

Abstract
2,2′,3,3′,6,6′-Hexachlorobiphenyl (PCB 136) is a chiral and highly neurotoxic PCB congener of environmental relevance. (+)-PCB 136 was previously shown to be enriched in tissues from mice treated with racemic PCB 136. We investigated the spectral interactions of (+)-, (-)- and (±)-PCB 136 with mouse and rat hepatic microsomal cytochrome P450 (P450) enzymes to test the hypothesis that enantioselective binding to specific P450 enzymes causes the enrichment of (+)-PCB 136 in vivo. Hepatic microsomes prepared from C57BL/6 mice or Long Evans rats treated with β-naphthoflavone or 3-methylcholanthrene, phenobarbital and dexamethasone (prototypical inducers of CYP1A, CYP2B and CYP3A, respectively) were used to determine first, if the (+)-PCB 136 atropisomer binds to hepatic microsomal P450 enzymes to a greater extent than does the (-)-PCB 136 atropisomer; and second, if P450 enzymes of one subfamily bind the two PCB 136 atropisomers more efficiently than do P450 enzymes of other subfamilies. Increasing concentrations of (+)-, (-)- or (±)-PCB 136 were added to hepatic microsomes and the difference spectrum and maximal absorbance change, a measure of PCB binding to P450 enzymes, were measured. A significantly larger absorbance change was observed with (+)-PCB 136 than with (-)-PCB 136 with all four hepatic microsomal preparations in mice and rats, indicating that (+)-PCB 136 interacted with microsomal P450 enzymes to a greater degree than did (-)-PCB 136. In addition, binding of the PCB 136 atropisomers was greatest in microsomes from PB-treated mice and rats, and was inhibited by CYP2B antibodies, indicating the involvement of CYP2B enzymes. Together these results suggest preferential binding of (+)-PCB 136 to P450 enzymes (such as CYP2B and CYP3A) in hepatic microsomes, an observation that may explain the enantioselective enrichment of the (+)-PCB 136 atropisomer in tissues of mice.
Introduction
Polychlorinated biphenyls (PCBs1) are a group of 209 semi-volatile, persistent organic pollutants with the general structure C12HnCl10-n (1). They were manufactured by batch chlorination of biphenyl and are still used in enclosed systems, such as capacitors and transformers. Individual PCB congeners have physicochemical properties that are determined by their degree of chlorination and their substitution pattern and differ by several orders of magnitude (2). PCBs are highly lipophilic, semi-volatile compounds and have excellent oral bioavailability. These properties led to worldwide contamination by PCBs and have resulted in considerable human exposure, especially via the diet. Individual PCB congeners cause adverse human health effects by different modes of action depending on their three-dimensional structure. Recent epidemiological studies have shown that exposure to PCB mixtures is linked to adverse developmental outcomes and neurotoxicity (3, 4). Based on structure activity studies, only PCB congeners with multiple ortho chlorine substituents are neurotoxic in vitro and in vivo.
Several highly neurotoxic PCB congeners with three or four ortho chlorine substituents exist as two stable rotational isomers (or atropisomers) and are chiral because of an unsymmetrical substitution pattern in both phenyl rings (5). Chiral PCB congeners are important constituents of both technical and environmental PCB mixtures. For example, the total concentration of chiral PCB congeners in Aroclors 1242 and Aroclor 1260 is 6% and 30% by weight, respectively (6). There is evidence that PCB atropisomers differ in their biological activities. Early studies have shown that pure PCB atropisomers differ in their potential to induce hepatic enzymes, such as cytochrome P450 (P450) enzymes and others (7). More recent studies have shown that PCB 84 atropisomers differ in their effect on protein kinase C translocation in cerebellar granule cells, a biological measure associated with neurotoxicity (8). There is also evidence that PCB 136 atropisomers enantioselectively activate ryanodine receptors RyR1 and RyR2 (9).
Pure PCB atropisomers not only cause adverse biological effects with different potencies, but are subject to enantioselective disposition processes due to enantioselective interactions with proteins. Several environmental studies have shown enantiomeric enrichment of chiral PCBs in fish, marine mammals and in humans (5). Enrichment of (+)-PCB 136 has also been reported in Arctic char (10) and eggs from predatory birds (11), whereas (-)-PCB 136 was enriched in rainbow trout (12, 13), wolverine (14) and Arctic fox (14). In contrast, neither atropisomer was enriched in the freshwater invertebrate, Mysis relicta (15), or in liver samples from bowhead whales (16) or whales from the Mediterranean Sea (17, 18). Similarly, laboratory studies investigating either individual PCB congeners or technical PCB mixtures have reported an enantiomeric enrichment in blood and tissues from mice (6, 19, 20) and rats (21). The extent of the enantiomeric enrichment depends on the route of administration (19) and the PCB dose (6). Moreover, increased enrichment of (+)-PCB 136 was observed in liver of mice that had been pretreated with β-naphthoflavone (NF), phenobarbital (PB), or dexamethasone (DEX) than corn oil (CO) (22) (Figure 1A).
Figure 1.
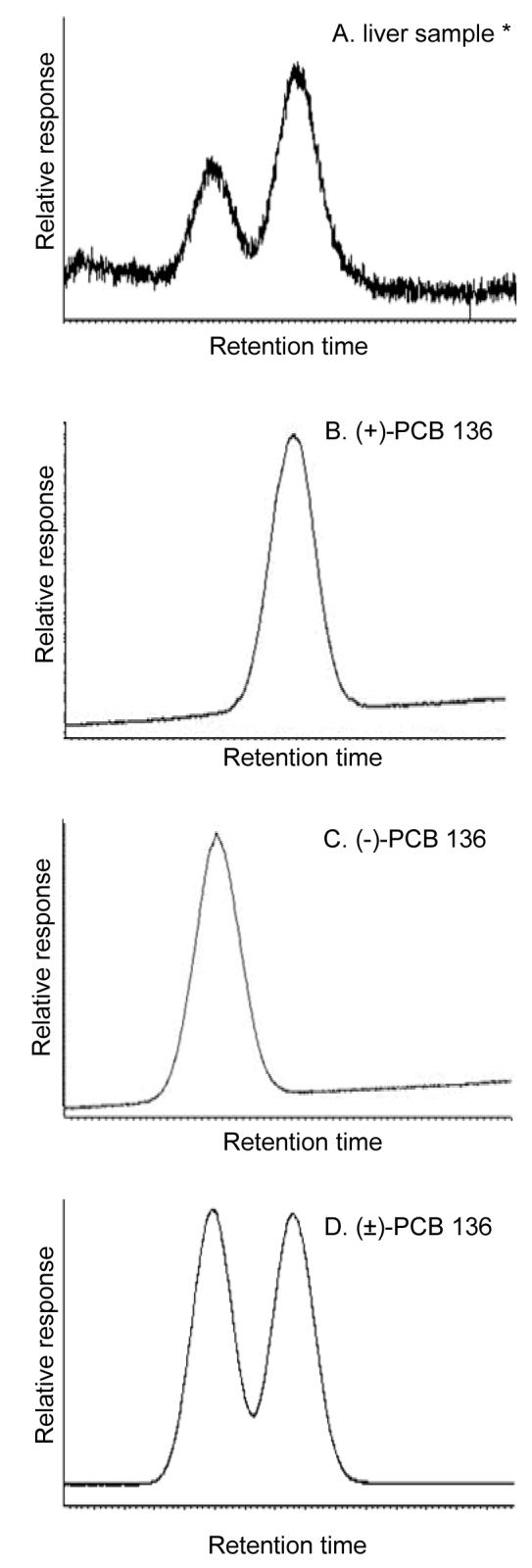
Separation of PCB 136 atropisomers using chiral gas chromatography. Representative chromatogram illustrating the enrichment of (+)-PCB 136 in the liver of a PB-pretreated mouse (22) (A) as well as chromatograms of pure (+)-PCB 136 atropisomer (B), pure (-)-PCB 136 atropisomer (C) and racemic PCB 136 standard (D) used in the spectral binding studies. The PCB atropisomers were separated using two serially connected Nucleodex P-PM columns as described in Experimental Procedures.
The enantioselective processes responsible for the enantiomeric enrichment of PCBs are poorly understood, but several studies suggested enantioselective biotransformation by P450 enzymes or binding to transport proteins (23-25). It has been shown that PCB 136 is rapidly metabolized by P450 enzymes in rodents (26-29). Thus, enantiomeric enrichment of (+)-PCB 136 in rodents may be due to the more rapid metabolism of (-)-PCB 136 by hepatic P450 enzymes. P450 enzymes differ in their ability to metabolize individual PCB congeners (30). The ability of PCBs to bind to P450 enzymes is influenced by the position of the chlorine substituents, which determines the degree of coplanarity of the phenyl rings. For instance, PCB congeners with four ortho chlorine substituents are poor substrates for hepatic CYP1A enzymes but have been shown to bind to microsomal CYP2B enzymes (30). However, it is not known if PCB atropisomers interact with P450 enzymes in an enantioselective manner. To investigate the hypothesis that enantioselective binding to specific P450 enzymes leads to enrichment of (+)-PCB 136 in vivo, we measured the spectral binding properties of PCB 136 atropisomers to hepatic microsomes prepared from mice and rats that had been pretreated with prototypical inducers of CYP1A, CYP2B and CYP3A enzymes (NF or 3-methylcholanthrene (MC) for CYP1A, PB for CYP2B, and DEX for CYP3A).
Experimental Procedures
Materials
A detailed description of the batch of (±)-PCB 136 (racemic mixture) used in this study was reported previously (31). Briefly, (±)-PCB 136 was synthesized by the Ullmann coupling reaction of 2,3,6-trichloro-1-iodobenzene and purified using charcoal to remove trace amounts of halogenated dibenzofurans. PCB 136 atropisomers were separated by reversed phase-high pressure liquid chromatography on two serially connected 4.6 × 250 mm Nucleodex β-PM columns (Macherey-Nagel, Düren, Germany), as described by Haglund (32). As shown in Figure 1, the enantiomeric purity of the PCB 136 atropisomers was determined using a HP6890 gas chromatograph equipped with a 63Ni μ-ECD detector and a Chirasil-Dex column (β-cyclodextrin chemically bonded with dimethylpolysiloxane, 25 m × 0.25 mm ID ×0.25 μm film thickness, Varian, Palo Alto, CA) (6, 19, 20). The enantiomeric fractions (EF) of (-)-PCB 136 and (+)-PCB 136 were 0.01 and 1.00, respectively (EF = peak area (+)-PCB 136/[peak area (+)-PCB 136 + peak area (-)-PCB 136] according to Harner et al. (33)). PCBs are hazardous and should be handled carefully.
Alkaline phosphatase-conjugated goat anti-rabbit F[ab’]2immunoglobulin G (IgG) was purchased from BioSource International (Camarillo, CA). Other chemicals were obtained from commercially available sources.
Animal treatment
Experiments involving mice were approved by the Institutional Animal Care and Use Committee at the University of Iowa. Female 8-week old C57BL/6 mice (n=104, average weight=18.2±1.1 g) were obtained from Harlan (Indianapolis, IN). Mice were allowed to acclimatize for one week before being randomly assigned to four treatment groups. Mice received intraperitoneal injections of NF in CO (n=27, 27 mg/kg/day), PB in saline (n=25, 102 mg/kg/day), DEX in CO (n=25, 50 mg/kg/day) or CO alone (n=27, 10 mL/kg/day) for 3 consecutive days (22). Mice were killed by CO2 asphyxiation followed by cervical dislocation 24 h after the last treatment. Livers were excised and immediately placed in ice-cold 0.05 M Tris-HCl buffer (pH 7.5) containing 0.15 M KCl.
Rats were housed at the University of British Columbia (Vancouver, British Columbia, Canada) and were cared for in accordance with the principles and guidelines of the Canadian Council on Animal Care. Adult male Long Evans rats were purchased from Charles River Laboratories (Montreal, Quebec, Canada) and were randomly assigned to four treatment groups with six animals per group. Rats received intraperitoneal injections of MC in CO (n=6, 25 mg/kg/day), PB in saline (n=6, 80 mg/kg/day), DEX in CO (n=6, 100 mg/kg/day) or CO alone (n=6, 2 mL/kg/day) for 3 consecutive days. Rats were killed by decapitation 24 h after the last treatment. Livers were excised and immediately placed in ice-cold 0.05 M Tris-HCl buffer (pH 7.5) containing 0.15 M KCl.
Preparation of hepatic microsomes
Mouse and rat hepatic microsomes were prepared by differential ultracentrifugation as described previously (34). Liver homogenates were prepared from individual livers and were then pooled for mice or rats from each treatment group. The pooled homogenates were spun at 9,000 g for 20 min, and the resulting supernatants were subjected to two consecutive centrifugations at 100,000 g for 60 min. The microsomal pellets were resuspended in 0.25 M sucrose and 1 mL aliquots were stored at -80°C. Protein concentrations were measured by the method of Lowry et al. (35) using bovine serum albumin as a standard. Total P450 concentrations were determined by the method of Omura and Sato (36) using an extinction coefficient of 91 mM-1cm-1.
Binding of PCB 136 to hepatic microsomal P450
The binding of (+)-, (-)- and (±)-PCB 136 to hepatic microsomal P450 enzymes was studied by recording the type I difference spectra at room temperature using an Aminco DW-2 dual wavelength/split beam spectrophotometer as described previously (37). Briefly, 0.99 mL of a hepatic microsomal preparation (diluted to 4 nmol total P450/mL in 0.1 M potassium phosphate buffer, pH 7.5) was added to both sample and reference cuvettes. After the baseline was recorded, 10 μl of (+)-, (-)- or (±)-PCB 136 dissolved in dimethyl sulfoxide (DMSO) was added to the sample cuvette, while 10 μl of DMSO was added to the reference cuvette to offset any contribution made by DMSO to the difference spectrum. DMSO alone (at 10 μl/mL) did not produce a type I difference spectrum. The difference spectrum was recorded with repetitive scanning between 350 and 500 nm. The absorbance change was determined as the difference between the spectral peak (λmax at approximately 390 nm) and trough (λmin at approximately 420 nm) and was expressed per nmol of P450. The apparent spectral dissociation constant (Ks app) and maximal absorbance change (ΔAmax) were calculated by nonlinear regression analysis using the Enzyme Kinetics Module of Sigma-Plot (Systat Software Inc., Ver. 10, San Jose, CA). The ΔAmax/Ks app parameter, which reflects the relative binding efficiency (38, 39), was calculated to facilitate comparisons.
Immunoblot analysis
Immunoblot analysis was used to measure protein levels of CYP1A1, CYP1A2, CYP2B1, CYP2B2, CYP2C11 and CYP3A enzymes in rat hepatic microsomes (37) and relative levels of immunoreactive CYP1A, CYP2B and CYP3A enzymes in mouse hepatic microsomes. Microsomal proteins were resolved by SDS-PAGE, then transferred onto nitrocellulose membranes and probed with rabbit anti-rat CYP1A2 polyspecific antiserum (1:1000 dilution), anti-CYP2B1 polyspecific IgG (2 μg/ml), anti-CYP2C11 monospecific IgG (10 μg/ml) or anti-CYP3A polyspecific IgG (5 μg/ml), at the concentrations indicated. Purified rat CYP1A1 (0.05-0.375 pmol/lane), CYP1A2 (0.025-0.25 pmol/lane), CYP2B1 (0.031-0.25 pmol/lane), CYP2C11 (0.1-0.4 pmol/lane) and rat recombinant CYP3A1 and CYP3A2 (0.125-0.375 pmol/lane) were applied onto the gels as calibration/reference standards in a volume of 20 μl/lane, at the concentration ranges indicated. The preparation of rat P450 enzymes and antibodies against rat P450 enzymes was described previously (37).
Antibody and serum inhibition studies
Serum was obtained from adult female New Zealand rabbits that had been immunized with purified, electrophoretically homogeneous CYP2B1, CY2C11 or CYP3A1. Each rabbit was immunized initially with 200 μl of one of the purified P450 enzymes emulsified in Freund’s complete adjuvant and administered subcutaneously. Three rabbits were immunized with each purified P450 enzyme. Four weeks later, the rabbits received a boost consisting of 50 μl of the P450 protein per rabbit as a 1:1 emulsion in Freund’s incomplete adjuvant administered by intramuscular injection. Thereafter, rabbits were boosted on a monthly basis by intravenous injection of the P450 protein (50 μl dissolved in phosphate-buffered saline, pH 7.4) into the ear vein, followed by collection of approximately 30 ml of blood from each rabbit 7 days after the boost. Blood from each bleeding was allowed to clot at 37°C and serum was removed and stored at -20°C. Control serum was obtained from rabbits that were not immunized.
Increasing volumes (0.05-0.5 mL) of serum were added to both sample and reference cuvettes containing hepatic microsomes (2 nmol P450/mL in 0.1 M potassium phosphate buffer, pH 7.5). The volume was brought up to 0.99 mL with phosphate buffered saline, where necessary. The microsomal samples were preincubated with the serum in the cuvettes at room temperature for 5 min, followed by addition of 10 μL of (+)-, (-)- or (±)-PCB 136 (2.5 mM in DMSO) to the sample cuvette and 10 μL of DMSO to the reference cuvette. The binding spectra were then recorded as described above.
Statistical analysis
Data are presented as the experimental mean ± SE where applicable. Differences between mean values were analyzed using open-source R statistical software (Ver. 2.5.1).
Results
Difference spectra using mouse hepatic microsomes
Addition of (+)-PCB 136, (-)-PCB 136, or PCB 136 racemate to hepatic microsomes prepared from NF-, PB-, DEX- or CO-treated female mice yielded type I difference spectra, with a λmaxat 385-390 nm and a λmin at 416-422 nm. Representative difference spectra for (+)- and (-)-PCB 136, at various concentrations, are presented in Figure 2. The effect of increasing PCB concentration on absorbance change can be seen more clearly in Figure 3. The magnitude of absorbance change varied with the microsomal preparation used and increased with increasing PCB concentration up to a concentration of 50-100 μM. A larger absorbance change was observed with (+)-PCB 136 than with (-)-PCB 136 with all four microsomal preparations at PCB concentrations greater than 25 μM.
Figure 2.

Type I difference spectra elicited by addition of (+)-PCB 136 (A) or (-)-PCB 136 (B) to hepatic microsomes from PB-treated mice (representative spectra of 3 determinations). Indicated concentrations of PCB 136 (in DMSO) were added in 10 μL quantities separately to 1 mL sample cuvettes containing microsomes (4 nmol CYP/mL) in 0.1 M potassium phosphate buffer (pH 7.5). Reference cuvettes received 10 μL of DMSO. Difference spectra were recorded separately for each PCB concentration as described in Experimental Procedures. Spectra were scanned and digitized using Engauge Digitizer software (http://digitizer.sourceforge.net/).
Figure 3.

Representative plots of the absorbance changes elicited by increasing concentrations of (+)-, (-)- or (±)-PCB 136 with mouse hepatic microsomes (3 experiments for PB and DEX, single experiment for NF and CO). Increasing amounts of PCB were added separately to 1 mL sample cuvettes containing hepatic microsomes, at 4 nmol P450/mL, prepared from NF- (A), PB- (B), DEX- (C), and CO-treated (D) mice. Difference spectra were recorded and ΔA values calculated as described in Experimental Procedures.
Values of maximal absorbance change (ΔAmax), apparent spectral affinity (Ks app) and relative binding efficiency (ΔAmax/Ks), were calculated for (+)-, (-)-, or (±)-PCB 136 and are presented in Table 1. These spectral parameters reflect the ability of PCB 136 atropisomers to bind to hepatic microsomal P450 enzymes. Maximal absorbance change values and relative binding efficiency values for both PCB 136 atropisomers and for racemic PCB 136 were much larger with hepatic microsomes prepared from PB-treated mice than NF- or CO-treated mice, suggesting that (+)-PCB 136 and (-)-PCB 136 are interacting to a greater extent with one or more P450 enzymes present in liver of PB-treated mice than with the P450 enzymes found in the liver of NF- or CO-treated mice. Moreover, ΔAmaxvalues were greater for (+)-PCB 136 than (-)-PCB 136 for all four microsomal preparations. However, there was no clear rank order in the relative binding efficiency of PCB 136 atropisomers within the four microsomal preparations. For instance, the relative binding efficiency of (-)-PCB 136 with hepatic microsomes from NF- or DEX-treated mice was greater than that obtained for (+)-PCB 136 and was smaller than that obtained for (+)-PCB 136 with hepatic microsomes from CO-treated mice.
Table 1.
Spectral constants and relative binding efficiencies for PCB 136 atropisomers with mouse hepatic microsomal P450 enzymesa
| Spectral parameter | (+)-PCB 136 | (-)-PCB 136 | (±)-PCB 136 |
|---|---|---|---|
| Hepatic microsomes from NF-treated miceb | |||
| ΔAmax (absorbance unit/nmol P450) | 0.011 | 0.005 | 0.008 |
| Ksapp (μM) | 9.2 | 2.9 | 4.2 |
| ΔAmax/Ksapp (absorbance unit/nmol P450/M) | 1200 | 1900 | 2000 |
| Hepatic microsomes from PB-treated micec | |||
| ΔAmax (absorbance unit/nmol P450) | 0.028 ± 0.002 | 0.020 ± 0.002 | 0.027 ± 0.001 |
| Ksapp (μM) | 8.2 ± 0.70 | 5.6 ± 1.0 | 7.0 ± 0.60 |
| ΔAmax/Ksapp (absorbance unit/nmol P450/M) | 3500 ± 450 | 3800 ± 480 | 3900 ± 430 |
| Hepatic microsomes from DEX-treated micec | |||
| ΔAmax (absorbance unit/nmol P450) | 0.026 ± 0.001 | 0.017 ± 0.001 | 0.020 ± 0.001 |
| Ksapp (μM) | 24 ± 5.4 | 7.6 ± 1.4 | 12 ± 2.8 |
| ΔAmax/Ksapp (absorbance unit/nmol P450/M) | 1200 ± 240 | 2400 ± 450 | 1800 ± 380 |
| Hepatic microsomes from CO-treated miceb | |||
| ΔAmax (absorbance unit/nmol P450) | 0.012 | 0.010 | 0.012 |
| Ksapp (μM) | 9.7 | 21 | 10 |
| ΔAmax/Ksapp (absorbance unit/nmol P450/M) | 1200 | 450 | 1200 |
Maximal absorbance change (ΔAmax), apparent spectral dissociation constant (Ksapp) and relative binding efficiency (ΔAmax/Ksapp) values were obtained for hepatic microsomes prepared from NF, PB, DEX and CO-treated female mice as described in Experimental Procedures.
Single measurement.
Mean ± SE of three determinations.
Difference spectra using rat hepatic microsomes
To determine if preferential binding of PCB 136 with hepatic microsomes from PB-treated mice and the enantiomeric differences observed are characteristic of other species, the spectral binding studies were repeated with rat hepatic microsomes. Similar results but larger absorbance changes were obtained when rat hepatic microsomes were used (Figure 4).
Figure 4.

Representative plots of absorbance changes elicited by increasing concentrations of (+)-, (-)- or (±)-PCB 136 with rat hepatic microsomes (3 separate experiments for PB, DEX and CO, single experiment for MC). Increasing amounts of PCB were added separately to 1 mL sample cuvettes containing hepatic microsomes at 4 nmol P450/mL prepared from MC- (A), PB- (B), DEX- (C), and CO-treated (D) rats. Difference spectra were recorded and ΔA values calculated as described in Experimental Procedures.
As shown in Table 2, the spectral parameters determined using rat hepatic microsomes and (+)-, (-)- or (±)-PCB 136 were comparable to those observed using mouse hepatic microsomes. The largest ΔAmax values were observed for PCB 136 atropisomers with microsomes from PB-treated rats, and the smallest ΔAmax values were observed for PCB 136 atropisomers with microsomes from MC-treated rats. The same effect was observed for relative binding efficiency (ΔAmax/Ks). As with the results obtained using mouse hepatic microsomes, there was no clear rank order in the relative binding efficiency of PCB 136 atropisomers within the four microsomal preparations.
Table 2.
Spectral constants and relative binding efficiencies for PCB 136 atropisomers with rat hepatic microsomal P450 enzymesa
| Spectral parameter | (+)-PCB 136 | (-)-PCB 136 | (±)-PCB 136 |
|---|---|---|---|
| Hepatic microsomes from MC-treated ratsb | |||
| ΔAmax (absorbance unit/nmol P450) | 0.012 | 0.010 | 0.011 |
| Ksapp (μM) | 13 | 6.9 | 8.8 |
| ΔAmax/Ksapp (absorbance unit/nmol P450/M) | 920 | 1500 | 1300 |
| Hepatic microsomes from PB-treated ratsc | |||
| ΔAmax (absorbance unit/nmol P450) | 0.060 ± 0.009 | 0.053 ± 0.006 | 0.058 ± 0.007 |
| Ksapp (μM) | 2.2 ± 0.47 | 3.8 ± 0.76 | 2.6 ± 0.48 |
| ΔAmax/Ksapp (absorbance unit/nmol P450/M) | 28000 ± 2800 | 14000 ± 1500 | 23000 ± 2100 |
| Hepatic microsomes from DEX-treated ratsc | |||
| ΔAmax (absorbance unit/nmol P450) | 0.036 ± 0.005 | 0.024 ± 0.003 | 0.032 ± 0.004 |
| Ksapp (μM) | 12 ± 2.4 | 16 ± 3.2 | 12 ± 2.8 |
| ΔAmax/Ksapp (absorbance unit/nmol P450/M) | 3100 ± 240 | 1600 ± 130 | 2800 ± 360 |
| Hepatic microsomes from CO-treated ratsc | |||
| ΔAmax (absorbance unit/nmol P450) | 0.046 ± 0.001 | 0.042 ± 0.001 | 0.044 ± 0.001 |
| Ksapp (μM) | 9.2 ± 1.6 | 10 ± 1.1 | 8.8 ± 1.2 |
| ΔAmax/Ksapp (absorbance unit/nmol P450/M) | 5300 ± 920 | 4000 ± 420 | 5300 ± 900 |
Maximal absorbance change (ΔAmax), apparent spectral dissociation constant (Ksapp) and relative binding efficiency (ΔAmax/Ksapp) values were obtained for hepatic microsomes prepared from MC, PB, DEX and CO-treated male rats as described in Experimental Procedures.
Single measurement.
Mean ± SE of three determinations.
P450 protein immunoquantitation and antibody inhibition
The contribution of individual P450 enzymes to PCB 136-P450 spectral interactions was assessed by determining hepatic levels of CYP1A, CYP2B and CYP3A proteins in the mouse and rat microsomal preparations used for the spectral binding study and by measuring difference spectra in the presence of P450 subfamily-specific polyclonal antibodies. As reported previously (37, 40-42) and shown in Tables 3 and 4, CYP1A enzymes were the predominant P450 enzymes in hepatic microsomes from NF-treated mice and MC-treated rats, CYP2B enzymes were the major P450 enzymes in microsomes from PB-treated mice and rats, and CYP3A enzymes were the predominant P450 enzymes in microsomes from DEX-treated mice and rats. In addition to CYP3A, relatively high CYP2B enzyme levels were measured in hepatic microsomes from DEX-treated mice.
Table 3.
Relative P450 protein levels in mouse hepatic microsomesa
| Treatment | Immunoreactive P450 protein levels | ||
|---|---|---|---|
| CYP1A2b | CYP2Bc | CYP3Ad | |
| NF | 9.5 ± 0.5 | 1.1 ± 0.1 | 1.0 ± 0.1 |
| PB | 2.1 ± 0.2 | 15.2 ± 1.1 | 5.3 ± 0.3 |
| DEX | 0.4 ± 0.1 | 14.9 ± 1.1 | 25.9 ± 1.0 |
| CO | 1.0 ± 0.1 | 1.0 ± 0.1 | 1.0 ± 0.2 |
P450 protein levels are expressed relative to those of CO-treated mice set at 1.00. The amount of immunoreactive P450 protein was calculated in terms of relative optical density units/mg of microsomal protein because purified or recombinant mouse P450 standards are not available. Values shown are the experimental mean ± SE of 3 or 4 determinations using pooled hepatic microsomes prepared from female mice that had been pretreated with NF, PB, DEX or CO. Purified or recombinant rat P450 enzymes were included on the gels as reference standards. Immunoreactive P450 protein bands were detected with antibodies directed against rat P450 enzymes as described in Experimental Procedures.
Only relative CYP1A2 levels are reported. CYP1A1 was not detected in hepatic microsomes from PB-, DEX- or CO-treated mice but was detected in hepatic microsomes from NF-treated mice. A relative CYP1A1 level was not determined for NF-treated mice due to the lack of a reference point.
The CYP2B level reported here corresponds to the immunoreactive band tentatively identified as CYP2B10 by reference to the electrophoretic mobility previously reported (52).
The CYP3A level reported here corresponds to the immunoreactive band tentatively identified as CYP3A11 by reference to the electrophoretic mobility previously reported (52).
Table 4.
P450 protein levels in rat hepatic microsomesa
| Treatment | P450 protein levels (pmol/nmol total P450) | |||||
|---|---|---|---|---|---|---|
| CYP1A1 | CYP1A2 | CYP2B1 | CYP2B2 | CYP2C11 | CYP3Ab | |
| MC | 370 ± 12 (9) | 220 ± 13 (12) | < 1 (2) | < 1 (2) | 62 (2) | 16 (2) |
| PB | < 1 (8) | 3 ± 0 (8) | 320 ± 21 (8) | 100 ± 6 (8) | 46 (2) | 70 (2) |
| DEX | < 1 (3) | 2 ± 0 (3) | 20 ± 0 (3) | 15 ± 1 (3) | 45 (2) | 130 (2) |
| CO | < 1 (6) | 7 ± 1 (6) | < 1 (6) | 2 ± 0 (6) | 120 (2) | 19 (2) |
Microsomes were prepared from pooled livers of male Long Evans rats that had been pretreated with MC, PB, DEX or CO. Individual P450 protein levels were determined by immunoblot analysis using purified or recombinant rat P450 enzymes as calibration standards, as described previously (37) and are expressed in terms of pmol/nmol total P450. Values shown are the experimental mean ± SE of the number of determinations indicated in parentheses. In some cases, the average value of two determinations is shown.
CYP3A1 levels are non-detectable in hepatic microsomes from MC- or CO-treated Long Evans rats but both CYP3A1 and CYP3A2 are induced after PB or DEX treatment (53). Microsomal CYP3A1 does not separate from CYP3A2 under the SDS-PAGE conditions used and CYP3A1 and CYP3A2 are visualized as a single band by immunoblot analysis (53). Consequently, the CYP3A protein band has been quantified as the sum of CYP3A1 and CYP3A2.
The relatively large binding efficiencies obtained with hepatic microsomes from PB-treated mice and rats and (+)- and (-)-PCB 136 (Tables 1 and 2) suggests that both atropisomers bind preferentially to CYP2B enzymes. To test this assumption, difference spectra produced by addition of (+)- or (-)-PCB136 to hepatic microsomes from PB-treated rats were measured in the presence of antisera against rat CYP2B, CYP2C and CYP3A enzymes. The magnitude of the absorbance change displayed by (+)-PCB136 or (-)-PCB 136 with hepatic microsomes from PB-treated rats was reduced by approximately 30% in the presence of CYP2B antisera at the largest volume tested, whereas CYP2C antisera or control antisera had little or no inhibitory effect (Figure 5). Incubation with CYP3A antisera also decreased the absorbance change by approximately 20% to 30% at the largest volume tested. When smaller volumes of serum were used, the inhibitory effect of CYP3A antisera was less than that of CYP2B antisera (Figure 5).
Figure 5.

Effect of anti-rat CYP2B, anti-rat CYP2C and anti-rat CYP3A sera on (+)-PCB 136 (A) and (-)-PCB 136-induced (B) absorbance changes in hepatic microsomes from PB-treated rats. Increasing amounts of rabbit anti-rat CYP2B, CYP2C or CYP3A sera or control rabbit serum were added separately to both sample and reference cuvettes containing hepatic microsomes at 2.04 nmol/mL in a final volume of 1 mL. Samples were preincubated with sera for 5 min before addition of the PCB 136 atropisomers and start of the spectral assay. The data are from a single experiment.
A similar effect was obtained with hepatic microsomes from DEX-treated rats (Figure 6). Incubation with CYP2B antisera produced the greatest reduction in the magnitude of absorbance change for (+)- and (-)-PCB 136 (Figure 6). CYP3A antisera decreased absorbance change for (+)-PCB 136, but had less effect on (-)-PCB 136 binding. Taken together, the antibody inhibition data suggest that (+)- and (-)-PCB 136 both bind preferentially to CYP2B and CYP3A enzymes.
Figure 6.

Effect of rabbit anti-rat CYP2B, anti-rat CYP2C and anti-rat CYP3A sera on (+)-PCB 136 (A) and (-)-PCB 136-induced (B) absorbance changes in hepatic microsomes from DEX-treated rats. Increasing amounts of rabbit anti-rat CYP2B, CYP2C or CYP3A sera or control rabbit serum were added separately to both sample and reference cuvettes containing hepatic microsomes at 2.04 nmol/mL in a final volume of 1 mL. Samples were preincubated with sera for 5 min before addition of the PCB 136 atropisomers and start of the spectral assay. The data are from a single experiment.
Discussion
The interaction of endogenous compounds and xenobiotics such as PCBs with hepatic microsomal or purified P450 enzymes produces spectral changes due to a disturbance of the coordination sphere of the heme iron. Specifically, formation of a P450 apoprotein-substrate complex with PCBs results in a type I difference spectra characterized by a peak at approximately 385 nm and a trough at 420 nm. Although the extent to which a spectral change caused by the binding of an individual PCB congener correlates with its rate of biotransformation to a hydroxylated PCB metabolite has not been proven conclusively (43-46), spectral changes allow us to assess the binding efficiencies of different PCB congeners with hepatic microsomal P450 preparations and thus, to establish structure-P450 binding relationships for different PCB congeners. Using this approach, Hrycay and Bandiera (37) recently showed that ortho-substituted PCBs (such as PCB 47, 52 and 54) preferentially bind to CYP2B and, to a lesser extent, to CYP3A enzymes in hepatic microsomes from PB-treated male rats. In the same study, it was demonstrated that PCB 54 bound preferentially to CYP3A and CYP2C enzymes in microsomes from control rats. Analogously, ortho-substituted 2-chlorobiphenyl (PCB 1) was also found to bind preferentially to hepatic CYP2B enzymes (47).
In the present study, PCB 136 atropisomers (i.e., (+)- and (-)-PCB 136) also exhibited concentration-dependent type I difference spectra with hepatic microsomes, independent of the species (mouse vs. rat) or the pretreatment (NF, PB, DEX, CO in mice and MC, PB, DEX, CO in rats). However, the magnitude of the absorbance change (ΔAmax) for both (+)- and (-)-PCB 136 was clearly influenced by pretreatment and decreased in the order PB > DEX > CO > NF in mice and PB > CO > DEX > MC in rats. The same rank order of binding to rat hepatic microsomes has been reported for PCB 54, a tetrachlorobiphenyl that, like PCB 136, has four ortho chlorine substituents (37). Similarly, the relative binding efficiency of (+)- and (-)-PCB 136 was much larger with hepatic microsomes from mice and rats that had been pretreated with PB compared to the other inducers. Overall, the spectral constants and binding efficiencies suggest that both PCB 136 atropisomers preferentially bind to CYP2B enzymes, which are the predominant P450 subfamily in microsomes prepared from PB- and, to a lesser extent, DEX-treated animals.
Inhibition studies were performed with selected rabbit anti-rat P450 serum samples to verify that PCB 136 atropisomers indeed bind to rat P450 enzymes. Analogous studies with mouse microsomes were not performed due to the unavailability of antibodies specific for mouse P450 enzymes. We focused on CYP2B and CYP3A enzymes in the inhibition studies because multi-ortho substituted PCBs (such as PCB 54) are known to interact with both P450 subfamilies (37), and because PCB 136 is known to be metabolized by CYP2B enzymes (48-50). Furthermore, experiments were performed only with microsomes from PB- and DEX-treated rats because, in agreement with previous results (37, 40-42), CYP2B is the major P450 enzyme in microsomes from PB-treated rats and CYP3A predominates in microsomes from DEX-treated rats (Table 4).
The antibody inhibition studies confirmed that PCB 136 atropisomers bind preferentially to CYP2B and CYP3A enzymes in microsomes from both PB- and DEX-treated rats. The observation that PCB 136 atropisomers bind efficiently to CYP2B enzymes is not surprising because, as mentioned above, PCB 136 is readily metabolized by this subfamily of P450 enzymes (48-50). The finding that PB induction leads to increased enantiomeric enrichment of (+)-PCB 136 in mice (22) further supports a role for CYP2B enzymes in the disposition of PCB 136 atropisomers. However, it is currently unclear if PCB 136 atropisomers are also metabolized by CYP3A enzymes, a question that warrants further investigation with purified P450 enzymes.
The most intriguing finding of our study was that (+)-PCB 136 appeared to interact to a greater degree than (-)-PCB 136 with microsomes from mice and rats, independent of the pretreatment, and is based on the absorbance changes observed with increasing PCB 136 atropisomer concentrations (see Figures 3 and 4) and the maximal absorbance changes (ΔAmax) reported in Tables 1 and 2. This finding suggests preferential binding of (+)-PCB 136 to P450 enzymes such as CYP2B and CYP3A present in the microsomal preparations. If (+)-PCB 136 is preferentially metabolized by CYP2B and possibly CYP3A enzymes, it is difficult to explain why (+)-PCB 136 is enriched in mouse tissues. However, a slightly analogous situation exists for a related group of compounds. Diliberto et al. found high levels of 2,3,7,8-tetrachlorodibenzo-p-dioxin and other laterally chlorinated dibenzo-p-dioxins and dibenzofurans in the liver of wild-type mice but not in the liver of CYP1A2 knockout mice (51). On the basis of the results, these investigators proposed that hepatic sequestration of laterally chlorinated dibenzo-p-dioxins and dibenzofurans is due to induction and binding of CYP1A2.
Analogous to the work by Diliberto et al. (51), the present study suggests two mechanisms that may be responsible for the enantiomeric enrichment of (+)-PCB 136 in tissues of these experimental animals. The first mechanism involves hepatic sequestration of (+)-PCB 136 due to enantioselective binding of the atropisomer to P450 enzymes. Similarly, the sequestration of dioxin-like PCB congeners is thought to be the result of binding to CYP1A2 enzymes (51). The second possible mechanism also involves preferential binding of the (+)-PCB 136 atropisomer to CYP2B enzymes, which results in a more rapid metabolism of the (-)-PCB 136 atropisomer due to the latter’s weaker binding to CYP2B enzymes. Further in vitro studies with purified P450 enzymes are needed to determine the binding efficiencies and rates of metabolism of both PCB 136 atropisomers, and these studies could be extended to other chiral PCB congeners to establish whether an inverse relationship exists between the binding of PCB substrates to, and their metabolism by, P450 enzymes.
Acknowledgments
The work was supported by the following grants ES05605, ES013661 and ES012475 from the National Institute of Environmental Health Sciences, NIH. S.M.B. wishes to acknowledge the Natural Sciences and Engineering Council of Canada (RGPIN 138733-01) for their financial support.
Abbreviations
- ΔA
absorbance change
- ΔAmax
maximal absorbance change
- CO
corn oil
- CYP
designates an individual cytochrome P450
- DEX
dexamethasone
- DMSO
dimethyl sulfoxide
- IgG
immunoglobulin G
- Ksapp
apparent spectral dissociation constant
- NF
β-naphthoflavone
- MC
3-methylcholanthrene
- P450
cytochrome P450
- PB
phenobarbital
- PCBs
polychlorinated biphenyls
- PCB 84
2,2′,3,3′,6-pentachlorobiphenyl
- PCB 136
2,2′,3,3′,6,6′-hexachlorobiphenyl.
References
- (1).Robertson LW, Hansen LG. PCBs: Recent Advances in Environmental Toxicology and Health Effects. University Press of Kentucky; Lexington: 2001. [Google Scholar]
- (2).Hornbuckle KC, Carlson DL, Swackhamer DL, Baker JE, Eisenreich SJ. Polychlorinated biphenyls in the Great Lakes. In: Hites R, editor. Handbook of Environmental Chemistry. Springer Verlag; Berlin Heidelberg: 2006. pp. 13–70. [Google Scholar]
- (3).Kodavanti PRS. Intracellular signaling and developmental neurotoxicity. In: Zawia NH, editor. Molecular Neurotoxicology: environmental agents and transcription-transduction coupling. CRC press; Boca Roton, FL: 2004. pp. 151–182. [Google Scholar]
- (4).Schantz SL, Widholm JJ, Rice DC. Effects of PCB exposure on neuropsychological function in children. Environ. Health Perspect. 2003;111:357–376. doi: 10.1289/ehp.5461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Lehmler H-J, Robertson LW. Atropisomers of PCBs. In: Robertson LW, Hansen LG, editors. PCBs: Recent Advances in Environmental Toxicology and Health Effects. University Press of Kentucky; Lexington: 2001. pp. 61–65. [Google Scholar]
- (6).Kania-Korwel I, Hornbuckle KC, Robertson LW, Lehmler HJ. Dose-dependent enantiomeric enrichment of 2,2′,3,3′,6,6′-hexachlorobiphenyl in female mice. Environ. Toxicol. Chem. 2007;27:299–305. doi: 10.1897/07-359R.1. [DOI] [PubMed] [Google Scholar]
- (7).Rodman LE, Shedlofsky SI, Mannschreck A, Puttmann M, Swim AT, Robertson LW. Differential potency of atropisomers of polychlorinated biphenyls on cytochrome P450 induction and uroporphyrin accumulation in the chick embryo hepatocyte culture. Biochem. Pharmacol. 1991;41:915–922. doi: 10.1016/0006-2952(91)90196-c. [DOI] [PubMed] [Google Scholar]
- (8).Lehmler H-J, Robertson LW, Garrison AW, Kodavanti PRS. Effects of PCB 84 enantiomers on [3H] phorbol ester binding in rat cerebellar granule cells and 45Ca2+-uptake in rat cerebellum. Toxicol. Lett. 2005;156:391–400. doi: 10.1016/j.toxlet.2004.12.011. [DOI] [PubMed] [Google Scholar]
- (9).Feng W, Lehmler H-J, Allen PD, Pessah IN. Chiral PCB 136 enantioselectively activates RyR1 and RyR2. Biophysical Society Meeting Abstracts. Biophys. J. Supplement. 2007;87A:403. Pos. [Google Scholar]
- (10).Wiberg K, Andersson PL, Berg H, Olsson P-E, Haglund P. The fate of chiral organochlorine compounds and selected metabolites in intraperitoneally exposed Arctic char (Salvelinus Alpinus) Environ. Toxicol. Chem. 2006;25:1465–1473. doi: 10.1897/05-329r.1. [DOI] [PubMed] [Google Scholar]
- (11).Gomara B, Gonzalez MJ. Enantiomeric fractions and congener specific determination of polychlorinated biphenyls in eggs of predatory birds from Donana National Park (Spain) Chemosphere. 2006;63:662–669. doi: 10.1016/j.chemosphere.2005.08.003. [DOI] [PubMed] [Google Scholar]
- (12).Buckman AH, Wong CS, Chow EA, Brown SB, Solomon KR, Fisk AT. Biotransformation of polychlorinated biphenyls (PCBs) and bioformation of hydroxylated PCBs in fish. Aquat. Toxicol. 2006;78:176–185. doi: 10.1016/j.aquatox.2006.02.033. [DOI] [PubMed] [Google Scholar]
- (13).Wong CS, Lau F, Clark M, Mabury SA, Muir DCG. Rainbow Trout (Oncorhynchus mykiss) can eliminate chiral organochlorine compound enantioselectively. Environ. Sci. Technol. 2002;36:1257–1262. doi: 10.1021/es0156791. [DOI] [PubMed] [Google Scholar]
- (14).Hoekstra PF, Braune BM, Wong CS, Wiliamson M, Elkin B, Muir DCG. Profile of persistent chlorinated contaminants, including selected chiral compounds, in wolverine (Gulo gulo) livers from Canadian Arctic. Chemosphere. 2003;53:551–560. doi: 10.1016/S0045-6535(03)00514-9. [DOI] [PubMed] [Google Scholar]
- (15).Warner NA, Wong CS. The freshwater invertebrate Mysis relicta can eliminate chiral organochlorine compounds enantioselectively. Environ. Sci. Technol. 2006;40:4158–4164. doi: 10.1021/es052166b. [DOI] [PubMed] [Google Scholar]
- (16).Hoekstra PF, Wong CS, O’Hara TM, Solomon KR, Mabury SA, Muir DCG. Enantiomer-specific accumulation of PCB atropisomers in the bowhead whale (Balaena mysticetus) Environ. Sci. Technol. 2002;36:1419–1425. doi: 10.1021/es015763g. [DOI] [PubMed] [Google Scholar]
- (17).Jimenez O, Jimenez B, Gonzales MJ. Isomer-specific polychlorinated biphenyl determination in cetaceans from the Mediterranean Sea: enantioselective occurrence of chiral polychlorinated biphenyl congeners. Environ. Toxicol. Chem. 2000;19:2653–2660. [Google Scholar]
- (18).Reich S, Jimenez B, Marsili L, Hernandez LM, Schurig V, Gonzales MJ. Congener specific determination and enantiomeric ratios of chiral polychlorinated biphenyl in striped dolphins (Stanella coeruleoalba) from Mediterranean Sea. Environ. Sci. Technol. 1999;33:1787–1793. [Google Scholar]
- (19).Kania-Korwel I, Shaikh N, Hornbuckle KC, Robertson LW, Lehmler H-J. Enantioselective disposition of PCB 136 (2,2′,3,3′,6,6′-hexachlorobiphenyl) in C57BL/6 mice after oral and intraperitoneal administration. Chirality. 2007;19:56–66. doi: 10.1002/chir.20342. [DOI] [PubMed] [Google Scholar]
- (20).Kania-Korwel I, Hornbuckle KC, Robertson LW, Lehmler HJ. Influence of dietary fat on the enantioselective disposition of 2,2′,3,3′,6,6′-hexachlorobiphenyl (PCB 136) in female mice. Food Chem. Toxicol. 2008;46:637–644. doi: 10.1016/j.fct.2007.09.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Kania-Korwel I, Garrison AW, Avants JK, Hornbuckle KC, Robertson LW, Sulkowski WW, Lehmler H-J. Distribution of chiral PCBs in selected tissues in the laboratory rat. Environ. Sci. Technol. 2006;40:3704–3710. doi: 10.1021/es0602086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Kania-Korwel I, Xie W, Hornbuckle KC, Robertson LW, Lehmler H-J. Enantiomeric enrichment of 2,2′,3,3′,6,6′-hexachlorobiphenyl (PCB 136) in mice after induction of CYP enzymes. Arch. Environ. Contam. Toxicol. 2008 doi: 10.1007/s00244-007-9111-4. in press. [DOI] [PubMed] [Google Scholar]
- (23).Harrad S, Ren J, Hazrati S, Robson M. Chiral signatures of PCB#s 95 and 149 in indoor air, grass, duplicate diets and human faeces. Chemosphere. 2006;63:1368–1376. doi: 10.1016/j.chemosphere.2005.09.027. [DOI] [PubMed] [Google Scholar]
- (24).Lehmler H-J, Price DJ, Garrison AW, Birge WJ, Robertson LW. Distribution of PCB 84 enantiomers in C57Bl/6 mice. Fresenius’ Environ. Bull. 2003;12:254–260. [Google Scholar]
- (25).Norström K, Eriksson J, Haglund J, Silvari V, Bergman A. Enantioselective formation of methyl sulfone metabolites of 2,2′,3,3′,4,6′-hexachlorobiphenyl in rat. Environ. Sci. Technol. 2006;40:7649–7655. doi: 10.1021/es061584t. [DOI] [PubMed] [Google Scholar]
- (26).Matthews HB, Tuey DB. The effect of chlorine position on the distribution and excretion of four hexachlorobiphenyl isomers. Toxicol. Appl. Pharmacol. 1980;53:377–388. doi: 10.1016/0041-008x(80)90351-8. [DOI] [PubMed] [Google Scholar]
- (27).Kato S, McKinney JD, Matthews HB. Metabolism of symmetrical hexachlorobiphenyl isomers in the rat. Toxicol. Appl. Pharma. 1980;53:389–398. doi: 10.1016/0041-008x(80)90352-x. [DOI] [PubMed] [Google Scholar]
- (28).Mizutani T, Hidaka K, Ohe T, Matsumoto M, Yamamoto K, Tajima K. Comparative study on accumulation and elimination of hexachlorobiphenyls and decachlorobiphenyl in mice. Bull. Environ. Contam. Toxicol. 1980;25:181–187. doi: 10.1007/BF01985508. [DOI] [PubMed] [Google Scholar]
- (29).Schnellmann R, Putnam C, Sipes I. Metabolism of 2,2′,3,3′,6,6′- hexachlorobiphenyl and 2,2′,4,4′,5,5′-hexachlorobiphenyl by human hepatic microsomes. Biochem. Pharmacol. 1983;32:3233–3239. doi: 10.1016/0006-2952(83)90209-5. [DOI] [PubMed] [Google Scholar]
- (30).Bandiera S. Cytochrome P450 enzymes as biomarkers of PCB exposure and modulators of toxicity. In: Robertson LW, Hansen LG, editors. PCBs: Recent Advances in Environmental Toxicology and Health Effects. University Press of Kentucky; Lexington: 2001. pp. 61–65. [Google Scholar]
- (31).Shaikh N, Parkin S, Lehmler H-J. The Ullmann coupling reaction: A new approach to tetraarylstannanes. Organometallics. 2006;25:4207–4214. [Google Scholar]
- (32).Haglund P. Isolation and characterization of polychlorinated biphenyl (PCB) atropisomers. Chemosphere. 1996;32:2133–2140. [Google Scholar]
- (33).Harner T, Wiberg K, Norstrom R. Enantiomer fractions are preferred to enantiomer ratios for describing chiral signatures in environmental analysis. Environ. Sci. Technol. 2000;34:218–220. [Google Scholar]
- (34).Thomas PE, Reik LM, Ryan DE, Levin W. Induction of two immunochemically related rat liver cytochrome P-450 isozymes, cytochromes P-450c and P- 450d, by structurally diverse xenobiotics. J. Biol. Chem. 1983;258:4590–4598. [PubMed] [Google Scholar]
- (35).Lowry OH, Rosenbrough NJ, Rarr AL, Randall RJ. Protein measurement with Folin Phenol reagent. J. Biol. Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- (36).Omura T, Sato R. The carbon monoxide-binding pigment of liver microsomes I. Evidence for its haemoprotein nature. J. Biol. Chem. 1964;239:2370–2378. [PubMed] [Google Scholar]
- (37).Hrycay EG, Bandiera SM. Spectral interactions of tetrachlorobiphenyls with hepatic microsomal cytochrome P450 enzymes. Chem. Biol. Interact. 2003;146:285–296. doi: 10.1016/j.cbi.2003.09.003. [DOI] [PubMed] [Google Scholar]
- (38).Vatsis KP, Coon MJ. Ipso-substitution by cytochrome P450 with conversion of p-hydroxybenzene derivatives to hydroquinone: evidence for hydroperoxo-iron as the active oxygen species. Arch. Biochem. Biophys. 2002;397:119–129. doi: 10.1006/abbi.2001.2665. [DOI] [PubMed] [Google Scholar]
- (39).Murray M, Reidy G. In vitro inhibition of hepatic drug oxidation by thioridazine. Kinetic analysis of the inhibition of cytochrome P-450 isoform-specific reactions. Biochem. Pharmacol. 1989;38:4359–4365. doi: 10.1016/0006-2952(89)90643-6. [DOI] [PubMed] [Google Scholar]
- (40).Lewis DFV. Cytochromes P450: structure, function and mechanism. Taylor and Francis; London: 1996. [Google Scholar]
- (41).Ryan DE, Levin W. Purification and characterization of hepatic microsomal cytochrome P-450. Pharmacol. Ther. 1990;45:153–239. doi: 10.1016/0163-7258(90)90029-2. [DOI] [PubMed] [Google Scholar]
- (42).Schenkman JB, Griem H. Cytochrome P450. Springer-Verlag; Berlin: 1993. [Google Scholar]
- (43).Schenkman JB, Frey I, Remmer H, Estabrook RW. Sex differences in drug metabolism by rat liver microsomes. Mol. Pharmacol. 1967;3:516–525. [PubMed] [Google Scholar]
- (44).Schenkman JB, Sligar SG, Cinti DL. Substrate interaction with cytochrome P-450. Pharmacol. Ther. 1981;12:43–71. doi: 10.1016/0163-7258(81)90075-9. [DOI] [PubMed] [Google Scholar]
- (45).Blanck H, Rein H, Sommer M, Ristau O, Smettan G, Ruckpaul K. Correlations between spin equilibrium shift, reduction rate, and N-demethylation activity in liver microsomal cytochrome P-450 and series of benzphetamine analogues as substrates. Biochem. Pharmacol. 1984;32:1683–1688. doi: 10.1016/0006-2952(83)90109-0. [DOI] [PubMed] [Google Scholar]
- (46).Tamburini PP, Gibson GG, Backes WL, Sligar SG, Schenkman JB. Reduction kinetics of purified rat liver cytochrome P-450. Evidence for sequential reaction mechanism dependent on the hemoprotein spin state. Biochemistry. 1984;23:4526–4533. doi: 10.1021/bi00315a004. [DOI] [PubMed] [Google Scholar]
- (47).Kennedy MW, Carpentier NK, Dymerski PD, Adams SM, Kaminsky LS. Metabolism of monochlorobiphenyl by hepatic microsomal cytochrome P-450. Biochem. Pharmacol. 1980;29:727–736. doi: 10.1016/0006-2952(80)90548-1. [DOI] [PubMed] [Google Scholar]
- (48).Waller SC, He YA, Harlow GR, He YQ, Mash EA, Halpert JR. 2,2′,3,3′,6,6′-hexachlorobiphenyl hydroxylation by active site mutants of cytochrome P450 2B1 and 2B11. Chem. Res. Toxicol. 1999;12:690–699. doi: 10.1021/tx990030j. [DOI] [PubMed] [Google Scholar]
- (49).Duignan D, Sipes I, Leonard T, Halpert J. Purification and characterization of the dog hepatic cytochrome P-450 isozyme responsible for the metabolism of 2,2′,4,4′,5,5′- hexachlorobiphenyl. Arch. Biochem. Biophys. 1987;255:290–303. doi: 10.1016/0003-9861(87)90396-1. [DOI] [PubMed] [Google Scholar]
- (50).Duignan D, Sipes IG, Ciaccio PJ, Halpert JR. The metabolism of xenobiotics and endogenous compounds by the constitutive dog liver cytochrome P450 PBD-2. Arch. Biochem. Biophys. 1988;267:106–115. doi: 10.1016/0003-9861(88)90035-5. [DOI] [PubMed] [Google Scholar]
- (51).Diliberto JJ, Burgin DE, Birnbaum LS. Effects of CYP1A2 on disposition of 2,3,7,8-tetrachlorodibenzo-p-dioxin, 2,3,4,7,8-pentachlorodibenzofuran, and 2,2′,4,4′,5,5′- hexachlorobiphenyl in CYP1A2 knockout and parental (C57BL/6N and 129/Sv) strains of mice. Toxicol. Appl. Pharmacol. 1999;159:52–64. doi: 10.1006/taap.1999.8720. [DOI] [PubMed] [Google Scholar]
- (52).Sakuma T, Takai M, Endo Y, Kuroiwa M, Ohara A, Jarukamjorn K, Honma R, Nemoto N. A novel female-specific member of the CYP3A gene subfamily in the mouse liver. Arch. Biochem. Biophys. 2000;377:153–162. doi: 10.1006/abbi.2000.1747. [DOI] [PubMed] [Google Scholar]
- (53).Jan Y-H, Mishin V, Busch CM, Thomas PE. Generation of specific antibodies and their use to characterize sex differences in four rat P450 3A enzymes following vehicle and pregnenolone 16α-carbonitrile treatment. Arch. Biochem. Biophys. 2006;446:101–110. doi: 10.1016/j.abb.2005.11.019. [DOI] [PubMed] [Google Scholar]