

Abstract
The PduO-type ATP:corrinoid adenosyltransferase from Lactobacillus reuteri (LrPduO) catalyzes the formation of the essential Co–C bond of adenosylcobalamin (coenzyme B12) by transferring the adenosyl group from co-substrate ATP to a transient Co1+corrinoid species generated in the enzyme active site. While PduO-type enzymes have previously been believed to be capable of adenosylating only Co1+cobalamin (Co1+Cbl−), our kinetic data obtained in this study provide in vitro evidence that LrPduO can in fact also utilize the incomplete corrinoid Co1+cobinamide (Co1+Cbi) as an alternative substrate. To explore the mechanism by which LrPduO overcomes the thermodynamically challenging reduction of its Co2+corrinoid substrates, we have examined how the enzyme active site alters the geometric and electronic properties of Co2+Cbl and Co2+Cbi+ by using electronic absorption, magnetic circular dichroism, and electron paramagnetic resonance spectroscopic techniques. Our data reveal that upon binding to LrPduO that was pre-incubated with ATP, both Co2+corrinoids undergo a partial (~40–50%) conversion to distinct paramagnetic Co2+species. The spectroscopic signatures of these species are consistent with essentially four-coordinate, square-planar Co2+ complexes, based on a comparison with the results obtained in our previous studies of related enzymes. Consequently, it appears that the general strategy employed by adenosyltransferases for effecting Co2+→Co1+ reduction involves the formation of an “activated” Co2+corrinoid intermediate that lacks any significant axial bonding interactions, so as to stabilize the redox-active, Co 3dz2-based molecular orbital.
Coenzyme B12, which is also known as adenosylcobalamin (AdoCbl1), provides one of very few known examples of a bio-organometallic species (1). AdoCbl contains a cobalt(III) ion that is equatorially ligated by four nitrogens of a tetrapyrrole macrocycle, termed the corrin ring, and axially coordinated by the 5′-carbon atom of an adenosyl (Ado) moiety on the “upper” face and a nitrogen atom from 5,6-dimethylimidazole (DMB) on the “lower” face (Figure 1) (2). The DMB is part of a nucleotide loop that is tethered to the corrin ring at C17; in adeonsylcobinamide (AdoCbi+), this loop is absent and the lower axial coordination site is occupied by a water molecule to complete a distorted octahedral ligand environment of the Co3+ ion (3).
Figure 1.
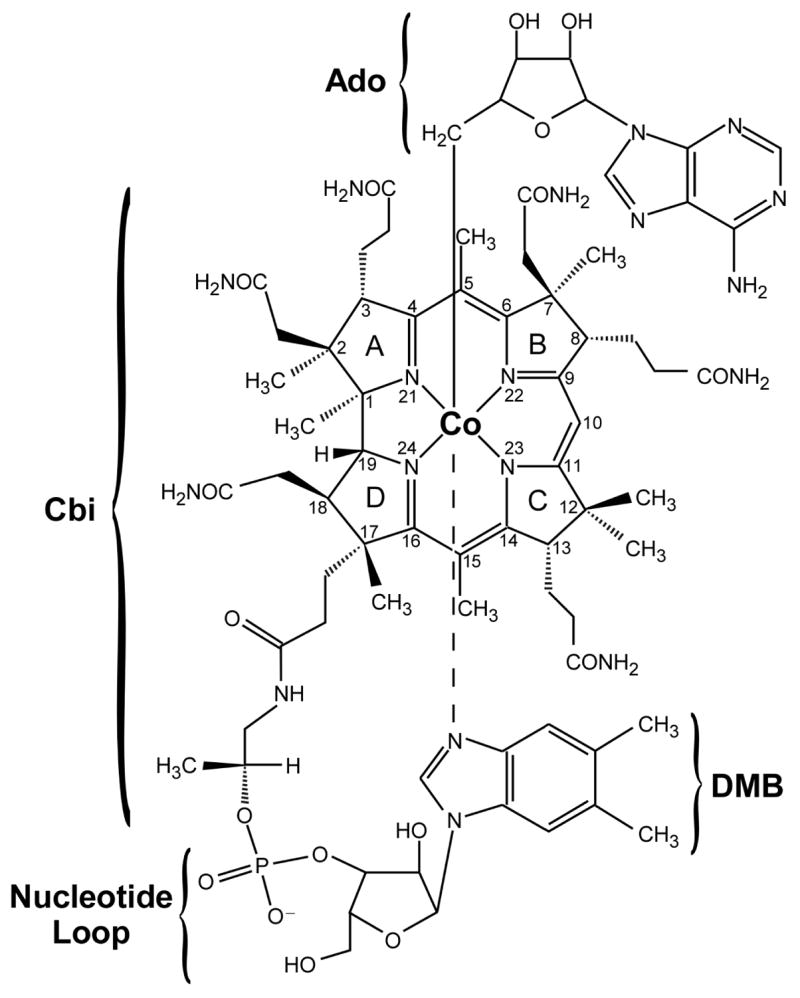
Schematic representation of the adenosylcobalamin cofactor (AdoCbl). In adenosylcobinamide (AdoCbi+), the nucleotide loop including the DMB base is absent and a water molecule occupies the lower axial position.
AdoCbl serves as a cofactor for numerous distinct enzymes that catalyze radical-induced rearrangement reactions (4–10), such as methylmalonyl-CoA mutase, glutamate mutase, diol dehydratase, and ethanolamine ammonia lyase, or ribonucleotide reduction, as exemplified by the ribonucleotide tri-phosphate reductase (11). Common to these enzymes is that the first step in their respective catalytic cycles involves homolytic cleavage of the cofactor’s Co–C bond to yield Co2+cobalamin (Co2+Cbl) and an organic radical centered on the 5′-carbon of the Ado moiety. The Ado• radical generated in this process then abstracts a hydrogen atom from substrate to initiate a protein-mediated rearrangement reaction that eventually leads to product formation and regeneration of the AdoCbl cofactor (12–14).
Eukaryotes lack the biosynthetic machinery needed to synthesize AdoCbl de novo. Instead, those eukaryotes that utilize AdoCbl in their metabolism (which does not include plants) convert exogenous cobalamins, such as vitamin B12 (in which the upper axial coordination site is occupied by a CN− moiety rather than Ado), to AdoCbl by using a class of enzymes termed adenosyltransferases (15, 16). The importance of these enzymes is highlighted by the fact that the malfunctioning of the human adenosyltransferase (hATR) can cause methylmalonic aciduria, an autosomal disorder that is often fatal in infants (17). Many prokaryotes also require AdoCbl; e.g., for the utilization of propanediol and ethanolamine to produce substrates for ATP production and as sources of carbon and nitrogen, respectively (7, 8). Some of these organisms are capable of synthesizing AdoCbl de novo in a process involving over 25 enzymes, which catalyze the stepwise conversion of 5-aminolevulinic acid to the complete cofactor via an intermediate named cobyric acid, a cobalamin precursor that possesses all of the necessary functionality except for the nucleotide loop and the axial Ado ligand (18).
Regardless of whether an organism synthesizes AdoCbl de novo or salvages incomplete corrinoids from its environment, both processes require two one-electron reduction steps to convert Co3+ to Co1+ and the formation of the essential Co–C bond by transferring the Ado group from a molecule of ATP to the transiently formed Co1+corrinoid species (19, 20). While previous studies revealed that the Co3+/2+corrinoid reduction potential is sufficiently positive to ensure that the Co3+corrinoid be converted to the Co2+ state in the reducing environment of the cytoplasm (21), the Co2+/1+corrinoid potential is lower than the midpoint potentials of putative in vivo reducing agents. For example, the Co2+/1+ reduction potential of aqueous Co2+Cbl (E° = −610 mV) is well below that of the semiquinone/reduced flavin couple of flavodoxin A (FldA, E° = −440 mV) (22–24). Despite this apparent thermodynamic dilemma, it has been shown that in the presence of adenosyltransferases, the Co2+→Co1+ reduction can be effected under physiologically relevant conditions (25–27).
Previously, our spectroscopic and computational studies of two representative members of the family of adenosyltransferases, CobA from Salmonella enterica and hATR, have provided definitive clues as to how these enzymes overcome the thermodynamically challenging task of reducing Co2+corrinoids to their Co1+ oxidation state (28, 29). Specifically, we have shown that upon binding of Co2+cobinamide (Co2+Cbi+) to CobA complexed with co-substrate ATP, the axially bound water molecule (partially) dissociates so as to generate an effectively four-coordinate square-planar Co2+ species. As a result of this (partial) ligand dissociation, the redox-active Co 3dz2 orbital that is oriented toward the axial coordination sites is significantly stabilized in energy, thereby raising the Co2+/1+ reduction midpoint potential by an estimated 250 mV (28). Remarkably, the same spectroscopic signatures characterizing this “activated” four-coordinate Co2+ species features have been found to develop upon binding of Co2+Cbl to hATR in the presence of ATP (29), even though hATR and CobA are structurally and evolutionary unrelated (7, 30, 31).
Recently, St. Maurice et al. have purified an hATR-type adenosyltransferase from Lactobacillus reuteri, termed LrPduO, and reported its X-ray crystal structure with ATP bound to the active site (32). In vivo and in vitro experiments have confirmed that LrPduO can convert Co1+Cbl− to AdoCbl and revealed that it can substitute for CobA in Salmonella enterica, implying that LrPduO possesses the ability to adenosylate both Co2+Cbl and Co2+Cbi+ (16, 32). In the present study, we have performed a detailed kinetic and spectroscopic characterization of LrPduO to obtain molecular-level insight into certain steps of the catalytic mechanism employed by this enzyme. Our kinetic parameters obtained for the reaction of Co1+Cbi with LrPduO/ATP confirm that cobinamide can serve as an alternative substrate for this adenosyltransferase, while our spectroscopic data indicate that LrPduO utilizes the same general strategy for promoting Co2+→Co1+ reduction as CobA and hATR; namely, to generate an effectively four-coordinate Co2+corrinoid species regardless of whether Co2+Cbl or Co2+Cbi+ is used as the substrate. Nonetheless, a comparison of the results obtained for LrPduO and those reported previously for hATR and CobA reveals small but notable differences with regards to the substrate specificity and degree of Co2+corrinoid activation.
MATERIALS AND METHODS
Cofactors and Chemicals
Aquacobalamin (H2OCbl+), dicyanocobinamide ((CN)2Cbi), and sodium borohydride (NaBH4) were purchased from Sigma and used as obtained. Diaquacobinamide ((H2O)2Cbi2+) was prepared by reducing (CN)2Cbi with NaBH4, loading the reaction mixture on a C18 SepPack column, washing with doubly distilled H2O, and eluting the product with methanol, as described in a previous report (28). Co2+Cbl and Co2+Cbi+ were prepared by adding NaBH4 to degassed aqueous solutions of H2OCbl+ and (H2O)2Cbi2+, respectively, in the presence of 60% (v/v) glycerol, and the progress of the conversion was monitored spectrophotometrically.
Protein Production and Purification
Recombinant PduO protein from Lactobacillus reuteri was obtained by overexpressing a pTEV3 plasmid transformed into Escherichia coli strain BL21(DE3). pTEV3 encodes the LrPduO protein with an N-terminal (His)6 tag (32, 33). This (His)6 tag was subsequently clipped off using rTEV protease, and the tag-free enzyme was purified using a HisTrap FF column (Amersham Biosciences), as described previously (32). Purified LrPduO was stored at −80 °C in Tris-HCl buffer (0.1 M, pH 8 at 4 °C) containing NaCl (0.5 M). Protein samples used for kinetic analyses additionally contained 10% (v/v) glycerol.
Kinetic Studies
Activity assays were performed as described previously (32) without modifications (final LrPduO concentration was 1 μg/mL). AdoCbi+ formation was monitored spectrophotometrically at 388 nm, which corresponds to the peak position of the dominant absorption feature of Co1+Cbi. The values of Vmax and Km were determined from plots of the initial velocity versus substrate concentration. All kinetic parameters were determined in duplicates; their values and uncertainties as reported in this paper correspond to the average of the two measurements and their standard deviations, respectively.
Sample Preparation
Solutions of LrPduO (in 50 mM Tris-HCl buffer, pH 8 containing 0.5 M NaCl) in the absence or presence of 3 mM MgATP were purged separately with Ar gas for 30 min at 4 °C before they were combined with the degassed free Co2+corrinoid solutions in a 0.9:1.0 Co2+corrinoid:LrPduO molar ratio in a sealed anaerobic vial (final enzyme concentration of 0.4 mM). The reaction mixtures, which contained 60% (v/v) glycerol, were then transferred anaerobically into the appropriate sample cells (previously purged with Ar gas) and immediately frozen in liquid N2.
Spectroscopy
Low-temperature electronic absorption (Abs) and magnetic circular dichroism (MCD) spectra were collected on a Jasco J-715 spectropolarimeter in conjunction with an Oxford Instruments SM-4000 8T magnetocryostat. All MCD spectra presented in this paper were obtained by taking the difference between spectra collected with the magnetic field oriented parallel and antiparallel to the light propagation axis to remove contributions from the natural CD and glass strain.
X-band EPR spectra were obtained by using a Bruker ESP 300E spectrometer in conjunction with an Oxford ESR 900 continuous flow liquid helium cryostat and an Oxford ITC4 temperature controller. The microwave frequency was measured with a Varian EIP model 625A CW frequency counter. All spectra were collected using a modulation amplitude of 10 G, a modulation frequency of 100 kHz, and a time constant of 41 ms. EPR spectral simulations were performed using the WEPR program developed by Dr. Frank Neese (34).
RESULTS AND ANALYSIS
Kinetic Studies
To explore whether LrPduO possesses the ability to adenosylate incompletely assembled corrinoids in addition to Co1+Cbl− (32), initial velocity kinetic measurements were performed using chemically reduced Co1+Cbi as the substrate. In these experiments, the concentration of Co1+Cbi was varied while that of the co-substrate ATP was kept at saturation. The apparent Km for Co1+Cbi (Km = 0.096 μM) was found to be similar to the one reported for Co1+Cbl− (Km = 0.13 μM). Likewise, the rates at which LrPduO can adenosylate Co1+Cbl− and Co1+Cbi are nearly indistinguishable (kcat = 2.0 and 2.4 × 10−2 s−1, respectively, see Table 1), indicating that, at least in vitro, the nucleotide loop is not required for the binding and adenosylation of the corrinoid substrate by LrPduO.
Table 1.
Kinetic parameters for the LrPduO catalyzed conversion of Co1+Cbl− and Co1+Cbi to AdoCbl and AdoCbi+, respectivelya
| Substrate | kcat (s−1) | Km (μM) | kcat/Km (M−1 s−1) |
|---|---|---|---|
| Co1+Cbl− | 2.4 ± 0.1 × 10−2 | 13 ± 1 × 10−2 | 1.8 ± 0.2 × 105 |
| Co1+Cbi | 2.0 ± 0.2 × 10−2 | 9.6 ± 1.4 × 10−2 | 2.1 ± 0.4 × 105 |
Data for Co1+Cbl- were taken from ref (32).
Co2+Cbl ↔ LrPduO Interactions
(A) Co2+Cbl + LrPduO
The low-temperature Abs spectra of Co2+Cbl in the absence and presence of LrPduO are nearly identical with respect to band positions and intensities (cf Figures 2A and 2B). Although the addition of LrPduO causes a minor blue-shift of the prominent feature in the visible spectral region (the α-band (35)) of the Co2+Cbl Abs spectrum, from 21010 to 21050 cm−1 (Table 2), it has no discernible effect on the position of the dominant Abs feature in the near-UV region at ~32150 cm−1 (note that the shoulder near 18520 cm−1 in the Co2+Cbl + LrPduO Abs spectrum is due to the presence of a small fraction of re-oxidized corrinoid in that sample).
Figure 2.
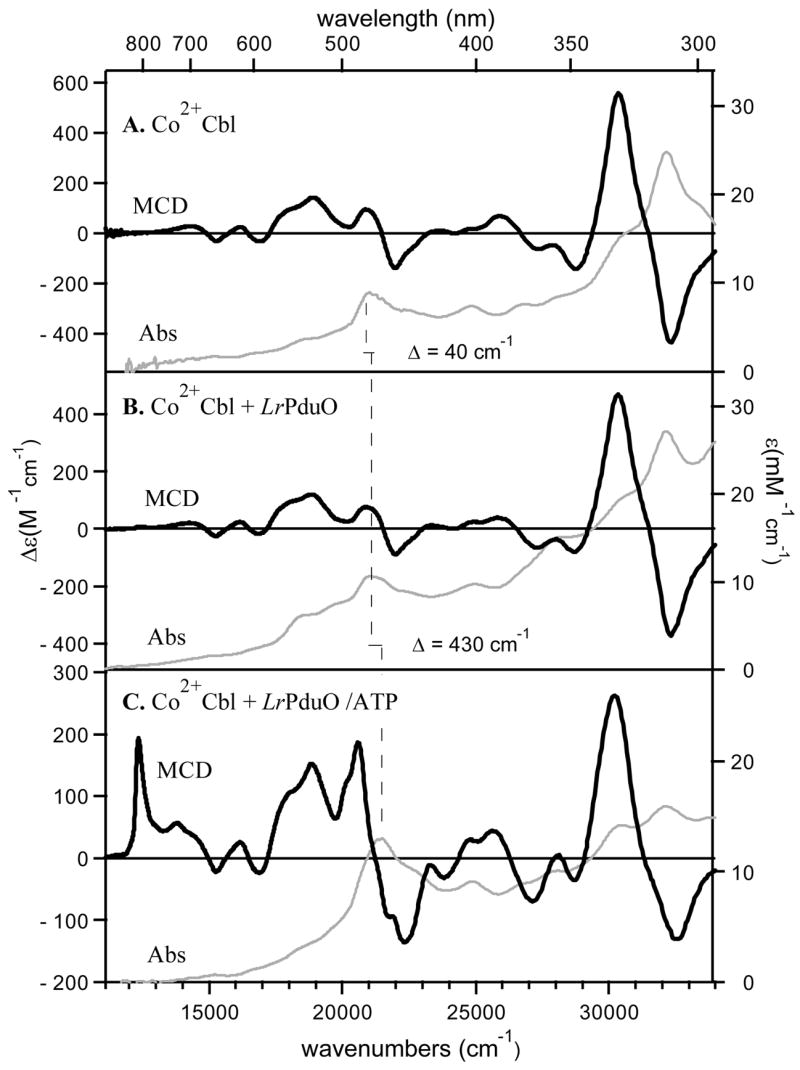
Abs (gray traces, right axis) and 7 T MCD (black traces, left axis) spectra collected at 4.5 K of (A) free Co2+Cbl, (B) Co2+Cbl in the presence of LrPduO, and (C) Co2+Cbl in the presence of the LrPduO/ATP complex. The peak position of the dominant Abs feature (the α-band) is indicated by the vertical solid line.
Table 2.
Changes in the α-band positions of Co2+Cbl and Co2+Cbi+ caused by the addition of LrPduO in the absence and presence of co-substrate ATP, as determined from the 4.5 K Abs spectra in Figures 2 and 4a
| Species | Free Corrinoid | + LrPduO | + LrPduO/ATP |
|---|---|---|---|
| Co2+Cbl | 21010 cm−1 (476.0 nm) | 21050 cm−1 (475.0 nm) | 21480 cm−1 (465.5 nm) |
| Co2+Cbi+ | 21250 cm−1 (470.5 nm) | 21250 cm−1 (470.5 nm) | 21480 cm−1 (465.5 nm) |
Note that a blue-shift of the α-band reflects a weakening of the axial ligand–Co2+corrinoid bonding interaction.
A complementary and considerably more sensitive probe of the interaction between Co2+Cbl and LrPduO is provided by low-temperature MCD spectroscopy. Because the low-energy region of the Co2+corrinoid MCD spectra is dominated by ligand-field (LF) transitions, this region is particularly valuable for monitoring changes in the Co2+ coordination environment (35). Hence, the fact that the MCD spectra of Co2+Cbl in the absence and presence of LrPduO are virtually identical (cf Figures 2A and 2B) rules out any major structural perturbations to the Co2+ center upon cofactor binding to the enzyme active site.
Consistent with our MCD results, the EPR spectra of Co2+Cbl in the absence and presence of LrPduO are nearly superimposable (cf Figures 3A and 3B). However, closer examination of the EPR spectrum of Co2+Cbl + LrPduO reveals the presence of a broad feature near 2600 G that has no counterpart in the spectrum of the free cofactor. This feature is characteristic of base-off Co2+Cbl (36, 37), a species in which a water molecule binds to the lower axial coordination site of the Co2+ center that was originally occupied by the DMB moiety in base-on Co2+Cbl (Figure 1). A quantitative analysis of the Co2+Cbl + LrPduO EPR spectrum reveals that this sample contains ~30% base-off Co2+Cbl. This finding correlates nicely with the slight blue-shift of the α-band that is observed in the Co2+Cbl Abs spectrum upon the addition of LrPduO (Figures 2A and 2B, Table 2). The g and 59Co hyperfine values A(Co) obtained from simulations of the EPR spectra presented in Figure 3 are summarized in Table 3.
Figure 3.
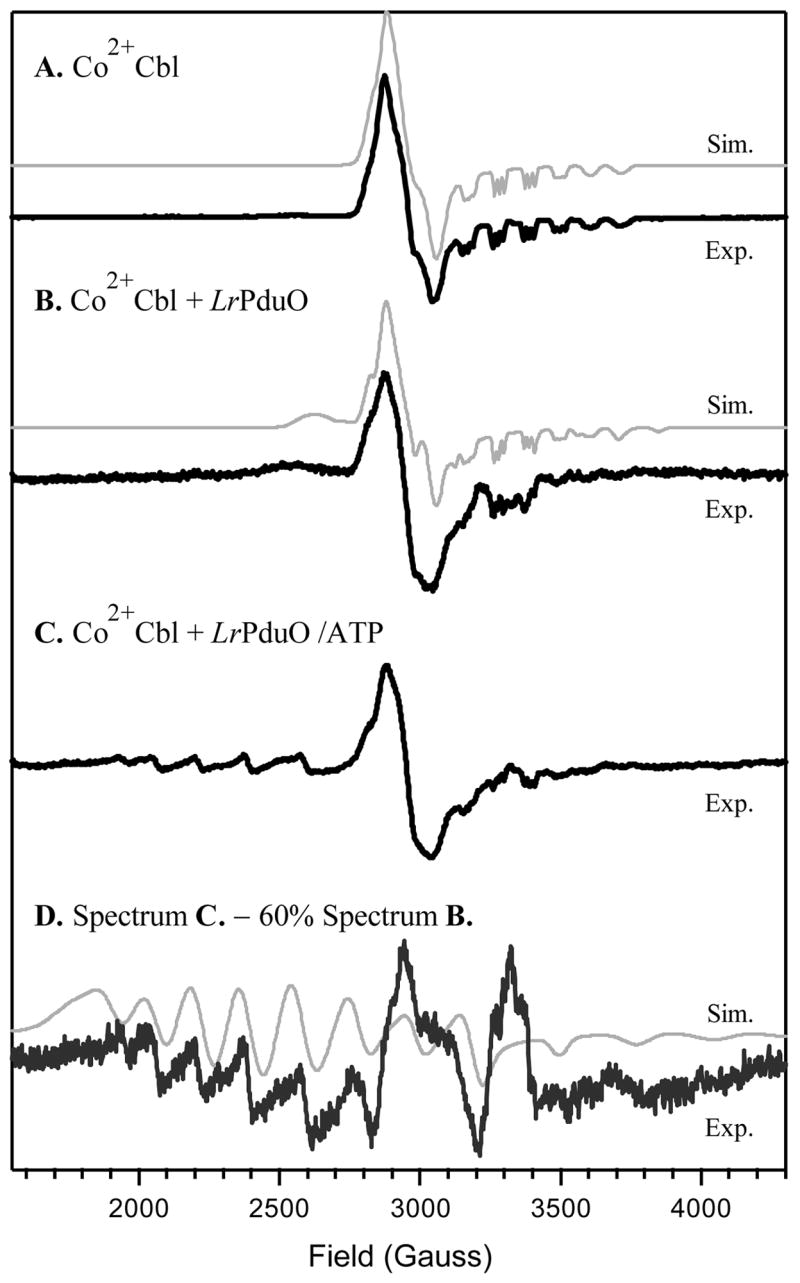
EPR spectra collected at 20 K of (A) free Co2+Cbl, (B) Co2+Cbl in the presence of LrPduO, and (C) Co2+Cbl in the presence of the LrPduO/ATP complex. Spectrum D was obtained by subtracting 60% of spectrum B from spectrum C. Spectral simulations (thin lines) were performed using the parameters provided in Table 3.
Table 3.
EPR g Values and 59Co Hyperfine Values A(Co) (in MHz) from the spectral simulations presented in Figures 3 and 5
| Species | Spectruma | g1 | g2 | g3 | A1(Co) | A2(Co) | A3(Co) |
|---|---|---|---|---|---|---|---|
| Free Co2+Cbl b | 3A | 2.00 | 2.23 | 2.28 | 305 | 30 | 40 |
| Co2+Cbl + LrPduO c | 3B | 2.00
2.00 |
2.23
2.34 |
2.28
2.37 |
305
400 |
30
220 |
40
220 |
| Co2+Cbl + LrPduO/ATP | 3D | 1.99 | 2.70 | 2.72 | 770 | 755 | 595 |
| Free Co2+Cbi+ b | 5A | 2.00 | 2.34 | 2.34 | 405 | 220 | 220 |
| Co2+Cbi+ + LrPduO | 5B | 2.00 | 2.34 | 2.37 | 400 | 220 | 220 |
| Co2+Cbi+ + LrPduO/ATP | 5D | 1.99 | 2.65 | 2.74 | 810 | 665 | 565 |
(B) Co2+Cbl + LrPduO/ATP
While the spectral changes in response to Co2+Cbl binding to substrate-free LrPduO are quite modest (see above), a rather significant blue-shift of the α-band is observed in the Abs spectrum of Co2+Cbl upon the addition of the LrPduO complexed with the co-substrate ATP (cf Figures 2B and 2C, Table 2). This shift occurs in parallel with a dramatic change to the MCD spectrum, most notably the appearance of new features at ~12340 and 20580 cm−1 (Figure 2C). Because the MCD feature at 12340 cm−1 has no discernible counterpart in the Abs spectrum, it can be attributed to a magnetic-dipole allowed Co2+ d→d transition (38, 39). An analogous low-energy MCD feature has been observed for Co2+Cbl bound to the hATR/ATP complex, where it was shown to reflect the presence of an effectively four-coordinate Co2+Cbl species (29). Our MCD data thus provide compelling evidence that a four-coordinate Co2+Cbl species is also formed when Co2+Cbl binds to the LrPduO/ATP complex. This conversion is not complete, however, as the negative features at 15250 and 16880 cm−1 and the positive features at 18000 and 18850 cm−1 in the MCD spectrum of Co2+Cbl + LrPduO/ATP (Figure 2C) are characteristic of base-on Co2+Cbl (see Figure 2A). This fraction of five-coordinate Co2+Cbl corresponds to either unbound corrinoid substrate or to an enzyme-bound species that has resisted conversion to the four-coordinate form.
In agreement with our MCD data presented in Figure 2, the EPR spectrum of Co2+Cbl + LrPduO/ATP (Figure 3C) reveals the presence of at least two distinct paramagnetic species, one exhibiting an EPR spectrum characteristic of base-on Co2+Cbl (see Figure 3A) and the other giving rise to the appearance of a series of widely spread resonances in the low-field region. To better resolve the features associated with the latter species, the suitably scaled (×0.6) Co2+Cbl + LrPduO EPR spectrum (Figure 3B) was subtracted from the composite spectrum in Figure 3C to obtain the trace shown in Figure 3D. As expected on the basis of our MCD data analysis (vide supra), the resulting spectrum is very similar to that reported for the four-coordinate Co2+Cbl species in samples of Co2+Cbl + hATR/ATP (29). In particular, the g2,3 and A(Co) values obtained from a fit of the EPR spectrum in Figure 3D are much larger than those of any five-coordinate Co2+Cbl species (Table 3), and are instead characteristic of an essentially square-planar Co2+ complex that lacks any significant axial bonding interactions (28, 40, 41).
Co2+Cbi+ ↔ LrPduO Interactions
(A) Co2+Cbi+ + LrPduO
The low-temperature Abs and MCD spectra of Co2+Cbi+ in the absence and presence of LrPduO are very similar to each other (cf Figures 4A and 4B). However, while the addition of LrPduO to the Co2+Cbi+ solution has no effect on the peak position of the α-band (Table 2), it causes a small but notable broadening of several features in the 16000 – 21000 cm−1 region of the MCD spectrum. Similarly, the EPR spectrum of Co2+Cbi+ is slightly perturbed when LrPduO is present, especially between 2800 and 3100 G (cf Figures 5A and 5B). Collectively, these data suggest that Co2+Cbi+ binds to LrPduO even in the absence of co-substrate ATP, but retains a five-coordinate Co2+ center with an axially bound water molecule. Interestingly, our simulation of the Co2+Cbi++ LrPduO EPR spectrum reveals that g2 ≠ g3 for this species, indicating that the Co2+ ion is in a ligand environment of rhombic symmetry, while for free Co2+Cbi+ an axially symmetric spectrum with g2 = g3 is observed (see Table 3).
Figure 4.
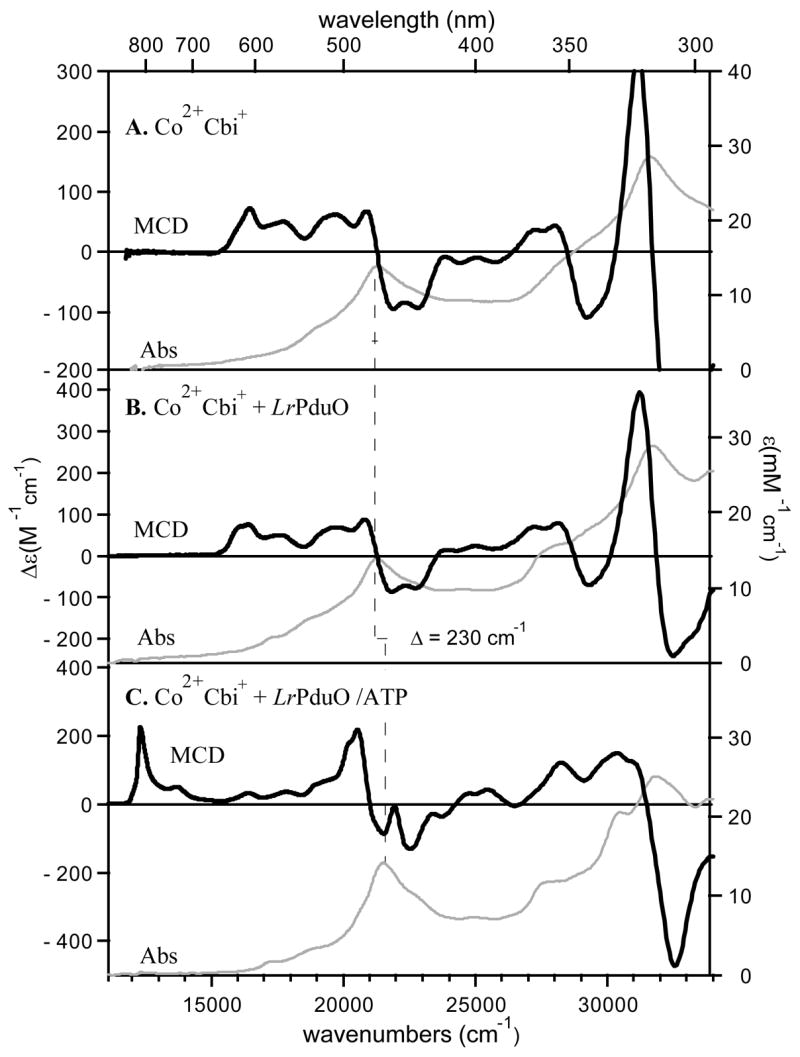
Abs (gray traces, right axis) and 7 T MCD (black traces, left axis) spectra collected at 4.5 K of (A) free Co2+Cbi+, (B) Co2+Cbi+ in the presence of LrPduO, and (C) Co2+Cbi+ in the presence of the LrPduO/ATP complex. The peak position of the dominant Abs feature (the α-band) is indicated by the vertical solid line.
Figure 5.
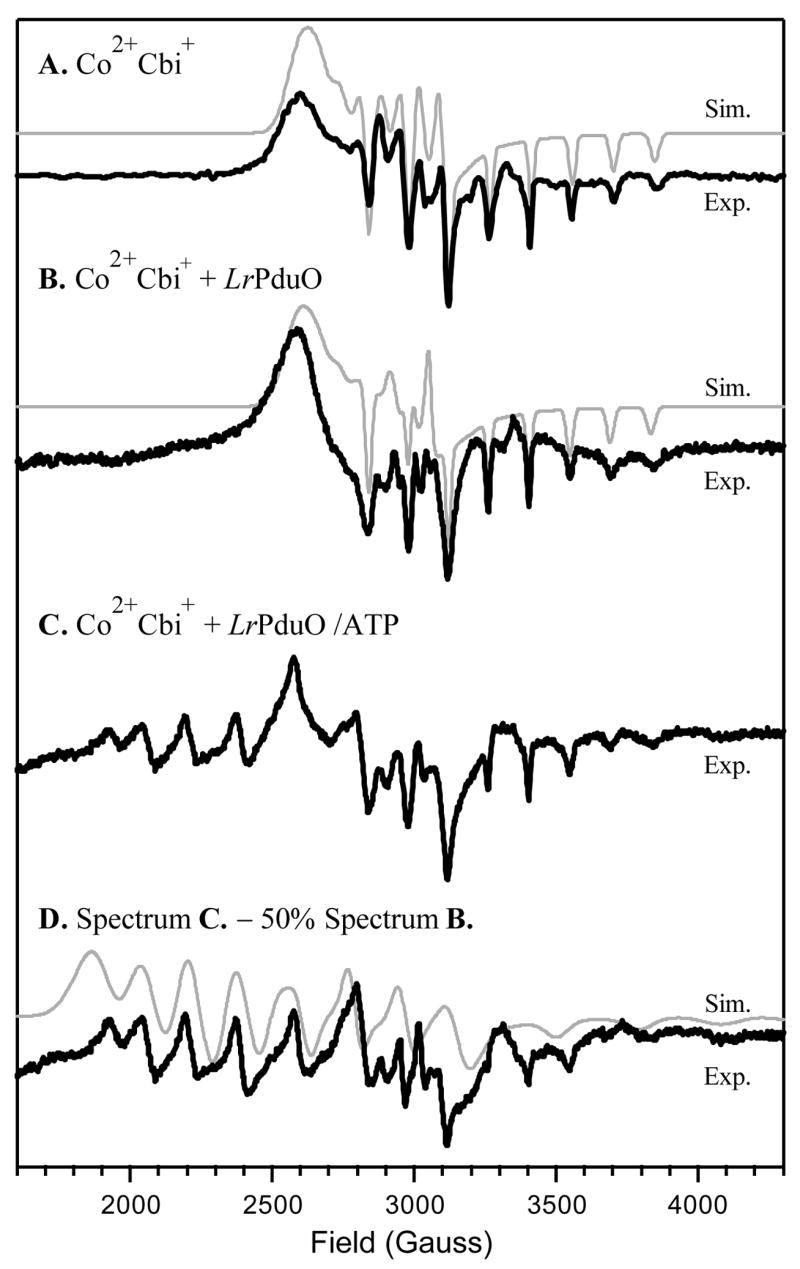
EPR spectra collected at 20 K of (A) free Co2+Cbi+, (B) Co2+Cbi+ in the presence of LrPduO, and (C) Co2+Cbi+ in the presence of the LrPduO/ATP complex. Spectrum D was obtained by subtracting 50% of spectrum B from spectrum C. Spectral simulations (thin lines) were performed using the parameters provided in Table 3.
(B) Co2+Cbi+ + LrPduO/ATP
As in the case of Co2+Cbl + LrPduO/ATP (Figure 2), a significant blue-shift of the α-band (Table 2) and the appearance of prominent MCD features at 12310 and 20530 cm−1 are observed when Co2+Cbi+ is incubated with LrPduO complexed with the co-substrate ATP (cf Figures 4A and 4C). Again, the conversion to a four-coordinate Co2+corrinoid species is incomplete, based on the observation of weak positive features in the 16000 – 20000 cm−1 region of the Co2+Cbl + LrPduO/ATP MCD spectrum that are characteristic of five-coordinate Co2+Cbi+ (Figure 4A).
As anticipated on the basis of our MCD data in Figure 4, the EPR spectrum of Co2+Cbi+ + LrPduO/ATP (Figure 5C) has contributions from two distinct paramagnetic species, an effectively four-coordinate Co2+Cbi+ species (responsible for the widely spread resonances in the low-field region) and five-coordinate Co2+Cbi+ possessing an axially bound water molecule (see Figure 5B). To obtain a pure spectrum of the four-coordinate Co2+Cbi+ fraction, the suitably scaled (×0.5) Co2+Cbi+ + LrPduO EPR spectrum (Figure 5B) was subtracted from the composite spectrum in Figure 5C. Although the resulting spectrum (Figure 5D) is qualitatively very similar to that exhibited by the four-coordinate Co2+Cbl fraction bound to the LrPduO/ATP complex (Figure 3D), the g2,3 and A(Co) values are in fact somewhat different (Table 3). This result suggests that even though the DMB moiety no longer serves as an axial ligand in the four-coordinate Co2+Cbl + LrPduO/ATP fraction, it nonetheless modulates the geometric and electronic structures of the Co2+ center to a small but noticeable degree, presumably via hydrogen-bonding to amino-acid residues near the enzyme active site.
DISCUSSION
LrPduO can adenosylate Co1+Cbl− and Co1+Cbi
Thus far, only the CobA-type adenosyltrasferases were believed to participate in the adenosylation of complete as well as incomplete corrinoids (42). However, the kinetic data obtained in this study provide evidence that in vitro a PduO-type adenosyltransferase can utilize incomplete corrinoids as alternative substrates, in support of a previous proposal that was based on in vivo evidence (32) Interestingly, LrPduO adenosylates Co1+Cbl− and Co1+Cbi with approximately the same catalytic efficiency, as judged on the basis of the similar kcat/Km values (Table 1). Additionally, under saturating ATP conditions the apparent Km of LrPduO for Co1+Cbi is nearly identical to that reported for Co1+Cbl− and at least 10-fold lower than for any previously reported adenosyltransferase (32). These results suggest that the nucleotide tail (Figure 1) is relatively unimportant for corrinoid binding to the enzyme active site and for product formation. It is important to note, though, that chemically reduced Co1+corrinoids were used in our activity assays. Thus our kinetic data do not rule out the possibility that the nucleotide tail may be required for allowing the physiological electron donor to accomplish the Co2+ → Co1+corrinoid reduction. However, such a requirement is quite unlikely, based on the close resemblance of our spectroscopic data obtained for Co2+Cbl and Co2+Cbi+ bound to the LrPduO/ATP complex (see below) and their obvious similarities with those reported for the CobA adenosyltransferase (28).
Interaction between Co2+corrinoids and LrPduO in the absence of co-subtrate ATP
The fact that the Abs and MCD spectra of Co2+Cbl and Co2+Cbi+ change only slightly when LrPduO is added (Figures 2–5) indicates that the geometric and electronic structures of these species are largely preserved in the presence of the enzyme. Nevertheless, small but noticeable changes are observed in each case, signaling that Co2+corrinoid binding to the enzyme active site does occur even in the absence of co-substrate ATP. Specifically, when a solution of Co2+Cbl is incubated with LrPduO, a significant fraction (~30%) of the cofactor converts to the base-off form, in which the Co2+ center is axially ligated by a water molecule instead of the DMB moiety (Figure 1) (35). Alternatively, the addition of LrPduO to a solution of Co2+Cbi+ causes a small rhombic splitting of the EPR signal (Table 3), possibly reflecting a perturbation of the equatorial ligand environment of the Co2+ ion; e.g., via hydrogen-bonding of active-site residues to the peripheral side chains of the corrin macrocycle.
Interaction between Co2+corrinoids and the LrPduO/ATP complex
While the spectroscopic properties of Co2+Cbl and Co2+Cbi+ are only weakly affected by the addition of LrPduO, rather drastic spectral changes are observed when the enzyme was pre-incubated with the co-substrate ATP (Figures 2–5). In each case, binding of the Co2+corrinoid substrate to the LrPduO/ATP complex leads to a substantial blue-shift of the dominant Abs feature in the visible spectral region, termed the α-band (Table 2), which corresponds to the lowest-energy corrin-centered π → π* transition (43–45). Previous studies revealed that the corrin π-based donor orbital involved in this transition also possesses some Co dz2 character, the extent of which is governed by the σ-donor strength of the axial ligand. As a result, the α-band of Co2+corrinoids shifts to higher energy with decreasing electron donating ability of the axial ligand (e.g., from Co2+Cbl to Co2+Cbi+, see Table 2) or a weekending of the axial bonding interaction (35, 46).
The large blue-shift of the α-band in response to Co2+Cbl and Co2+Cbi+ binding to the LrPduO/ATP complex is accompanied by drastic changes to the corresponding MCD spectra, most notably the appearance of a prominent low-energy feature at ~12340 cm−1 (Figures 2 and 4), as well as to their EPR data, in particular a large increase in the g2, g3, and A(Co) values (Table 3). Collectively, these spectral perturbations are consistent with a sizeable stabilization of the singly occupied Co 3dz2-based molecular orbital (28) owing to a significant weakening of the axial ligand–Co2+corrinoid bonding interaction, so as to generate essentially square-planar Co2+ species in the enzyme active site. Although these “activated” forms of Co2+Cbl and Co2+Cbi+ bound to the LrPduO/ATP complex are best described as possessing an effectively four-coordinate Co2+ center that lacks any significant axial bonding interactions, the corresponding MCD and EPR spectra (Figures 2–5) exhibit small but notable differences. Specifically, when the alternative substrate Co2+Cbi+ is used instead of Co2+Cbl, the prominent MCD feature in the near-IR spectral region displays a minor red-shift (by ~30 cm−1, Table 4) and the EPR g and A(Co) values become more rhombic (Table 3). These observations suggest that even though the DMB moiety is displaced from the Co2+ ion in the Co2+Cbl + LrPduO/ATP complex, it may still play a role in organizing the enzyme active site for maximum catalytic activity (e.g., by promoting Co2+ → Co1+ reduction).
Table 4.
Peak position of the prominent MCD feature in the near-IR spectral region observed for Co2+corrinoids bound to several alkyltransferase/ATP complexes
The spectroscopic data obtained in this work nicely complement the information that has recently been obtained from structural and kinetic studies of LrPduO. First, Mera et al. prepared numerous variants of LrPduO that were characterized by using kinetic and X-ray crystallographic techniques to develop a better understanding of the function of several conserved residues in the enzyme active site (47). Importantly, these studies revealed that substrate inhibition occurs at subsaturating concentrations of ATP, in strong support of an ordered binding scheme according to which the enzyme must bind co-substrate ATP prior to the corrinoid substrate. Second, in the course of this investigation, St. Maurice et al. succeeded in obtaining an X-ray crystal structure (at 1.9 Å resolution) of Co2+Cbl bound to the LrPduO/ATP complex (48). As expected on the basis of our spectroscopic data reported here, this structure provides visual evidence for the formation of a base-off, four-coordinate Co2+Cbl intermediate in the catalytic cycle of LrPduO. This structure also offers clues as to how the enzyme imposes such a unique coordination environment on the Co2+corrioind substrate. Specifically, it shows that the N-terminus of LrPduO that is disordered in the substrate-free enzyme (32) becomes ordered upon ATP-binding, thereby inducing a distinct active-site conformation that is poised for Co2+corrinoid binding (48). Moreover, a conserved Phe residue moves to within 3.8 Å of the Co2+ ion so as to promote dissociation of the axially bound 22 solvent ligand that would normally be present in base-off Co2+Cbl. Although not obvious from the LrPduO crystal structures, our MCD and EPR data indicate that the formation of such a four-coordinate Co2+Cbl intermediate leads to a significant stabilization of the redox-active Co 3dz2 orbital that is oriented toward the axial coordination sites, thereby raising the Co2+/1+ reduction midpoint potential by an estimated 250 mV (28).
Comparison to other adenosyltransferases
The involvement of unprecedented, essentially four-coordinate Co2+corrinoid intermediates in the catalytic cycles of adenosyltransferases has originally been proposed on the basis of spectroscopic and computational studies of CobA from Salmonella enterica and the human enzyme hATR (28, 29). As expected by taking into consideration that hATR is a PduO-type adenosyltransferase, Co2+Cbl interacts similarly with this enzyme and LrPduO. Specifically, while binding of Co2+Cbl to hATR in the absence of ATP leads to a partial (~40%) conversion to the base-off form, a significant blue-shift of the α-band and the appearance of the MCD and EPR spectroscopic signatures characteristic of an approximately four-coordinate Co2+corrinoid species are observed when hATR was pre-incubated with co-substrate ATP (29). However, the prominent low-energy feature in the MCD spectrum of Co2+Cbl + hATR/ATP is blue-shifted by 240 cm−1 relative to that of Co2+Cbl + LrPduO/ATP (Table 4), signifying a slightly stronger residual axial bonding interaction in the former species. Moreover, a comparison of the corresponding EPR spectra reveals that the conversion from the five- to the four-coordinate Co2+Cbl species is more complete in the case of hATR (~90%) than for LrPduO (~40%). Collectively, these results suggest that a stronger interaction could exist between the dissociated DMB moiety of the “activated” Co2+Cbl species and the enzyme active site in hATR than in LrPduO. It is tempting to speculate, then, that due to this increased interaction with the nucleotide loop, hATR may exhibit an increased substrate specificity and thus be unable to adenosylate incompletely assembled corrinoids, such as Co2+Cbi+. Although to our knowledge the range of possible corrinoid substrates for hATR has not yet been established, it is interesting to note that the PduO-type adenosyltransferase from S. enterica appears to be specific for AdoCbl synthesis, despite its high sequence similarity with LrPduO (49).
In the case of CobA, for which the natural substrate is a Co2+Cbl precursor that lacks the nucleotide loop and thus more closely resembles Co2+Cbi+ (16, 50), MCD and EPR spectroscopic studies revealed that in the Co2+Cbi+ + CobA/ATP complex, a predominant fraction (~70%) of four-coordinate Co2+corrinoid species is formed (28). When the alternative substrate Co2+Cbl is added to the CobA/ATP complex, however, the conversion to a similar four-coordinate Co2+ species is largely suppressed (to ~10%). While hATR and CobA are thus relatively specific for their respective substrates, the results obtained in this study reveal that a sizeable a fraction of a four-coordinate Co2+corrinoid species is formed regardless of whether Co2+Cbl or Co2+Cbi+ binds to the LrPduO/ATP complex (40% and 50%, respectively). Although this low substrate-specificity of LrPduO is consistent with our kinetic data reported in the Results and Analysis section (Table 1), this finding is rather unexpected, because among the three types of adenosyltransferases found in Salmonella enterica, CobA has been presumed to be the only enzyme capable of adenosylating incomplete corrionid species (7, 8, 16, 28). Yet, the nearly identical positions of the prominent near-IR features in the MCD spectra of Co2+Cbi+ + LrPduO/ATP and Co2+Cbi+ + CobA/ATP (Table 4) suggest that Co2+Cbi+ does in fact interact similarly with the LrPduO/ATP and CobA/ATP complexes. Despite these similarities, a more complete conversion to a four-coordinate Co2+Cbi+ species occurs in the latter enzyme (50% and 70% for LrPduO and CobA, respectively).
Although small differences exist among the spectroscopic signatures of the formally four-coordinate Co2+corrinoid species that have now been identified in the active sites of various adenosyltransferases (28, 29), the general strategy of these enzymes evidently involves the formation of an essentially square-planar Co2+corrinoid intermediate, so as to reduce the thermodynamic barrier for the reduction to the Co1+ state through stabilization of the redox-active, Co 3dz2-based molecular orbital that is oriented along the axial coordination sites of the Co2+ ion. Remarkably, for each adenosyltransferase studied to date (28, 29), this “activated” Co2+corrinoid intermediate is formed in high yield only in the presence of co-substrate ATP, which provides a mechanism for protecting the enzyme active site from being attacked by the transiently formed Co1+corrinoid “supernucleophile”. Collectively, the insights gained in this study and the recent structural and kinetic characterizations of LrPduO provide an excellent basis for further investigations into the fascinating bio-organometallic reaction catalyzed by PduO-type adenosyltransferases.
Footnotes
This work was supported in part by the National Science Foundation grant MCB-0238530 (to T.C.B.) and the National Institutes of Health grant R01-GM40313 (to J.C.E.-S.). P.E.M was supported in part by National Research Service Award 5F31GM081979-02 from the National Institute of General Medical Sciences (NIGMS).
Abbreviations: Abs, electronic absorption; AdoCbl, adenosylcobalamin; AdoCbi+, adenosylcobinamide; H2OCbl+, aquacobalamin; Cbl, cobalamin; cbi, cobinamide; CNCbl, cyanocobalamin; (H2O)2Cbi2+, diaquacobinamide; (CN)2Cbi, dicyanocobinamide; DMB, dimethylbenzimidazole; EPR, electron paramagnetic resonance; hATR, human adenosyltransferase; LrPduO, Lactobacillus reuteri PduO; MCD, magnetic circular dichroism; NaBH4, sodium borohydride.
References
- 1.Butler PA, Kräutler B. Topics in organometallic chemistry. Vol. 17. Springer; Berlin, Heidelberg: 2006. [Google Scholar]
- 2.Lenhert PG. The structure of vitamin B12 VII. The X-ray analysis of the vitamin B12 coenzyme. Proceedings of the Royal Society of London Series A. 1968;303:45–84. [Google Scholar]
- 3.Chowdhury S, Banerjee R. Role of the dimethylbenzimidazole tail in the reaction catalyzed by coenzyme B12-dependent methylmalonyl-CoA mutase. Biochemistry. 1999;38:15287–15294. doi: 10.1021/bi9914762. [DOI] [PubMed] [Google Scholar]
- 4.Banerjee R. Radical peregrinations catalyzed by coenzyme B12-dependent enzymes. Biochemistry. 2001;40:6191–6198. doi: 10.1021/bi0104423. [DOI] [PubMed] [Google Scholar]
- 5.Chowdhury S, Banerjee R. Thermodynamic and kinetic characterization of Co–C bond homolysis catalyzed by coenzyme B12-dependent methylmalonyl-CoA mutase. Biochemistry. 2000;39:7998–8006. doi: 10.1021/bi992535e. [DOI] [PubMed] [Google Scholar]
- 6.Marsh ENG, Ballou DP. Coupling of cobalt-carbon bond homolysis and hydrogen atom abstraction in adenosylcobalamin-dependent glutamate mutase. Biochemistry. 1998;37:11864–11872. doi: 10.1021/bi980512e. [DOI] [PubMed] [Google Scholar]
- 7.Johnson CLV, Pechonick E, Park SD, Havemann GD, Leal NA, Bobik TA. Functional genomic, biochemical, and genetic characterization of the Salmonella pduO gene, an ATP:cob(I)alamin adenosyltransferase gene. J Bacteriol. 2001;183:1577–1584. doi: 10.1128/JB.183.5.1577-1584.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buan NR, Suh S-J, Escalante-Semerena JC. The eutT gene of Salmonella enterica encodes an oxygen-labile, metal-containing ATP:corrinoid adenosyltransferase enzyme. J Bacteriol. 2004;186:5708–5714. doi: 10.1128/JB.186.17.5708-5714.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thoma NH, Evans PR, Leadlay PF. Protection of radical intermediates at the active site of adenosylcobalamin-dependent methylmalonyl-CoA mutase. Biochemistry. 2000;39:9213–9221. doi: 10.1021/bi0004302. [DOI] [PubMed] [Google Scholar]
- 10.Gerfen GJ, Licht S, Willems J-P, Hoffman BM, Stubbe J. Electron paramagnetic resonance investigations of a kinetically competent intermediate formed in ribonucleotide reduction: evidence for a thiyl radical-cob(II)alamin interaction. J Am Chem Soc. 1996;118:8192–8197. [Google Scholar]
- 11.Brown KL, Zou X. Thermolysis of coenzymes B12 at physiological temperatures: activation parameters for cobalt–carbon bond homolysis and a quantitative analysis of the perturbation of the homolysis equilibrium by the ribonucleoside triphosphate reductase from Lactobacillus leichmannii. Journal of Inorganic Biochemistry. 1999;77:185–195. doi: 10.1016/s0162-0134(99)00190-7. [DOI] [PubMed] [Google Scholar]
- 12.Banerjee R. Radical carbon skeleton rearrangements: catalysis by coenzyme B12-dependent mutases. Chem Rev. 2003;103:2083–2094. doi: 10.1021/cr0204395. [DOI] [PubMed] [Google Scholar]
- 13.Banerjee R, Ragsdale SW. The many faces of vitamin B12: catalysis by cobalamin-dependent enzymes. Annu Rev Biochem. 2003;72:209–247. doi: 10.1146/annurev.biochem.72.121801.161828. [DOI] [PubMed] [Google Scholar]
- 14.Marsh ENG, Drennan CL. Adenosylcobalamin-dependent isomerases: new insight into structure and mechanism. Curr Opin Chem Biol. 2001;5:499–505. doi: 10.1016/s1367-5931(00)00238-6. [DOI] [PubMed] [Google Scholar]
- 15.Fenton WA, Rosenberg LE. The defect in the CblB class of human methylmalonic aciduria: deficiency of cob(I)alamin adenosyltransferase activity in extracts of cultured fibroblasts. Biochemical and Biophysical Research Communications. 1981;98:283–289. doi: 10.1016/0006-291x(81)91900-8. [DOI] [PubMed] [Google Scholar]
- 16.Escalante-Semerena JC, Suh S-J, Roth JR. CobA function is required for both de novo cobalamin biosynthesis and assimilation of exogenous corrinoids in Salmonella typhimurium. J Bacteriol. 1990;172:273–280. doi: 10.1128/jb.172.1.273-280.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dobson CM, Wai T, Leclerc D, Kadir H, Narang M, Lerner-Ellis JP, Hudson TJ, Rosenblatt DS, Gravel RA. Identification of the gene responsible for the cblB complementation group of vitamin B12-dependent methylmalonic aciduria. Hum Mol Gen. 2002;11:3361–3369. doi: 10.1093/hmg/11.26.3361. [DOI] [PubMed] [Google Scholar]
- 18.Warren MJ, Raux E, Schubert HL, Escalante-Semerena JC. The biosynthesis of adenosylcobalamin (vitamin B12) Natural Product Reports. 2002;19:390–412. doi: 10.1039/b108967f. [DOI] [PubMed] [Google Scholar]
- 19.Walker GA, Murphy S, Huennekens FM. Enzymatic conversion of vitamin B12a to adenosyl-B12: evidence for the existence of two separate reducing systems. Arch Biochem Biophysics. 1969;134:95–102. doi: 10.1016/0003-9861(69)90255-0. [DOI] [PubMed] [Google Scholar]
- 20.Lawrence AD, Deery E, McLean KJ, Munro AW, Pickersgill RW, Rigby SE, Warren MJ. Identification, characterization and structure/function analysis of a corrin reductase involved in adenosylcobalamin biosynthesis. J Biol Chem. 2008 doi: 10.1074/jbc.M710431200. in press. [DOI] [PubMed] [Google Scholar]
- 21.Fonseca MV, Escalante-Semerena JC. Reduction of cob(III)alamin to cob(II)alamin in Salmonella enterica Serovar Typhimurium LT2. J Bacteriol. 2000;182:4304–4309. doi: 10.1128/jb.182.15.4304-4309.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Olteanu H, Wolthers KR, Munro AW, Scrutton NS, Banerjee R. Kinetic and thermodynamic characterization of the common polymorphic variants of human methionine synthase reductase. Biochemistry. 2004;43:1988–1997. doi: 10.1021/bi035910i. [DOI] [PubMed] [Google Scholar]
- 23.Lexa D, Saveant J-M. The electrochemistry of vitamin B12. Acc Chem Res. 1983;16:235–243. [Google Scholar]
- 24.Mciver L, Leadbeater C, Campopiano DJ, Baxter RL, Daff SN, Chapman SK, Munro AW. Characterisation of flavodoxin NADP+ oxidoreductase and flavodoxin; key components of electron transfer in Escherichia coli. Eur J Biochem. 1998;257:557–585. doi: 10.1046/j.1432-1327.1998.2570577.x. [DOI] [PubMed] [Google Scholar]
- 25.Fonseca MV, Escalante-Semerena JC. An in vitro reducing system for the enzymic conversion of cobalamin to adenosylcobalamin. J Biol Chem. 2001;276:32101–32108. doi: 10.1074/jbc.M102510200. [DOI] [PubMed] [Google Scholar]
- 26.Sampson EM, Johnson CLV, Bobik TA. Biochemical evidence that the pduS gene encodes a bifunctional cobalamin reductase. Microbiology. 2005;151:1169–1177. doi: 10.1099/mic.0.27755-0. [DOI] [PubMed] [Google Scholar]
- 27.Leal NA, Olteanu H, Banerjee R, Bobik TA. Human ATP:cob(I)alamin adenosyltransferase and its interaction with methionine synthase reductase. J Biol Chem. 2004;279:47536–47542. doi: 10.1074/jbc.M405449200. [DOI] [PubMed] [Google Scholar]
- 28.Stich TA, Buan NR, Escalante-Semerena JC, Brunold TC. Spectroscopic and computational studies of the ATP:corrinoid adenosyltransferase (CobA) from Salmonella enterica: insights into the mechanism of adenosylcobalamin biosynthesis. J Am Chem Soc. 2005;127:8710–8719. doi: 10.1021/ja042142p. [DOI] [PubMed] [Google Scholar]
- 29.Stich TA, Yamanishi M, Banerjee R, Brunold TC. Spectroscopic evidence for the formation of a four-coordinate Co2+cobalamin species upon binding to the human ATP:cobalamin adenosyltransferase. J Am Chem Soc. 2005;127:7660–7661. doi: 10.1021/ja050546r. [DOI] [PubMed] [Google Scholar]
- 30.Leal NA, Park SD, Kima PE, Bobik TA. Identification of the human and bovine ATP:cob(I)alamin adenosyltransferase cDNAs based on complementation of a bacterial mutant. J Biol Chem. 2003;278:9227–9234. doi: 10.1074/jbc.M212739200. [DOI] [PubMed] [Google Scholar]
- 31.Dobson CM, Wai T, Leclerc D, Wilson A, Wu X, Dore C, Hudson T, Rosenblatt DS, Gravel RA. Identification of the gene responsible for the cblA complementation group of vitamin B12-responsive methylmalonic aciduria based on analysis of prokaryotic gene arrangements. Proc Natl Acad Sci USA. 2002;99:15554–15559. doi: 10.1073/pnas.242614799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.StMaurice M, Mera PE, Taranto MP, Sesma F, Escalante-Semerena JC, Rayment I. Structural characterization of the active site of the PduO-type ATP:co(I)rrinoid adenosyltransferase from Lactobacillus reuteri. J Biol Chem. 2007;282:2596–2605. doi: 10.1074/jbc.M609557200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rocco CJ, Dennison KL, Klenchin VA, Rayment I, Escalante-Semerena JC. Construction and use of new cloning vectors for the rapid isolation of recombinant proteins from Escherichia coli. Plasmid. 2008 doi: 10.1016/j.plasmid.2008.01.001. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Neese, F. (1997), University of Konstanz.
- 35.Stich TA, Buan NR, Brunold TC. Spectroscopic and computational studies of Co2+corrinoids: spectral and electronic properties of the biologically relevant base-on and base-off forms of Co2+cobalamin. J Am Chem Soc. 2004;126:9735–9749. doi: 10.1021/ja0481631. [DOI] [PubMed] [Google Scholar]
- 36.Pilbrow JR. In: B12. Dolphin D, editor. Wiley; New York: 1982. pp. 431–462. [Google Scholar]
- 37.Doorslaer SV, Jeschke G, Epel B, Goldfarb D, Eichel R-A, Kräutler B, Schweiger A. Axial solvent coordination in “base-off” cob(II)alamin and related Co(II)-corrinates revealed by 2D-EPR. J Am Chem Soc. 2003;125:5915–5927. doi: 10.1021/ja021218j. [DOI] [PubMed] [Google Scholar]
- 38.Johnson MK. In: Physical methods in bioinorganic chemistry. Lawrence Que J, editor. University Science Books; Sausalito: 2000. pp. 233–285. [Google Scholar]
- 39.Schellman JA. Circular dichroism and optical rotation. Chem Rev. 1975;75:323–331. [Google Scholar]
- 40.Ozarowski A, Lee HM, Balch AL. Crystal environments probed by EPR spectroscopy. Variations in the EPR spectra of CoII(octaethylporphyrin) doped in crystalline diamagnetic hosts and a reassessment of the electronic structure of four-coordinate cobalt(II) J Am Chem Soc. 2003;125:12606–12614. doi: 10.1021/ja030221f. [DOI] [PubMed] [Google Scholar]
- 41.Assour JM, Kahn WK. Electron spin resonance of α- and β-cobalt phthalocyanine. J Am Chem Soc. 1965;87:207–212. [Google Scholar]
- 42.Suh S-J, Escalante-Semerena JC. Purification and initial characterization of the ATP:corrinoid adenosyltransferase encoded by the cobA gene of Salmonella typhimurium. J Bacteriol. 1995;177:921–925. doi: 10.1128/jb.177.4.921-925.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kuhn H, Drexhage KH, Martin H. The light absorption of vitamin B12. Proceedings of the Royal Society of London Series. 1965;A288:348–351. [Google Scholar]
- 44.Firth RA, Hill HAO, Pratt JM, Williams RJP, Jackson WR. The 31 circular dichroism and absorption spectra of some vitamin B12 derivatives. Biochemistry. 1967;6:2178–2189. doi: 10.1021/bi00859a040. [DOI] [PubMed] [Google Scholar]
- 45.Day P. A theory of the optical properties of vitamin B12 and its derivatives. Theoret Chim Acta. 1967;7:328–341. [Google Scholar]
- 46.Stich TA, Brooks AJ, Buan NR, Brunold TC. Spectroscopic and computational studies of Co3+-corrinoids: spectral and electronic properties of the B12 cofactors and biologically relevant precursors. J Am Chem Soc. 2003;125:5897–5914. doi: 10.1021/ja029328d. [DOI] [PubMed] [Google Scholar]
- 47.Mera PE, StMaurice M, Rayment I, Escalante-Semerena JC. Structural and functional analyses of the human-type corrinoid adenosyltransferase (PduO) from Lactobacillus reuteri. Biochemistry. 2007;46:13829–13836. doi: 10.1021/bi701622j. [DOI] [PubMed] [Google Scholar]
- 48.StMaurice M, Mera PE, Park K, Brunold TC, Escalante-Semerena JC, Rayment I. Structural characterization of a human-type corrinoid adenosyltransferase confirms that coenzyme B12 is synthesized through a four-coordinate intermediate. Biochemistry. 2008 doi: 10.1021/bi800132d. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Johnson CL, Pechonick E, Park SD, Havemann GD, Leal NA, Bobik TA. Functional genomic, biochemical, and genetic characterization of the SalmonellapduO gene, an ATP:cob(I)alamin adenosyltransferase gene. J Bacteriol. 2001;183:1577–1584. doi: 10.1128/JB.183.5.1577-1584.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.O’Toole GA, Escalante-Semerena JC. cobU-dependent assimilation of nonadenosylated cobinamide in cobA mutants of Salmonella typhimurium. J Bacteriol. 1993;175:6328–6336. doi: 10.1128/jb.175.19.6328-6336.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]