

Abstract
Flavonoids are polyphenolic compounds widely distributed in the plant kingdom. Compelling research indicates that flavonoids have important roles in cancer chemoprevention and chemotherapy possibly due to biological activities that include action through anti-inflammation, free radical scavenging, modulation of survival/proliferation pathways, and inhibition of the ubiquitin-proteasome pathway. Plant polyphenols including the green tea polyphenol, (-)-epigallocatechin gallate or (-)-EGCG, and the flavonoids apigenin, luteolin, quercetin, and chrysin have been shown to inhibit proteasome activity and induce apoptosis in human leukemia cells. However, biotransformation reactions to the reactive hydroxyl groups on polyphenols could reduce their biological activities. Although methylated polyphenols have been suggested to be metabolically more stable than unmethylated polyphenols, the practical use of methylated polyphenols as a cancer preventative agent warrants further investigation. In the current study, methylated and unmethylated flavonoids were studied for their proteasome-inhibitory and apoptosis-inducing abilities in human leukemia HL60 cells. Methylated flavonoids displayed sustained bioavailability and inhibited cellular proliferation by arresting cells in the G1 phase. However, they did not act as proteasome inhibitors in either an in vitro system or an in silico model and only weakly induced apoptosis. In contrast, unmethylated flavonoids exhibited inhibition of the proteasomal activity in intact HL60 cells, accumulating proteasome target proteins and inducing caspase activation and poly (ADP-ribose) polymerase cleavage. We conclude that methylated flavonoids lack potent cytotoxicity against human leukemia cells and most likely have limited ability as chemopreventive agents.
Keywords: Methylated flavonoids, Proteasome inhibitors, Apoptosis, Cell cycle progression
Interest in the potential cancer chemopreventive and therapeutic properties of diet-derived compounds such as plant polyphenols has increased in recent years. A subgroup of polyphenols, flavonoids, contains a flavone backbone with hydroxylations at various positions. These compounds can be found in a number of fruits and vegetables including celery, apples, onions, parsley, capsicum pepper, etc. [Ramos, 2007], and have been shown to possess anti-cancer activities. The mechanisms by which flavonoids impart their anti-cancer effects are varied and may include action through anti-inflammation [Choi et al., 2004], free radical scavenging [Sim et al., 2007], modulation of survival and proliferation pathways [Lee et al., 2005; Lee et al., 2004], and inhibition of the ubiquitin-proteasome pathway [Chen et al., 2007; Chen et al., 2005].
Although numerous studies indicate positive anti-cancer effects, the bioavailability and therefore efficacy of ingested flavonoids may be limited due to conjugation reactions (methylation, sulfonation, and glucuronidation) to the free hydroxyl groups surrounding the molecules [Manach and Donovan, 2004]. Exogenously derived methylated flavonoids have been shown to promote oral bioavailability [Wen and Walle, 2006a; Wen and Walle, 2006b] but direct evidence to indicate whether these compounds promote apoptotic cell death in cancer cells remains unclear. Furthermore, the effects of methylated flavonoids on proteasome activity have not been examined.
The eukaryotic proteasome is a large multi-catalytic, multi-subunit protease complex possessing at least three distinct activities, which are associated with three different β subunits, respectively: chymotrypsin-like (with β 5 subunit), trypsin-like (with β 2 subunit), and peptidyl-glutamyl peptide-hydrolyzing-like (PGPH- or caspase-like; with β 1 subunit) [Groll et al., 1997]. Inhibition of the chymotrypsin-like, but not the trypsin-like, activity of the proteasome has been found to be associated with induction of tumor cell apoptosis [An et al., 1998; Lopes et al., 1997]. By examining a broad range of cell culture models, it has been found that proteasome inhibitors rapidly induce tumor cell apoptosis, selectively activate the cell death program in cancer or oncogene-transformed, but not normal or untransformed cells, and are able to trigger apoptotic death in human cancer cells that are resistant to various anticancer agents [Adams, 2003; Almond and Cohen, 2002; Dou and Li, 1999; Dou et al., 2003; Goldberg, 1995].
We have previously observed cytotoxicity associated with proteasome inhibition by the hydroxylated flavonoids apigenin, luteolin, quercetin, and chrysin in leukemia cells but not in non-transformed cells [Chen et al., 2007; Chen et al., 2005]. Proteasome inhibition leads to accumulation of proteasome target proteins (such as IκBα) and subsequent induction of apoptosis in human cancer cell lines, apparent by activation of caspases and cleavage of poly(ADP-ribose) polymerase (PARP) [Chen et al., 2007; Chen et al., 2005]. In this work, we analyzed fully methylated flavonoids for their ability to impart cytotoxicity to human leukemia HL60 cells and whether cell death could be correlated to proteasomal-inhibitory activity. We found that methylated flavonoids inhibit tumor cell proliferation, but have little to no effect on proteasomal inhibition or apoptosis induction. These findings correlate with the notion that methylated flavonoids could increase bioavailability. However, due to their limited ability to induce apoptosis, the usefulness of methylated flavonoids as cancer therapeutic or preventative agents remains to be further studied.
Materials & Methods
Materials
Bisbenzimide Hoechst No. 33258 stain, dimethylsulfoxide (DMSO), apigenin, luteolin, quercetin, chrysin and other chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA). Methylated flavonoids were purchased from Indofine (Hillsborough, NJ, USA). 3-[4,5-dimethyltiazol-2-yl]-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium salt (MTS) was purchased from Promega (Madison, WI, USA) RPMI 1640, penicillin, and streptomycin were purchased from Invitrogen (Carlsbad, CA). The fluorogenic peptide substrate Suc-LLVY-AMC (for the proteasomal chymotrypsin-like) and N-acetyl- DEVD-AMC (for caspase-3/-7 activity) was from Calbiochem (San Diego, CA, USA). Mouse monoclonal antibody against human PARP was purchased from BIOMOL International LP (Plymouth Meeting, PA). Mouse monoclonal antibody against ubiquitin, rabbit polyclonal antibody against IκBα, goat polyclonal antibody against actin, and secondary antibodies were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA).
Cell culture and cell extract preparation
Human promyelocytic leukemia HL60 cells were grown in RPMI 1640 supplemented with 10% fetal bovine serum, 100 units/mL of penicillin, and 100 μg/mL of streptomycin. Cells were grown at 37 °C in a humidified incubator with an atmosphere of 5% CO2. A whole cell extract was prepared as previously described [An and Dou, 1996].
Cell proliferation/viability assay
The MTS assay was used to determine the effects of flavonoids on proliferation of leukemia HL60 cells by plating 100 μl of 200,000 cells/ml in a 96-well plate. Cells were allowed to recover for 24 h, followed by the addition of each flavonoid at varying concentrations and incubated at 37 °C. At the end of each time point, 20 μl of MTS solution was added to the wells and incubated for 2 to 4 h. The presence of the bioreduced colored formazan product was measured by absorbance at 490 nm using a Victor 3 Multilabel Counter (PerkinElmer, Boston, MA, USA). All samples were assayed in triplicate in three independent experiments, and the mean value for each experiment was calculated. The results are displayed as mean (± standard deviation) and are expressed as a percentage of the control, which was considered to be 100%.
Trypan blue assay and apoptotic morphology changes
The trypan blue dye exclusion assay was used to ascertain cell death in HL60 cells treated with each compound at 1, 25, or 50 μM for 4 or 24 h. Cell morphology was assessed using phase-contrast microscopy as described previously [Kuhn et al., 2005; Smith et al., 2002].
Caspase-3 activity assay
HL60 cells were treated with each compound as indicated, harvested, and lysed as described previously [Nam et al., 2001; Smith et al., 2002]. Whole cell extracts (20 μg) were incubated with Ac-DEVD-AMC (40 μM) fluorogenic substrate at 37 °C in 100 μl of assay buffer (50 mM Tris-HCL, pH 7.5) for 2.5 h. After incubation, production of hydrolyzed 7-amino-4-methylcoumarin (AMC) groups was measured using a Victor 3 Multilabel Counter with an excitation filter of 380 nm and an emission filter of 460 nm as described previously [An et al., 1998]. All samples were assayed in triplicate in three independent experiments, and the mean value for each experiment was calculated.
Cellular and nuclear morphology analysis
A Zeiss (Thornwood, NY) Axiovert 25 microscope was used for all microscopic fluorescence imaging for cellular and nuclear morphology by Hoechst staining, as previously described [Daniel et al., 2005].
Proteasome activity assay
HL60 cells were treated with the indicated compounds, harvested, and lysed. Suc-Leu-Leu-Val-Tyr-AMC (20 μM) was then incubated with the prepared cell lysates for 2.5 h and the chymotrypsin-like activity was measured as described previously [An et al., 1998]. All samples were assayed in triplicate in three independent experiments and the mean value for each experiment was calculated.
Western blot analysis
HL60 cells were treated, harvested and lysed. Cell lysates (50 μg) were separated by SDS-PAGE and transferred to a nitrocellulose membrane, followed by visualization using the enhanced chemiluminescence kit (Amersham Biosciences, Piscataway, NJ), as previously described [Chen et al., 2005].
Cell cycle analysis
Cell cycle analysis was performed using the BD Biosciences Pharmingen, APO-DIRECT™ kit (San Diego, CA). Cells were treated, harvested, fixed and stained according to the protocol. Briefly at each time point cells were collected, washed twice with PBS, and fixed in 70% ethanol for 24 h at -20 °C. The cells were then centrifuged, re-suspended in 1 ml of sample buffer containing propidium iodide (1× PBS, 50 mg propidium iodide, 1 mg/ml glucose, and 100 U/ml ribonuclease A) and incubated at room temperature for 30 min in the dark. Propidium iodide staining, indicative of G0/G1, S, and G2/M phase distribution of the cell population, was visualized with FACScan (Becton Dickinson Immunocytometry, CA) and cell cycle distribution was determined by ModFit LT cell cycle analysis software (Verity Software, Topsham, ME). The cell cycle distribution is presented as the percentage of cells containing G0/G1, S, and G2/M DNA content.
Computational Modeling and nucleophilic susceptibility analysis
Molecules were constructed using the CAChe Workstation (Fujitsu, inc.) as described previously [Smith et al., 2004]. The AutoDock 3.0 suite of programs, was used for the docking calculations and the output from AutoDock was rendered with PyMOL as described previously [Smith et al., 2004].
RESULTS
Methylated flavonoids inhibit proliferation of leukemia HL60 cells
The chemical structures of various flavonoids and their methylated counterparts are depicted in Figure 1A. To determine the biological effects that methylation imparts on these flavonoids, an MTS cell proliferation/viability assay was performed using increasing concentrations of each compound. A 24 h treatment of HL60 cells with the indicated flavonoids revealed that cell proliferation was inhibited by both unmethylated and methylated compounds, most notably at the highest concentrations (Fig. 1B). Apigenin and methylated apigenin (me-apigenin) were the most potent compounds at 50 μM and inhibited cell proliferation by 67 and 75%, respectively (Fig. 1B).
Fig. 1.

Flavonoids inhibit cell proliferation and induce apoptosis. A. Chemical structures of flavonoids and their fully methylated counterparts. B. Leukemia HL60 cells were treated for 24 h with 1, 25, or 50 μM of the indicated flavonoid. An MTS proliferation assay was performed and is described in the MATERIALS AND METHODS. C. Leukemia HL60 cells were treated for 24 h with 50 μM of the indicated flavonoid followed by trypan blue dye exclusion analysis. D. HL60 cells were treated for 24 h with 1, 25, or 50 μM of the indicated flavonoid followed by detection of caspase-3/-7 activity, described in the MATERIALS AND METHODS. Error bars, SD; *, p-value < 0.05; **, p-value < 0.01.
Methylated flavonoids weakly induce apoptotic cell death
Because of the pronounced inhibition of cellular proliferation observed (Fig. 1B), we determined if methylated flavonoids could induce cell death by performing a trypan blue dye exclusion assay. HL60 cells were treated with increasing concentrations (up to 50 μM) of each pair of unmethylated and methylated compounds. After 24 h treatment, the four methylated flavonoids were found to induce only around 10% cell death at the highest concentration (50 μM) compared to the DMSO-treated cells while the four unmethylated counterparts induced cell death from ∼60% (quercetin) to ∼80% (apigenin) at the same concentration (Fig. 1C and data not shown).
To investigate whether the failure of methylated flavonoids to induce cell death is due to their failure to activate the apoptotic program, we performed a caspase-3 activity assay with aliquots of the treated HL60 cells from the same experiment (Fig. 1D). We found that treatment with 50 μM of the methylated flavonoids exhibited very limited caspase-3 activity, inducing only up to 2-fold increase in caspase-3 activity, while the same treatment with the unmethylated flavonoids displayed pronounced caspase-3 activation by up to 13-fold, compared to the control-treated HL60 cells (Fig. 1D).
To further define the limited apoptosis-inducing ability of methylated flavonoids, Western blot analysis, Hoechst staining and flow cytometry were also performed. Western blot analysis revealed that the methylated flavonoids induced little cleaved PARP (p85 fragment, Fig. 2A) even at the highest concentration tested (50 μM) after 24 h. In contrast, increasing PARP cleavage coincided with increasing concentrations of the unmethylated flavonoids after 24 h treatment (Fig. 2A). Likewise, Hoechst staining revealed that brightly stained, punctate, apoptotic nuclei predominated in HL60 cells treated with 50 μM of unmethylated flavonoids, compared to those treated with methylated flavonoids (Fig. 2B).
Fig. 2.

Unmethylated flavonoids induce apoptosis. A. HL60 cells were treated for 24 h with increasing concentrations of unmethylated and methylated flavonoids (apigenin, Api; methylated apigenin MeApi; luteolin, Lut; methylated luteolin, MeLut; quercetin, Quer; methylated quercetin, MeQuer; chrysin, Chry, methylated chrysin, MeChry). Cells were harvested and the cell extracts were utilized for Western blot analysis. Antibody specific for PARP indicated the presence of the full length p116 fragment and the cleaved p85 fragment. B. HL60 cells were treated for 24 h with 50 μM of indicated unmethylated and methylated flavonoids and nuclei that were punctuate or granular and bright were shown (Magnification 100×).
A third measure of apoptosis, by flow cytometry, revealed that treatment with 50 μM of methylated flavonoids for 24 h did not appreciably increase the subG1 population (1.3-3.1%, Table 1), compared to the control (1.3%, Table 1). However, an increase in the fraction of subG1 cells was observed in HL60 cells after 24 h exposure to 50 μM of the unmethylated flavonoids (18.8-39.3%, Table 1) compared to the DMSO control. Similar results were obtained in all other treatments with unmethylated vs. methylated flavonoids (Table 1). These results demonstrate that while methylated flavonoids apparently inhibit cellular proliferation, they are poor inducers of apoptosis in human leukemia HL60 cells especially when compared to unmethylated flavonoids.
Table 1.
Flow cytometry analysis for the SubG1 cell population
| SubG1 Population (% total cells) | |
|---|---|
| DMSO | 1.3 |
| Apigenin | 36.0 |
| Me-Apigenin | 3.1 |
| Luteolin | 32.6 |
| Me-Luteolin | 1.3 |
| Quercetin | 39.3 |
| Me-Quercetin | 1.7 |
| Chrysin | 18.8 |
| Me-Chrysin | 2.8 |
Methylated flavonoids do not inhibit cellular proteasome activity in intact leukemia HL60 cells
Inhibition of the proteasomal chymotrypsin-like, but not trypsin-like activity is associated with apoptosis induction in cancer cells [An et al., 1998; Lopes et al., 1997]. We have previously shown that the flavonoids apigenin, luteolin, quercetin, and chrysin are able to inhibit proteasomal chymotrypsin-like activity and induce apoptosis in leukemia cells [Chen et al., 2007; Chen et al., 2005]. Therefore, we hypothesized that the failure of methylated flavonoids to induce apoptosis is due to their inability to inhibit the cellular proteasomal chymotrypsin-like activity. To test this hypothesis, HL60 cells were treated with increasing concentrations of each methylated flavonoid, using their unmethylated counterparts as controls. After each treatment, proteins were extracted and used for measuring proteasomal activity. In cells treated with the methylated flavonoids, proteasomal chymotrypsin-like activity was only inhibited 10% or less after 24 h treatment, even when a 50 μM concentration was used (Fig. 3A). In contrast, the unmethylated flavonoid treatments at 50 μM inhibited the proteasomal chymotrypsin-like activity by as much as 73% after 24 h treatment (such as Apigenin; Fig. 3A). Inhibition of proteasomal activity should cause accumulation of ubiquitinated proteins and natural proteasome targets, such as IκBα [An et al., 1998; Chen et al., 1996; Li and Dou, 2000]. We have previously reported that in association with proteasomal inhibition, an ubiquitinated form of IκBα protein with a molecular weight of about 56 kDa, is accumulated [Chen et al., 2005]. While cells treated for 24 h with 50 μM of the unmethylated flavonoids revealed an ubiquitinated form of IκBα (indicated by an arrow), the methylated flavonoids did not appear to have such an effect (Fig. 3B).
Fig. 3.

Dose-dependent proteasome inhibition by flavonoids. A. HL60 cells were treated with DMSO or 1, 25, or 50 μM of the indicated flavonoids for 24 h. Chymotrypsin-like activity is represented as a percentage of the solvent (DMSO) control. B. Western blot analysis was performed using reserved aliquots from the chymotrypsin-like activity assay. A ubiquitinated form of IκBα was observed at approximately 56 kD and is indicated by an arrow. Actin was used as a loading control. Error bars, SD; *, p-value < 0.05; **, p-value < 0.01.
Methylated flavonoids affect cellular proliferation but not apoptosis in a time-dependent manner
To confirm the proliferation-inhibitory activity of methylated flavonoids, 50 μM of the methylated and unmethylated flavonoids was used to examine the effects of each compound on HL60 proliferation after multiple time points. After exposure to each compound for 16 and 24 the MTS assay revealed that both the methylated and unmethylated flavonoids were able to inhibit cellular proliferation (Fig. 4A). However, the unmethylated compounds generally displayed greater potency. Therefore, we performed a more detailed kinetic analysis to determine whether methylated flavonoids might elicit apoptosis-inducing activity at even earlier time points. Various assays were utilized to compare the effects of HL60 cells after treatment for 2, 4, 6, 8, 16, and 24 h with 50 μM of each compound. After each treatment, cells were harvested and proteins extracted to measure their potential for caspase-3/-7 activation and for PARP cleavage. Me-apigenin exhibited very weak caspase-3/-7 activation at all the time points tested while apigenin appeared to peak in caspase-3/-7 activity at 8 h (Fig. 4B). The processes of apoptosis were confirmed by the presence of PARP cleavage that dominated in the apigenin-treated cells compared to treatment with me-apigenin (Fig. 4C).
Fig. 4.
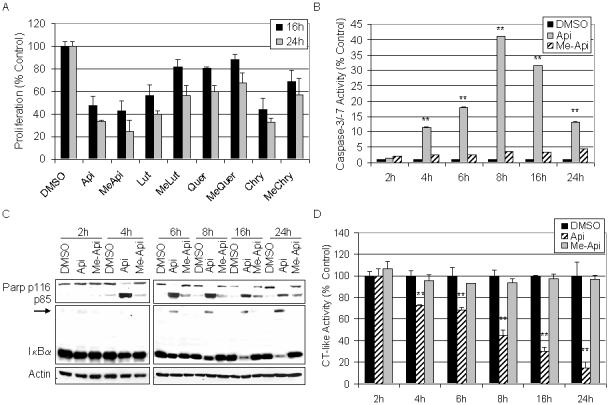
Kinetic evaluation of flavonoids on cell proliferation, apoptosis, and proteasome inhibition. A. HL60 cells were plated in 96-well plates and treated with 50 μM of the indicated flavonoid for 16 and 24 h before an MTS assay was performed. B-D. HL60 cells were treated with 50 μM of apigenin, me-apigenin, or the DMSO control for the indicated time point. Cells were harvested and proteins extracted, followed by assaying caspase-3/-7 activity (B), Western blotting (C) and the proteasomal chymotrypsin-like activity (D). Error bars, SD; *, p-value < 0.05; **, p-value < 0.01.
The chymotrypsin-like activity in the HL60 cells was also examined in this kinetic experiment to determine if the small amount of caspase-3/-7 (Fig. 4B) and PARP cleavage (Fig. 4C) observed in the me-apigenin-treated cells was a consequence of proteasome inhibition. Protein aliquots from the same treatment were tested for the proteasomal chymotrypsin-like activity. While me-apigenin did not effectively inhibit proteasome activity, a time-dependent decrease in proteasome activity, as much as 82% inhibition, was evident in the apigenin-treated cells (Fig. 4D). Proteasome inhibition was confirmed by Western blot analysis that showed accumulation of ubiquitinated IκBα in the apigenin-treated cells only (Fig. 4C).
Methylated flavonoids do not inhibit the chymotrypsin-like activity of a purified 20S proteasome
The unmethylated flavonoids, apigenin, luteolin, quercetin, and chrysin, have been shown to inhibit chymotrypsin-like activity against a purified 20S proteasome with IC50 values less than 5 μM [Chen et al., 2007; Chen et al., 2005; Table 2]. We then examined the proteasome-inhibitory potency of methylated flavonoids against a purified 20S proteasome using the unmethylated flavonoids as controls. The IC50 of methylated flavonoids against chymotrypsin-like activity was greater than 50 μM (Table 2). This finding, in combination with the lack of proteasome inhibition in intact HL60 cells (Figs. 3-4), indicates that methylated flavonoids do not possess proteasome-inhibitory activity.
Table 2.
Inhibition of 20S proteasome activity by unmethylated and methylated flavonoids
| Flavonoid | IC50 (μM)‡ |
|---|---|
| Apigenin | 1.8 (± 0.03)§ |
| MeApi | >50 |
| Luteolin | 1.5 (± 0.01)€ |
| MeLut | >50 |
| Quercetin | 3.5 (± 0.05)§ |
| MeQuer | >50 |
| Chrysin | 4.9 (± 0.15)€ |
| MeChry | >50 |
Results were obtained from 3 independent experiments performed in triplicate.
Methylation on flavonoids results in an unstable proteasome-inhibitory pose
The carbonyl carbon in the C ring of unmethylated flavonoids (Fig. 1A) has been shown to confer their proteasome-inhibitory potencies [Chen et al., 2005]. Since methylation significantly worsened the IC50 of the flavonoids (Table 2), we examined whether methylation has any effect on the potential for these compounds to undergo nucleophilic attack by Thr 1 within the β5 subunit of the proteasome. We found that methylation had no apparent effect on the nucleophilic susceptibility of the carbonyl carbon of apigenin and me-apigenin (Fig. 5A). Likewise, all other flavonoids and their methylated counterparts showed similar nucleophilic susceptibility (data not shown). This suggests that the loss of proteasome-inhibitory activity in methylated flavonoids may not be due to changes in their nucleophilic susceptibility but in the manner with which the compounds are oriented within the proteasomal β5 subunit.
Fig. 5.

The nucleophilic susceptibility and docking poses of unmethylated and methylated flavonoids. A. Molecular orbital energy analysis is demonstrated by drawing an electron density isosurface colored by nucleophilic susceptibility. The red center signifies the highest area of susceptibility. B. Apigenin and methylated apigenin are represented by the stick structure (see also Fig. 1 for description) and the colors are representative of atom type (carbon, gray; oxygen, red, methyl, light blue). The dotted line in yellow represents the distance in angstroms to Thr 1 of β5 subunit and is indicative of potential nucleophilic attack. The dotted lines in light blue represent potential hydrogen bond formations with surrounding amino acids in the S1 pocket. The pose shown represents the most frequently adopted conformation of the molecule.
To test this idea, apigenin and me-apigenin were docked to the proteasomal β5 subunit and cluster analysis was performed. The predictability of binding was determined based on the number of times the molecule adopts an inhibitory pose out of 100 docks. Similar to what we reported previously, apigenin docked with 82 inhibitory poses (82%) placing the carbonyl carbon in a suitable position to undergo nucleophilic attack (Fig 5B, left). However, me-apigenin docks with only 63 poses (63%) placing the carbonyl carbon in such a suitable position to undergo nucleophilic attack (Fig 5B, right). Therefore, the apigenin docking demonstrated higher predictability than me-apigenin. All other methylated and unmethylated flavonoids were examined using the same analysis. Similar results were obtained in that the unmethylated flavonoids were able to adopt an inhibitory pose with higher predictability than the methylated flavonoids (data not shown).
The docking pose most adopted by apigenin places the carbonyl carbon at an appropriate distance for nucleophilic attack, 2.96 Å (Fig. 5B, left) [Daniel et al., 2006]. Likewise, me-apigenin adopted a pose that places the carbonyl carbon within the optimal distance for nucleophilic attack, 3.20 Å from Thr 1 (Fig. 5B, right). Apigenin possesses the ability to form hydrogen bonds (due to exposed hydroxyl groups) with surrounding amino acids e.g., tyrosine 170 and Serine 96, within the β5-subunit, strengthening its potential to bind tightly (Fig. 5B, left). On the contrary, the probability that me-apigenin binds tightly to Thr1 is diminished based on its inability to form the stabilizing hydrogen bonds with nearby amino acids (Fig. 5B, right). For this reason, me-apigenin most likely lacks potent proteasome-inhibitory potential.
An analysis of the remaining unmethylated and methylated polyphenols revealed that all unmethylated flavonoids (luteolin, quercetin, and chrysin) were able to adopt a pose that placed the carbonyl carbon within the optimal distance for nucleophilic attack by Thr1 (data not shown). The unmethylated flavonoids were able to form hydrogen bonds with various surrounding amino acids within the β5-subunit, strengthening their potential for binding and inhibition. In contrast, the methylated flavonoids me-luteolin and me-chrysin found a majority pose in which the carbonyl carbon was within the optimal distance for nucleophilic attack by Thr1, but me-quercetin did not (data not shown). Compounded with the notion that hydrogen bonding with the surrounding amino acids of the β5 subunit can not take place in all the methylated flavonoids, me-quercetin appears to have lost a predictable binding pose for proteasome inhibition (data not shown).
Methylated flavonoids induce G0/G1 cell cycle arrest in HL60 cells
We have shown that although methylated flavonoids inhibited proliferation of leukemia HL60 cells (Figs. 1 and 4), the molecular basis was not due to the induction of apoptosis, as shown by lack of caspase-3/-7 activation, PARP cleavage, Hoechst positivity, and subG1 population (Figs. 1-4 and Table 1). To determine the cause for inhibited cellular proliferation by methylated flavonoids, HL60 cells were treated for 24 and 48 h with 50 μM of apigenin and me-apigenin and examined by flow cytometry for cell cycle analysis (Fig. 6). We found that, compared to the control-treated cells, me-apigenin increased the G0/G1 population of cells by 19% after 24 h and by 46% after 48 h (Fig. 6). This provides an explanation, at least in part, for the reduction in cell proliferation/viability observed in Figures 1 and 4. In contrast, apigenin’s mechanism of action appears to be through apoptosis induction, confirmed by the significantly increased subG1 population, especially at 48 h (50.0%; Fig. 6B).
Fig. 6.

Effect of methylated and unmethylated flavonoids on the cell cycle progression of HL60 cells. HL60 cells were treated with 50 μM of apigenin, me-apigenin or DMSO control for the indicated time. Cells were collected and labeled with PI and analyzed by DNA flow cytometry. Experiments were performed in duplicate and gave similar results.
DISCUSSION
The pervasiveness of drug resistance extends to virtually every known disseminated cancer. Therefore, pursuing molecular targets and novel, nontoxic cancer therapeutics has become increasingly important. Numerous studies indicate that cancer and transformed cells are significantly more sensitive to proteasome inhibition than normal, non-transformed cells [Adams, 2003; Almond and Cohen, 2002; Delic et al., 1998; Dou and Li, 1999; Kuhn et al., 2005; Landis-Piwowar et al., 2005], providing relevance for proteasome inhibitors as potential novel anti-cancer drugs.
Our previous studies have suggested that polyphenolic compounds, including flavonoids, from natural plant sources are innocuous toward normal cells, while they could inhibit the proteasome in cancer cells [Chen et al., 2007; Chen et al., 2005]. However, flavonoids are unstable under physiological conditions and could be rapidly degraded or metabolized, through interactions with the hydroxyl groups that surround the molecules [Manach and Donovan, 2004]. Although the methylated flavonoids examined in this work are synthetic compounds, their presence has been shown in nature, e.g., me-apigenin present in citrus [Mizuno et al., 1991]. Therefore, understanding the effects of methylated flavonoids on cell proliferation/viability could potentially be useful in chemopreventive strategies.
Methylation has been suggested as a measure to enhance the entry of flavonoids into cells, thereby preventing their degradation [Wen and Walle, 2006b]. Additionally, because of their increased stability, these compounds have been proposed to be more potent chemopreventive agents [Walle et al., 2007]. The data presented in this work indicate that methylated flavonoids possess a different mechanism of action compared to unmethylated flavonoids. While unmethylated flavonoids induce apoptosis through proteasome inhibition, methylated flavonoids do not induce apoptosis or proteasome inhibition and act mostly through inducing G0/G1 cell cycle arrest. Therefore, methylated flavonoids could have some efficacy as chemopreventive agents, but as anti-cancer therapeutics the efficacy of these compounds is questionable.
The properties of methylated and unmethylated tea polyphenols in leukemia Jurkat T cells were examined previously [Landis-Piwowar et al., 2007]. In the current study, we hypothesized that methylated flavonoids would display reduced apoptosis inducing abilities as a consequence of reduced proteasome-inhibitory activity. To test this notion, HL60 cells were treated in dose-dependent and kinetic assays with methylated and unmethylated flavonoids. The anti-proliferative effects were determined using an MTS assay and indicated that both unmethylated and methylated flavonoids possess the capacity to reduce proliferation of HL60 cells in a dose-dependent manner (Fig. 1C). When the 50 μM treatment was examined after multiple time points extended to 48 h, the methylated flavonoid, me-apigenin, was found to produce results most closely correlated with its unmethylated counterpart, apigenin (Fig. 4A). In contrast, other methylated flavonoids, me-luteolin, me-quercetin, and me-chrysin, appeared to reverse their inhibitory activities by 48 h (data not shown). This reversal in inhibitory activity could potentially arise from intracellular detoxification of the methylated compounds by sulfonation/glucuronidation [Lu et al., 2003], thus decreasing their potential to impart long-lasting effects.
Since the methylated flavonoids appeared adept at inhibiting cell proliferation, the mechanism to explain the reduced cell numbers was examined. Apoptotic events predominated after treatment with unmethylated, but not methylated compounds in both dose- and time-dependent analysis and therefore, inhibited cellular proliferation was found not to be a consequence of apoptosis by the methylated flavonoids (Figs. 2 and 4D).
To ascertain whether the limited apoptosis induction by methylated flavonoids was a function of reduced proteasome inhibitory activity, a number of proteasome activity assays were examined. HL60 cells treated in dose-dependent and kinetic experiments were found to display substantial proteasome inhibition after treatment with only unmethylated flavonoids (Figs. 3 and 4). In addition, ubiquitination of the proteasome target protein IκBα was not observed after treatment with the methylated flavonoids (Figs. 3 and 4). Furthermore, incubation with a purified 20S proteasome revealed that methylated flavonoids were inactive as inhibitors of chymotrypsin-like activity (Table 2).
In silico analysis of methylated and unmethylated flavonoids indicated that both contained a nucleophilic site of attack, the carbonyl carbon (Fig. 5 and data not shown), and that methylated flavonoids were able to adopt a pose that positioned the carbonyl carbon in appropriate proximity to Thr1 in the β5 subunit of the proteasome (Fig. 5 and data not shown). However, stabilization within the β5 subunit is theoretically reduced by the deficiency in hydrogen bonding of methylated flavonoids with surrounding amino acid residues. These results point to the potential failings of methylated flavonoids and the rationale for unmethylated flavonoids as proteasome inhibitors.
A strong correlation between the times of proteasome inhibition and apoptosis induction was observed after treatment with the unmethylated flavonoids. By 4 h, proteasome activity was inhibited by approximately 30% (Fig. 4C) and caspase-3 activity (Fig. 4B) had risen to 10-fold above the DMSO control before peaking at 8 h. Ubiquitinated IκBα was not observed until 6 h (Fig. 4D), but the full length fragment was decreased by 4 h and PARP cleavage was clear (Fig. 4D), indicating that apoptosis was strongly stimulated.
Methylated flavonoids also induced PARP cleavage albeit to a lesser extent than their unmethylated counterparts (Fig. 4C). Ban et al. reported that the compound 2,3-dihydro-3,5-dihydroxy-6-methyl-4H-pyranone (DMMP) is an important constituent of onions that confers anti-cancer activities through NF-kB inhibition [Ban et al., 2007]. Likewise, the apoptotic events observed in Figure 4C by the methylated flavonoids may be a result of NF-kB inhibition independent of proteasome inhibition.
In an attempt to determine the rationale for the decrease in cell proliferation, flow cytometry for cell cycle analysis was performed. Me-apigenin exhibited the strongest measure of arrest in the G0/G1 population of cells compared to the control cells with an increase of 19 and 46% after 24 h and 48 h treatment, respectively (Fig. 6). These findings are similar to those obtained by Walle et al., in which methylated flavonoids induced an increase in the G0/G1 population of oral squamous cell carcinoma SCC-9 cells [Walle et al., 2007]. Similarly, Jiang et al., examined the effects of DU145 cells treated with a methylated selenium compound and also observed G1 arrest that was associated with increase p27kip1 and p21cip1 expression [Jiang et al., 2002]. However, continued investigation into the mechanisms by which methylated flavonoids induce cell cycle arrest is warranted.
The findings reported here indicate that methylated flavonoids lack proteasome-inhibitory and apoptosis-inducing abilities while apparently inhibiting the growth, or at least viability, of human leukemia cells. Although various reports have indicated that methylated flavonoids exhibit increased bioavailability [Wen and Walle, 2006a; Wen and Walle, 2006b], the mechanisms for potential cancer prevention or therapeutics do not appear to be a function of proteasome inhibition and apoptosis induction. On the other hand, unmethylated flavonoids are apparently profoundly cytotoxic to HL60 cells and provide additional evidence for their use as chemopreventive and possibly therapeutic agents. Although beyond the scope of the current study, a future investigation should elucidate the molecular mechanism by which methylated flavonoids inhibit tumor cell proliferation in order to evaluate the potential of the methylated compounds as cancer therapeutic or preventative agents.
Acknowledgments
Contract grant sponsor: National Cancer Institute
Contract grant numbers: 1R01CA120009; 5R03CA112625
References
- Adams J. Potential for proteasome inhibition in the treatment of cancer. Drug Discov Today. 2003;8:307–15. doi: 10.1016/s1359-6446(03)02647-3. [DOI] [PubMed] [Google Scholar]
- Almond JB, Cohen GM. The proteasome: a novel target for cancer chemotherapy. Leukemia. 2002;16:433–43. doi: 10.1038/sj.leu.2402417. [DOI] [PubMed] [Google Scholar]
- An B, Dou QP. Cleavage of retinoblastoma protein during apoptosis: an interleukin 1 beta-converting enzyme-like protease as candidate. Cancer Res. 1996;56:438–42. [PubMed] [Google Scholar]
- An B, Goldfarb RH, Siman R, Dou QP. Novel dipeptidyl proteasome inhibitors overcome Bcl-2 protective function and selectively accumulate the cyclin-dependent kinase inhibitor p27 and induce apoptosis in transformed, but not normal, human fibroblasts. Cell Death Differ. 1998;5:1062–75. doi: 10.1038/sj.cdd.4400436. [DOI] [PubMed] [Google Scholar]
- Ban JO, Hwang IG, Kim TM, Hwang BY, Lee US, Jeong HS, Yoon YW, Kimz DJ, Hong JT. Anti-proliferate and pro-apoptotic effects of 2,3-dihydro-3,5-dihydroxy-6-methyl-4H-pyranone through inactivation of NF-kappaB in human colon cancer cells. Arch Pharm Res. 2007;30(11):1455–63. doi: 10.1007/BF02977371. [DOI] [PubMed] [Google Scholar]
- Chen D, Chen MS, Cui QC, Yang H, Dou QP. Structure-proteasome-inhibitory activity relationships of dietary flavonoids in human cancer cells. Front Biosci. 2007;12:1935–45. doi: 10.2741/2199. [DOI] [PubMed] [Google Scholar]
- Chen D, Daniel KG, Chen MS, Kuhn DJ, Landis-Piwowar KR, Dou QP. Dietary flavonoids as proteasome inhibitors and apoptosis inducers in human leukemia cells. Biochem Pharmacol. 2005;69:1421–32. doi: 10.1016/j.bcp.2005.02.022. [DOI] [PubMed] [Google Scholar]
- Chen ZJ, Parent L, Maniatis T. Site-specific phosphorylation of IkappaBalpha by a novel ubiquitination-dependent protein kinase activity. Cell. 1996;84:853–62. doi: 10.1016/s0092-8674(00)81064-8. [DOI] [PubMed] [Google Scholar]
- Choi JS, Choi YJ, Park SH, Kang JS, Kang YH. Flavones mitigate tumor necrosis factor-alpha-induced adhesion molecule upregulation in cultured human endothelial cells: role of nuclear factor-kappa B. J Nutr. 2004;134:1013–9. doi: 10.1093/jn/134.5.1013. [DOI] [PubMed] [Google Scholar]
- Daniel KG, Chen D, Orlu S, Cui QC, Miller FR, Dou QP. Clioquinol and pyrrolidine dithiocarbamate complex with copper to form proteasome inhibitors and apoptosis inducers in human breast cancer cells. Breast Cancer Res. 2005;7:R897–908. doi: 10.1186/bcr1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel KG, Landis-Piwowar KR, Chen D, Wan SB, Chan TH, Dou QP. Methylation of green tea polyphenols affects their binding to and inhibitory poses of the proteasome beta5 subunit. Int J Mol Med. 2006;18:625–32. [PubMed] [Google Scholar]
- Delic J, Masdehors P, Omura S, Cosset JM, Dumont J, Binet JL, Magdelenat H. The proteasome inhibitor lactacystin induces apoptosis and sensitizes chemo- and radioresistant human chronic lymphocytic leukaemia lymphocytes to TNF-alpha-initiated apoptosis. Br J Cancer. 1998;77:1103–7. doi: 10.1038/bjc.1998.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou QP, Li B. Proteasome inhibitors as potential novel anticancer agents. Drug Resist Updat. 1999;2:215–223. doi: 10.1054/drup.1999.0095. [DOI] [PubMed] [Google Scholar]
- Dou QP, Smith DM, Daniel KG, Kazi A. Interruption of tumor cell cycle progression through proteasome inhibition: implications for cancer therapy. Prog Cell Cycle Res. 2003;5:441–6. [PubMed] [Google Scholar]
- Goldberg AL. Functions of the proteasome: the lysis at the end of the tunnel. Science. 1995;268:522–3. doi: 10.1126/science.7725095. [DOI] [PubMed] [Google Scholar]
- Groll M, Ditzel L, Lowe J, Stock D, Bochtler M, Bartunik HD, Huber R. Structure of 20S proteasome from yeast at 2.4 A resolution. Nature. 1997;386:463–71. doi: 10.1038/386463a0. [DOI] [PubMed] [Google Scholar]
- Jiang C, Wang Z, Ganther H, Lu J. Distinct effects of methylseleninic acid versus selenite on apoptosis, cell cycle, and protein kinase pathways in DU145 human prostate cancer cells. Mol Cancer Ther. 2002;1(12):1059–66. [PubMed] [Google Scholar]
- Kuhn D, Lam WH, Kazi A, Daniel KG, Song S, Chow LM, Chan TH, Dou QP. Synthetic peracetate tea polyphenols as potent proteasome inhibitors and apoptosis inducers in human cancer cells. Front Biosci. 2005;10:1010–23. doi: 10.2741/1595. [DOI] [PubMed] [Google Scholar]
- Landis-Piwowar KR, Kuhn DJ, Wan SB, Chen D, Chan TH, Dou QP. Evaluation of proteasome-inhibitory and apoptosis-inducing potencies of novel (-)-EGCG analogs and their prodrugs. Int J Mol Med. 2005;15:735–42. [PubMed] [Google Scholar]
- Landis-Piwowar KR, Wan SB, Wiegand RA, Kuhn DJ, Chan TH, Dou QP. Methylation suppresses the proteasome-inhibitory function of green tea polyphenols. J Cell Physiol. 2007;213:252–60. doi: 10.1002/jcp.21124. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Wang CJ, Kuo HC, Chou FP, Jean LF, Tseng TH. Induction apoptosis of luteolin in human hepatoma HepG2 cells involving mitochondria translocation of Bax/Bak and activation of JNK. Toxicol Appl Pharmacol. 2005;203:124–31. doi: 10.1016/j.taap.2004.08.004. [DOI] [PubMed] [Google Scholar]
- Lee LT, Huang YT, Hwang JJ, Lee AY, Ke FC, Huang CJ, Kandaswami C, Lee PP, Lee MT. Transinactivation of the epidermal growth factor receptor tyrosine kinase and focal adhesion kinase phosphorylation by dietary flavonoids: effect on invasive potential of human carcinoma cells. Biochem Pharmacol. 2004;67:2103–14. doi: 10.1016/j.bcp.2004.02.023. [DOI] [PubMed] [Google Scholar]
- Li B, Dou QP. Bax degradation by the ubiquitin/proteasome-dependent pathway: involvement in tumor survival and progression. Proc Natl Acad Sci U S A. 2000;97:3850–5. doi: 10.1073/pnas.070047997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes UG, Erhardt P, Yao R, Cooper GM. p53-dependent induction of apoptosis by proteasome inhibitors. J Biol Chem. 1997;272:12893–6. doi: 10.1074/jbc.272.20.12893. [DOI] [PubMed] [Google Scholar]
- Lu H, Meng X, Yang CS. Enzymology of methylation of tea catechins and inhibition of catechol-O-methyltransferase by (-)-epigallocatechin gallate. Drug Metab Dispos. 2003;31:572–9. doi: 10.1124/dmd.31.5.572. [DOI] [PubMed] [Google Scholar]
- Manach C, Donovan JL. Pharmacokinetics and metabolism of dietary flavonoids in humans. Free Radic Res. 2004;38:771–85. doi: 10.1080/10715760410001727858. [DOI] [PubMed] [Google Scholar]
- Mizuno M, Iinuma M, Ohara M, Tanaka T, Iwamasa M. Chemotaxonomy of the genus Citrus based on polymethoxyflavones. Chemical and Pharmaceutical Bulletin. 1991;39:945–949. [Google Scholar]
- Nam S, Smith DM, Dou QP. Ester bond-containing tea polyphenols potently inhibit proteasome activity in vitro and in vivo. J Biol Chem. 2001;276:13322–30. doi: 10.1074/jbc.M004209200. [DOI] [PubMed] [Google Scholar]
- Ramos S. Effects of dietary flavonoids on apoptotic pathways related to cancer chemoprevention. J Nutr Biochem. 2007;18:427–42. doi: 10.1016/j.jnutbio.2006.11.004. [DOI] [PubMed] [Google Scholar]
- Sim GS, Lee BC, Cho HS, Lee JW, Kim JH, Lee DH, Pyo HB, Moon DC, Oh KW, Yun YP, Hong JT. Structure activity relationship of antioxidative property of flavonoids and inhibitory effect on matrix metalloproteinase activity in UVA-irradiated human dermal fibroblast. Arch Pharm Res. 2007;30:290–8. doi: 10.1007/BF02977608. [DOI] [PubMed] [Google Scholar]
- Smith DM, Daniel KG, Wang Z, Guida WC, Chan TH, Dou QP. Docking studies and model development of tea polyphenol proteasome inhibitors: applications to rational drug design. Proteins. 2004;54:58–70. doi: 10.1002/prot.10504. [DOI] [PubMed] [Google Scholar]
- Smith DM, Wang Z, Kazi A, Li LH, Chan TH, Dou QP. Synthetic analogs of green tea polyphenols as proteasome inhibitors. Mol Med. 2002;8:382–92. [PMC free article] [PubMed] [Google Scholar]
- Walle T, Ta N, Kawamori T, Wen X, Tsuji PA, Walle UK. Cancer chemopreventive properties of orally bioavailable flavonoids--methylated versus unmethylated flavones. Biochem Pharmacol. 2007;73:1288–96. doi: 10.1016/j.bcp.2006.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen X, Walle T. Methylated flavonoids have greatly improved intestinal absorption and metabolic stability. Drug Metab Dispos. 2006a;34:1786–92. doi: 10.1124/dmd.106.011122. [DOI] [PubMed] [Google Scholar]
- Wen X, Walle T. Methylation protects dietary flavonoids from rapid hepatic metabolism. Xenobiotica. 2006b;36:387–97. doi: 10.1080/00498250600630636. [DOI] [PubMed] [Google Scholar]