

Abstract
1,1-Bis(3′-indolyl)-1-(p-substituted phenyl)methanes (C-DIMs) activate the orphan receptors peroxisome proliferator-activated receptor γ (PPARγ) and Nur77 and induce receptor-dependent and -independent apoptotic pathways in colon and other cancer cells. Structure-activity studies show that the p-bromo (DIM-C-pPhBr) and p-fluoro (DIM-C-pPhF) analogs, which exhibit minimal activation of Nur77 and PPARγ, induce expression of CCAAT/enhancer-binding protein homologous protein (CHOP/GADD153) in colon cancer cells. Moreover, among a series of bromo and fluoro C-DIM analogs, their induction of CHOP was dependent on the position of the phenyl substituents (para ≥ meta ≥ ortho) and required a free indole group. DIM-C-pPhBr and DIM-C-pPhF not only induced CHOP but also activated death receptor 5 (CHOP dependent), cleavage of caspase 8 and poly (ADP ribose) polymerase (PARP) that is consistent with activation of the extrinsic pathway of apoptosis. These responses were associated with the activation of c-jun N-terminal kinase (JNK) pathway since inhibition of JNK inhibited induction of the extrinsic apoptotic pathway by these C-DIMs. However, in contrast to classical inducers of endoplasmic reticulum (ER) stress such as tunicamycin and thapsigargin, the C-DIM compounds did not induce glucose-related protein 78 that is a marker of ER stress. Proapoptotic and anticarcinogenic effects were also observed in athymic nude mice bearing RKO cell xenografts and treated with 30 mg/kg/day DIM-C-pPhBr and this was accompanied by increased JNK phosphorylation in the tumors. Thus, the anticarcinogenic activity of DIM-C-pPhBr in colon cancer cells and tumors is related to a novel ER stress-independent activation of JNK.
Introduction
The endoplasmic reticulum (ER) is a multifunctional membrane-structured subcellular component that is important for lipid and membrane biogenesis and vesicle trafficking and is the site for the synthesis and folding of many classes of proteins (1,2). The ER is highly susceptible to various stressors including those that affect redox potential, nutrient status and calcium ion levels, and this can result in aberrant protein folding and accumulation of cytotoxic protein aggregates. To counteract the effects of ER stress and restore cellular homeostasis, cells respond by activating the unfolded protein response which initiates several pathways that decrease unfolded proteins and restore ER function (2–6). The failure of a cell to counteract the condition of ER stress can lead to activation of apoptosis (6–8). Both normal and cancer cells undergo ER stress, and several anticancer drugs induce persistent ER stress resulting in apoptosis (9–17). Drugs that induce ER stress can also synergistically enhance activity of other anticancer drugs (16). For example, bortezomib (Velcade or PS-341) is a boronic acid dipeptide and proteasome inhibitor that has been approved for treatment of multiple myeloma. Bortezomib exhibits multiple activities associated with its anticancer effects, and recent reports suggest that activation of ER stress is an important proapoptotic pathway induced by this compound in pancreatic cancer cells and tumors (15,16).
Studies in this laboratory have identified a series of ring-substituted diindolylmethanes (DIMs) and 1,1-bis(3′-indolyl)-1-(p-substituted phenyl)methanes (C-DIMs) that are cytotoxic to cancer cells and inhibit tumor growth in vivo (11,18–28). C-DIM compounds containing para-trifluoromethyl (DIM-C-pPhCF3), t-butyl (DIM-C-pPhtBu), phenyl (DIM-C-pPhC6H5) and cyano (DIM-C-pPhCN) activate peroxisome proliferator-activated receptor γ (PPARγ), whereas the methoxy and unsubstituted analogs activate nerve growth factor-induced-Bα (Nur77), an orphan nuclear receptor. Both PPARγ-active and Nur77-active C-DIM compounds induce both receptor-dependent and -independent growth inhibitory and proapoptotic pathways in cancer cells and tumors including activation of ER stress in both pancreatic and ovarian cancer cell lines (11,26).
Materials and methods
Reagents and antibodies
C-DIMs were synthesized in this laboratory from the condensation of indole or substituted indole plus a substituted benzaldehyde derivative and confirmed by gas chromatography–mass spectrometry as described previously (18). Antibodies for poly (ADP ribose) polymerase (PARP) (sc-7150), CHOP (sc-793), β-tubulin (sc-33749), glucose-related protein 78 (GRP78) (sc-13968) and c-Jun small inhibitory RNA (sc-44201) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies for cleaved PARP (#9541), cleaved caspase 8 (#9496), cleaved caspase 3 (#9664), phospho-c-jun N-terminal kinase (JNK) (#9251), JNK (#9252), phospho-c-Jun (#9161), c-Jun (#9162) and death receptor 5 (DR5) (#3696) were obtained from Cell Signaling Technology (Danvers, MA). Kinase inhibitors H89, wortmannin, LY 294002, SB 203580, GF-109203X, PD98059, SP600125, thapsigargin and tunicamycin were purchased from EMD Calbiochem (Darmstadt, Germany). Plasmid extraction kits were obtained from Qiagen (Santa Clarita, CA). Lipofectamine 2000 transfection reagent was purchased from Invitrogen (Carlsbad, CA). Reporter lysis buffer and luciferase reagent for luciferase assays were purchased from Promega (Madison, WI). The GRP78 promoter–luciferase construct was kindly provided by Dr K. Park (Center for Molecular Medicine, Sungkyunkwan University, Seoul, Korea). The CHOP promoter constructs were provided by Dr Pierre Fafournoux (Saint Genes Champanelle, France) and the DR5 constructs were from Dr H.G.Wang (Moffitt Cancer, Tampa, FL).
Cell culture and treatment
RKO and SW480 cells (American Type Culture Collection, Manassas, VA) were maintained in Dulbecco’s modified Eagle’s medium/Ham’s F-12 (Sigma, St. Louis, MO) supplemented with 0.22% sodium bicarbonate, 5% fetal bovine serum and 1× antibiotic/antimycotic solution (Sigma) in a humid atmosphere of 5% CO2. Cells were trypsinized and suspended in medium, and cell numbers were determined with a Z1 Dual Coulter Particle Counter (Beckman Coulter, Fullerton, CA). Equal number of cells were seeded in 96-well, 6-well or 12-well plates and allowed to attach overnight. Cells were treated with vehicle [dimethyl sulfoxide (DMSO)] or compounds of various concentrations diluted in Dulbecco’s modified Eagle’s medium/Ham’s F-12 without phenol red and supplemented with 2.5% charcoal-stripped fetal bovine serum.
WST-1 cell proliferation assay
RKO cells were seeded in 96-well plates at a density of 2 × 103 per well and then treated with DMSO or various compounds. The WST-1 assay was carried out according to the manufacturer’s instructions. The absorbance of each sample was analyzed with FLUOstar OPTIMA Elisa reader (Offenburg, Germany) at 450 with 620 nm as the reference wavelength. All experiments were determined in triplicates and repeated at least two times and results are expressed as means ± SDs for each treatment group.
Transfection and luciferase assay
RKO cells were seeded in 12-well plates at 80% confluence and allowed to attach overnight. Various amounts of DNA [i.e. β-gal DNA (0.1 μg), pCHOP (0.4 μg), pGRP78 (0.4 μg) and pDR5 (0.4 μg)] or small inhibitory RNA (100 nM) were transfected with LipofectAMINE 2000 (Invitrogen) according to the manufacturer’s instruction. After incubation for 5–6 h, the transfection mix was replaced with Dulbecco’s modified Eagle’s medium/F12 media without phenol red containing either vehicle (DMSO) or the indicated compounds for 20–24 h. Cells were then lysed with 100 μl of 1× reporter lysis buffer and 30 μl of cell extracts were subjected to luciferase and β-galactosidase assays. Luciferase and β-gal activities were measured with a multifunctional microplate reader (FLUOstar OPTIMA) and luciferase activities were normalized to β-galactosidase. Results are expressed as means ± SDs for at least three independent determinations for each treatment group.
Western blot analysis
Whole-cell lysates were extracted with high-salt lysis buffer [50 mmol/l N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, 0.5 mol/l sodium chloride, 1.5 mmol/l magnesium chloride, 1 mmol/l ethyleneglycol-bis(aminoethylether)-tetraacetic acid, 10% (vol/vol) glycerol, 1% Triton X-100 and 5 μl/ml of Protease Inhibitor Cocktail (Sigma)] and quantified with Bio-Rad Protein Assay (Hercules, CA). Equal amounts of protein from each treatment group were separated on a sodium dodecyl sulfate–polyacrylamide gel and then transferred to polyvinylidene difluoride membrane (Immobilon-P, Millipore Corp., Bedford, MA). The polyvinylidene difluoride membrane was then blocked with 5% milk in buffer containing 1.576 g/l Tris, 8.776 g/l sodium chloride and 0.5 ml/l Tween 20 and probed with primary antibodies, followed by incubation with horseradish peroxidase-conjugated secondary antibodies as indicated. For protein knockdown experiments, the small inhibitory RNA was transfected for 36–48 h before isolation of whole-cell lysates. Western blots are representatives of at least three independent experiments.
Immunostaining
Cells were fixed in 4% paraformaldehyde/phosphate-buffered saline (PBS) for 10 min and treated with 0.3% Triton X-100/Tween (0.1%)–PBS for 10 min. After incubation with 10% normal goat serum (Vector Laboratories, Burlingame, CA)/Tween (0.1%)–PBS for 1 h, cells were probed with anti-phospho-JNK antibody (1:100) and fluorescein isothiocyanate-conjugated secondary antibody (1:200; Vector Laboratories). The fluorescence signal was then detected by Zeiss LSM 510 confocal microscope. Colon tumor tissue sections were deparaffinized by standard methods and then subjected to antigen retrieval with 0.1% pepsin in 0.01 N HCl at room temperature for 10 min followed by treatment with 0.1% H2O2 to block endogenous peroxidase activity. Sections were incubated with p-JNK antibody (1:100) at 4°C overnight after blocking with normal goat serum at room temperature for 1 h. After washing with PBS, sections were incubated with biotinylated goat anti-rabbit IgG at room temperature for 30 min. Biotin density was visualized with Vectastain Elite ABC kit (Vector Laboratories) and 3,3′-diaminobenzidine (Biogenex Laboratories, San Ramon, CA) as the chromagen following the manufacturer’s protocol. The sections were then counterstained with hematoxylin and dehydrated for future observation.
Xenograft studies in athymic mice
Male athymic nude mice (NCI-nu) were purchased from the Animal Production Area of the National Cancer Institute Frederick Cancer Research and Development Center (Frederick, MD). The mice were housed and maintained under specific pathogen-free conditions in facilities approved by the American Association for Accreditation of Laboratory Animal Care and in accordance with current regulations and standards of the United States Department of Agriculture, United States Department of Health and Human Services. The mice were used in accordance with institutional guidelines when they were 8- to 12-week-olds. To produce tumors, RKO cells were harvested from subconfluent cultures by a brief exposure to 0.25% trypsin and 0.02% ethylenediaminetetraacetic acid. Trypsinization was stopped with medium containing 10% fetal bovine serum, and the cells were washed once in serum-free medium and resuspended in Hanks’ balanced salt solution. Only suspensions consisting of single cells with >90% viability were used for the injections. A xenograft was established by subcutaneous injection of the cells (7 × 106) into the flanks of individual mice. Tumors were allowed to grow for 6 days until they were palpable. Mice were then randomized into two groups of five mice per group and dosed by oral gavage in corn oil or 30 mg/kg/day DIM-C pPhBr for 24 days. The mice were weighed, and tumor size was measured every fourth day with calipers to permit calculation of tumor volumes: V = LW2/2, where L and W were length and width, respectively. Final body, organ and tumor weights were determined at the end of the dosing regimen, and both organ and tumor blocks were obtained for histopathologic analysis.
Chromatin immunoprecipitation assay
RKO cells (5 × 106 cells) were treated with DMSO or various compounds for 12 and 24 h and cells were then fixed with 1% formaldehyde, and the cross-linking reaction was stopped by addition of 0.125 M glycine. After washing twice with PBS, cells were scraped and pelleted. Collected cells were hypotonically lysed, and nuclei were collected. Nuclei were then sonicated to desired chromatin length (∼500 bp). The chromatin was precleared by addition of protein A-conjugated beads (PIERCE, Rockford, IL) and then incubated at 4°C for 1 h with gentle agitation. The beads were pelleted, and the precleared chromatin supernatant was immunoprecipitated with antibodies to IgG or CHOP at 4°C overnight. The protein–antibody complexes were collected by addition of protein A-conjugated beads at room temperature for 1 h, beads were extensively washed and protein–DNA cross-links were reversed. DNA was purified by phenol extract/ethanol precipitation followed by polymerase chain reaction amplification. The DR5 primers were 5′-AGGTTAGTTCCGGTCCCTTC-3′ (forward) and 5′-CAACTGCAAATTCCACCACA-3′ (reverse); and they amplified a 111 bp region of the human DR5 promoter, which contained a CHOP-binding site: GAGGATTGCGTTG. The positive control primers were 5′-TACTAGCGGTTTTACGGGCG-3′ (forward) and 5′-TCGAACAGGAGGAGCAGAGAGCGA-3′ (reverse), and they amplified a 167 bp region of human glyceraldehyde-3-phosphate dehydrogenase gene. The negative control primers were 5′-ATGGTTGCCACTGGGGATCT-3′ (forward) and 5′-TGCCAAAGCCTAGGGGAAGA-3′ (reverse), and amplified a 174 bp region of genome DNA between human glyceraldehyde-3-phosphate dehydrogenase and chromosome condensation-related SMC-associated protein 1 genes. Polymerase chain reaction products were resolved on a 2% agarose gel in the presence of 1:10 000 CYBR gold.
Results
Activation of ER stress (PARP cleavage and CHOP induction) by a series of substituted C-DIM analogs (Figure 1A) was investigated in RKO and SW480 colon cancer cells (Figure 1B and C). Treatment of RKO cells for 12 h with 7.5 or 15 μM concentrations of C-DIM analogs resulted in a structure-dependent activation of PARP cleavage and CHOP induction with the most active compound being the PPARγ-active C-DIMs and the cyano analog (which is also PPARγ active) along with the p-bromo-substituted (DIM-C-pPhBr) and p-fluoro-substituted (DIM-C-pPhF) analogs (Figure 1B and C). All these compounds induced both CHOP and PARP cleavage, whereas the p-methoxy, p-dimethylamino, p-hydroxy, p-methyl, p-chloro, p-carboxymethyl, p-iodo and the unsubstituted (H) C-DIMs exhibited minimal induction of PARP cleavage and lower induction of CHOP after 12 h. We also investigated the effects of DIM on ER stress in RKO cells and did not observe induction of CHOP or PARP cleavage using 15 or 30 μM concentrations (Figure 1B). Results observed for C-DIMs in SW480 and RKO cells were similar with the exception of the p-cyano analog (DIM-C-pPhCN) that induced high levels of PARP cleavage even after treatment for only 12 h, suggesting that this compound may activate other apoptotic pathways in SW480 cells (Figure 1C).
Fig. 1.

Structure-dependent induction of CHOP. (A) Structure of C-DIMs. Structure-dependent activation of CHOP and PARP cleavage by C-DIMs in RKO (B) and SW480 (C) colon cancer cells. Cells were treated with either DMSO (D) or various C-DIM compounds at 15 μM for 12 h and changes in protein expression were determined by western blot analysis as described in Materials and Methods.
DIM-C-pPhF and DIM-C-pPhBr only minimally activate PPARγ or Nur77 (18,19,25,28) and were used to further investigate their time-dependent induction of CHOP. Figure 2A illustrates structures of the para (p), meta (m) and ortho (o) fluoro- and bromophenyl C-DIMs and the 2-methyl (2Me) and N-methyl (NMe) derivatives of both DIM-C-pPhF and DIM-C-pPhBr. The order of potency for induction of CHOP in SW480 and RKO cells by the ortho-, meta- and para-fluoro or bromo isomers was para > meta > ortho, showing that the para-substituted DIM-C-pPhF and DIM-C-pPhBr analogs were the most active (Figure 2B). Further substitution of these compounds with a 2-methyl substituent on the indole ring did not affect their induction of CHOP, whereas the N-methyl derivatives of DIM-C-pPhBr and DIM-C-pPhF did not induce CHOP. Thus, the presence of the free indole group was necessary for induction of CHOP by DIM-C-pPhF and DIM-C-pPhBr in SW480 and RKO cells.
Fig. 2.

Structure-dependent activation of CHOP by DIM-C-pPhBr and DIM-C-pPhF analogs. (A) Structure of DIM-C-PhBr and DIM-C-PhF analogs. Structure-dependent (B), dose-dependent (C) and time-dependent (D) activation of CHOP in colon cancer cells. Cells were treated with either DMSO (D) or various C-DIM compounds at 15 μM for 12 h and changes in protein expression were determined by western blot analysis as described in Materials and Methods. Protein levels were measured with Image J and normalized to β-tubulin or compared with a non-specific (NS) band (Figure 2D). CHOP was non-detectable in cells treated with DMSO (Figure 2D).
The concentration-dependent (Figure 2C) and time-dependent (Figure 2D) induction of CHOP expression by two active and two relatively inactive analogs of the bromo- and fluoro-substituted C-DIMs was also determined in RKO cells. Treatment of RKO cells with 7.5, 12.5 and 15 μM DIM-C-pPhBr/DIM-C-pPhF (p) and their corresponding 2-methyl analogs (active compounds) for 12 h induced CHOP protein at concentrations of 7.5 and 12.5 μM (Figure 2B). In contrast, the N-methyl derivatives of DIM-C-pPhBr and DIM-C-pPhF did not induce CHOP expression at concentrations as high as 30 μM. The ortho-bromo (DIM-C-oPhBr) and ortho-fluoro (DIM-C-oPhF) compounds exhibited minimal activity except for 15 μM DIM-C-oPhBr that clearly induced CHOP expression. Fifteen micromolar concentrations of active and inactive C-DIM compounds were also used to determine their time-dependent induction of CHOP in RKO cells (Figure 2D). The active DIM-C-pPhBr and DIM-C-pPhF compounds and their corresponding 2-methyl analogs induced CHOP protein expression as early as 2 h after treatment, whereas the N-methyl analogs were inactive. Interestingly, the ortho-fluoro- and -bromo-substituted analogs also induced CHOP expression; however, both the timing and levels of induction were different from the more potent para-substituted DIM-C-pPhBr and DIM-C-pPhF.
The structure-dependent effects of the bromo- and fluoro-substituted C-DIMs on RKO and SW480 cell viability was investigated using the WST assay. Treatment of RKO and SW480 cells with 15 μM DIM-C-pPhF and DIM-C-pPhBr and their corresponding 2-methyl-substituted analogs caused a 40–60% decrease in cell viability, whereas the N-methyl-substituted compounds were inactive (Figure 3A). These results paralleled the relative potencies of the same C-DIM compounds as inducers of CHOP. The ortho-substituted C-DIMs (DIM-C-oPhBr and DIM-C-oPhF) were approximately equipotent with the para isomers with respect to decreasing cell viability (Figure 3A), even though induction of CHOP (and PARP cleavage) was minimal in colon cancer cells after treatment for 12 h (Figure 2A). However, induction of CHOP and DR5 by the ortho isomers was observed at later time points (24 h) (data not shown), and these structure-dependent temporal differences are currently being investigated.
Fig. 3.

Activation of proapoptotic pathways by C-DIMs in colon cancer cells. (A) C-DIMs decreased cell survival rates. RKO and SW480 cells were treated with various C-DIMs (15 μM) or DMSO (D) for 24 h and cell viability was measured with WST-1 assay as described in Materials and Methods. Results are expressed as means ± SDs for three replicate determinations for each treatment group and significantly (P < 0.05) decreased activity is indicated by *. Cell survival for DMSO treatment was set at 100%. Activation of PARP and caspase cleavage, induction of GRP78, DR5 and CHOP and phosphorylation of JNK in RKO and SW480 cells treated with 15 μM C-DIM analogs for 24 h (B) and comparison of DIM-C-pPhBr, DIM-C-pPhF with the classical ER stress activators, Tg and Tm (C). Colon cancer cells were treated with 15 μM C-DIMs, 10 μM Tg and 10 μg/ml Tm for 24 h and whole-cell lysates were analyzed by western blot as described in Materials and Methods. (D) Effects of caspase inhibitors on DIM-C-pPhBr-induced PARP cleavage in RKO cells. Cells were pretreated with 10 μM Z-IETD-FMK or Z-VAD-FMK for 1 h and then cotreated with DIM-C-pPhBr for 24 h. Whole-cell lysates were analyzed by western blot analysis as described in Materials and Methods.
The effects of DIM-C-pPhBr and DIM-C-pPhF and their corresponding inactive N-methyl derivatives on induction of proapoptotic responses were determined in RKO and SW480 cells treated with 15 μM DIM-C-pPhBr and DIM-C-pPhF for 24 h (Figure 3B). Caspase-dependent PARP cleavage and cleavage of caspases 8 and 3 were induced by both compounds, whereas the N-methyl analogs were inactive in this assay. Induction of ER stress by C-DIMs in other cancer cells was accompanied by induction of the stress response gene GRP78; however, in RKO and SW480 cells, neither DIM-C-pPhBr nor DIM-C-pPhF induced GRP78, although both compounds induced CHOP and DR5 as reported previously (11,26). In addition, C-DIM-induced cleavage of ATF-6, another marker of ER stress, was also not observed in these colon cancer cell lines (data not shown). The N-methyl compounds did not induce GRP78, DR5 or CHOP. In contrast, both DIM-C-pPhBr and DIM-C-pPhF but not their N-methyl derivatives induced phosphorylation of JNK and jun but did not affect expression of JNK or jun proteins (Figure 3B). In Figure 3C, the effects of DIM-C-pPhBr and DIM-C-pPhF are compared with the classical ER stress activators Thapsigargin (Tg) and Tunicamycin (Tm) on induction of GRP78/CHOP/DR5, activation of the JNK pathway and caspase-dependent PARP cleavage in RKO cells. All compounds induced CHOP and DR5 expression and cleavage of PARP, caspases 8 and 3; however, after the 24 h treatment period, PARP cleavage induced by Tm was less than observed for the other three compounds. The major difference between Tm–Tg and the C-DIM compounds was the induction of ER stress-dependent GRP78 expression by Tg and Tm but not by DIM-C-pPhF or DIM-C-pPhBr. Activation of the extrinsic apoptotic pathway is supported by the induction of caspase 8 cleavage (Figure 3B and C) and by caspase inhibitor studies (Figure 3D). Both the caspase 8 and pancaspase inhibitors Z-IETD-FMK and Z-VAD-FMK, respectively, significantly inhibited DIM-C-pPhBr- and DIM-C-pPhF-induced PARP cleavage in RKO cells.
CHOP-dependent activation of DR5 was important for activation of the extrinsic pathway of apoptosis by C-DIMs (11,26) and, in RKO cells, DIM-C-pPhBr and Tg also induced CHOP and DR5 expression. In cells transfected with a construct containing a −991 to –7 DR5 promoter insert (pDR5), both DIM-C-pPhBr and Tg induced luciferase activity (Figure 4A). Moreover, in a chromatin immunoprecipitation assay, we also showed that in RKO cells treated with DIM-C-pPhBr, DIM-C-pPhF or Tg, there was recruitment of CHOP to the DR5 promoter (Figure 4B). As a positive control for the chromatin immunoprecipitation assay, we showed that the transcription factor TFIIB was bound to the glyceraldehyde-3-phosphate dehydrogenase start site but not to exon 1 of the chromosome condensation-related SMC-associated protein 1 gene (Figure 4B).
Fig. 4.
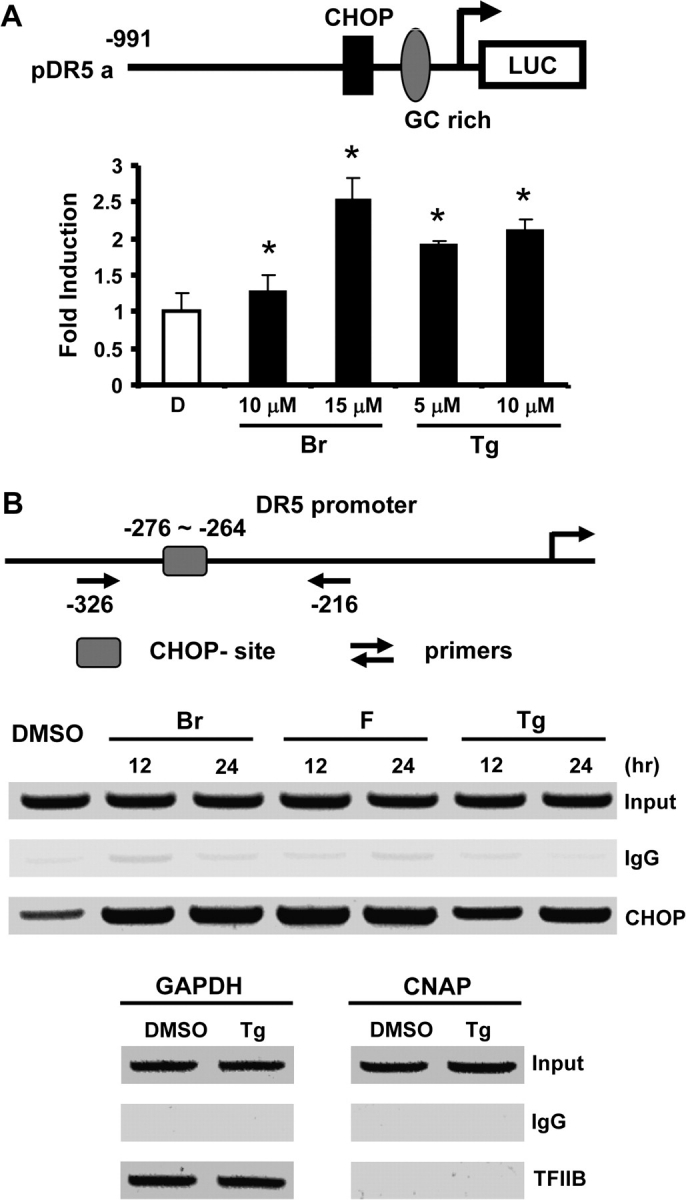
CHOP-dependent induction of DR5 by DIM-C-pPhBr, DIM-C-pPhF and Thapsigargin in RKO cells. (A) DIM-C-pPhBr and Tg induced transactivation in RKO cells transfected with pDR5 constructs. Cells were transfected with pDR5-Luc construct and then treated with 10–15 μM DIM-C-pPhBr or 5–10 μM Tg for 24 h. Whole-cell lysates were analyzed for luciferase activity as described in Material and Methods. Results are expressed as means ± SDs for three replicates for each treatment group and significant (P < 0.05) induction is indicated by an asterisk. (B) Increased CHOP recruitment to the DR5 promoter as determined in a chromatin immunoprecipitation assay. Cells were treated with 15 μM DIM-C-pPhF, DIM-C-pPhBr or 5 μM Tg for 12 or 24 h, and recruitment of CHOP to the DR5 promoter was determined by chromatin immunoprecipitation assay as described in Materials and Methods. The interaction of TFIIB with the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) promoter but not genomic DNA between the GAPDH and chromosome condensation-related SMC-associated protein 1 genes served as positive and negative controls for the chromatin immunoprecipitation assay.
Since C-DIMs did not induce the GRP78 marker of ER stress, the effects of DIM-C-pPhBr and DIM-C-pPhF on JNK activation were investigated in RKO cells. DIM-C-pPhBr and DIM-C-pPhF activated JNK phosphorylation in RKO cells within 1 h after treatment, and this persisted for up to 24 h (Figure 5A). Cells treated with 15 μM DIM-C-pPhBr were immunostained for JNK phosphorylation [Figure 5Ba and d] and with propidium iodide to show nuclear staining [Figure 5Bb and e]. Phospho-JNK and propidium iodide staining directly overlapped, showing that phospho-JNK is localized to the nucleus. Enhanced phosphorylation of JNK and c-jun in RKO cells treated with DIM-C-pPhBr or DIM-C-pPhF was inhibited by cotreatment with the JNK inhibitor SP600125, and this inhibitor also blocked induction of CHOP and PARP cleavage by the C-DIM compounds (Figure 5C). ER stress-independent activation of JNK may involve other kinases including apoptosis signal-regulating kinase-1 and mitogen-activated protein kinase kinase 4 (29); however, DIM-C-pPhBr and DIM-C-pPhF did not enhance phosphorylation of these kinases in RKO cells (Figure 5D). A recent study also reported that phorbol ester-dependent activation of JNK phosphorylation involved protein kinase C (30,31), and we therefore determined the effects of PKC and other kinase inhibitors on the induction on JNK phosphorylation by C-DIM compounds (Figure 5D). The protein kinase C inhibitor GF-109203X did not inhibit C-DIM-induced phosphorylation of JNK; moreover, inhibitory effects were also not observed in cells cotreated with inhibitors of phosphatidylinositol-3-kinase (wortmannin and LY 294002), p38mitogen-activated protein kinase (SB 294002) or p42MAPK (PD98059).
Fig. 5.

Induction of CHOP and apoptosis by C-DIMs mediated through activation of JNK. (A) Time-dependent activation of JNK phosphorylation by DIM-C-pPhBr in RKO cells. Cells were treated with 15 μM DIM-C-pPhBr and collected at indicated times, and whole-cell lysates were analyzed by western blots as described in Materials and Methods. (B) Immunostaining for phospho-JNK. Cells were treated with DMSO or 15 μM DIM-C-pPhBr for 24 h and stained for phospho-JNK or nucleus with propidium iodide as described in Materials and Methods. (C) Inhibition of JNK and c-Jun phosphorylation, induction of CHOP and PARP cleavage by SP600125 in RKO cells. Cells were pretreated with 20 μM SP600125 for 1 h and then cotreated with 15 μM of the indicated compounds for 24 h, and whole-cell lysates were analyzed by western blots as described in Materials and Methods. (D) Effects of DIM-C-pPhBr alone and in combination with various kinase inhibitors on phosphorylation of apoptosis signal-regulating kinase-1, mitogen-activated protein kinase kinase 4 and JNK. Cells were treated with 15 μM DIM-C-pPhBr for 30–960 min or pretreated with kinase inhibitors, H89 (10 μM), Wortmannin (5 μM), LY 294002 (20 μM), SB 203580 (10 μM), GF-109203X (5 μM), PD98059 (5 and 20 μM) for 1 h and then treated with 15 μM DIM-C-pPhBr for 24 h. Whole-cell lysates were analyzed by western blot analysis as described in Materials and Methods.
The effects of DIM-C-pPhBr (30 mg/kg) on tumor growth and weight were investigated in athymic nude mice bearing RKO cells as xenografts. The results show that DIM-C-pPhBr inhibited tumor volume (Figure 6A) and weight (Figure 6B) and also induced apoptosis in tumors as indicated in results of the terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling assay (Figure 6C). Immunostaining of the tumors also showed that phospho-JNK was enhanced in tumors from treated versus non-treated mice (Figure 6C). These results complement the in vitro studies (Figure 5B) demonstrating that DIM-C-pPhBr-dependent activation of JNK and the extrinsic pathway of apoptosis are major factors in the anticarcinogenic activity of this compound in colon cancer. We also examined body or organ weights and observed minimal differences in treated versus untreated animals and histopathology did not detect compound-induced toxicities.
Fig. 6.
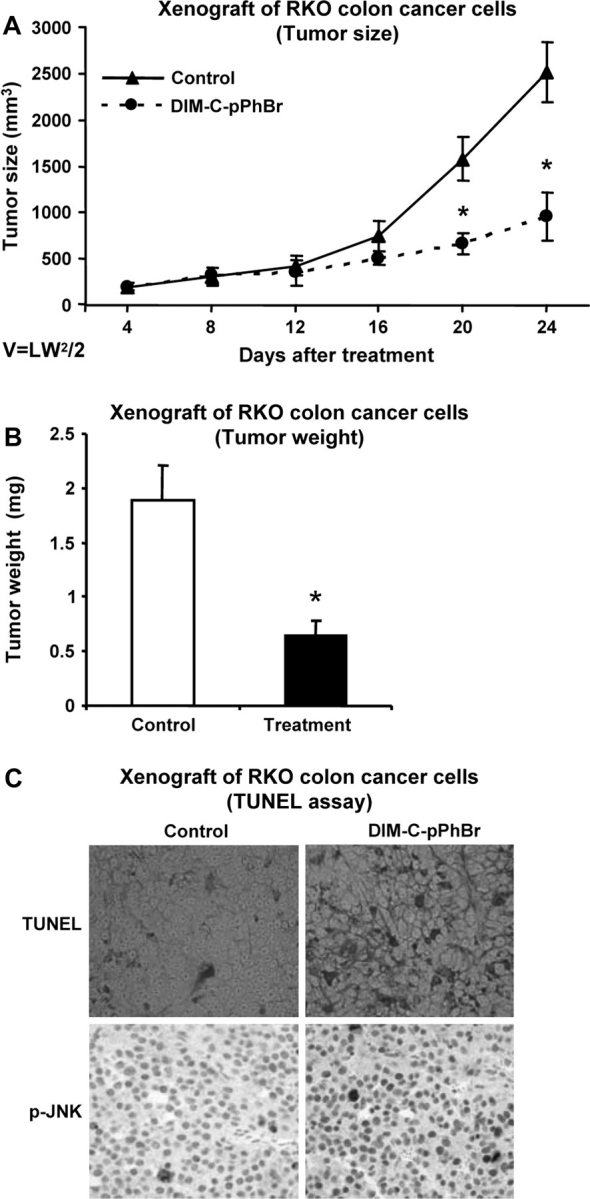
Inhibition of tumor growth by DIM-C-pPhBr. Male athymic nude mice bearing RKO cell xenografts were treated with DIM-C-pPhBr, and tumor volumes (A) and weight (B) were determined as described in Materials and Methods. The compound was administered daily (30 mg/kg/day) in corn oil by oral gavage, and corn oil served as a solvent control. (C) Tumor sections were stained for apoptosis terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling and phospho-JNK as described in Materials and Methods.
Discussion
Cells are constantly stressed through changes in nutrient and oxygen levels and other stressors and cellular homeostasis are maintained, in part, through activation of ER stress that triggers both survival and death pathways (1–8). Persistent ER stress invariably leads to cell death and there is evidence that some anticancer drugs and other therapeutic agents can act, in part, through activation of ER stress (9–17,32). The accumulation of unfolded proteins through ER stress can also lead to other human diseases including Parkinson’s disease, cystic fibrosis and other neurodegenerative disorders (33–37). ER stress can lead to cell survival or apoptosis through activation of several pathways and typically anticancer drugs preferentially induce cell death; however, these effects are dependent on the specific agent and cell/tumor context. Previous studies in this laboratory showed that DIM and 5,5′-dibromo-diindolylmethane induced ER stress and apoptosis in pancreatic cancer cells and PPARγ-active C-DIMs induced similar responses in ovarian cancer cells (11,26). In ovarian cancer cells, the C-DIMs activated GRP78, a hallmark of ER stress, and CHOP which in turn induced constitutively active DR5- and caspase 8-dependent apoptosis. In contrast, Nur77-active C-DIMs induce apoptosis through receptor-dependent activation of tumor necrosis factor-related apoptosis-inducing ligand.
In this study, we first investigated the structure-dependent activation of CHOP and PARP cleavage by a series of different p-substituted C-DIM analogs in RKO and SW480 cells (Figure 1). The PPARγ-active C-DIMs containing p-trifluoromethyl, p-phenyl, p-t-butyl and p-cyano groups induced CHOP expression and PARP cleavage within 12 h after treatment. The p-cyano-substituted analog was the most active compound in SW480 cells. In contrast, DIM that causes ER stress in some cell lines (11,38) was inactive in SW480 cells at concentrations as high as 30 μM. At least two C-DIMs, namely DIM-C-pPhBr and DIM-C-pPhF that exhibit minimal activation of Nur77 or PPARγ (18,19,25,28), induced CHOP and PARP cleavage in both colon cancer cell lines. These analogs were used as models for subsequent studies in order to minimize any Nur77- or PPARγ-dependent proapoptotic responses induced by C-DIMs. The structural determinants important for activation of CHOP were initially investigated by varying the position of the Br or F substituents on the phenyl ring (para, meta or ortho) and by comparing the activity of the 2- and 1-methylindole derivatives (Figure 2A). There were some structure-dependent differences and similarities between and among the DIM-C-PhBr and DIM-C-PhF analogs. The ortho- and meta-fluoro isomers were less active than the corresponding bromo isomers, whereas the para- and meta-bromo isomers exhibited comparable activities and were more active than the ortho isomer. DIM-C-pPhBr and DIM-C-pPhF and the corresponding 2-methyl derivatives exhibited similar activities as inducers of CHOP; however, their corresponding 1-methyl(N-methyl) derivatives were inactive (Figure 2B and C). The 12 h time course study illustrated in Figure 2D shows that both DIM-C-pPhF and DIM-C-pPhBr and their 2-methyl derivatives induce CHOP within 2–4 h after treatment, whereas minimal induction of CHOP was observed for the N-methyl isomers and similar results were obtained for induction of PARP cleavage and other putative ER stress-dependent responses (Figure 3B and C). These data complement a similar structure-dependent loss of metabolic activity in the WST assay (Figure 3A) demonstrating that the free indole group is a critical structural feature required for the cytotoxicity of these C-DIM analogs in colon cancer cells.
Previous studies have demonstrated that C-DIMs activate GRP78 and the JNK pathway resulting in induction of CHOP, DR5 and the extrinsic apoptotic pathway (11,26). DIM-C-pPhF and DIM-C-pPhBr but not the corresponding N-methyl derivatives induce CHOP (Figure 2B) that is recruited to the DR5 promoter (Figure 4B) and this results in induction of DR5/DR5 promoter (Figures 3B and 4A) and induction of apoptosis (Figure 3B–D). Moreover, activation of caspase-dependent PARP cleavage by the C-DIMs is inhibited by the caspase 8 inhibitor Z-IETD-FMK (Figure 4D). Both Tg and C-DIMs induce JNK phosphorylation CHOP and DR5 expression and caspase 8-dependent PARP cleavage in colon cancer cells (Figure 3D); however, Tg also induces GRP78, which is a hallmark of ER stress. Tm also induces GRP78 and apoptosis (weakly); however, only minimal activation of JNK was observed for this compound (Figure 3D). The observation that C-DIMs do not induce GRP78 (Figure 3B and C) or ATF-6 cleavage (data not shown) suggests that the major proapoptotic pathways induced by these compounds are JNK-dependent and ER stress-independent activation of CHOP, DR5 and downstream caspases.
We also investigated the in vivo effects of DIM-C-pPhBr on inhibition of tumor growth and induction of apoptosis in nude mice bearing RKO cell xenografts (Figure 6). These results complement the in vitro studies and showed that DIM-C-pPhBr inhibited tumor volumes and weight and increased apoptosis (terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling assay) in the tumors. Moreover, immunostaining of tumors from mice treated with DIM-C-pPhBr showed that phospho-JNK was increased (Figure 6C) and this correlated with enhanced phospho-JNK expression in RKO cells treated with the same compound (Figure 5B).
In summary, our results show that DIM-C-pPhF and DIM-C-pPhBr are cytotoxic and induce apoptosis in colon cancer cells and in tumors through ER stress-independent activation of JNK, whereas the induction of these same responses in pancreatic and ovarian cancer cells were ER stress dependent (11,26). Although ER stress-induced phosphorylation of JNK through activation of inositol-requiring gene 1 has been well characterized (6–8), the mechanisms associated with increased JNK phosphorylation by DIM-C-pPhBr in colon cancer cells are unclear. Preliminary studies show that DIM-C-pPhF and DIM-C-pPhBr do not enhance phosphorylation of ASK-1 and MKK4 (Figure 5D), kinases upstream from JNK. Thus, the proapoptotic and antitumorigenic effects of DIM-C-pPhBr and DIM-C-pPhF in colon cancer cells are associated with enhanced JNK phosphorylation that appears to be independent of activation of classical markers of ER stress. Our current research is focused on determining the mechanism of C-DIM-dependent activation of JNK and identifying more proximal intracellular targets for these compounds. In addition, since in vivo treatment with C-DIMs results in minimal toxic side effects, we are also investigating clinical applications of C-DIMs alone or in combination with other drugs for colon cancer chemotherapy.
Funding
National Institutes of Health (ES09106 and CA108718); Texas Agricultural Experiment Station.
Acknowledgments
Conflict of Interest Statement: None declared.
Glossary
Abbreviations
- ASK1
apoptosis signal-regulating kinase-1
- C-DIM
1,1-Bis(3′-indolyl)-1-(p-substituted phenyl)methane
- DIM
diindolylmethane
- DMSO
dimethyl sulfoxide
- DR5
death receptor 5
- ER
endoplasmic reticulum
- GRP78
glucose-related protein 78
- JNK
c-jun N-terminal kinase
- PARP
poly (ADP-ribose) polymerase
- PBS
phosphate-buffered saline
- PPARγ
peroxisome proliferator-activated receptor γ
- Tg
Thapsigargin
- Tm
Tunicamycin
References
- 1.Alberts B, et al. Molecular Biology of the Cell. New York: Garland Science; 2002. [Google Scholar]
- 2.Schroder M, et al. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 3.Mori K. Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell. 2000;101:451–454. doi: 10.1016/s0092-8674(00)80855-7. [DOI] [PubMed] [Google Scholar]
- 4.Kaufman RJ. Stress signaling from the lumen of the endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev. 1999;13:1211–1233. doi: 10.1101/gad.13.10.1211. [DOI] [PubMed] [Google Scholar]
- 5.Ma Y, et al. The unfolding tale of the unfolded protein response. Cell. 2001;107:827–830. doi: 10.1016/s0092-8674(01)00623-7. [DOI] [PubMed] [Google Scholar]
- 6.Boyce M, et al. Cellular response to endoplasmic reticulum stress: a matter of life or death. Cell Death Differ. 2006;13:363–373. doi: 10.1038/sj.cdd.4401817. [DOI] [PubMed] [Google Scholar]
- 7.Szegezdi E, et al. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006;7:880–885. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ma Y, et al. The role of the unfolded protein response in tumour development: friend or foe? Nat. Rev. Cancer. 2004;4:966–977. doi: 10.1038/nrc1505. [DOI] [PubMed] [Google Scholar]
- 9.Mahboobi S, et al. Bis(1H-2-indolyl)methanones as a novel class of inhibitors of the platelet-derived growth factor receptor kinase. J. Med. Chem. 2002;45:1002–1018. doi: 10.1021/jm010988n. [DOI] [PubMed] [Google Scholar]
- 10.Lee AH, et al. Proteasome inhibitors disrupt the unfolded protein response in myeloma cells. Proc. Natl Acad. Sci. USA. 2003;100:9946–9951. doi: 10.1073/pnas.1334037100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abdelrahim M, et al. 3,3′-Diindolylmethane (DIM) and derivatives induce apoptosis in pancreatic cancer cells through endoplasmic reticulum stress-dependent upregulation of DR5. Carcinogenesis. 2006;27:717–728. doi: 10.1093/carcin/bgi270. [DOI] [PubMed] [Google Scholar]
- 12.Shiraishi T, et al. Tunicamycin enhances tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in human prostate cancer cells. Cancer Res. 2005;65:6364–6370. doi: 10.1158/0008-5472.CAN-05-0312. [DOI] [PubMed] [Google Scholar]
- 13.Tsutsumi S, et al. Endoplasmic reticulum stress response is involved in nonsteroidal anti-inflammatory drug-induced apoptosis. Cell Death Differ. 2004;11:1009–1016. doi: 10.1038/sj.cdd.4401436. [DOI] [PubMed] [Google Scholar]
- 14.Fribley A, et al. Proteasome inhibitor PS-341 induces apoptosis through induction of endoplasmic reticulum stress-reactive oxygen species in head and neck squamous cell carcinoma cells. Mol. Cell. Biol. 2004;24:9695–9704. doi: 10.1128/MCB.24.22.9695-9704.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nawrocki ST, et al. Bortezomib inhibits PKR-like endoplasmic reticulum (ER) kinase and induces apoptosis via ER stress in human pancreatic cancer cells. Cancer Res. 2005;65:11510–11519. doi: 10.1158/0008-5472.CAN-05-2394. [DOI] [PubMed] [Google Scholar]
- 16.Nawrocki ST, et al. Bortezomib sensitizes pancreatic cancer cells to endoplasmic reticulum stress-mediated apoptosis. Cancer Res. 2005;65:11658–11666. doi: 10.1158/0008-5472.CAN-05-2370. [DOI] [PubMed] [Google Scholar]
- 17.Hagg M, et al. Induction of endoplasmic reticulum stress by ellipticine plant alkaloids. Mol. Cancer Ther. 2004;3:489–497. [PubMed] [Google Scholar]
- 18.Qin C, et al. A new class of peroxisome proliferator-activated receptor γ (PPARγ) agonists that inhibit growth of breast cancer cells: 1,1-bis(3′-indolyl)-1-(p-substitutedphenyl)methanes. Mol. Cancer Ther. 2004;3:247–259. [PubMed] [Google Scholar]
- 19.Chintharlapalli S, et al. 1,1-Bis(3′-indolyl)-1-(p-substitutedphenyl)methanes induce peroxisome proliferator-activated receptor γ-mediated growth inhibition, transactivation and differentiation markers in colon cancer cells. Cancer Res. 2004;64:5994–6001. doi: 10.1158/0008-5472.CAN-04-0399. [DOI] [PubMed] [Google Scholar]
- 20.Hong J, et al. Peroxisome proliferator-activated receptor γ-dependent activation of p21 in Panc-28 pancreatic cancer cells involves Sp1 and Sp4 proteins. Endocrinology. 2004;145:5774–5785. doi: 10.1210/en.2004-0686. [DOI] [PubMed] [Google Scholar]
- 21.Contractor R, et al. A novel ring-substituted diindolylmethane 1,1-bis[3′-(5-methoxyindolyl)]-1-(p-t-butylphenyl)methane inhibits ERK activation and induces apoptosis in acute myeloid leukemia. Cancer Res. 2005;65:2890–2898. doi: 10.1158/0008-5472.CAN-04-3781. [DOI] [PubMed] [Google Scholar]
- 22.Chintharlapalli S, et al. 1,1-Bis(3′-indolyl)-1-(p-substitutedphenyl)methanes are peroxisome proliferator-activated receptor gamma agonists but decrease HCT-116 colon cancer cell survival through receptor-independent activation of early growth response-1 and NAG-1. Mol. Pharmacol. 2005;68:1782–1792. doi: 10.1124/mol.105.017046. [DOI] [PubMed] [Google Scholar]
- 23.Kassouf W, et al. Inhibition of bladder tumor growth by 1,1-bis(3′-indolyl)-1-(p-substitutedphenyl)methanes: a new class of peroxisome proliferator-activated receptor γ agonists. Cancer Res. 2006;66:412–418. doi: 10.1158/0008-5472.CAN-05-2755. [DOI] [PubMed] [Google Scholar]
- 24.Chintharlapalli S, et al. 1,1-Bis(3′-indolyl)-1-(p-substituted phenyl)methanes inhibit colon cancer cell and tumor growth through PPARγ-dependent and PPARγ-independent pathways. Mol. Cancer Ther. 2006;5:1362–1370. doi: 10.1158/1535-7163.MCT-06-0002. [DOI] [PubMed] [Google Scholar]
- 25.Chintharlapalli S, et al. Activation of Nur77 by selected 1,1-Bis(3′-indolyl)-1-(p-substituted phenyl)methanes induces apoptosis through nuclear pathways. J. Biol. Chem. 2005;280:24903–24914. doi: 10.1074/jbc.M500107200. [DOI] [PubMed] [Google Scholar]
- 26.Lei P, et al. 1,1-Bis(3′-indolyl)-1-(p-substituted phenyl)methanes inhibit ovarian cancer cell growth through peroxisome proliferator-activated receptor-dependent and independent pathways. Mol. Cancer Ther. 2006;5:2324–2336. doi: 10.1158/1535-7163.MCT-06-0184. [DOI] [PubMed] [Google Scholar]
- 27.Chintharlapalli S, et al. 2-Cyano-3,12-dioxoolean-1,9-dien-28-oic acid and related compounds inhibit growth of colon cancer cells through peroxisome proliferator-activated receptor γ-dependent and -independent pathways. Mol. Pharmacol. 2005;68:119–128. doi: 10.1124/mol.105.011437. [DOI] [PubMed] [Google Scholar]
- 28.Cho SD, et al. Nur77 agonists induce proapoptotic genes and responses in colon cancer cells through nuclear receptor-dependent and independent pathways. Cancer Res. 2007;67:674–683. doi: 10.1158/0008-5472.CAN-06-2907. [DOI] [PubMed] [Google Scholar]
- 29.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 30.Ikezoe T, et al. JNK interacting protein 1 (JIP-1) protects LNCaP prostate cancer cells from growth arrest and apoptosis mediated by 12-0-tetradecanoylphorbol-13-acetate (TPA) Br. J. Cancer. 2004;90:2017–2024. doi: 10.1038/sj.bjc.6601834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gonzalez-Guerrico AM, et al. Phorbol ester-induced apoptosis in prostate cancer cells via autocrine activation of the extrinsic apoptotic cascade: a key role for protein kinase C delta. J. Biol. Chem. 2005;280:38982–38991. doi: 10.1074/jbc.M506767200. [DOI] [PubMed] [Google Scholar]
- 32.Silva AM, et al. Salicylates trigger protein synthesis inhibition in a protein kinase R-like endoplasmic reticulum kinase-dependent manner. J. Biol. Chem. 2007;282:10164–10171. doi: 10.1074/jbc.M609996200. [DOI] [PubMed] [Google Scholar]
- 33.Imai Y, et al. Parkin suppresses unfolded protein stress-induced cell death through its E3 ubiquitin-protein ligase activity. J. Biol. Chem. 2000;275:35661–35664. doi: 10.1074/jbc.C000447200. [DOI] [PubMed] [Google Scholar]
- 34.Imai Y, et al. An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell. 2001;105:891–902. doi: 10.1016/s0092-8674(01)00407-x. [DOI] [PubMed] [Google Scholar]
- 35.Imaizumi K, et al. The unfolded protein response and Alzheimer's disease. Biochim. Biophys. Acta. 2001;1536:85–96. doi: 10.1016/s0925-4439(01)00049-7. [DOI] [PubMed] [Google Scholar]
- 36.Iwahashi H, et al. Synergistic anti-apoptotic activity between Bcl-2 and SMN implicated in spinal muscular atrophy. Nature. 1997;390:413–417. doi: 10.1038/37144. [DOI] [PubMed] [Google Scholar]
- 37.Tamatani M, et al. ORP150 protects against hypoxia/ischemia-induced neuronal death. Nat. Med. 2001;7:317–323. doi: 10.1038/85463. [DOI] [PubMed] [Google Scholar]
- 38.Sun S, et al. Endoplasmic reticulum stress as a correlate of cytotoxicity in human tumor cells exposed to diindolylmethane in vitro. Cell Stress Chaperones. 2004;9:76–87. doi: 10.1379/CSC-2R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]