreover, in both ECs and fibroblasts, defects in FAK-null cell proliferation were ameliorated by p53 inactivation. Although FAK activation of pro-survival pathways such as the phosphatidylinositol 3'-kinase-AKT can impact and inhibit p53 activation72, 73, we found that FAK-mediated inhibition of p53 was through a novel pathway that involved FAK FERM-mediated nuclear translocation, binding of p53, and the promotion of murine double minute-2 (Mdm2)-dependent p53 ubiquitination and turnover74. Importantly, this FAK-FERM-mediated survival pathway did not require intrinsic FAK activity.
FAK FERM Regulation of p53, A Novel Survival Pathway
For FAK-null ECs and fibroblasts, growth in culture is facilitated by co-inactivation of p5361, 62. Analysis of embryos revealed that loss of FAK resulted in p53 activation and the up-regulated expression of cell cycle progression inhibitors such as p21(Cip/WAF1) (Fig. 2A). Elevated expression of p53 and p21 did not result in increased apoptosis, but was associated with a block in mesenchymal cell proliferation in the embryo74. Interestingly, deletion of β1 integrin in mammary glands was also associated with increased p21 levels and reduced cell proliferation75. Importantly, deletion of p21 facilitated FAK-null fibroblast growth in culture and allowed for analyses of how FAK could regulate endogenous p53 through add-back experiments74. Re-expression of the FAK FERM domain alone acted to reduce p53 levels and p53 transcriptional activity. Although FAK binding to p53 has been proposed to inhibit p53 transcriptional activity76, we found that the MG132 proteosomal inhibitor blocks FAK FERM regulation of p53. Further experiments showed that FAK FERM functioned as a scaffold with the binding of p53 and Mdm2 to different lobes of the FERM domain. p53 inactivation by FAK required FAK FERM F1 lobe binding to p53, FERM F2 lobe-mediated nuclear translocation, and FERM F3 lobe for connections to Mdm2 and proteasomal degradation. This novel FAK FERM mediated survival pathway is different from canonical FAK functions in two ways, it is a FAK kinase independent event and the direct regulation of p53 requires FAK nuclear localization.
Figure 2.
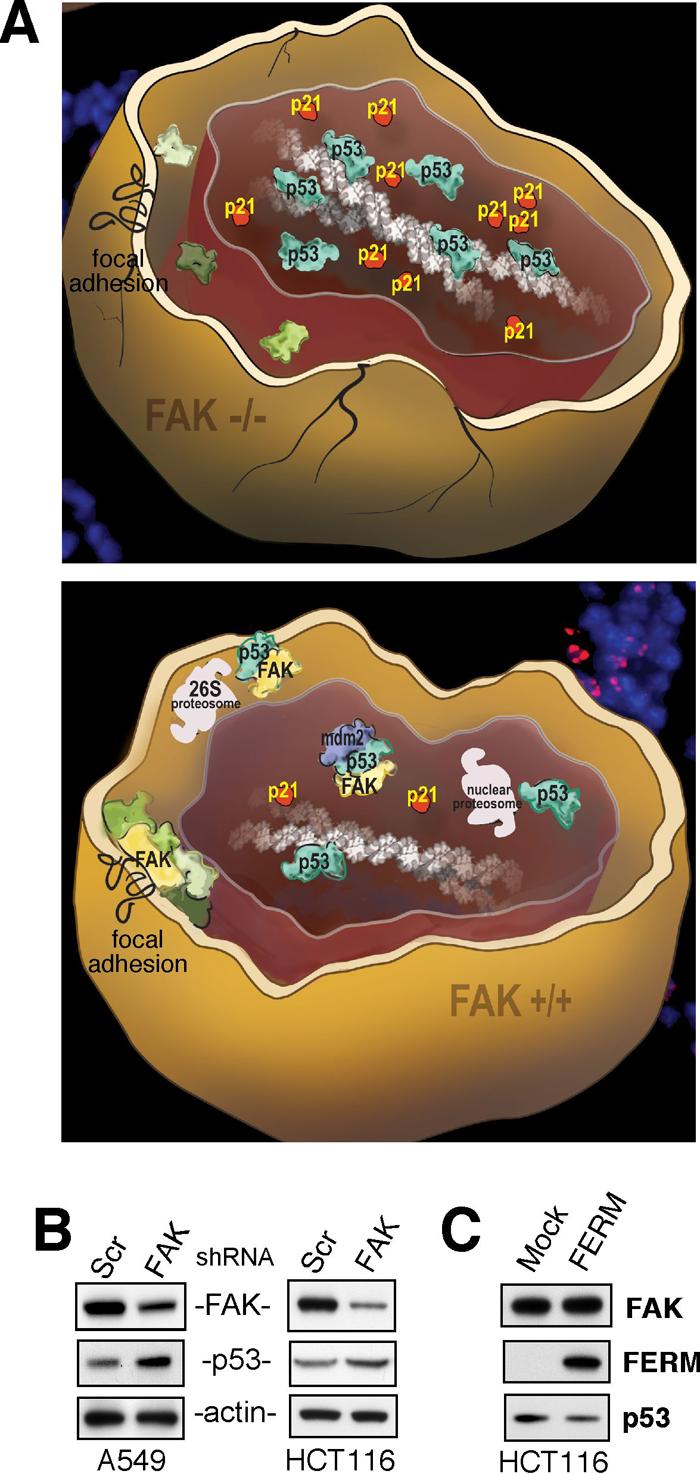
FAK FERM regulation of p53 during development and in carcinoma cells. (A) Model of how loss of FAK affects mesenchymal cell survival during development. Depicted are FAK-null (FAK−/−, top) and normal (FAK+/+, bottom) mouse cells where knockout of FAK leads to early embryonic lethality and FAK−/− cells exhibit elevated tumor suppressor p53 activation, leading to enhanced p21(Cip/WAF1) expression, and a p21-dependent block in cell proliferation. Elevated p53 activity in FAK−/− cells inhibits proliferation, but does not directly promote apoptosis. In normal cells, FAK signaling occurs at sites of integrin clustering at the plasma membrane (focal adhesions) as well as FAK translocation to the nucleus upon conditions of reduced adhesion or cellular stress. FAK-mediated control of p53 and cell survival involves FAK FERM-mediated nuclear translocation, p53 binding, and FAK FERM-enhanced murine double minute-2 (Mdm2) ubiquitination leading to p53 proteosomal degradation. This regulatory connection between FAK and p53 is dependent on the FAK FERM domain but is independent of FAK kinase activity. (B) FAK expression was reduced by short-hairpin RNA (shRNA) interference in A549 lung or HCT116 colorectal carcinoma cells and compared to scrambled (Scr) shRNA-expressing controls. FAK knockdown was associated with increased p53 protein levels as determined by anti-FAK, p53, and actin immunoblotting. (C) Recombinant adenovirus was used to over-express the FAK FERM region in HCT116 carcinoma cells and this resulted in decreased p53 levels compared to Mock-infected cells as determined by anti-FAK, Myc-tag for FERM, and p53 immunoblotting.
FAK FERM-mediated Nuclear Localization
There have been several reports of FAK in the nucleus based upon immunofluorescent staining patterns or the distribution of proteolytic breakdown products of FAK in nuclear versus cytoplasmic extracts77-79. Although it remains unclear as to whether proteolytic products of FAK have biological activity, addition of the nuclear export inhibitor leptomycin B to cells facilitates the accumulation of full-length FAK in the nucleus74, 80. This indicates that FAK is shuttling in and out of the nucleus in cells. GFP-fusion protein studies showed that FAK nuclear targeting is mediated by the FERM domain76, 81 and this activity is selectively-observed for other FERM domain-containing proteins82, 83. Although FAK can become sumoylated within the FERM domain84, this is not required for nuclear FAK accumulation74. The nuclear localization motif within FAK FERM is localized to the F2 lobe and is comprised of a basic residue cluster (K190, K191, K216, K218, R221, and K222) separated by primary sequence but contiguous at the distal tip of the F2 lobe74. FAK mutations (R177A/ R178A) that are likely to disrupt FAK FERM domain folding also inhibit FERM-mediated nuclear translocation. What remain unknown are the controlling events that promote the import and facilitate the export of FAK from the nucleus. We found that stress stimuli such as staurosporine or the disruption of cell adhesion triggered increased FAK nuclear accumulation independent of FAK activation74. For Pyk2, over-expression85 or neuronal depolarization is associated with nuclear accumulation86. Interestingly, calcineurin (protein phosphatase 2B) was found to be important for Pyk2 nuclear translocation86 and rapid FAK nuclear accumulation was associated with staurosporine-associated loss of FAK tyrosine phosphorylation74. As FAK and Pyk2 are phosphorylated on multiple serine/threonine residues as well as tyrosine4, it will be interesting to determine whether FAK or Pyk2 nuclear accumulation is regulated by a site-specific phosphorylation event.
FAK-p53 Survival Signaling in Cancer Cells
There is now abundant evidence that FAK expression is elevated in many human cancers87. Although p53 is mutationally-inactivated in many tumors, approximately 50% of cancers remain p53-positive88. In these tumors, secondary events may keep p53 activation levels low. In p53-positive A549 lung and HCT116 colon carcinoma cells, knockdown of FAK results in elevated p53 levels (Fig. 2B). In addition, FAK FERM over-expression can reduce steady-state p53 levels in HCT116 colon carcinoma cells (Fig. 2C). As increased FAK expression is often found in early-stage tumors, our results support the hypothesis that a FAK FERM-mediated cell survival pathway may be functional in tumor cells. During tumor progression and metastasis, a key event facilitating the spread of cells from the primary tumor mass is the ability to grow and survive in an anchorage-independent manner. In most cases, this does not involve integrin signaling. We speculate that under these conditions, a tumor cell that expresses higher levels of FAK may be more resistant to apoptosis by the ability of non-integrin-associated FAK to translocate to the nucleus and prevent excessive p53 activation.
In culture, FERM domain over-expression can enhance breast cancer survival57 and it can reduce p53-mediated apoptosis76. However, it remains to be determined as to whether the FAK FERM domain alone functions to promote tumor progression. Interestingly, FAK and phospho-397-reactive FAK was detected within the nuclear region of colorectal, esophageal, pancreatic, and mammary cancers compared to normal tissue by immunostaining89. As mRNA analyses reveal that alternative-spliced transcripts encompassing the N-terminal FERM domain without the FAK kinase or C-terminal regions are present within neuronal cells90, it will be interesting to determine whether these FAK FERM domain transcripts are also expressed during tumor progression.
FAK Inhibitors in Cancer Therapy
In mouse tumor models, conditional knockout of FAK blocks skin and breast tumor progression91, 92. This is associated with increased cell apoptosis, and it remains unclear if apoptosis is connected to p53 activation upon loss of FAK. In xenotropic mouse tumor studies, knockdown of FAK expression in p53-null tumor cells does not affect cell growth in culture, but results in tumors that are smaller and less vascularized93. In a syngeneic mouse tumor model, knockdown of FAK prevented spontaneous breast to lung metastasis94. FAK re-expression in both models showed that intrinsic FAK kinase activity was needed to promote both tumor growth-angiogenesis and tumor metastasis. FAK signaling was connected to increased production of vascular endothelial growth factor (VEGF) and urokinase plasminogen activator (uPA), known factors involved in tumor progression95, 96. These knockdown and re-expression studies emphasizing the requirement for FAK kinase activity serve as proof-of-principal for the development of small molecule inhibitors of FAK catalytic activity as anti-tumor agents.
Both Novartis and Pfizer have developed small molecule ATP-competitive inhibitors to FAK (Fig. 3A). Novartis TAC-544 and TAE-226 exhibit nanomolar inhibitory activity toward FAK. TAE-226 inhibits FAK, Pyk2 as well as other PTKs such as the insulin-like growth factor I receptor and has anti-tumor activity in glioma97 and ovarian cancer models98. In addition to inhibiting FAK tyrosine phosphorylation, TAE-226 blocked cell proliferation in culture, prevented cell invasion through Matrigel, and increased apoptosis in xenotropic tumor models98, 99. Notably, TAE-226 exhibits synergistic effects with docetaxel in promoting ovarian carcinoma tumor regression with altered levels of angiogenesis, invasion, and apoptosis98. Interestingly, TAE-226 inhibits tumor cell proliferation in a dose-dependent manner in culture and it remains unclear as to whether this is due to the inhibition of FAK or some other target.
Figure 3.

Small molecule inhibitors of FAK and effects on basic fibroblast growth factor (bFGF) induced angiogenesis. (A) Compounds developed by Novartis (TAC-544 and TAE-226) or Pfizer (PF-228 and PF-562,271) inhibit FAK activity in vitro at low nanomolar concentrations and can reduce FAK Y397 phosphorylation in cells at sub-micromolar concentrations. TAC-544 and PF-228 show specificity toward FAK versus Pyk2 and the dual FAK-Pyk2 inhibitors (TAE-226 and PF-562,271) can act to reduce tumor growth in mouse models 98, 99, 101. (B) Angiogenesis in the chicken chorioallantoic membrane (CAM) assay is stimulated by bFGF addition and inhibited by administration of PF-562,271. Data represent average number of vessel branch points +/− SEM (n= 12). (C) Representative images of chicken CAMs after saline, bFGF, or bFGF plus PF-562,271 addition. Blood vessels are dark with backlit illumination of the CAM. Scale bar is 0.5 mm.
In parallel, Pfizer has developed a related compound (PF-228) that shows nanomolar inhibitory activity toward FAK and specificity to FAK compared to other PTKs100. Notably, PF-228 did not inhibit fibroblast or prostate carcinoma cell proliferation or induce apoptosis in culture at concentrations that inhibited FAK Y397 phosphorylation. This result is consistent with PF-228 being more selective toward FAK than TAE-226, and supports our hypothesis that FAK kinase activity is not essential for cell growth-proliferation as this is mediated through FAK FERM regulation of p53. In functional cell-based assays, PF-228 had potent effects in blocking cell motility in part through decreased focal adhesion turnover100. However, one interesting finding with compounds such as TAC-544 and PF-228 that inhibit FAK but not Pyk2, is that there is a potential for rapid compensatory Pyk2 kinase activation in cells with inhibited FAK36. Although it remains unclear how or why this occurs, the potential biological impact of Pyk2 in promoting RhoGTPase activation and triggering focal adhesion formation37 should be carefully evaluated when using these compounds with cells. Although it remains unclear as to whether pharmacological inhibition of FAK in vivo may lead to the activation of Pyk2, FAK knockout studies have documented this occurrence36 and the co-targeting of both FAK and Pyk2 with a small molecule inhibitor may be needed to insure the complete inhibition of FAK-associated signaling pathways.
To this end, Pfizer has also developed a related diaminopyrimindine-type compound (PF562,271) that inhibits FAK and Pyk2 and shows a high degree of selectivity in the inhibition of these PTKs (Fig. 3A)101. The crystal structure shows that PF-562,271 makes unique contacts with the DFG loop of the FAK kinase domain which may impart its selectivity for FAK. Twice-daily administration of PF-562,271 resulted in the inhibition of tumor growth in a dose-dependent manner in PC3M (prostate), BxPC3 (pancreatic), LoVo (colon), U87MG (glioblastoma),and H460 lung xenotropic tumor models101. Tumors treated with PF-562,271 showed reduced microvascular density and increased TUNEL staining, but how or why this occurred were not revealed. One possibility is that FAK inhibition prevents the production of angiogenic growth factors from tumors. This is supported by FAK knockdown and re-expression studies showing that kinase-inactive FAK did not facilitate the angiogenic switch as did tumor cells re-expressing normal FAK93. As FAK signaling is important for the production of tumor-associated VEGF and uPA expression, the inhibition of FAK is likely to affect the production to pro-survival, angiogenic, or invasive factors.
Alternately or potentially concurrently, small molecule inhibitors of FAK may function as anti-angiogenic drugs with effects on the tumor stroma. To the end, we found that PF-562,271 blocked bFGF-stimulated blood vessel angiogenesis as performed in chicken chorioallantoic membrane assays (Fig. 3B and C). Low level administration of PF-262,271 potently blocked blood vessel sprouting without detectable changes in vascular leakage (Fig. 3C). As FAK expression is elevated in tumor-associated endothelial cells102, a FAK inhibitor may selectively act on blood vessels that are undergoing remodeling during the processes of angiogenesis. Importantly, Pfizer PF-562,271 is being evaluated in human trials and has shown low levels of drug toxicity30. Moreover, potential efficacy responses (46% decline in tumor mass) were observed in one patent with ovarian carcinoma and other patients exhibit signs of disease stabilization upon initiating PF-562,271 treatment30. Clearly, these early studies bode well for the future of FAK inhibitors in the treatment of cancer.
Conclusions
It is an interesting time to study integrin signaling and the functions of the FAK and Pyk2 kinases in these events. The development of small molecule inhibitors to FAK will allow for continued studies of how FAK functions to promote tumor progression, and through the use of conditional knockout animals combined with inhibitor studies, we might be able to glean insights into whether FAK signaling within the tumor or stroma is the key event in promoting tumor progression. Further, it will be exciting to determine whether FAK FERM-mediated regulation of p53 might act in a kinase independent manner to promote tumor survival. As results in ovarian carcinoma have shown that inhibition of FAK can synergize with existing therapies such as docetaxel to enhance tumor regression, and it is becoming clear that FAK activity is not essential for promoting cell survival and proliferation in the absence of p53 activity, further studies are likely to provide important information as to whether inhibitors of FAK may show increased anti-tumor efficacy when used in combination to cytostatic therapies such as cisplatin or radiation treatments.
Material and Methods
RNA interference, virus production, and immunoblotting
Human FAK and scrambled shRNA cloned into pLentiLox 3.7 were used as described94. Stable shRNA-expressing cell lines were generated by sorting for green-fluorescent protein that is expressed from a second promoter within pLentiLox. Adenoviral Myc-tagged FAK FERM (residues 1-402) were cloned into pCMV-Shuttle, adenovirus produced using the AdEasy system (Stratagene), and target cells infected with 50 plaque forming units of virus per cell as described74. A549 lung and HCT116 colon carcinoma cells were obtained from ATCC. Cell lysates were made in modified RIPA cell lysis buffer as described94, and anti-FAK (4.47) from Millipore, anti-Myc-tag (9E10) from Covance, anti-β-actin (AC-17) from Sigma, and anti-p53 (DO-1) from Santa Cruz were used for immunoblotting.
Chicken chorioallantoic membrane (CAM) angiogenesis
Windows were cut in the shell of 10-day-old fertilized chicken eggs, the CAM was lowered with forceps, and sterile filter discs were placed onto the CAM surface as described103. 10 μl of 10 ng/μl bFGF was added to the disc, the shell sealed with tape, and eggs incubated in a humidified incubator at 38°C. After 24 h, 10 μl of PF-562,271 (100 pmol/μl) in dimethylsulfoxide-saline or an equal amount of vehicle was added to the filter disc and the eggs resealed. After 72 h, filter discs with the associated CAM tissue were excised, placed into petri dishes, and blood vessel branch points were imaged and counted using a dissecting microscope (Zeiss M2-Bio equipped with an AxioCam MRM CCD camera). PF-562,271 was synthesized and purity evaluated by mass spectroscopy as described36.
Acknowledgments
S.T. Lim was supported in part by an American Heart Association (AHA) postdoctoral fellowship (0725169Y). This work was supported by NIH grants to Dwayne Stupack (CA107263) and to David Schlaepfer (CA102310). D. Schlaepfer is an Established Investigator of the AHA (0540115N).
Abbreviations
- bFGF
basic fibroblast growth factor
- CAM
chicken chorioallantoic membrane
- ECM
extracellular matrix
- EC
endothelial cell
- FAK
focal adhesion kinase
- FAT
focal adhesion targeting
- FERM
band 4.1, ezrin, radixin, moesin homology
- FN
fibronectin
- FRNK
FAK related non-kinase
- PTK
protein-tyrosine kinase
- Mdm-2
murine double minute-2
- MEFs
mouse embryonic fibroblasts
- SH
Src-homology
- shRNA
short-hairpin RNA
- TNFα
tumor necrosis factor α
- uPA
urokinase plasminogen activator
- VEGF
vascular endothelial growth factor
- WT
wildtype
References
- 1.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–87. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 2.Guo W, Giancotti FG. Integrin signalling during tumour progression. Nat Rev Mol Cell Biol. 2004;5:816–26. doi: 10.1038/nrm1490. [DOI] [PubMed] [Google Scholar]
- 3.Larsen M, Artym VV, Green JA, Yamada KM. The matrix reorganized: extracellular matrix remodeling and integrin signaling. Curr Opin Cell Biol. 2006;18:463–71. doi: 10.1016/j.ceb.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 4.Reddig PJ, Juliano RL. Clinging to life: cell to matrix adhesion and cell survival. Cancer Metastasis Rev. 2005;24:425–39. doi: 10.1007/s10555-005-5134-3. [DOI] [PubMed] [Google Scholar]
- 5.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–25. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 6.Assoian RK, Schwartz MA. Coordinate signaling by integrins and receptor tyrosine kinases in the regulation of G1 phase cell-cycle progression. Curr Opin Genet Dev. 2001;11:48–53. doi: 10.1016/s0959-437x(00)00155-6. [DOI] [PubMed] [Google Scholar]
- 7.Schwartz MA, Assoian RK. Integrins and cell proliferation: regulation of cyclin-dependent kinases via cytoplasmic signaling pathways. J Cell Sci. 2001;114:2553–60. doi: 10.1242/jcs.114.14.2553. [DOI] [PubMed] [Google Scholar]
- 8.Stupack DG, Teitz T, Potter MD, Mikolon D, Houghton PJ, Kidd VJ, Lahti JM, Cheresh DA. Potentiation of neuroblastoma metastasis by loss of caspase-8. Nature. 2006;439:95–9. doi: 10.1038/nature04323. [DOI] [PubMed] [Google Scholar]
- 9.White DE, Muller WJ. Multifaceted roles of integrins in breast cancer metastasis. J Mammary Gland Biol Neoplasia. 2007;12:135–42. doi: 10.1007/s10911-007-9045-5. [DOI] [PubMed] [Google Scholar]
- 10.Kuphal S, Bauer R, Bosserhoff AK. Integrin signaling in malignant melanoma. Cancer Metastasis Rev. 2005;24:195–222. doi: 10.1007/s10555-005-1572-1. [DOI] [PubMed] [Google Scholar]
- 11.Woodward WA, Sulman EP. Cancer stem cells: markers or biomarkers? Cancer Metastasis Rev. 2008 doi: 10.1007/s10555-008-9130-2. [DOI] [PubMed] [Google Scholar]
- 12.Miranti CK, Brugge JS. Sensing the environment: a historical perspective on integrin signal transduction. Nat Cell Biol. 2002;4:E83–90. doi: 10.1038/ncb0402-e83. [DOI] [PubMed] [Google Scholar]
- 13.Romer LH, Birukov KG, Garcia JG. Focal adhesions: paradigm for a signaling nexus. Circ Res. 2006;98:606–16. doi: 10.1161/01.RES.0000207408.31270.db. [DOI] [PubMed] [Google Scholar]
- 14.Berrier AL, Yamada KM. Cell-matrix adhesion. J Cell Physiol. 2007;213:565–73. doi: 10.1002/jcp.21237. [DOI] [PubMed] [Google Scholar]
- 15.Zaidel-Bar R, Cohen M, Addadi L, Geiger B. Hierarchical assembly of cell-matrix adhesion complexes. Biochem Soc Trans. 2004;32:416–20. doi: 10.1042/BST0320416. [DOI] [PubMed] [Google Scholar]
- 16.Buccione R, Orth JD, McNiven MA. Foot and mouth: podosomes, invadopodia and circular dorsal ruffles. Nat Rev Mol Cell Biol. 2004;5:647–57. doi: 10.1038/nrm1436. [DOI] [PubMed] [Google Scholar]
- 17.Weaver AM. Invadopodia: specialized cell structures for cancer invasion. Clin Exp Metastasis. 2006;23:97–105. doi: 10.1007/s10585-006-9014-1. [DOI] [PubMed] [Google Scholar]
- 18.Schwartz MA. Integrin signaling revisited. Trends Cell Biol. 2001;11:466–70. doi: 10.1016/s0962-8924(01)02152-3. [DOI] [PubMed] [Google Scholar]
- 19.Parsons JT. Focal adhesion kinase: the first ten years. J Cell Sci. 2003;116:1409–16. doi: 10.1242/jcs.00373. [DOI] [PubMed] [Google Scholar]
- 20.Hanks SK, Ryzhova L, Shin NY, Brabek J. Focal adhesion kinase signaling activities and their implications in the control of cell survival and motility. Front Biosci. 2003;8:982–96. doi: 10.2741/1114. [DOI] [PubMed] [Google Scholar]
- 21.Mitra SK, Schlaepfer DD. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr Opin Cell Biol. 2006;18:516–23. doi: 10.1016/j.ceb.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 22.Rozengurt E. Mitogenic signaling pathways induced by G protein-coupled receptors. J Cell Physiol. 2007;213:589–602. doi: 10.1002/jcp.21246. [DOI] [PubMed] [Google Scholar]
- 23.Sieg DJ, Hauck CR, Ilic D, Klingbeil CK, Schaefer E, Damsky CH, Schlaepfer DD. FAK integrates growth-factor and integrin signals to promote cell migration. Nat Cell Biol. 2000;2:249–56. doi: 10.1038/35010517. [DOI] [PubMed] [Google Scholar]
- 24.Schlaepfer DD, Hou S, Lim ST, Tomar A, Yu H, Lim Y, Hanson DA, Uryu SA, Molina J, Mitra SK. Tumor necrosis factor-alpha stimulates focal adhesion kinase activity required for mitogen-activated kinase-associated interleukin 6 expression. J Biol Chem. 2007;282:17450–9. doi: 10.1074/jbc.M610672200. [DOI] [PubMed] [Google Scholar]
- 25.Cox BD, Natarajan M, Stettner MR, Gladson CL. New concepts regarding focal adhesion kinase promotion of cell migration and proliferation. J Cell Biochem. 2006;99:35–52. doi: 10.1002/jcb.20956. [DOI] [PubMed] [Google Scholar]
- 26.Mitra SK, Hanson DA, Schlaepfer DD. Focal adhesion kinase: in command and control of cell motility. Nat Rev Mol Cell Biol. 2005;6:56–68. doi: 10.1038/nrm1549. [DOI] [PubMed] [Google Scholar]
- 27.Cance WG, Harris JE, Iacocca MV, Roche E, Yang X, Chang J, Simkins S, Xu L. Immunohistochemical analyses of focal adhesion kinase expression in benign and malignant human breast and colon tissues: correlation with preinvasive and invasive phenotypes. Clin Cancer Res. 2000;6:2417–23. [PubMed] [Google Scholar]
- 28.Lark AL, Livasy CA, Dressler L, Moore DT, Millikan RC, Geradts J, Iacocca M, Cowan D, Little D, Craven RJ, Cance W. High focal adhesion kinase expression in invasive breast carcinomas is associated with an aggressive phenotype. Mod Pathol. 2005;18:1289–94. doi: 10.1038/modpathol.3800424. [DOI] [PubMed] [Google Scholar]
- 29.McLean GW, Carragher NO, Avizienyte E, Evans J, Brunton VG, Frame MC. The role of focal-adhesion kinase in cancer - a new therapeutic opportunity. Nat Rev Cancer. 2005;5:505–15. doi: 10.1038/nrc1647. [DOI] [PubMed] [Google Scholar]
- 30.Parsons JT, Slack-Davis J, Tilghman R, Roberts WG. Focal adhesion kinase: targeting adhesion signaling pathways for therapeutic intervention. Clin Cancer Res. 2008;14:627–32. doi: 10.1158/1078-0432.CCR-07-2220. [DOI] [PubMed] [Google Scholar]
- 31.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–34. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 32.Sieg DJ, Ilic D, Jones KC, Damsky CH, Hunter T, Schlaepfer DD. Pyk2 and Src-family protein-tyrosine kinases compensate for the loss of FAK in fibronectin-stimulated signaling events but Pyk2 does not fully function to enhance FAK− cell migration. EMBO J. 1998;17:5933–47. doi: 10.1093/emboj/17.20.5933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klingbeil CK, Hauck CR, Hsia DA, Jones KC, Reider SR, Schlaepfer DD. Targeting Pyk2 to beta 1-integrin-containing focal contacts rescues fibronectin-stimulated signaling and haptotactic motility defects of focal adhesion kinase-null cells. J Cell Biol. 2001;152:97–110. doi: 10.1083/jcb.152.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Avraham H, Park SY, Schinkmann K, Avraham S. RAFTK/Pyk2-mediated cellular signalling. Cell Signal. 2000;12:123–33. doi: 10.1016/s0898-6568(99)00076-5. [DOI] [PubMed] [Google Scholar]
- 35.Orr AW, Murphy-Ullrich JE. Regulation of endothelial cell function BY FAK and PYK2. Front Biosci. 2004;9:1254–66. doi: 10.2741/1239. [DOI] [PubMed] [Google Scholar]
- 36.Weis SM, Lim ST, Lutu-Fuga KM, Barnes LA, Chen XL, Göthert JR, Shen TL, Guan JL, Schlaepfer DD, Cheresh DA. Compensatory role for Pyk2 during angiogenesis in adult mice lacking endothelial cell FAK. J Cell Biol. 2008;181:43–50. doi: 10.1083/jcb.200710038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lim Y, Lim ST, Tomar A, Gardel M, Bernard-Trifilo JA, Chen XL, Uryu SA, Canete-Soler R, Zhai J, Lin H, Schlaepfer WW, Nalbant P, Bokoch G, Ilic D, Waterman-Storer C, Schlaepfer DD. PyK2 and FAK connections to p190Rho guanine nucleotide exchange factor regulate RhoA activity, focal adhesion formation, and cell motility. J Cell Biol. 2008;180:187–203. doi: 10.1083/jcb.200708194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schlaepfer DD, Hauck CR, Sieg DJ. Signaling through focal adhesion kinase. Prog Biophys Mol Biol. 1999;71:435–78. doi: 10.1016/s0079-6107(98)00052-2. [DOI] [PubMed] [Google Scholar]
- 39.Wu L, Bernard-Trifilo JA, Lim Y, Lim ST, Mitra SK, Uryu S, Chen M, Pallen CJ, Cheung NK, Mikolon D, Mielgo A, Stupack DG, Schlaepfer DD. Distinct FAK-Src activation events promote alpha5beta1 and alpha4beta1 integrin-stimulated neuroblastoma cell motility. Oncogene. 2008 doi: 10.1038/sj.onc.1210770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Girault JA, Labesse G, Mornon JP, Callebaut I. The N-termini of FAK and JAKs contain divergent band 4.1 domains. Trends Biochem Sci. 1999;24:54–7. doi: 10.1016/s0968-0004(98)01331-0. [DOI] [PubMed] [Google Scholar]
- 41.Diakowski W, Grzybek M, Sikorski AF. Protein 4.1, a component of the erythrocyte membrane skeleton and its related homologue proteins forming the protein 4.1/FERM superfamily. Folia Histochem Cytobiol. 2006;44:231–48. [PubMed] [Google Scholar]
- 42.Ceccarelli DF, Song HK, Poy F, Schaller MD, Eck MJ. Crystal structure of the FERM domain of focal adhesion kinase. J Biol Chem. 2006;281:252–9. doi: 10.1074/jbc.M509188200. [DOI] [PubMed] [Google Scholar]
- 43.Chan PY, Kanner SB, Whitney G, Aruffo A. A transmembrane-anchored chimeric focal adhesion kinase is constitutively activated and phosphorylated at tyrosine residues identical to pp125FAK. J Biol Chem. 1994;269:20567–74. [PubMed] [Google Scholar]
- 44.Schlaepfer DD, Hunter T. Evidence for in vivo phosphorylation of the Grb2 SH2-domain binding site on focal adhesion kinase by Src-family protein-tyrosine kinases. Mol Cell Biol. 1996;16:5623–33. doi: 10.1128/mcb.16.10.5623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cooper LA, Shen TL, Guan JL. Regulation of focal adhesion kinase by its amino-terminal domain through an autoinhibitory interaction. Mol Cell Biol. 2003;23:8030–41. doi: 10.1128/MCB.23.22.8030-8041.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lietha D, Cai X, Ceccarelli DF, Li Y, Schaller MD, Eck MJ. Structural basis for the autoinhibition of focal adhesion kinase. Cell. 2007;129:1177–87. doi: 10.1016/j.cell.2007.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Poullet P, Gautreau A, Kadare G, Girault JA, Louvard D, Arpin M. Ezrin interacts with focal adhesion kinase and induces its activation independently of cell-matrix adhesion. J Biol Chem. 2001;276:37686–91. doi: 10.1074/jbc.M106175200. [DOI] [PubMed] [Google Scholar]
- 48.Dunty JM, Gabarra-Niecko V, King ML, Ceccarelli DF, Eck MJ, Schaller MD. FERM domain interaction promotes FAK signaling. Mol Cell Biol. 2004;24:5353–68. doi: 10.1128/MCB.24.12.5353-5368.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cai X, Lietha D, Ceccarelli DF, Karginov AV, Rajfur Z, Jacobson K, Hahn KM, Eck MJ, Schaller MD. Spatial and temporal regulation of focal adhesion kinase activity in living cells. Mol Cell Biol. 2008;28:201–14. doi: 10.1128/MCB.01324-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jacamo RO, Rozengurt E. A truncated FAK lacking the FERM domain displays high catalytic activity but retains responsiveness to adhesion-mediated signals. Biochem Biophys Res Commun. 2005;334:1299–304. doi: 10.1016/j.bbrc.2005.07.034. [DOI] [PubMed] [Google Scholar]
- 51.Toutant M, Costa A, Studler JM, Kadare G, Carnaud M, Girault JA. Alternative splicing controls the mechanisms of FAK autophosphorylation. Mol Cell Biol. 2002;22:7731–43. doi: 10.1128/MCB.22.22.7731-7743.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hagel M, George EL, Kim A, Tamimi R, Opitz SL, Turner CE, Imamoto A, Thomas SM. The adaptor protein paxillin is essential for normal development in the mouse and is a critical transducer of fibronectin signaling. Mol Cell Biol. 2002;22:901–15. doi: 10.1128/MCB.22.3.901-915.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Iwahara T, Akagi T, Fujitsuka Y, Hanafusa H. CrkII regulates focal adhesion kinase activation by making a complex with Crk-associated substrate, p130Cas. Proc Natl Acad Sci U S A. 2004;101:17693–8. doi: 10.1073/pnas.0408413102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dunty JM, Schaller MD. The N termini of focal adhesion kinase family members regulate substrate phosphorylation, localization, and cell morphology. J Biol Chem. 2002;277:45644–54. doi: 10.1074/jbc.M201779200. [DOI] [PubMed] [Google Scholar]
- 55.Kohno T, Matsuda E, Sasaki H, Sasaki T. Protein-tyrosine kinase CAKbeta/PYK2 is activated by binding Ca2+/calmodulin to FERM F2 alpha2 helix and thus forming its dimer. Biochem J. 2008;410:513–23. doi: 10.1042/BJ20070665. [DOI] [PubMed] [Google Scholar]
- 56.Serrels B, Serrels A, Brunton VG, Holt M, McLean GW, Gray CH, Jones GE, Frame MC. Focal adhesion kinase controls actin assembly via a FERM-mediated interaction with the Arp2/3 complex. Nat Cell Biol. 2007;9:1046–56. doi: 10.1038/ncb1626. [DOI] [PubMed] [Google Scholar]
- 57.Kurenova E, Xu LH, Yang X, Baldwin AS, Jr., Craven RJ, Hanks SK, Liu ZG, Cance WG. Focal adhesion kinase suppresses apoptosis by binding to the death domain of receptor-interacting protein. Mol Cell Biol. 2004;24:4361–71. doi: 10.1128/MCB.24.10.4361-4371.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen SY, Chen HC. Direct interaction of focal adhesion kinase (FAK) with Met is required for FAK to promote hepatocyte growth factor-induced cell invasion. Mol Cell Biol. 2006;26:5155–67. doi: 10.1128/MCB.02186-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.George EL, Georges-Labouesse EN, Patel-King RS, Rayburn H, Hynes RO. Defects in mesoderm, neural tube and vascular development in mouse embryos lacking fibronectin. Development. 1993;119:1079–91. doi: 10.1242/dev.119.4.1079. [DOI] [PubMed] [Google Scholar]
- 60.George EL, Baldwin HS, Hynes RO. Fibronectins are essential for heart and blood vessel morphogenesis but are dispensable for initial specification of precursor cells. Blood. 1997;90:3073–81. [PubMed] [Google Scholar]
- 61.Ilic D, Furuta Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N, Nomura S, Fujimoto J, Okada M, Yamamoto T, Aizawa S. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. 1995;377:539–44. doi: 10.1038/377539a0. [DOI] [PubMed] [Google Scholar]
- 62.Ilic D, Kovacic B, McDonagh S, Jin F, Baumbusch C, Gardner DG, Damsky CH. Focal adhesion kinase is required for blood vessel morphogenesis. Circ Res. 2003;92:300–7. doi: 10.1161/01.res.0000055016.36679.23. [DOI] [PubMed] [Google Scholar]
- 63.Schlaepfer DD, Mitra SK, Ilic D. Control of motile and invasive cell phenotypes by focal adhesion kinase. Biochim Biophys Acta. 2004;1692:77–102. doi: 10.1016/j.bbamcr.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 64.Shen T-L, Park AYJ, Alcaraz A, Peng X, Jang I, Koni P, Flavell RA, Gu H, Guan J-L. Conditional knockout of focal adhesion kinase in endothelial cells reveals its role in angiogenesis and vascular development in late embryogenesis. J Cell Biol. 2005;169:941–52. doi: 10.1083/jcb.200411155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Braren R, Hu H, Kim YH, Beggs HE, Reichardt LF, Wang R. Endothelial FAK is essential for vascular network stability, cell survival, and lamellipodial formation. J Cell Biol. 2006;172:151–62. doi: 10.1083/jcb.200506184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Qi JH, Claesson-Welsh L. VEGF-induced activation of phosphoinositide 3-kinase is dependent on focal adhesion kinase. Exp Cell Res. 2001;263:173–82. doi: 10.1006/excr.2000.5102. [DOI] [PubMed] [Google Scholar]
- 67.Haskell H, Natarajan M, Hecker TP, Ding Q, Stewart J, Jr., Grammer JR, Gladson CL. Focal adhesion kinase is expressed in the angiogenic blood vessels of malignant astrocytic tumors in vivo and promotes capillary tube formation of brain microvascular endothelial cells. Clin Cancer Res. 2003;9:2157–65. [PubMed] [Google Scholar]
- 68.Xu LH, Yang X, Craven RJ, Cance WG. The COOH-terminal domain of the focal adhesion kinase induces loss of adhesion and cell death in human tumor cells. Cell Growth Differ. 1998;9:999–1005. [PubMed] [Google Scholar]
- 69.Hauck CR, Hsia DA, Puente XS, Cheresh DA, Schlaepfer DD. FRNK blocks v-Src-stimulated invasion and experimental metastases without effects on cell motility or growth. EMBO J. 2002;21:6289–302. doi: 10.1093/emboj/cdf631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.van Nimwegen MJ, Verkoeijen S, van Buren L, Burg D, van de Water B. Requirement for focal adhesion kinase in the early phase of mammary adenocarcinoma lung metastasis formation. Cancer Res. 2005;65:4698–706. doi: 10.1158/0008-5472.CAN-04-4126. [DOI] [PubMed] [Google Scholar]
- 71.Bryant P, Zheng Q, Pumiglia K. Focal adhesion kinase controls cellular levels of p27/Kip1 and p21/Cip1 through Skp2-dependent and -independent mechanisms. Mol Cell Biol. 2006;26:4201–13. doi: 10.1128/MCB.01612-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Xia H, Nho RS, Kahm J, Kleidon J, Henke CA. Focal Adhesion Kinase Is Upstream of Phosphatidylinositol 3-Kinase/Akt in Regulating Fibroblast Survival in Response to Contraction of Type I Collagen Matrices via a {beta}1 Integrin Viability Signaling Pathway. J Biol Chem. 2004;279:33024–34. doi: 10.1074/jbc.M313265200. [DOI] [PubMed] [Google Scholar]
- 73.Feng J, Tamaskovic R, Yang Z, Brazil DP, Merlo A, Hess D, Hemmings BA. Stabilization of Mdm2 via decreased ubiquitination is mediated by protein kinase B/Akt-dependent phosphorylation. J Biol Chem. 2004;279:35510–7. doi: 10.1074/jbc.M404936200. [DOI] [PubMed] [Google Scholar]
- 74.Lim ST, Chen XL, Lim Y, Hanson DA, Vo TT, Howerton K, Larocque N, Fisher SJ, Schlaepfer DD, Ilic D. Nuclear FAK promotes cell proliferation and survival through FERM-enhanced p53 degradation. Mol Cell. 2008;29:9–22. doi: 10.1016/j.molcel.2007.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li N, Zhang Y, Naylor MJ, Schatzmann F, Maurer F, Wintermantel T, Schuetz G, Mueller U, Streuli CH, Hynes NE. b1 integrins regulate mammary gland proliferation and maintain the integrity of mammary alveoli. EMBO J. 2005;24:1942–53. doi: 10.1038/sj.emboj.7600674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Golubovskaya VM, Finch R, Cance WG. Direct Interaction of the N-terminal domain of focal adhesion kinase with the N-terminal transactivation domain of p53. J Biol Chem. 2005;280:25008–21. doi: 10.1074/jbc.M414172200. [DOI] [PubMed] [Google Scholar]
- 77.Levkau B, Herren B, Koyama H, Ross R, Raines EW. Caspase-mediated cleavage of focal adhesion kinase pp125FAK and disassembly of focal adhesions in human endothelial cell apoptosis. J Exp Med. 1998;187:579–86. doi: 10.1084/jem.187.4.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lobo M, Zachary I. Nuclear localization and apoptotic regulation of an amino-terminal domain focal adhesion kinase fragment in endothelial cells. Biochem Biophys Res Commun. 2000;276:1068–74. doi: 10.1006/bbrc.2000.3547. [DOI] [PubMed] [Google Scholar]
- 79.Jones G, Machado J, Jr., Merlo A. Loss of focal adhesion kinase (FAK) inhibits epidermal growth factor receptor-dependent migration and induces aggregation of N-terminal FAK in the nuclei of apoptotic glioblastoma cells. Cancer Res. 2001;61:4978–81. [PubMed] [Google Scholar]
- 80.Jones G, Stewart G. Nuclear import of N-terminal FAK by activation of the FcepsilonRI receptor in RBL-2H3 cells. Biochem Biophys Res Commun. 2004;314:39–45. doi: 10.1016/j.bbrc.2003.12.055. [DOI] [PubMed] [Google Scholar]
- 81.Stewart A, Ham C, Zachary I. The focal adhesion kinase amino-terminal domain localises to nuclei and intercellular junctions in HEK 293 and MDCK cells independently of tyrosine 397 and the carboxy-terminal domain. Biochem Biophys Res Commun. 2002;299:62–73. doi: 10.1016/s0006-291x(02)02547-0. [DOI] [PubMed] [Google Scholar]
- 82.Kressel M, Schmucker B. Nucleocytoplasmic transfer of the NF2 tumor suppressor protein merlin is regulated by exon 2 and a CRM1-dependent nuclear export signal in exon 15. Human molecular genetics. 2002;11:2269–78. doi: 10.1093/hmg/11.19.2269. [DOI] [PubMed] [Google Scholar]
- 83.Batchelor CL, Woodward AM, Crouch DH. Nuclear ERM (ezrin, radixin, moesin) proteins: regulation by cell density and nuclear import. Exp Cell Res. 2004;296:208–22. doi: 10.1016/j.yexcr.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 84.Kadare G, Toutant M, Formstecher E, Corvol J-C, Carnaud M, Boutterin M-C, Girault J-A. PIAS1-mediated sumoylation of focal adhesion kinase activates its autophosphorylation. J Biol Chem. 2003:47434–40. doi: 10.1074/jbc.M308562200. [DOI] [PubMed] [Google Scholar]
- 85.Aoto H, Sasaki H, Ishino M, Sasaki T. Nuclear translocation of cell adhesion kinase beta/proline-rich tyrosine kinase 2. Cell Struct Funct. 2002;27:47–61. doi: 10.1247/csf.27.47. [DOI] [PubMed] [Google Scholar]
- 86.Faure C, Corvol JC, Toutant M, Valjent E, Hvalby O, Jensen V, El Messari S, Corsi JM, Kadare G, Girault JA. Calcineurin is essential for depolarization-induced nuclear translocation and tyrosine phosphorylation of PYK2 in neurons. J Cell Sci. 2007;120:3034–44. doi: 10.1242/jcs.009613. [DOI] [PubMed] [Google Scholar]
- 87.Gabarra-Niecko V, Schaller MD, Dunty JM. FAK regulates biological processes important for the pathogenesis of cancer. Cancer Metastasis Rev. 2003;22:359–74. doi: 10.1023/a:1023725029589. [DOI] [PubMed] [Google Scholar]
- 88.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–10. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 89.Murata T, Naomoto Y, Yamatsuji T, Okawa T, Shirakawa Y, Gunduz M, Nobuhisa T, Takaoka M, Sirmali M, Nakajima M, Ohno Y, Tanaka N. Localization of FAK is related with colorectal carcinogenesis. Int J Oncol. 2008;32:791–6. [PubMed] [Google Scholar]
- 90.Corsi JM, Rouer E, Girault JA, Enslen H. Organization and post-transcriptional processing of focal adhesion kinase gene. BMC Genomics. 2006;7:198. doi: 10.1186/1471-2164-7-198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.McLean GW, Komiyama NH, Serrels B, Asano H, Reynolds L, Conti F, Hodivala-Dilke K, Metzger D, Chambon P, Grant SG, Frame MC. Specific deletion of focal adhesion kinase suppresses tumor formation and blocks malignant progression. Genes Dev. 2004;18:2998–3003. doi: 10.1101/gad.316304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lahlou H, Sanguin-Gendreau V, Zuo D, Cardiff RD, McLean GW, Frame MC, Muller WJ. Mammary epithelial-specific disruption of the focal adhesion kinase blocks mammary tumor progression. Proc Natl Acad Sci U S A. 2007;104:20302–7. doi: 10.1073/pnas.0710091104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mitra SK, Mikolon D, Molina JE, Hsia DA, Hanson DA, Chi A, Lim ST, Bernard-Trifilo JA, Ilic D, Stupack DG, Cheresh DA, Schlaepfer DD. Intrinsic FAK activity and Y925 phosphorylation facilitate an angiogenic switch in tumors. Oncogene. 2006;25:5969–84. doi: 10.1038/sj.onc.1209588. [DOI] [PubMed] [Google Scholar]
- 94.Mitra SK, Lim ST, Chi A, Schlaepfer DD. Intrinsic focal adhesion kinase activity controls orthotopic breast carcinoma metastasis via the regulation of urokinase plasminogen activator expression in a syngeneic tumor model. Oncogene. 2006;25:4429–40. doi: 10.1038/sj.onc.1209482. [DOI] [PubMed] [Google Scholar]
- 95.Weis SM, Cheresh DA. Pathophysiological consequences of VEGF-induced vascular permeability. Nature. 2005;437:497–504. doi: 10.1038/nature03987. [DOI] [PubMed] [Google Scholar]
- 96.Duffy MJ. The urokinase plasminogen activator system: role in malignancy. Curr Pharm Des. 2004;10:39–49. doi: 10.2174/1381612043453559. [DOI] [PubMed] [Google Scholar]
- 97.Shi Q, Hjelmeland AB, Keir ST, Song L, Wickman S, Jackson D, Ohmori O, Bigner DD, Friedman HS, Rich JN. A novel low-molecular weight inhibitor of focal adhesion kinase, TAE226, inhibits glioma growth. Mol Carcinog. 2007;46:488–96. doi: 10.1002/mc.20297. [DOI] [PubMed] [Google Scholar]
- 98.Halder J, Lin YG, Merritt WM, Spannuth WA, Nick AM, Honda T, Kamat AA, Han LY, Kim TJ, Lu C, Tari AM, Bornmann W, Fernandez A, Lopez-Berestein G, Sood AK. Therapeutic efficacy of a novel focal adhesion kinase inhibitor TAE226 in ovarian carcinoma. Cancer Res. 2007;67:10976–83. doi: 10.1158/0008-5472.CAN-07-2667. [DOI] [PubMed] [Google Scholar]
- 99.Liu TJ, LaFortune T, Honda T, Ohmori O, Hatakeyama S, Meyer T, Jackson D, de Groot J, Yung WK. Inhibition of both focal adhesion kinase and insulin-like growth factor-I receptor kinase suppresses glioma proliferation in vitro and in vivo. Mol Cancer Ther. 2007;6:1357–67. doi: 10.1158/1535-7163.MCT-06-0476. [DOI] [PubMed] [Google Scholar]
- 100.Slack-Davis JK, Martin KH, Tilghman RW, Iwanicki M, Ung EJ, Autry C, Luzzio MJ, Cooper B, Kath JC, Roberts WG, Parsons JT. Cellular characterization of a novel focal adhesion kinase inhibitor. J Biol Chem. 2007;282:14845–52. doi: 10.1074/jbc.M606695200. [DOI] [PubMed] [Google Scholar]
- 101.Roberts WG, Ung E, Whalen P, Cooper B, Hulford C, Autry C, Richter D, Emerson E, Lin J, Kath J, Coleman K, Yao L, Martinez-Alsina L, Lorenzen M, Berliner M, Luzzio M, Patel N, Schmitt E, LaGreca S, Jani J, Wessel M, Marr E, Griffor M, Vajdos F. Antitumor activity and pharmacology of a selective focal adhesion kinase inhibitor, PF-562,271. Cancer Res. 2008;68:1935–44. doi: 10.1158/0008-5472.CAN-07-5155. [DOI] [PubMed] [Google Scholar]
- 102.Weis S, Shintani S, Weber A, Kirchmair R, Wood M, Cravens A, McSharry H, Iwakura A, Yoon YS, Himes N, Burstein D, Doukas J, Soll R, Losordo D, Cheresh D. Src blockade stabilizes a Flk/cadherin complex, reducing edema and tissue injury following myocardial infarction. J Clin Invest. 2004;113:885–94. doi: 10.1172/JCI20702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Storgard C, Mikolon D, Stupack DG. Angiogenesis Assays in the Chick CAM. Methods Mol Biol. 2005;294:123–36. doi: 10.1385/1-59259-860-9:123. [DOI] [PubMed] [Google Scholar]