

Abstract
This study probes the impact of electronic asymmetry of diiron(I) dithiolato carbonyls. Treatment of Fe2(S2CnH2n)(CO)6-x(PMe3)x compounds (n = 2, 3) with NOBF4 gave the derivatives [Fe2(S2CnH2n)(CO)5-x(PMe3)x(NO)]BF4 (x = 1, 2, 3) which are electronically unsymmetrical due to the presence of a single NO+ ligand. Whereas the mono phosphine derivative is largely undistorted, the bis PMe3 derivatives are distorted such that the CO ligand on the Fe(CO)(PMe3)(NO)+ subunit is semibridging. Two isomers of [Fe2(S2C3H6)(CO)3(PMe3)2(NO)]BF4 were characterized spectroscopically and crystallographically. Each isomer features electron-rich [Fe(CO)2PMe3] and electrophilic [Fe(CO)(PMe3)(NO)]+ subunits. These species are in equilibrium with an unobserved isomer that reversibly binds CO (ΔH = −35 kJ/mol, ΔS = −139 J/mol•K) to give the symmetrical adduct [Fe2(S2C3H6)(μ-NO)(CO)4(PMe3)2]BF4. In contrast to Fe2(S2C3H6)(CO)4(PMe3)2, the bis(PMe3) nitrosyls readily undergo CO-substitution to give the (PMe3)3 derivatives. The nitrosyl complexes reduce at potentials that are ~1 V milder than their carbonyl counterparts. DFT calculations, specifically NBO values, reinforce the electronic resemblance of the nitrosyl complexes with the corresponding mixed-valence diiron complexes. Unlike other diiron dithiolato carbonyls, these species undergo reversible reductions at mild conditions. The results show that the novel structural and chemical features associated with mixed valence diiron dithiolates – the so-called Hox models - can be replicated in the absence of mixed-valency by introducing electronic asymmetry.
Introduction
Modeling of the hydrogenase enzymes complements biophysical studies, providing insights into the mechanism and spectroscopy relevant to this important family of biocatalysts.1,2 The [FeFe]-hydrogenases are particularly attractive for modeling studies because the Fe2(SR)2(CO)6-xLx core resembles well known organoiron complexes that are easily assembled with a variety of ligands and thiolates.3,4 Variation of the coligands L and the thiolate SR offers the prospect for tuning the redox potentials and basicity of this diiron center. These variations in turn can affect the catalytic roperties of the diiron center, which undergoes three characteristic reactions – the reversible binding of CO, 1-electron redox, and the catalytic interconversion of protons and dihydrogen. Although hundreds of derivatives of Fe2(SR)2(CO)6-xLx have been studied as models for the [FeFe]-hydrogenase, no (FeI)2 compound to date exhibits the single most distinctive structural feature of the enzyme - the “rotated” nature of the distal iron center (Scheme 1). Previously we reported a method to induce rotation requiring strong Lewis acids, a highly basic diiron center, but resulting species were thermally unstable.5
Scheme 1.

Structure of the Diiron Active Site of the [FeFe]-Hydrogenases ( X = CH2, NH, or O).
The rotated structure is observed in the crystallography of the two active states, as revealed by the crystallographic studies of enzymes obtained from C. pasteurianum and D. desulfuricans.6 The rotated geometry contrasts with the idealized C2v structure and has recently been replicated in models for the Hox state, which feature a mixedvalence diiron core.7,8 The geometry of the diiron site in the Hred state is unclear – it is either rotated diiron(I) or diiron(II) core with a terminal hydride ligand.9 From the mechanistic perspective, and relevant to understanding the hydrogenases as well as exploiting the diiron dithiolate platform for other applications, the rotated structure is of special significance because it provides a well-defined site for the binding of substrates.1
Many different types of ligands have been installed at one or more of the six terminal sites on the diiron dithiolate framework, including, in approximate order of their abundance, phosphines,10 phosphites,11 thioethers and bridging thiolates,12,13 isocyanides,14 carbenes,15 arsines,11 and alkenes.16 Prior to this work, strong acceptor ligands had not been installed on the diiron(I) dithiolato platform, although electrophiles were found to add to the Fe-Fe bond.17 In this work, we have examined the ability of NO+ to alter the electronic structure of the diiron dithiolate carbonyls. As one of strongest acceptor ligands in inorganic chemistry,18 NO+ often forms complexes that are isoelectronic and isostructural with CO compounds.We show in this work, however, NO+ forms diiron dithiolates that are not isostructural with the corresponding carbonyl complexes, and these nitrosyl complexes display enhanced Lewis acidity. Although iron nitrosyl thiolates have attracted intense interest,19 the derivatives described below are novel. Closest to the present complexes are the Roussin esters Fe2(SR)2(NO)4,20 but substituted derivatives of these spin-paired 34e- diiron complexes have not been well developed.21
Results
Synthesis of [Fe2(S2CnH2n)(CO)5-x(PMe3)x(NO)]BF4
Employing NOBF4 as an NO+ source, PMe3-substituted diiron dithiolato carbonyls were found to readily undergo nitrosylation to afford monosubstituted products in good yields (Scheme 1). Thus, CH2Cl2 solutions of the complexes Fe2(S2CnH2n)(CO)6-x(PMe3)x were found to react efficiently with suspensions of NOBF4 over the course of hours at 0 °C for x = 1 and 2. In this way, we prepared [Fe2(S2CnH2n)(CO)4(PMe3)(NO)]BF4 for n = 2 (1) and 3 (1’) and [Fe2(S2CnH2n)(CO)3(PMe3)2(NO)]BF4 for n = 2 (2) and 3 (2’) (eq 1). The IR spectra of 1 and 2 displayed νCO bands ~50 cm−1 higher in energy relative to their precursors. Each displayed a strong νNO band in the range ~1825 - 1760 cm−1.
| (1) |
Solutions of the mono and bisphosphines were stable for hours, eventually converting to mixtures containing the trisphosphine derivatives [Fe2(S2CnH2n)(CO)2(PMe3)3(NO)]BF4 (see below) as well as other unidentified species.
Crystallographic Results
The structures 1, 2, 2’, and 3’ were verified crystallographically. As expected, the FeI 2(SR)2 core is complemented with six terminal ligands.22 The iron-ligand distances in 1, 2, 2’, and 3’ are unremarkable, and carbonyl and nitrosyl ligands were distinguished by the Fe-NO distances, which were observed to be ~0.15 Å shorter than the Fe-CO distances (Figure 1–Figure 3).
Figure 1.

Structure of 1 with thermal ellipsoids set at 35%. Disordered anions and hydrogen atoms have been omitted for clarity. Key bond distances (Å): Fe1-Fe2, 2.5529(9); Fe1-N1, 1.669(4), 1.67; Fe1-P1, 2.2971(13); Fe1-C1, 1.821(5); Fe2-C5, 1.804(4); Fe2-C6, 1.804(5); Fe2-C7: 1.794(5).
Figure 3.

Structure of 2’ba with thermal ellipsoids set at 35%. Of the two molecules in the asymmetric unit, the one with the smaller Ψ value (62.1°) is displayed. Side—on (left) and end—on (right) views. Hydrogen atoms, disordered methyl groups on P4, disorder in C13 and O6, anion, and solvate have been omitted for clarity. Key bond distances (Å): Fe3-Fe4: 2.5433(12), Fe3-N2: 1.616(6), Fe3-P3: 2.253(2), Fe3-C13: 1.778(9), Fe4-C13: 2.323(9), Fe4-C17: 1.747(7), Fe4-C18: 1.786(7), Fe4-P4: 2.273(4).
The striking aspect of the structures is the distortion of the Fe2L6 framework from the usual4 eclipsed quasi-C2v geometry (Scheme 2). Especially for 2 and 2’, the Fe(CO)(PMe3)(NO)+ subunits are rotated so as to orient a CO ligand into a semibridging position. This distortion is manifested in a more acute FeCO-FeNO-CO bond angle, Ψ, where FeCO and FeNO refers to the NO-free and NO-bearing iron atoms, respectively. Whereas mixed-valence diiron (I/II) dithiolato carbonyls display semi-bridging carbonyls with Ψ < 80° (see Table 1), for diiron(I) dithiolato carbonyls Ψ falls within the narrow range from 95 to 105°, e.g., for Fe2(S2C3H6)(CO)4(PMe3)2 (103.6°).23 For previously reported (NO+-free) diiron dithiolates, this value varies only slightly (± 5°) depending on the size and chelating nature of the other ligands (see Table 1). Previously reported unsymmetrically substituted derivatives usually feature unremarkable structures.24,25 One exception is the triphos complex.22 Because of its rotated nature, the Fe(CO)(PMe3)(NO)+ center adopts an approximate T-shaped orientation.
Scheme 2.

Idealized Frameworks for C2v Structure (left, Fe-CO vectors twisted for clarity) and the T-Shaped Rotated Framework (right).
Table 1.
Fe-Fe-CO Bond Angles for Various Compounds.
Compound 1 crystallized with two molecules in its asymmetric unit, the Ψ values both being ~91° (Figure 1). These are less than typical (~100°) values but by the definition of Crabtree26 correspond to terminal carbonyls. Otherwise, the structure is not unusual: the ligands are eclipsed and the Fe(CO)3 subunit is unaffected.
Two separate rotamers of the bisphosphine propanedithiolate 2’ crystallized. Each is severely distorted. For one rotamer, labeled 2’ap, the phosphine on the Fe(CO)2(PMe3) center occupies an apical site, and Ψ is 76° (Figure 2).
Figure 2.

Structure of 2’ap with thermal ellipsoids set at 35%. Side-on (left) and end-on (right) views. Hydrogen atoms, disordered methyl groups on P2, the anion, and solvate have been omitted for clarity. Key bond distances (Å): Fe1-Fe2, 2.5806(5); Fe1-N1, 1.648(3); Fe1-P1, 2.2622(8); Fe1-C1, 1.794(3); Fe2-C5, 1.778(3); Fe2-C6, 1.777(3); Fe2-P2, 2.2231(8).
In the second rotamer, 2’ba, the phosphine ligand on the Fe(CO)2(PMe3) center is basal, and the Fe(CO)(PMe3)(NO)+ center is even more distorted than in 2’ap. The shift of the PMe3 ligand from the apical to the basal site on the Fe(CO)2PMe3 center induces the bridging CO ligand into a more symmetrical position. Two molecules were found in the asymmetric unit: Ψ = ~62° and 67°, i.e. these are bent semi-bridging carbonyls (Figure 3). The two distinct structures of 2’ba indicate some flexibility for the semi-bridging carbonyl parallel to the Fe-Fe vector. Because of the increased rotation of the [Fe(CO)(PMe3)(NO)]+ subunit (versus that seen in 2’ap), the vacant coordination site on FeNO is particularly open and more closely resembles the diiron coordination environment in the Hox state and models thereof.7,8 Although semi-bridging carbonyls are present in the solid state, a band for νμ–CO is not apparent in the solution IR spectra of bulk samples of 2’ because 2’ba is a minority component. However, a νμ–CO is observed in the solid-state IR spectrum of single crystals of 2’.
The framework of ethanedithiolate 2 resembles that for 2’ap (see supplemental information). Only a single isomer was observed both in the solid state and in solution. The Ψ value is somewhat larger (~80°, i.e., FeCO is less bent) than for the propanedithiolate, consistent with the expected diminished steric influence of the smaller dithiolate.5
Given that two rotamers of 2’ crystallized, questions arose about the possibility that either 2’ap or 2’ba could be exceptional crystals that are not significantly represented in bulk samples. This issue was addressed through powder X-ray diffraction studies. Powder diffraction patterns for 2’ap and 2’ba were calculated from the single crystal data. The powder diffraction data for bulk samples were shown to consist of these two isomers as well as a third component. 2’ap was found to be the majority component, although calculations (below) suggest that 2’ba is ~3 kcal/mol lower in energy. That third component corresponds to species (one or more) obtained when single crystalline 2’ desolvates. The main point, however, is that of the solvates, polymorphs 2’ap and 2’ba are significant components of the solid mixture and are not statistical outliers.
Further substitution by phosphines occurs at the FeNO subunit to yield 3’ (see supporting information). Aside from a slightly distorted Fe(PMe3)2(NO)+ subunit containing a FeNO-FeCO-P bond angle of 96°, the structure is typical.
DNMR Studies
Variable temperature NMR studies indicated that the propanedithiolate 2’ consists of two major isomers in solution, as anticipated from the crystallography. At 150 °C, the 31P NMR spectrum consisted of two well resolved singlets (see supporting info). Upon cooling the sample, these peaks significantly broadened and decoalesced into a total of 4 peaks. Coalescence occurred at 25 and 10 °C, corresponding to the interconversion of 2’ap and 2’ba with an estimated activation energy in the range 51 – 52 kJ/mol. Below -60 °C, further splitting is observed due to the slow equilibration of the bridging propanedithiolate, as seen for non—nitrosylated Fe2(S2C3H6)(CO)4(PMe3)2.30
The 31P NMR spectrum of the ethanedithiolate 2 displayed only two singlets over the temperature range of 20 to −90 °C (Figure 4). Thus, it appears that only the propanedithiolate exists as energetically distinct rotamers.
Figure 4.

202 MHz 31P NMR spectra of 2’ (left) and 2 (right) at various temperatures (CD2Cl2 solutions).
Carbonylation of [Fe2(S2CnH2n)(CO)3(PMe3)2(NO)]+
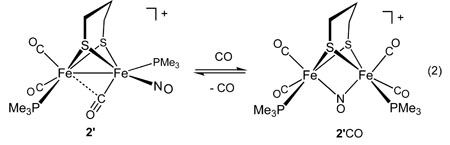
Unlike other diiron(I) dithiolates, 2’ and 2 are Lewis acidic - both reversibly add CO at low temperatures. In the IR spectrum of the adduct 2’CO, νNO absorbs at 1513 cm−1, about 200 cm−1 below the region for terminal nitrosyl ligands in this series. The spectra for the carbonylation exhibit isosbestic behavior, indicative of an equilibrium between two principal species (see Figure 5). Compounds 1 and 1’ did not display similar behaviour, however, 31P NMR indicated that 3’ also formed an adduct with CO at low temperature (see Supplementary Information).
Figure 5.

IR spectra recorded at intervals over the course of 10 min for the carbonylation of 2’ (CH2Cl2 solution, −80 °C). Notice the diminution of the 1797s cm−1 band for 2’ and the concomitant appearance of the band at 1513 cm−1 in 2’CO assigned to νμ-NO.
The IR spectra of 2’CO are consistent with the presence of a bridging nitrosyl ligand. This assignment is supported by the low temperature 31P NMR spectrum, which indicated equivalent phosphine ligands. Both isomers of 2’ formed the same adduct. The 200 cm−1 shift of the νNO band, also predicted by DFT calculations, is consistent with the relocation of the NO ligand from a terminal to a bridging position. Binding of CO by 2’ occurs with ΔH = −35 kJ/mol and ΔS = −139 J/mol•K, consistent with an associative process (eq 2, Figure 6). Despite the demonstrated affinity of 2’ for CO, solutions of 2’ did not appear to react with H2. Furthermore no H-D exchange was observed when solutions of 2’ and H2 were treated with MeOD in the presence of noncoordinating base.
Figure 6.

Plot of ln(Keq) vs 1/T (K) for 0.022 M 2’ (CH2Cl2) under 1 atm CO.
Like 2’, ethanedithiolate 2 displays a similar affinity toward CO. In a competition study, a solution that was equimolar in 2 and 2’ was treated with 1 atm of CO. Upon cooling to −80°C both CO adducts were observed, but 2’CO was favored by ~7 fold relative to 2CO. The adducts 2CO and 2’CO presumably occur as intermediates in the synthesis of 2’ from the reaction of NO+ and [Fe2(S2CnH2n)(CO)4(PMe3)2.
Labeling with 13CO has proven to be an effective tool to probe the carbonylation of models for Hox and Hox CO,31,32 but the present system is advantageous because the diamagnetism enabled analysis by 13C NMR spectroscopy. Exposure of 2’ to 13CO at −80 °C resulted primarily in a single isotopomers of 2’CO and singly labeled 2’. In 2’13CO, the label is proposed to be located at one of the two equivalent apical sites. The selectively labeled 2’ is proposed to arise by dissociation of 12CO from one of these two equivalent apical sites. Upon thermal equilibration of the sample, the −80 °C spectrum displayed five 31P-coupled doublets; two for 2’CO and three for the major isomer of 2’ (see supplementary information). If instead the 13CO atmosphere was rigorously removed above the sample followed by thermal equilibration, the remaining carbonyls also became enriched.
When equimolar amounts of 1’ and 2’ were pressurized with 13CO at −80 °C, the 31P NMR spectra indicated carbonylation exclusively at 2’, as expected since 1’ exhibits no Lewis acidity. Upon thermal equilibration, the 13C NMR spectrum showed that 2’ alone was enriched. Similarly, when equimolar amounds of 2’ and 3’ were equilibrated under an atmosphere of 13CO, enrichment was only observed at 2’.
Reactions of PMe3 with [Fe2(S2CnH2n)(CO)3(PMe3)2(NO)]+
In contrast to the nucleophilicity of Fe2(S2C3H6)(CO)4(PMe3)2, the nitrosyl compounds 2’ and 2 were found to be electrophilic. For example, 2’ is not visibly protonated by triflic acid. For example, they react readily with PMe3 to afford the corresponding trisphosphines [Fe2(S2CnH2n)(CO)2(PMe3)3(NO)]BF4, 3 and 3’. Two intermediates for these conversions were detected by in-situ IR spectroscopy. An initial adduct was observed immediately upon the addition of the phosphine to a CH2Cl2 solution of the diphosphine complex at −80 °C. IR spectra showed that the νNO is most affected (Fig. 7). The IR spectrum resembles that for the above mentioned adducts 2’CO and 2CO, implicating the formation of a bridging NO complex. The νNO is ~25 cm−1 lower in energy.
Figure 7.

IR spectra of 2’ (CH2Cl2 solution) at −78 °C (a), following the addition of one equiv of PMe3 (b), upon warming to −45 °C for 10 min. (c), and upon warming to room temperature for 3 h (d). For peak maxima, see Experimental Section.
The monophosphines 1 and 1’ were also found to readily undergo substitution by PMe3 to give 2 and 2’, respectively, as well as other nucleophiles (see supporting information). In these cases, IR measurements suggest that the nucleophile initially adds to the Fe(CO)3 subunit via a metastable intermediate containing a bridging CO, although this aspect was not pursued.
Electrochemistry
Compounds 1–2 exhibit two one-electron reduction steps (Table 2), but they oxidize only at very positive potentials (>1.3 V versus Ag|AgCl). For 1 and 1’, the first reductions are reversible, and the second reductions are only reversible at slow scan rates. Although ΔνNO is much larger than ΔνCO, the effect of 1e reduction is smaller than that seen for the 1e reduction of mononuclear iron nitrosyls.33 For 2’, the first reduction is quasi-reversible (ipa/ipc ≈ 0.7) at moderate scan rates (100 mV/s) and the second reduction is irreversible. The behavior of 2 is similar, although the first reduction is less reversible.
Table 2.
Reduction Potentials (V) of 1, 2, 3a and Related Non-Nitrosylated Derivatives
The reductions were determined to be 1—electron processes by comparing the ipa with the ipc of the oxidation of Fe2(S2C3H6)(CO)4(dppv), which is known to be one— electron process.34 Thus, in CH2Cl2 solution, compounds 1—3 are first reduced at the mild potentials of −0.36 (1) to −1.02 V (3) (potentials versus Ag|AgCl). Highlighting the electron—acceptor character of these nitrosyl complexes, the potentials for the second reduction of the diphosphine—derivatives 2—2’ are less negative than the first reduction step of the corresponding tetracarbonyl parents. Reduction of 2’ is about 0.1 V milder than that of 2, indicating the expected effect of the dithiolate—bridge on the redox properties of the diiron derivatives.5 In contrast to the behavior of compounds 1—2, related complexes lacking NO+ display reversible oxidations and a single poorly reversible reduction at highly negative potentials.35,36
DFT Calculations
DFT has accurately reproduced the experimental structure of 2’ (Figure S27, Supporting Information). The two isomers characterized by X-ray diffraction (2’ba and 2’ap) correspond to the two most stable isomeric forms computed by DFT. However, several other isomers are within a few kcal/mol (see Supporting Information). To quantitatively evaluate the effect of the NO+ ligand on the electronic structure of 2’ap and 2’ba, NBO charges (Table 3) have been computed and compared to the corresponding values obtained for the optimized structures of the isoelectronic carbonyl, Fe2(S2C3H6)(CO)4(PMe3)2 (ap and ba isomers). Related calculations for the oxidized species [Fe2(S2C3H6)(CO)4(PMe3)2]+ proved especially relevant. These species adopt “rotated” structures and are considered electronic models for the Hox state of the active site.34
Table 3.
NBO Charges of 2’ap, 2’ba, Fe2(S2C3H6)(CO)4(PMe3)2 (ap and ba isomer), and [Fe2(S2C3H6)(CO)4(PMe3)2]+ (ap and ba isomer) Computed at RI—BP86/def—TZVP Level of Theory.
| Complex | FeCO | FeNO |
|---|---|---|
| 2’ba | −0.22 | 0.03 |
| 2’ap | −0.19 | 0.03 |
| Fe2(S2C3H6)(CO)4(PMe3)2 | −0.19 to −0.18a | −0.19 to −0.21a |
| [Fe2(S2C3H6)(CO)4(PMe3)2]+ | −0.19 to −0.21a | −0.02 to −0.00a |
The range of values correspond to the results for different isomers (see Supporting Information).
Although absolute values of NBO charges must be interpreted cautiously,38 it is evident that the charge densities on the Fe atoms are far from formal oxidation states, reflecting the covalent character of the complex. Most importantly, the NBO charges on the Fe atoms in 2’ba and 2’ap, which formally are Fe(I)Fe(I) species, are more similar to the corresponding values calculated for the mixed-valence complex7 [Fe2(S2C3H6)(CO)4(PMe3)2]+ than in the subferrous Fe2(S2C3H6)(CO)4(PMe3)2 species. In particular, it can be concluded that the FeNO atom in [Fe2(S2C3H6)(CO)3(PMe3)2(NO)]+ (both isomers) have Fe(II)-like character. This observation conforms with the experimentally characterized Lewis acidity of the 2’ complex (see above) and its structural dissimilarity to FeIFeI diiron dithiolates.7
Computed IR bands for the 2’ isomers (2’ap and 2’ba) are collected in Table 4. The calculations are consistent with 2’ap being the major isomer in solution. Experimentally, signals corresponding to 2’ba are not observed, in part because νNO is intrinsically weaker and the νCO bands overlap with those for 2’ap. The lowest energy band always corresponds to νNO, whereas the other bands are assigned as νCO. Notably, moving from 2’ap to 2’ba the νμ-CO band shifts by almost 100 cm−1, due to the shortening of the C(O)—FeCO distance in 2’ba (2.78 versus 2.33 Å). The computed C(O)—FeCO distance in 2’ba could be underestimated due to the very flat potential energy surface corresponding to slight C(O)—FeCO distance modifications, as indicated also by the crystallographic results which range from 2.33 to 2.47 Å.
Table 4.
Vibrational Frequencies and Intensities of the 2’ap and 2’ba Computed at the RI—BP86/def—TZVP Level of Theory.
| 2’ap νNO and νCO (rel int.) | 2’ba νNO and νCO (rel. int.) | observed (CH2Cl2 soln) |
|---|---|---|
| 1814 (843) | 1817 (795) | 1787 |
| 1975 (289) | 1880 (418) | |
| 1978 (490) | 1988 (532) | 1981 |
| 2030 (780) | 2028 (559) | 2033 |
Calculations of the vibrational frequencies also supports the structure assigned to 2’CO (see above). For example, the μ-NO group is calculated to be 1522 cm−1, which reasonably matches the experimental value of 1513 cm−1.
Regarding the structure of 2’CO, DFT calculations indicate that the symmetrical μ-NO species is significantly more stable than other isomers (Figure 8), in agreement with the experimental evidence. Previous computational results from Schaefer and King indicate a preference for structures with bridging NO (relative to bridging CO) in electron-rich late transition metal complexes.39 The enthalpy of carbonylation for 2’ba, −12.2 kcal/mol (Table 5), is also consistent with the experimental value.
Figure 8.

DFT optimized structures of the possible isomers of 2’CO. Atom color scheme: oxygen, red; nitrogen, blue; carbon, green; iron, cyan; sulfur, yellow; and phosphorus, purple.
Table 5.
Binding Energies (kcal mol−1) for 2’ba + CO → 2’CO Computed at RI-BP86/def-TZVP Level of Theory.
| Reaction | ΔE |
|---|---|
| 2’ba + CO → axial-PMe3/μ-CO | 23.4 |
| 2’ba + CO → axial-PMe3/μ-NO | 4.3 |
| 2’ba + CO → equatorial-cis-PMe3/μ-CO | −0.3 |
| 2’ba + CO → equatorial-cis-PMe3/μ-NO | −9.4 |
| 2’ba + CO → equatorial-trans-PMe3/μ-CO | −1.6 |
| 2’ba + CO → equatorial-trans-PMe3/μ-NO | −12.2 |
Calculations predict that a high energy species (35 kcal/mol) features PMe3 groups in apical positions with a bent NO ligand (Fe-N-O angle 123°). Interestingly, the Fe-Fe distance contracts to 2.591 Å. This in silico experiment demonstrates, in effect, the competition between bending of FeNO and Fe---Fe bonding. In other geometries, including the lowest energy species, the Fe---Fe distances are non-bonding: 2.987 (apical-PMe3/μ-CO), 3.342 (equatorial-PMe3/μ-CO), and 2.968 Å (equatorial-PMe3/μ-NO).
We were surprised to observe bridging nitrosyl ligands in the adducts 2CO and 2’CO because the precursor complexes featured semi-bridging carbonyls that were expected to be occupy the bridging position. Calculations indicate enhanced stability for the adducts with μ-NO ligands and basal phosphines. Two isomerization pathways are plausible and very similar, involving turnstile rotations of the FeNO center. Only one isomer of CO adduct is observed. We did not consider alternative mechanisms involving the binding of CO directly to 2’ba, immediately followed by isomerization of NO into the bridging position, because the binding energies are unfavorable.
Discussion
The starting complexes in this work, species of the type Fe2(SR)2(CO)6-xLx, are well known, having been well developed even in the 1970’s.40 Because diiron(I) dithiolato carbonyls are structurally similar to the active site of the [FeFe]—hydrogenases, hundreds of derivatives have been described in recent years.4 Although slight deviations from idealized symmetry are encountered with bulky or constraining ligands (see Table 1), this paper describes the first derivatives where FeIFeI structures deviate strongly from the well established C2v motif.41,42 Our results demonstrate that electronic asymmetry imposed by ligands can cause very substantial geometric distortions otherwise induced by redox (Figure 9).7,8 These distortions in turn impact the reactivity of these diiron compounds and further indicate the versatility of the Fe2(SR)2(CO)6-xLx platform.4
Figure 9.

Overlay of (order from left to right): 2’ba (the examples in the asymmetric unit), 2’ap, 2, and 1 (thermal ellipsoids set at 10% and the anions; H atoms, and phosphine methyls omitted).
The electronic asymmetry is particularly acute in the diphosphines [Fe2(S2CnH2n)(CO)3(PMe3)2(NO)]+, wherein the Fe(CO)(PMe3)(NO)+ center is electrophilic and the Fe(CO)2(PMe3) center is electron—rich. As seen previously,5 the positioning of the central methylene group on the propanedithiolate accentuates the asymmetry by nonbonding interactions with ligands occupying the apical sites. As supported also by DFT calculations, the new compounds approximate the structure of the Hox state of the diiron site of the [FeFe]-hydrogenases (Figure 10), possibly because these compounds simulate the electronic asymmetry of seen Hox and its models.43 Not only do [Fe2(S2CnH2n)(CO)x(PMe3)2(NO)]+ (x = 3, 4) resemble the Hox, the binding of CO gives a derivative that, like HoxCO, that is symmetrized. Recall that binding of CO to valence localized Hox gives valence delocalized HoxCO.7,31,34,44 Unlike the Hox/HoxCO system and its models, which interconvert 33e−/35e− species, the 34e− nitrosyl cations rearrange before forming the corresponding 36e− adducts with CO.
Figure 10.

Overlay of 2’ba with the diiron portion of the Hox state from C. pasteurianum.45 The molecule of 2’ba with a smaller Ψ value was selected.
Associated with their novel structures, the new nitrosyl derivatives exhibit properties rarely seen in related complexes:
1) Lewis acidity. Unlike the many previously reported 34e diiron(I) dithiolato complexes, the species [Fe2(S2CnH2n)(CO)3(PMe3)2(NO)]+ are Lewis acidic.
2) Interconversions involving μ-NO ligands. The binding of CO provides a rare example of the interconversion of terminal and bridging NO ligands. Relatively few examples are known for complexes with μ—NO ligands,46 but for dimetallic mixed CO-NO complexes NO bridges more often than CO.39,47
3) Mild reduction potentials. Nitrosyl derivatives of the diiron(I) dithiolates reduce at potentials approximately 1 V milder than for related complexes.48 For example Fe2(S2C2H4)(CO)4(PMe3)2 reduces at ~−1.5 V vs Ag|AgCl, whereas the nitrosyl derivatives reduce at ~−0.5 V. The effect of replacing CO by NO+ is equivalent to protonation of the related CO derivative.36,49
4) Susceptibility toward ligand substitution. Diiron(I) dithiolates resist polysubstitution by phosphine ligands, at least in the absence of chelate effects.25,29 The presence of the nitrosyl allows the preparation of [Fe2(S2C3H6)(CO)2(PMe3)3(NO)]+. This finding further establishes that the resistance of Fe2(S2C3H6)(CO)4(PMe3)2 toward substitution is not due to steric factors, but is the result of its diminished electrophilicity. The diiron center in the enzymes is bound to three terminal donor ligands: two cyanides and one 4Fe-4S cluster.13
5) High rotational barriers. The isolation of two rotamers separated by substantial (~12 kcal/mol) barriers, as in the case of 2’ap and 2’ba, is unprecedented within the chemistry of diiron(I) dithiolato carbonyls. High barriers to isomerization are implicated for mixed-valence diiron dithiolates which also feature semibridging CO ligands.34 A “turnstile” mechanism42,50 is assumed to describe the interconversion of 2’ap and 2’ba (Scheme 4).
Scheme 4.

View down the Fe-Fe axis, showing the proposed pathway for interconversion of 2’ba and 2’ap.
Experimental Section
Procedures and materials have recently been described.25 NOBF4 was sublimed at 200 °C (0.02 mm Hg).51
[Fe2(S2C2H4)(CO)4(PMe3)(NO)]BF4, 1
At −30 °C, a mixture of 0.350 g (0.83 mmol) of Fe2(S2C2H4)(CO)5(PMe3) and 0.095 g (0.81 mmol) of NOBF4, in 20 mL of CH2Cl2 gave an intensely red solution over the course of 6 h. After concentrating the reaction to ~2 mL, the product precipitated upon addition of ~50 mL of hexanes and stirring for several minutes. The product was recrystallized by extraction into ~2 mL of CH2Cl2 followed by the addition of 50 mL of hexanes. Yield: 0.303 g (72%). Layering of a CH2Cl2 solution of 1 with hexanes afforded red single crystals after several days at −30 °C. 500 MHz 1H NMR (CD2Cl2): δ 3.23 (ddd, JH-H = 4.3, 8.2, 12.5, 1H, SCH2), 3.07 (ddd, JH-H = 4.5, 8.0, 17.3, 1H, SCH2), 2.94 (ddd, JH-H = 4.4, 8.0, 17.1, 1H, SCH2), 2.88 (ddd, JH-H = 4.4, 8.0, 17,1, 1H, SCH2), 1.85 (d, JP-H = 11.6, 9H, PMe3). 202 MHz 31P NMR (CD2Cl2, 20 °C): δ 21.4 (s). (CD2Cl2, −75 °C): δ 23.3 (s). IR (CH2Cl2): νCO = 2091, 2042; νNO = 1824 cm−1. ESI-MS: m/z = 421.9 (M+). Anal. Calcd for C9H13BF4Fe2NO5P2S2 (found): C, 21.25 (20.96); H, 2.49 (2.49); N, 2.75 (2.69).
[Fe2(S2C3H6)(CO)4(PMe3)(NO)]BF4, 1’
This compound was prepared as for 1 starting from 0.189 g (0.36 mmol) of Fe2(S2C3H6)(CO)5(PMe3) and 0.048 g (0.41 mmol) of NOBF4. Yield: 0.131 g (58%). 500 MHz 1H NMR (CD2Cl2): δ 2.72 (m, 2H, SCH2), 2.56 (m, 2H, SCH2), 2.15 (m, 1H, CH2), 2.03 (m, 1H, CH2), 1.84 (d, JP-H = 11.4, 9H, PMe3). 202 MHz, 31P NMR (CD2Cl2, 20 °C): 30.0 (s), (CD2Cl2, −80 °C): 32.3 (s), 24.9 (s). IR (CH2Cl2): νCO = 2089, 2036; νNO = 1813 cm−1. ESI-MS: m/z = 421.9 (M+). Anal. Calcd for C9H13BF4Fe2NO5P2S2 (found): C, 22.97 (23.33); H, 2.89 (2.68); N, 2.68 (2.63).
[Fe2(S2C2H4)(CO)3(PMe3)2(NO)]BF4, 2
To a mixture of 0.400 g (0.85 mmol) of Fe2(S2C2H4)(CO)4(PMe3)2 and 0.100 g (0.86 mmol) of NOBF4, cooled to 0 °C, was added 10 mL of CH2Cl2. After 3h, the dark brown-colored reaction mixture was concentrated in vacuo and precipitated upon addition of 50 mL hexanes. Recrystallization from 1:6 CH2Cl2:hexane mixtures provided analytically pure product. Yield: 0.435 g (91%). Layering of a CH2Cl2 solution of the product with hexanes followed by cooling at −30 °C, afforded deep red single crystals. 1H NMR (CD2Cl2): δ 2.98 (m, 2H, SCH2), 2.82 (m, 1H, SCH), 2.74 (m, 1H, SCH), 1.75 (d, JP-H = 11.2, 9H, PMe3), 1.69 (d, JP-H = 9.8, PMe3). 31P NMR (CD2Cl2, 20 °C): δ 31.2 (bs), 28.4 (bs); (CD2Cl2, −90 °C): δ 34.5 (s), 34.0 (s). IR (CH2Cl2): νCO = 2035, 1980; νNO = 1793. ESI-MS: m/z = 470.1 (M+). Anal. Calcd for C11H22BF4Fe2NO4P2S2 (found): C, 23.73 (23.78); H, 3.98 (4.26); N, 2.53 (2.54).
[Fe2(S2C3H6)(CO)3(PMe3)2(NO)]BF4, 2’
The preparation was modeled after that for 2 from 0.504 g (1.05 mmol) of Fe2(S2C3H6)(CO)4(PMe3)2 and 0.121 g (1.04 mmol) of NOBF4. Yield: 0.382 g (64%). Layering of a CH2Cl2 solution of the product with hexanes followed by cooling to −30 °C, afforded dark brown needles. 1H NMR (CD2Cl2): δ 2.58 (m, 2H, SCH2), 2.45 (m, 2H, SCH2), 1.10 (m, 2H, CH2), 1.78 (d, JP-H = 11.6, 9H, PMe3), 1.72 (d, JP-H = 10.1, 9H, PMe3). 31P NMR (CD2Cl2, 20 °C): δ 33.7 (bs), ~19 (bs, v. br); (CD2Cl2, −40 °C): 37.2 (s), 33.8 (s), 31.2 (s), 13.1 (s); (CD2Cl2, −90 °C): δ 41.4 (s), 38.7 (s), 36.4 (s), 34.0 (s), 32.0 (s), 14.6 (s), 13.4 (s). Integrations indicate that the δ34.0 peak consists of two overlapping signals. IR (CH2Cl2): νCO = 2033, 1981, νNO = 1787. ESI-MS: m/z = 484.0 (M+). Anal. Calcd for C12H24BF4Fe2NO4P2S2 (found): C, 25.25 (25.07); H, 4.24 (4.20); N, 2.45 (2.40).
[Fe2(S2C3H6)(CO)2(PMe3)3(NO)]BF4, 3’
To a solution of 0.141 g (0.25 mmol) [Fe2(S2C3H6)(CO)3(PMe3)2(NO)]BF4 in 10 mL of CH2Cl2, cooled to −78 °C, was added 1.27 mL of a 0.193 M solution of PMe3 (0.25 mmol) in CH2Cl2. The solution was warmed to room termperature and maintained at that temperature until all of the intermediate ([Fe2(S2C3H6)(μ-CO)(CO)2(PMe3)3(NO)]BF4) had converted to 3’ (~ 2.5 h). The red product precipitated from solution upon addition of 50 mL of hexanes followed by removal of approximately 1/6 of the total volume in vacuo. Yield: 0.115 g (75%). Layering of a CH2Cl2 solution of the product with hexanes, followed by cooling this mixture to −30 °C, afforded dark red cubic single crystals after several days. 1H NMR (CD2Cl2): δ 2.41 (m, 2H, SCH2), 2.18 (m, 1H, CH2), 2.11 (m, 2H, 2H, SCH2), 1.76 (d, JP-H = 8.9, PMe3), 1.71 (m, 1H, CH2), 1.60 (s, 18H, PMe3). 31P NMR (CD2Cl2, 20 °C): δ 20.8 (s, 1P, Fe(CO)2(PMe3)), 6.4 (s, 2P, Fe(PMe3)2(NO)); (CD2Cl2, −40 °C): δ 20.8 (s, 1P, Fe(CO)2(PMe3)), 7.5 (s, 2P, Fe(PMe3)2(NO)). IR (CH2Cl2): νCO = 1991, 1939; νNO = 1755. ESI-MS: m/z = 532.3 (M+). Anal. Calcd for C14H33BF4Fe2NO3P3S2 (found): C, 27.17 (27.26); H, 4.24 (5.67); N, 2.26 (2.48). CV (CH2Cl2, vs Ag|AgCl): Epc = −0.93, −1.49, Epa = 1.02, 1.20 (reductions and oxidations were irreversible). During the synthesis of 3’, in situ IR spectra (ReactIR 4000, Mettler Toledo) (CH2Cl2, −80 °C): νCO = 2034, 1984; νNO = 1486. IR (CH2Cl2, −45 °C): νCO = 1953; νμ-CO = 1984; νNO = 1760.
CO Binding Experiments
In a typical experiment, onto ~8 mg of the diiron complex in a J. Young NMR tube was distilled 0.7 mL of CD2Cl2, and the solution was frozen in an isopentane/N2 bath and the tube was evacuated. The tube was then pressurized with 1 atm of CO. After cooling to the appropriate temperature within the spectrometer, the reaction mixture was allowed to equilibrate until no significant changes were observed (typically ~30–60 min). The probe temperature was calibrated with a methanol standard.
[2’CO]BF4: 31P NMR (CD2Cl2, −80 °C): δ 21.2 (s, 1P). IR (CH2Cl2, −80 °C): νCO = 2038, 2003; νNO = 1513.
[2CO]BF4: 31P NMR (CD2Cl2, −80 °C): δ 22.1 (s, 1P). IR (CH2Cl2, −80 °C): νCO = 2038, 2011, 1999; νNO = 1498.
[3’CO]BF4: (CD2Cl2, −80 °C): δ 19.5 (s, 1P), 19.3 (s, 1P), 18.2 (s, 1P), 18.1 (s, 2P), 17.5 (s, 1P). The low temperature 31P NMR spectrum indicated that [3’CO]BF4 consists of two isomers; one with 3 inequivalent phosphines, and the other containing 2 equivalent and 1 inequivalent phosphine.
Isotopic Labeling Experiments
J. Young NMR tubes were pressurized to ~0.3 atm of 13CO, sealed, thawws in a CH2Cl2/N2 slush bath and then immediately transferred to a spectrometer, where the probe was cooled to the appropriate temperature. A series of spectra were collected containing some combination of the following peaks: 13C NMR (CD2Cl2, −80 °C): δ 222.2 (d, JC-P = 25.1, 2’), 211.3 (d, JC-P = 12.0, 2’), 210.8 (d, JC-P = 10.4, 2’(CO)ap), 205.9 (d, JC-P = 20.7, 2’), 203.3 (d, JC-P = 16.5, 2’(CO)ba).
The initial spectrum at −80 °C showed a doublet at δ 210.8 and a weak signal at δ 205.9.
The sample was then warmed briefly by ejecting it followed after 5 s by reinjecting it into the −80 °C probe. The resultant spectrum showed signals at δ 210.8 and 205.9 of comparable intensity.
The sample was then ejected, warmed at 20 °C for ~5 min and then reinserting into the −80 °C probe, the spectrum feature all five signals listed above.
The sample was ejected, warmed to 20 °C for 5 min, and then vented to air. After cooling back to −80 °C, the spectrum showed peaks at δ 222.2, 211.3, and 205.9.
Alternatively, the sample could be subjected to several freeze/pump/thaw cycles using a CH2Cl2/N2 slush bath. The spectrum (−80 °C) consisted of a major signal at δ 205.9 and weaker signals at δ 222.2 and 211.3.
DNMR Measurements
With peaks at −30 °C labeled from left to right as A, B, C, and D: Coalescence of peaks A and C occurs at Tc ≈ 10 °C with Δν = 560 Hz. The coalescence of peaks B and D occurs at Tc ≈ 25 °C with Δν = 2700 Hz.
These value correspond to the activation free energies of 51.4 and 52.4 kJ/mol 52
X-ray Powder Diffraction
Powder diffraction patterns for 2ap and 2ba were calculated from the single crystal data using Topas, Version 3, by Bruker AXS.
DFT Calculations
DFT calculations were carried out using the BP86 functional53 and a valence triple-ζ basis set with polarization on all atoms (TZVP),54 a level of theory which has been found to give reliable results for analogous organometallic compounds.55 Stationary points of the energy hypersurface have been located by means of energy gradient techniques and a full vibrational analysis has been carried out to further characterize each stationary point. Partial atomic charges have been computed according to the Natural Bond Orbital (NBO) scheme.56
Supplementary Material
Supporting Information Available: Spectra, voltammograms, and CIFs for crystal structures.
Scheme 3.

Proposed Pathways for the Carbonylation of 2’.
Acknowledgement
This research was supported by the National Institutes of Health and the Petroleum Research Fund. MTO thanks the NIH Chemistry-Biology Interface program for support.
Contributor Information
Luca De Gioia, Email: luca.degioia@unimib.it.
Thomas B. Rauchfuss, Email: rauchfuz@uiuc.edu.
References
- 1.De Lacey AL, Fernández VM, Rousset M, Cammack R. Chem. Rev. 2007;107:4304–4330. doi: 10.1021/cr0501947. [DOI] [PubMed] [Google Scholar]
- 2.Fontecilla-Camps JC, Volbeda A, Cavazza C, Nicolet Y. Chem. Rev. 2007;107:4273–4303. doi: 10.1021/cr050195z. [DOI] [PubMed] [Google Scholar]; Vignais PM, Billoud B. Chem. Rev. 2007;107:4206–4272. doi: 10.1021/cr050196r. [DOI] [PubMed] [Google Scholar]
- 3.Liu X, Ibrahim SK, Tard C, Pickett CJ. Coord. Chem. Rev. 2005;249:1641–1652. [Google Scholar]
- 4.Hogarth G. In: Comprehensive Organometallic Chemistry III. Mingos RHCaDMP., editor. Elsevier: Amsterdam; 2007. [Google Scholar]
- 5.Justice AK, Zampella G, De Gioia L, Rauchfuss TB. Chem. Commun. 2007:2019–2021. doi: 10.1039/b700754j. [DOI] [PubMed] [Google Scholar]
- 6.Nicolet Y, Lemon BJ, Fontecilla-Camps JC, Peters JW. Trends Biochem. Sci. 2000;25:138–143. doi: 10.1016/s0968-0004(99)01536-4. [DOI] [PubMed] [Google Scholar]; Pandey AS, Harris TV, Giles LJ, Peters JW, Szilagyi RK. J. Am. Chem. Soc. 2008;130:4533–4540. doi: 10.1021/ja711187e. [DOI] [PubMed] [Google Scholar]
- 7.Justice AK, Rauchfuss TB, Wilson SR. Angew. Chem., Int. Ed. 2007;46:6152–6154. doi: 10.1002/anie.200702224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu T, Darensbourg MY. J. Am. Chem. Soc. 2007;129:7008–7009. doi: 10.1021/ja071851a. [DOI] [PubMed] [Google Scholar]
- 9.van der Vlugt JI, Rauchfuss TB, Whaley CM, Wilson SR. J. Am. Chem. Soc. 2005;127:16012–16013. doi: 10.1021/ja055475a. [DOI] [PubMed] [Google Scholar]
- 10.Arabi MS, Mathieu R, Poilblanc RJ. Organomet. Chem. 1979;177:199–209. [Google Scholar]
- 11.de Beer JA, Haines RJ. J. Organomet. Chem. 1972;36:297–312. [Google Scholar]
- 12.Song L-C, Yang Z-Y, Bian H-Z, Hu Q-M. Organometallics. 2004;23:3082–3084. [Google Scholar]
- 13.Tard C, Liu X, Ibrahim SK, Bruschi M, De Gioia L, Davies SC, Yang X, Wang L-S, Sawers G, Pickett CJ. Nature. 2005;433:610–614. doi: 10.1038/nature03298. [DOI] [PubMed] [Google Scholar]
- 14.Nehring JL, Heinekey DM. Inorg. Chem. 2003;42:4288–4292. doi: 10.1021/ic034334b. [DOI] [PubMed] [Google Scholar]; Boyke CA, Rauchfuss TB, Wilson SR, Rohmer M-M, Bénard M. J. Am. Chem. Soc. 2004;126:15151–15160. doi: 10.1021/ja049050k. [DOI] [PubMed] [Google Scholar]
- 15.Capon J-F, El Hassnaoui S, Gloaguen F, Schollhammer P, Talarmin J. Organometallics. 2005;24:2020–2022. [Google Scholar]; Tye JW, Lee J, Wang HW, Mejia-Rodriguez R, Reibenspies JH, Hall MB, Darensbourg MY. Inorg. Chem. 2005;44:5550–5552. doi: 10.1021/ic050402d. [DOI] [PubMed] [Google Scholar]
- 16.Lawrence JD, Li H, Rauchfuss TB. Chem. Commun. 2001:1482–1483. [Google Scholar]
- 17.Arabi MS, Mathieu R, Poilblanc R. Inorg. Chim. Acta. 1977;23:L17–L18. [Google Scholar]; Arabi MS, Mathieu R, Poilblanc R. Inorg. Chim. Acta. 1979;34:L207–L208. [Google Scholar]; Bonnet JJ, Mathieu R, Poilblanc R, Ibers JA. J. Am. Chem. Soc. 1979;101:7487–7496. [Google Scholar]
- 18.Mariusz MM, Artur MA. Organometallics. 2007;25:6576–6580. [Google Scholar]; Richter-Addo GB, Legzdins P. Metal Nitrosyls. New York: Oxford University Press; 1992. [Google Scholar]
- 19.Butler AR, Megson IL. Chem. Rev. 2002;102:1155–1165. doi: 10.1021/cr000076d. [DOI] [PubMed] [Google Scholar]; Wasser IM, de Vries S, Moënne-Loccoz P, Schröder I, Karlin KD. Chem. Rev. 2002;102:1201–1234. doi: 10.1021/cr0006627. [DOI] [PubMed] [Google Scholar]; Tsai M-L, Liaw W-F. Inorg. Chem. 2006;45:6583–6585. doi: 10.1021/ic0608849. [DOI] [PubMed] [Google Scholar]; Conradie J, Quarless DA, Jr, Hsu H-F, Harrop TC, Lippard SJ, Koch SA, Ghosh A. J. Am. Chem. Soc. 2007;129:10446–10456. doi: 10.1021/ja0719982. [DOI] [PubMed] [Google Scholar]; Harrop TC, Song D, Lippard SJ. J. Am. Chem. Soc. 2006;128:3528–3529. doi: 10.1021/ja060186n. [DOI] [PubMed] [Google Scholar]; Harrop TC, Song D, Lippard SJ. J. Inorg. Biochem. 2007;101:1730–1738. doi: 10.1016/j.jinorgbio.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 20.Rauchfuss TB, Weatherill TD. Inorg. Chem. 1982;21:827–831. [Google Scholar]; Conrado CL, Bourassa JL, Egler C, Wecksler S, Ford PC. Inorg. Chem. 2003;42:2288–2293. doi: 10.1021/ic020309e. [DOI] [PubMed] [Google Scholar]
- 21.Chen H-W, Lin C-W, Chen C-C, Yang L-B, Chiang M-H, Liaw W-F. Inorg. Chem. 2005;44:3226–3232. doi: 10.1021/ic049105j. [DOI] [PubMed] [Google Scholar]; Lee C-M, Chen C-H, Chen H-W, Hsu J-L, Lee G-H, Liaw W-F. Inorg. Chem. 2005;44:6670–6679. doi: 10.1021/ic050108l. [DOI] [PubMed] [Google Scholar]
- 22.Adam FI, Hogarth G, Richards I, Sanchez BE. Dalton Trans. 2007:2495–2498. doi: 10.1039/b706123b. [DOI] [PubMed] [Google Scholar]
- 23.Zhao X, Georgakaki IP, Miller ML, Yarbrough JC, Darensbourg MY. J. Am. Chem. Soc. 2001;123:9710–9711. doi: 10.1021/ja0167046. [DOI] [PubMed] [Google Scholar]
- 24.Duan LL, Wang M, Li P, Na Y, Wang N, Sun LC. Dalton Trans. 2007:1277–1283. doi: 10.1039/b616645h. [DOI] [PubMed] [Google Scholar]
- 25.Justice AK, Zampella G, De Gioia L, Rauchfuss TB, van der Vlugt JI, Wilson SR. Inorg. Chem. 2007;46:1655–1664. doi: 10.1021/ic0618706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crabtree RH, Lavin M. Inorg. Chem. 1986;25:805–812. [Google Scholar]
- 27.Lyon EJ, Georgakaki IP, Reibenspies JH, Darensbourg MY. Angew. Chem., Int. Ed. Engl. 1999;38:3178–3180. [PubMed] [Google Scholar]
- 28.Li P, Wang M, He C, Li G, Liu X, Chen C, Åkermark B, Sun L. Eur. J. Inorg. Chem. 2005:2506–2513. [Google Scholar]
- 29.Hogarth G, Richards I. Inorg. Chem. Commun. 2007;10:66–70. [Google Scholar]
- 30.Georgakaki IP, Thomson LM, Lyon EJ, Hall MB, Darensbourg MY. Coord. Chem. Rev. 2003:238–239. 255–266. [Google Scholar]
- 31.Justice AK, Nilges M, De Gioia L, Rauchfuss TB, Wilson SR, Zampella GJ. Am. Chem. Soc. 2008;130:5293–5301. doi: 10.1021/ja7113008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thomas CM, Liu T, Hall MB, Darensbourg MY. Chem. Commun. 2008:1563–1565. doi: 10.1039/b719559a. [DOI] [PubMed] [Google Scholar]
- 33.Sellmann D, Blum N, Heinemann FW, Hess BA. Chem. Eur. J. 2001;7:1874–1880. doi: 10.1002/1521-3765(20010504)7:9<1874::aid-chem1874>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]; Serres RG, Grapperhaus CA, Bothe E, Bill E, Weyhermuller T, Neese F, Wieghardt K. J. Am. Chem. Soc. 2004;126:5138–5153. doi: 10.1021/ja030645+. [DOI] [PubMed] [Google Scholar]
- 34.Justice AK, De Gioia L, Nilges MJ, Rauchfuss TB, Wilson SR, Zampella G. Inorg. Chem. 2008 doi: 10.1021/ic8007552. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Felton GAN, Vannucci AK, Chen J, Lockett LT, Okumura N, Petro BJ, Zakai UI, Evans DH, Glass RS, Lichtenberger DL. J. Am. Chem. Soc. 2007;129:12521–12530. doi: 10.1021/ja073886g. [DOI] [PubMed] [Google Scholar]; Borg SJ, Behrsing T, Best SP, Razavet M, Liu X, Pickett CJ. J. Am. Chem. Soc. 2004;126:16988–16999. doi: 10.1021/ja045281f. [DOI] [PubMed] [Google Scholar]; Borg SJ, Tye JW, Hall MB, Best SP. Inorg. Chem. 2007;46:384–394. doi: 10.1021/ic061211t. [DOI] [PubMed] [Google Scholar]
- 36.Gloaguen F, Lawrence JD, Rauchfuss TB, Bénard M, Rohmer M-M. Inorg. Chem. 2002;41:6573–6582. doi: 10.1021/ic025838x. [DOI] [PubMed] [Google Scholar]
- 37.Mejia-Rodriguez R, Chong D, Reibenspies JH, Soriaga MP, Darensbourg MY. J. Am. Chem. Soc. 2004;126:12004–12014. doi: 10.1021/ja039394v. [DOI] [PubMed] [Google Scholar]
- 38.Reed AE, Curtiss LA, Weinhold F. Chem. Rev. 1988;88:899–926. [Google Scholar]
- 39.Wang H, Xie Y, King RB, Schaefer HF., III Inorg. Chem. 2008;46:1836–1847. doi: 10.1021/ic0620541. [DOI] [PubMed] [Google Scholar]
- 40.Zhao X, Georgakaki IP, Miller ML, Mejia-Rodriguez R, Chiang C-Y, Darensbourg MY. Inorg. Chem. 2002;41:3917–3928. doi: 10.1021/ic020237r. [DOI] [PubMed] [Google Scholar]; Fauvel K, Mathieu R, Poilblanc R. Inorg. Chem. 1976;15:976–978. [Google Scholar]
- 41.Dahl LF, Wei CH. Inorg. Chem. 1963;2:328–333. [Google Scholar]
- 42.Winter A, Zsolnai L, Huttner GZ. Naturforsch. 1982;37b:1430–1436. [Google Scholar]
- 43.Nicolet Y, Piras C, Legrand P, Hatchikian CE, Fontecilla-Camps JC. Structure. 1999;7:13–23. doi: 10.1016/s0969-2126(99)80005-7. [DOI] [PubMed] [Google Scholar]
- 44.Silakov A, Reijerse EJ, Albracht SPJ, Hatchikian EC, Lubitz W. J. Am. Chem. Soc. 2007;129:11447–11458. doi: 10.1021/ja072592s. [DOI] [PubMed] [Google Scholar]
- 45.Peters JW, Lanzilotta WN, Lemon BJ, Seefeldt LC. Science. 1998;282:1853–1858. doi: 10.1126/science.282.5395.1853. [DOI] [PubMed] [Google Scholar]
- 46.Paul PP, Tyeklar Z, Farooq A, Karlin KD, Liu S, Zubieta J. J. Am. Chem. Soc. 1990;112:2430–2432. [Google Scholar]
- 47.Hayton TW, Legzdins P, Sharp WB. Chem. Rev. 2002;102:935–991. doi: 10.1021/cr000074t. [DOI] [PubMed] [Google Scholar]
- 48.Cheah MH, Borg SJ, Best SP. Inorg. Chem. 2007;46:1741–1750. doi: 10.1021/ic0623361. [DOI] [PubMed] [Google Scholar]
- 49.Gloaguen F, Lawrence JD, Rauchfuss TB. J. Am. Chem. Soc. 2001;123:9476–9477. doi: 10.1021/ja016516f. [DOI] [PubMed] [Google Scholar]
- 50.Adams RD, Cotton FA, Cullen WR, Hunter DL, Mihichuk L. Inorg. Chem. 1975;14:1395–1399. [Google Scholar]; Lyon EJ, Georgakaki IP, Reibenspies JH, Darensbourg MY. J. Am. Chem. Soc. 2001;123:3268–3278. doi: 10.1021/ja003147z. [DOI] [PubMed] [Google Scholar]
- 51.Mocella MT, Okamoto MS, Barefield EK. Syn. React. Inorg. Metal-Org. Chem. 1974;4:69–90. [Google Scholar]
- 52.Nelson JH. Nuclear Magnetic Resonance Spectroscopy. NY: Prentice Hall; 2003. [Google Scholar]
- 53.Becke AD. J. Chem. Phys. 1986;84:4524–4529. [Google Scholar]; Perdew JP. P. Phys. Rev. 1986;B33:8882. [Google Scholar]
- 54.Schaefer A, Huber C, Ahlrichs R. J. Chem. Phys. 1994;100:5829–5835. [Google Scholar]
- 55.Niu S, Hall MB. Chem. Rev. 2000;100:353. doi: 10.1021/cr980404y. [DOI] [PubMed] [Google Scholar]; Bruschi M, Zampella G, Fantucci P, De Gioia L. Coord. Chem. Rev. 2005;249:1620. doi: 10.1021/ja0508424. [DOI] [PubMed] [Google Scholar]; Siegbahn PEM, Tye JW, Hall MB. Chem. Rev. 2007;107:4414–4435. doi: 10.1021/cr050185y. [DOI] [PubMed] [Google Scholar]
- 56.Reed AE, Weinstock RB, Weinhold F. J. Chem. Phys. 1985;83:735–746. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Available: Spectra, voltammograms, and CIFs for crystal structures.