

Abstract
L-methionine (Met) has been implicated in parenteral nutrition-associated cholestasis in infants and, at high levels, causes liver toxicity by mechanisms that are not clear. In this study, Met toxicity was characterized in freshly-isolated male and female mouse hepatocytes incubated with 5-30 mM Met for 0 to 5 h. In male hepatocytes, 20 mM Met was cytotoxic at 4 h as indicated by trypan blue exclusion and LDH leakage assays. Cytotoxicity was preceded by GSH depletion at 3 h without GSSG formation. Exposure to 30 mM Met resulted in increased cytotoxicity and GSH depletion. Interestingly, female hepatocytes were resistant to Met-induced cytotoxicity at these concentrations and showed increased cellular GSH levels compared to hepatocytes exposed to medium alone. The effects of aminooxyacetic acid (AOAA), an inhibitor of Met transamination, and 3-deazaadenosine (3-DA), an inhibitor of the Met transmethylation pathway enzyme S-adenosylhomocysteine hydrolase on Met toxicity in male hepatocytes were then examined. Addition of 0.2 mM AOAA partially blocked Met-induced GSH depletion and cytotoxicity whereas 0.1 mM 3-DA potentiated Met-induced toxicity. Exposure of male hepatocytes to 0.3 mM 3-methylthiopropionic acid (3-MTP), a known Met transamination metabolite, resulted in cytotoxicity and cellular GSH depletion similar to that observed with 30 mM Met whereas incubations with D-methionine resulted in no toxicity. Female hepatocytes were less sensitive to 3-MTP toxicity than males which may partially explain their resistance to Met toxicity. Collectively, these results suggest that Met transamination and not transmethylation plays a major role in Met toxicity in male mouse hepatocytes.
Introduction
L-Methionine (Met), while necessary for life, is also regarded as the most toxic amino acid of those normally involved in protein synthesis (Benevanga, 1974). Rats given excessive dietary Met had increased markers of lipid peroxidation in the liver after six months (Mori and Hirayama, 2000). Guinea pigs injected with toxic levels of Met developed fatty liver with hepatocytic ATP depletion, nucleolar disaggregation, and RNA synthesis inhibition (Shinozuka et al., 1971; Cox et al., 1973). Rabbits infused with Met alone or with the Met-containing total parenteral nutrition (TPN) solution given to infants developed nearly identical choleostatic liver damage indicating that Met toxicity may contribute to TPN cholestasis in infants (Moss et al., 1999). Additionally, male Met adenosyltransferase 1A (MAT1A) knockout mice exhibited an 8-fold elevation of plasma Met levels associated with an increased sensitivity to oxidative stress and liver injury in comparison with wild-type littermates (Lu et al., 2001).
Many studies have provided insights into the potential mechanisms of Met-induced liver toxicity. Several metabolites from the Met transmethylation (TM) pathway have been studied for their toxicological contributions (Figure 1). Met TM begins with the ATP-dependent conversion of Met to S-adenosylmethionine (SAM), an important biological methyl donor molecule (Finkelstein, 1990). ATP depletion from excessive SAM formation as well as the accumulation of SAM itself have been linked to Met-induced hepatotoxicity, however the evidence is largely correlative (Hardwick et al., 1970; Regina et al., 1993). Methyl group donation from SAM results in the formation of S-adenosylhomocysteine (SAH) which is subsequently hydrolyzed to homocysteine (Hcy) (Finkelstein, 1990). Elevated levels of Hcy, which may be converted to the lysine-reactive metabolite Hcy thiolactone, have been clearly linked to arteriosclerosis and are an independent risk factor for cardiovascular disease in humans (Chwatko and Jakubowski, 2005). The toxicological role of Hcy and/or Hcy thiolactone in Met-induced liver toxicity, however, has not been investigated.
Figure 1.

Schematic of the Met TM and TA pathways and the points of inhibition of AOAA and 3-DA. Bolded metabolites have been of particular toxicological interest.
Alternative to Met TM, the Met transamination (TA) pathway has also been investigated for its involvement in Met toxicity (Figure 1). Met TA is catalyzed by multiple transaminases including glutamine transaminase K and L and results in the formation of 2-keto-4-methylthiobutyric acid (KMB) (Cooper, 1989; Scislowski and Pickard, 1993). KMB is then oxidatively decarboxylated by branched chain 2-oxo acid dehydrogenase complex to form 3-methylthiopropionic acid (3-MTP) (Steele and Benevenga, 1978; Jones and Yeaman, 1986). 3-MTP is further metabolized to the highly toxic and volatile molecules methanethiol, hydrogen sulfide and dimethylsulfide (Steele and Benevenga, 1979; Blom et al., 1988a; Gahl et al., 1988; Tangermen et al., 2000). Methanethiol has been shown to inhibit enzymes involved in protection from peroxidative damage in a similar fashion to Met (Finkelstein and Benevenga, 1986). Furthermore, rats fed 3-MTP-spiked chow exhibited markedly similar toxicological symptoms as rats fed chow spiked with excess Met which included growth retardation and hemolytic anemia (Steele et al., 1979). Taken together, these studies suggest that Met TA may also be involved in Met-induced liver toxicity.
In summary, the Met TM and TA pathways have both been implicated as toxicologically relevant pathways, but their relative roles in the liver toxicity associated with hypermethionemia remain unclear. The primary purpose of this study was to assess and compare the relative toxicological roles of Met TM and TA in freshly-isolated male or female mouse hepatocytes exposed to high levels of Met through the use of inhibitors of these pathways. Met TA was inhibited using 0.2 mM aminooxyacetic acid (AOAA) which was previously shown to inhibit Met TA in vitro (Mitchell and Benevenga, 1978). The formation of the Met TM metabolites Hcy and Hcy thiolactone was inhibited using 0.1 mM 3-deazaadenosine (3-DA), a known inhibitor of SAH hydrolase (Garcia-Trevijano et al., 2000). Trypan blue exclusion, LDH leakage, GSH and GSSG levels, and Caspase 3/7 activation were measured to characterize and quantitate the Met-induced cytotoxic and biochemical effects. The toxicity of D-methionine (D-Met) and 3-MTP was also characterized using these endpoints to gain further insight into the mechanism of Met-induced toxicity.
Methods
Chemicals
Trypsin inhibitor (type II-O), collagenase (Type IV), L-methionine (Met), D-methionine (D-Met), aminooxyacetic acid (AOAA), 3-deazaadenosine (3-DA), reduced glutathione (GSH), glutathione disulfide (GSSG), GSH reductase, 5,5’-dithio-bis(2-nitrobenzoic acid) (DTNB), pyruvate, NADH, NADPH, 2-vinylpyridine, 5-sulfosalicylic acid (SSA), ethylenediaminetetraacetic acid (EDTA), and triton-X-100 were obtained from Sigma Chemical Co. (St. Louis, MO). 3-Methylthiopropionic acid (3-MTP) was purchased from Lancaster (Pelham, NH). Hank’s balanced salt solution (HBSS) was obtained from Gibco (Grand Island, NY). Dulbecco’s modified Eagle’s Medium (DMEM) (1x) with 4500 mg/L glucose, and sodium pyruvate but without L-glutamine, methionine, and cystine was purchased from HyClone (Logan, UT). All other chemicals and reagents were of the highest quality commercially available.
Animals
Male and female B6C3F1 mice (7-11 weeks old) were purchased from Jackson Laboratories (Bar Harbor, ME). Mice were maintained on a 12-h light/dark cycle and were allowed feed and water ad libitum. Hepatocytes were isolated at the same time of day to minimize the effects of circadian variation on GSH levels and other enzymatic processes of interest. Hepatocytes were isolated using the two-step EDTA / collagenase perfusion method as described by Kemper et al. (2000) except that livers were perfused with 0.6 mg / mL collagenase solution. The collagenase solution was also centrifuged at 39000 g for 30 min to remove non-soluble components. Initial cell yield and viability were determined by trypan blue exclusion using a hemacytometer. Only hepatocytes with an initial overall viability of greater than 85% after isolation were used in experiments. Cells were then diluted to a concentration of 1 × 106 cells / mL in DMEM and maintained on ice until use.
Cell Incubations
Incubations were carried out in 24-mL vials with screw caps fitted with Teflon-faced septa. Samples of suspended hepatocytes (2.5 mL of 1 × 106 cells/mL) were transferred to the vials. Vial samples were purged with 95% O2 / 5% CO2 (carbogen) before incubation at 37°C with gentle shaking (140 rpm). Following a 4 min preincubation, 131.5 μL of substrate dissolved in DMEM was added to each 2.5 mL cell sample resulting in a final concentration of 5-30 mM Met. For D-Met, the final concentration was 30 mM. For 3-MTP, the final concentration was 0.3 mM. For inhibitor studies 118.4 μL of substrate solution was added followed by 13.1 μL of inhibitor dissolved in DMEM resulting in final concentrations of 0.1 mM for 3-DA and 0.2 mM for AOAA. Samples were then repurged with carbogen and incubated for 0-5 h. Cell incubations were terminated by being placed on ice. After gentle mixing, aliquots were collected for biochemical and toxicological analysis.
Determination of Cell Viability
Trypan blue exclusion was measured by adding 12 μL of cell sample to 12 μL 0.4% trypan blue. Viability was then determined using a hemacytometer. LDH leakage was analyzed by a method adapted from Cummings et al. (2000). The percentage viability was defined as: (LDH in solubilized cell sample) / (LDH in solubilized cell sample + LDH in cell medium sample).
Measurement of apoptosis
Apoptosis was measured using the Caspase-Glo 3/7 luminescence assay as described by Promega (Madison, WI). Briefly, 50 μL Caspase 3/7 reagent was added to 50 μL of cell sample. The reaction was allowed to proceed at room temperature for 30 min before luminescence was measured. Background luminescence from the medium in each sample was also measured and subtracted from the value obtained with cells in medium to obtain the final corrected value. Dimethyl sulphoxide (5% solution) was used as a positive control.
Quantitation of GSH and GSSG
Samples were obtained to measure intracellular and medium levels of GSH and GSSG. Briefly, 500 μL of cell sample was centrifuged at 50 g for 2 min. An aliquot of the supernatant (200 μL) was then added to 800 μL of 5% SSA to be used for analysis of GSH and GSSG levels in the medium. The cell pellet was then washed with 1 mL of phosphate-buffered saline, pH=7.4. Following centrifugation at 50 g for 2 min, the supernatant was removed from the pellet and 1.25 mL of 5% SSA was added. The resulting solution was transferred to a clean microcentrifuge tube and stored at -80°C until analysis. GSH and GSSG levels were measured using the enzymatic recycling method of Tietze (1969). All samples were first centrifuged at 10000 g for 5 min. “Total GSH” (GSH+GSSG) levels and GSSG levels alone were then determined as described by Gunnarsdottir and Elfarra (2003). Standard curves were run in all experiments to allow for quantitation of the results. The concentration of reduced GSH in a sample was determined by subtracting the molar amount of GSH equivalents coming from GSSG from the molar amount of total GSH calculated from the standard curve.
Statistics
Statistical analyses were carried out using the SigmaStat software program (SPSS Inc., Chicago, IL). Comparisons of means were done by paired t-test or analysis of variance. Post hoc comparisons were carried out using the Student-Newman-Keul method. Significance level was set at p≤0.05. All data are expressed as mean ± S.D.
Results
To characterize the toxicity of Met in freshly-isolated male and female mouse hepatocytes, cell viability along with cellular and medium GSH/GSSG levels were monitored in hepatocytes exposed to medium alone or medium containing increasing concentrations of Met for 0-5 h at 37°C.
Male hepatocytes exposed to 30 mM Met had decreased cell viability as determined by trypan blue exclusion (Figure 2A) and LDH leakage (Figure 2C) starting at 3 h compared to hepatocytes exposed to medium alone. Additionally, Met-exposed male hepatocytes had depleted intracellular GSH levels starting at 2 h compared to hepatocytes incubated in medium alone (Figure 3A). Incubations with 30 mM D-Met resulted in no cytotoxicity or GSH depletion (Figure 4). Exposure of male hepatocytes to 20 mM Met led to GSH depletion by 3 h (Figure 3A) and cytotoxicity at 4 h (Figure 2A and 2C) indicating a dose-dependent response. Incubations with 5 and 10 mM Met resulted in no detectable cytotoxicity. No caspase 3/7 activation was detected at any time point or Met concentration. Interestingly, female hepatocytes exposed to 30 mM Met showed no cytotoxic effects (Figure 2B and 2D) and significantly higher GSH levels at 2 and 3 h (Figure 3B) compared to hepatocytes exposed to medium alone. Medium GSH levels in Met-exposed male and female hepatocytes were lower than hepatocytes exposed to medium alone starting at 2 and 3 h, respectively (Figure 3C and 3D). Cellular and medium GSSG levels in Met-exposed hepatocytes of both genders were similar to or lower than levels in hepatocytes exposed to medium alone (data not shown).
Figure 2.

Time course of the cell viability of male (n=4) or female (n=3) hepatocytes exposed to medium alone or medium spiked with Met as determined by TB exclusion (A, B) and LDH leakage (C, D). The symbol * indicates values that were significantly lower than cells incubated with medium alone (*p<0.05, **p<0.01).
Figure 3.

Time course of intracellular (A, B) and medium (C, D) GSH levels of male (n=7 for 20 mM Met, n=4 for 30 mM Met) or female (n=3) hepatocytes exposed to medium alone or medium spiked with Met. The symbol * indicates values that were significantly different than cells incubated with medium alone (*p<0.05, **p<0.01).
Figure 4.
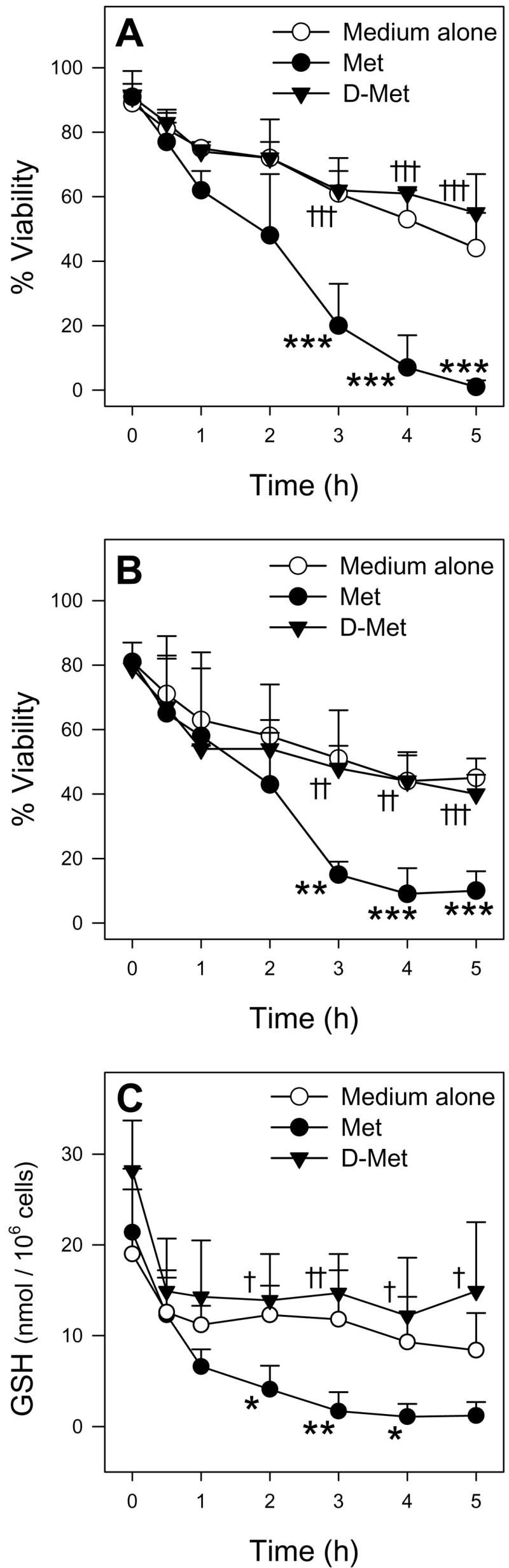
Time course of TB exclusion viability (A), LDH leakage viability (B), and intracellular GSH levels (C) of male hepatocytes (n=3) exposed to medium alone, medium spiked with 30 mM Met, or medium spiked with 30 mM D-Met. The symbol * indicates values that were significantly lower than cells incubated with medium alone (*p<0.05, **p<0.01, ***p<0.001). The symbol † indicates values that were significantly higher than cells incubated with Met († p<0.05, † † p<0.01, † † † p<0.001)
To assess the role of Met TA in Met toxicity in male mouse hepatocytes, the effect of the transaminase inhibitor AOAA was tested. Addition of 0.2 mM AOAA ameliorated Met-induced GSH depletion compared to cells exposed to 30 mM Met alone (Figure 5A). Moreover, AOAA protected male hepatocytes from Met-induced cytotoxicity at 3, 4, and 5 h leading to a nearly two-fold increase in cell viability at each of these time points compared to cells exposed to only 30 mM Met (Figure 6A and 6C).
Figure 5.

The effect of 0.2 mM AOAA (n=4) and 0.1 mM 3-DA (n=3) on intracellular GSH levels in male hepatocytes exposed to 30 mM Met for 2 h. The symbol * indicates values that were significantly lower than cells incubated with medium alone (*p<0.05, **p<0.01, ***p<0.001). The symbol † indicates values that were significantly higher than cells incubated with Met alone (p<0.05).
Figure 6.

Time course of the effect of 0.2 mM AOAA (n=4) and 0.1 mM 3-DA (n=3) on the cell viability of male hepatocytes exposed to medium alone or medium spiked with 30 mM Met as determined by TB exclusion (A, B) and LDH leakage (C, D). The symbol * indicates values that were significantly lower than cells incubated with medium alone (*p<0.05, **p<0.01, ***p<0.001). The symbol † indicates values that were significantly different than cells incubated with Met alone († p<0.05, † † p<0.01, † † † p<0.001).
To assess the role of Met TM and homocysteine formation in Met toxicity in male mouse hepatocytes, the effect of the inhibitor 3-DA was examined. In contrast to AOAA, addition of 0.1 mM 3-DA to Met-exposed cells seemed to increase the magnitude of GSH depletion compared to hepatocytes exposed to Met alone at 2 h (Figure 5B), though the results were not statistically significant (p=0.12). 3-DA also potentiated Met-induced cytotoxicity resulting in a significant decrease in cell viability starting at 2 h (Figure 6B and 6D). At 3 h and 4 h, the viability of cells exposed to 30 mM Met and 0.1 mM 3-DA was nearly half that of cells exposed to Met alone.
Since the above studies indicated that Met TA was an important pathway for Met toxicity, the toxicity of 3-MTP, a known human Met TA metabolite, was investigated. Exposure of male hepatocytes to 0.3 mM 3-MTP resulted in toxicity that was similar in profile to that of 30 mM Met including trypan blue and LDH cytotoxicity starting at 3 h (Figure 7A and 7C respectively) preceded by GSH depletion starting at 2 h (Figure 7E) without concomitant GSSG formation (data not shown). The toxicity of 3-MTP was also assessed in female hepatocytes to further investigate potential mechanisms responsible for the gender-dependent toxicity of Met. Exposure of female hepatocytes to 0.3 mM 3-MTP resulted in GSH depletion at 3 h (Figure 7F), but little to no cytotoxicity (Figure 7B and 7D).
Figure 7.

Time course of TB exclusion viability (A, B), LDH leakage viability (C, D), and intracellular GSH levels (E, F) of male (n=4) and female (n=4) hepatocytes exposed to medium alone or medium spiked with 0.3 mM 3-MTP. The symbol * indicates values that were significantly lower than cells incubated with medium alone (p<0.05).
Discussion
The use of selective inhibitors for the Met TA and TM pathways, the comparison of D-Met and 3-MTP hepatotoxicity with that of Met, and the observed gender differences in Met and 3-MTP toxicological sensitivity in mouse hepatocytes allowed for significant insights to be gained regarding in vitro Met hepatotoxicity in mice. Freshly-isolated hepatocytes provided a closer representation of in vivo conditions than immortalized cell lines while permitting more controlled manipulation of Met exposure levels and key metabolic pathways than would have been possible in vivo.
In male hepatocytes, exposure to as low as 20 mM Met resulted in dose-dependent GSH depletion without increased formation of GSSG that preceded necrotic cell death. Addition of AOAA decreased both the Met-induced GSH depletion and cytotoxicity while addition of 3-DA potentiated these effects. The Met TA metabolite 3-MTP caused nearly identical toxicity as Met at 100-fold lower concentrations. Taken together, these results suggest a major role for Met TA in Met hepatotoxicity. Additional support for this hypothesis is provided by the lack of hepatotoxicity of D-Met which is a poor substrate for L-amino acid-specific transaminases. Transamination of D-Met can be accomplished by D-amino acid oxidase, but this enzyme is not present in mouse liver (Konno et al., 1997).
The Met-induced depletion of GSH without GSSG formation in male hepatocytes suggested that GSH was conjugating with reactive metabolites. Thus, Met TA may be involved in the production of GSH-reactive electrophiles. Further metabolism of 3-MTP has been investigated by Blom et al. (1988a) who found that methanethiol, dimethyl sulfide, and methanethiol-mixed disulfides formed from 3-MTP metabolism in human and rat hepatocytes. Gahl et al. (1988) also detected increased levels of mixed disulfides (CH3S-SX) in a human with hepatic MAT deficiency. Thus, it is likely that volatile sulfur molecules formed in the Met TA pathway react with free GSH to form mixed GSH disulfides. The excessive formation of both protein and non-protein mixed disulfides in the cell may result in cellular damage and cytotoxicity.
The potentiation of Met toxicity by 3-DA indicated that formation of Hcy via the Met TM pathway did not play a major role in Met hepatotoxicity and suggested that Met TM acted as a detoxification pathway at these Met levels. Exposure to 3-DA is known to result in increased SAM and SAH levels and inhibition of methylation reactions in liver (Duerre, 1988; Prytz and Aarbakke, 1990). The buildup of SAM results in feedback inhibition of MAT activity (Finkelstein, 1990). Thus, more Met may be available for Met TA which may explain the increased hepatotoxicity detected in this study. It cannot be ruled out, however, that a toxic buildup of cellular SAM or SAH also played a role in the potentiation of Met hepatotoxicity by 3-DA.
The finding that female hepatocytes were completely resistant to Met hepatotoxicity and had higher cellular GSH levels after Met exposure was interesting and unexpected. Increases in cellular GSH levels in Met-exposed female hepatocytes may be due to increased GSH synthesis via cysteine formation from the Met TM pathway (Wang et al., 1997). Met has also been shown to inhibit cellular GSH efflux by 50-70% in Met-exposed rat hepatocytes, possibly by allosteric inhibition of the GSH transport system, resulting in higher cellular GSH levels compared to control cells (Aw et al., 1986). Consistent with this finding, we detected lower medium and higher cellular GSH levels at 3 h in Met-exposed female hepatocytes compared to hepatocytes exposed to medium alone. While similar processes may occur in male hepatocytes, any cellular gains in GSH levels appear to be overwhelmed by GSH losses secondary to Met TA and formation of mixed disulfides.
Gender differences in 3-MTP toxicological sensitivity allowed for some insights to be gained regarding potential mechanisms of gender-dependent Met hepatotoxicity. Female hepatocytes were much less sensitive to 3-MTP-induced cytotoxicity compared to male hepatocytes, but were only slightly less sensitive to 3-MTP-induced GSH depletion. These results suggest that cellular events independent of GSH depletion may play a role in gender differences in 3-MTP and Met cytotoxicity. Other mechanisms may also be involved in eliciting gender dependent Met hepatotoxicity and GSH depletion. For example, male and female differences in cellular Met uptake and efflux could result in sensitivity differences to Met hepatotoxicity. Met is known to be transported by both the “A” and “L” amino acid transport systems. Hissin and Hilf (1979) reported that 17β-estradiol inhibited system “A” transport of proline in vitro in breast cancer cells demonstrating that sex-hormones can modulate amino acid transport systems utilized by Met. While the effect of 17β-estradiol on liver amino acid transport is not known, lower levels of Met uptake in female hepatocytes compared to males could contribute to their resistance to Met hepatotoxicity. Gender differences in Met metabolism such as increased Met TM and/or decreased Met TA in females could also contribute to the observed toxicological differences. In support of this hypothesis, Met-dosed female mice had higher levels of SAM present in the liver after 15 min compared to Met-dosed males (Dever and Elfarra, 2006). Further investigations are required to examine potential gender differences in 3-MTP metabolism and rates of Met TM and Met TA in mouse hepatocytes.
While gender differences in Met hepatotoxicity have not been previously reported, interspecies differences were identified by Shinozuka et al. (1971) who found that guinea pigs were more sensitive than rats to Met-induced liver toxicity after i.p. injection of a single high dose of Met. Interestingly, however, guinea pigs of both sexes were used while in rats, only females were dosed. Our finding that female mouse hepatocytes were resistant to Met hepatotoxicity complicates the conclusions of that study since male rats were not tested and it is unknown whether rats would also exhibit gender-dependent toxicity following exposure to excess Met.
In humans, the toxicological significance of the Met TA pathway remains unclear. Typical Met levels in hypermethionemic humans (0.5-2.5 mM) are significantly lower than the concentrations of Met required to elicit cytotoxicity in mouse hepatocytes in the present study (20 mM) suggesting that they may not be sufficient to cause liver injury (Gahl et al., 1988; Chou, 2000). However, Met toxicity has been implicated in TPN-cholestasis, a condition characterized by liver damage and high plasma Met levels (1-1.5 mM) in infants as well as decreased hepatic GSH levels in neonatal rats (Heyman et al., 1984; Moss et al., 1999). Furthermore, the Met TM pathway has been shown to be immature and not fully functional in infants (Balistreri et al., 1983). Taken together, these observations suggest that Met TA could play a toxicological role in TPN-cholestasis.
Gender differences in Met metabolism in humans have also been reported. Higher levels of Met transamination products were detected in premenopausal women given oral Met compared to men of similar age given oral Met (Blom et al., 1988b). It was suggested that increased Met TA in healthy humans may actually function as a protective pathway by preventing formation of Hcy and decreasing the long-term cardiovascular risk correlated with high levels of Hcy.
In conclusion, male freshly-isolated mouse hepatocytes, but not female, exhibited GSH depletion followed by cytotoxicity when exposed to high levels of Met. The toxicity was at least partially mediated by Met TA, but not TM. Evidence has been provided that gender differences in metabolism and detoxification of 3-MTP may contribute to gender-specific Met cytotoxicity in male hepatocytes, but other mechanisms are likely involved. Further studies are required to elucidate the exact causes of gender differences in Met toxicity in mouse hepatocytes which may serve as a useful model for human Met metabolism and hepatotoxicity.
Acknowledgments
This research is supported by NIH R01 DK44295 and T32-ES-007015. A preliminary report of this data was presented at the Society of Toxicology meeting held in Seattle, WA on March 15-20, 2008.
Abbreviations used are
- Met
L-methionine
- D-Met
D-methionine
- AOAA
aminooxyacetic acid
- 3-DA
3-deazaadenosine
- SAM
S-adenosyl-L-methionine
- SAH
S-adenosyl-L-homocysteine
- Hcy
homocysteine
- ATP
adenosine triphosphate
- MAT
methionine adenosyltransferase
- 3-MTP
3-methylthiopropionic acid
- TM
transmethylation
- TA
transamination
- TB
trypan blue
- LDH
lactate dehydrogenase
- EDTA
ethylenediaminetetraacetic acid
- DMEM
Dulbecco’s modified Eagle’s Medium
- HBSS
Hank’s balanced salt solution
- SSA
5-sulfosalicylic acid
- GSH
glutathione
- GSSG
glutathione disulfide
- DTNB
5,5’-dithio-bis(2-nitrobenzoic acid)
- TPN
total parenteral nutrition
References
- Aw TY, Ookhtens M, Kaplowitz N. Mechanism of inhibition of glutathione efflux by methionine from isolated rat hepatocytes. Am J Physiol. 1986;251:G354–G361. doi: 10.1152/ajpgi.1986.251.3.G354. [DOI] [PubMed] [Google Scholar]
- Balistreri WF, Heubi JE, Suchy FJ. Immaturity of the enterohepatic circulation in early life: factors predisposing to “physiologic” maldigestion and cholestasis. J Pediatr Gastroenterol Nutr. 1983;2:346–354. [PubMed] [Google Scholar]
- Benevenga NJ. Toxicities of methionine and other amino acids. J Agr Food Chem. 1974;22:2–9. doi: 10.1021/jf60191a036. [DOI] [PubMed] [Google Scholar]
- Blom HJ, van den Elzen JP, Yap SH, Tangerman A. Methanethiol and dimethylsulfide formation from 3-methylthiopropionate in human and rat hepatocytes. Biochim Biophys Acta. 1988a;972:131–136. doi: 10.1016/0167-4889(88)90111-5. [DOI] [PubMed] [Google Scholar]
- Blom HJ, Boers GH, van den Elzen JP, van Roessel JJ, Trijbels JM, Tangerman A. Differences between premenopausal women and young men in the transamination pathway of methionine catabolism, and the protection against vascular disease. Eur J Clin Invest. 1988b;18:633–638. doi: 10.1111/j.1365-2362.1988.tb01279.x. [DOI] [PubMed] [Google Scholar]
- Chou JY. Molecular genetics of hepatic methionine adenosyltransferase deficiency. Pharmacol Ther. 2000;85:1–9. doi: 10.1016/s0163-7258(99)00047-9. [DOI] [PubMed] [Google Scholar]
- Chwatko G, Jakubowski H. Urinary excretion of homocysteine-thiolactone in humans. Clin Chem. 2005;51:408–415. doi: 10.1373/clinchem.2004.042531. [DOI] [PubMed] [Google Scholar]
- Cooper AJL. Methionine transamination in vivo. Biochem J. 1989;262:689–691. doi: 10.1042/bj2620689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox R, Martin JT, Shinozuka H. Studies on acute methionine toxicity. II. Inhibition of ribonucleic acid synthesis in guinea pig liver by methionine and ethionine. Lab Invest. 1973;29:54–59. [PubMed] [Google Scholar]
- Cummings BS, Zangar RC, Novak RF, Lash LH. Cytotoxicity of trichloroethylene and S-(1, 2-dichlorovinyl)-L-cysteine in primary cultures of rat renal proximal tubular and distal tubular cells. Toxicology. 2000;150:83–98. doi: 10.1016/s0300-483x(00)00252-3. [DOI] [PubMed] [Google Scholar]
- Dever JT, Elfarra AA. In vivo metabolism of L-methionine in mice: Evidence for stereoselective formation of methionine-d-sulfoxide and quantitation of other major metabolites. Drug Metab Dispos. 2006;34:2036–2043. doi: 10.1124/dmd.106.012104. [DOI] [PubMed] [Google Scholar]
- Duerre JA. Effect of 3-deazaadenosine on methionine metabolism in isolated perfused livers. Biochem Cell Biol. 1988;66:1032–1039. doi: 10.1139/o88-119. [DOI] [PubMed] [Google Scholar]
- Finkelstein A, Benevenga NJ. The effect of methanethiol and methionine toxicity on the activities of cytochrome c oxidase and enzymes involved in protection from peroxidative damage. J Nutr. 1986;116:204–215. doi: 10.1093/jn/116.2.204. [DOI] [PubMed] [Google Scholar]
- Finkelstein JD. Methionine metabolism in mammals. J Nutr Biochem. 1990;1:228–237. doi: 10.1016/0955-2863(90)90070-2. [DOI] [PubMed] [Google Scholar]
- Gahl WA, Bernardini I, Finkelstein JD, Tangerman A, Martin JJ, Blom HJ, Mullen KD, Mudd SH. Transsulfuration in an adult with hepatic methionine adenosyltransferase deficiency. J Clin Invest. 1988;81:390–397. doi: 10.1172/JCI113331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Trevijano ER, Latasa MU, Carretero MV, Berasain C, Mato JM, Avila MA. S-adenosylmethionine regulates MAT1A and MAT2A gene expression in cultured rat hepatocytes: a new role for S-adenosylmethionine in the maintenance of the differentiated status of the liver. FASEB J. 2000;14:2511–2518. doi: 10.1096/fj.00-0121com. [DOI] [PubMed] [Google Scholar]
- Gunnarsdottir S, Elfarra AA. Distinct tissue distribution of metabolites of the novel glutathione-activated thiopurine prodrugs cis-6-(2-acetylvinylthio)purine and trans-6-(2-acetylvinylthio)guanine and 6-thioguanine in the mouse. Drug Metab Dispos. 2003;31:718–726. doi: 10.1124/dmd.31.6.718. [DOI] [PubMed] [Google Scholar]
- Hardwick DF, Applegarth DA, Cockcroft DM, Ross PM, Calder RJ. Pathogenesis of methionine-induced toxicity. Metabolism. 1970;19:381–391. doi: 10.1016/0026-0495(70)90135-6. [DOI] [PubMed] [Google Scholar]
- Heyman MB, Tseng HC, Thaler MM. Total parenteral nutrition decreases hepatic glutathione concentration in weanling rats. Hepatology. 1984;4:1049. [Google Scholar]
- Hissin PJ, Hilf R. Effects of estrogen to alter amino acid transport in R3230AC mammary carcinomas and its relationship to insulin action. Cancer Res. 1979;39:3381–3387. [PubMed] [Google Scholar]
- Jones SMA, Yeaman SJ. Oxidative decarboxylation of 4-methylthio-2-oxobutyrate by branched-chain 2-oxo acid dehydrogenase complex. Biochem J. 1986;237:621–623. doi: 10.1042/bj2370621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu SC, Alvarez L, Huang ZZ, Chen L, An W, Corrales FJ, Avila MA, Kanel G, Mato JM. Methionine adenosyltransferase 1A knockout mice are predisposed to liver injury and exhibit increased expression of genes involved in proliferation. Proc Natl Acad Sci. 2001;98:5560–5565. doi: 10.1073/pnas.091016398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemper RA, Krause RJ, Elfarra AA. Metabolism of butadiene monoxide by freshly isolated hepatocytes from mice and rats: Different partitioning between oxidative, hydrolytic, and conjugation pathways. Drug Metab Dispos. 2001;29:830–836. [PubMed] [Google Scholar]
- Konno R, Sasaki M, Asakura S, Fukui K, Enami J, Niwa A. D-amino-acid oxidase is not present in the mouse liver. Biochim Biophys Acta. 1997;1335:173–181. doi: 10.1016/s0304-4165(96)00136-5. [DOI] [PubMed] [Google Scholar]
- Martinez-chantar ML, Corrales FJ, Martinez-Cruz LA, Garcia-Trevijano ER, Huang Z, Chen L, Kanel G, Avila MA, Mato JM, Lu SC. Spontaneous oxidative stress and liver tumors in mice lacking methionine adenosyltransferase 1A. FASEB J. 2002;16:1292–1294. doi: 10.1096/fj.02-0078fje. [DOI] [PubMed] [Google Scholar]
- Mitchell AD, Benevenga NJ. The role of transamination in methionine oxidation in the rat. J Nutr. 1978;108:67–78. doi: 10.1093/jn/108.1.67. [DOI] [PubMed] [Google Scholar]
- Mori N, Hirayama K. Long-term consumption of methionine-supplemented diet increases iron and lipid peroxide levels in rat liver. J Nutr. 2000;130:2349–2355. doi: 10.1093/jn/130.9.2349. [DOI] [PubMed] [Google Scholar]
- Moss LR, Haynes AL, Pastuszyn A, Glew RH. Methionine infusion reproduces liver injury of parenteral nutrition cholestasis. Pediatr Res. 1999;45:664–668. doi: 10.1203/00006450-199905010-00009. [DOI] [PubMed] [Google Scholar]
- Prytz PS, Aarbakke J. Differential cell cycle perturbation by transmethylation inhibitors. Biochem Pharm. 1990;39:203–206. doi: 10.1016/0006-2952(90)90666-9. [DOI] [PubMed] [Google Scholar]
- Regina M, Korhonen V, Smith T, Alakuijala L, Eloranta T. Methionine toxicity in the rat in relation to hepatic accumulation of S-adenosylmethionine: Prevention by dietary stimulation of the hepatic transsulfuration pathway. Arch Biochem Biophys. 1993;300:598–607. doi: 10.1006/abbi.1993.1083. [DOI] [PubMed] [Google Scholar]
- Scislowski PWD, Pickard K. Methionine transamination – metabolic function and subcellular compartmentation. Mol Cell Biochem. 1993;129:39–45. doi: 10.1007/BF00926574. [DOI] [PubMed] [Google Scholar]
- Shinozuka H, Estes LW, Farber E. Studies on acute methionine toxicity. I. Nucleolar disaggregation in guinea pig hepatic cells with methionine or ethionine and its reversal with adenine. Amer J Path. 1971;64:241–249. [PMC free article] [PubMed] [Google Scholar]
- Steele RD, Barber TA, Lalich J, Benevenga NJ. Effects of dietary 3-methylthiopropionate on metabolism, growth, and hematopoiesis in the rat. J Nutr. 1979;109:1739–1751. doi: 10.1093/jn/109.10.1739. [DOI] [PubMed] [Google Scholar]
- Steele RD, Benevenga NJ. The metabolism of 3-methylthiopropionate in rat liver homogenates. J Biol Chem. 1979;254:8885–8890. [PubMed] [Google Scholar]
- Tangerman A, Wilcken B, Levy HL, Boers GHJ, Mudd SH. Methionine transamination in patients With homocystinuria due to cystathionine ß-synthase deficiency. Metabolism. 2000;49:1071–1077. doi: 10.1053/meta.2000.7709. [DOI] [PubMed] [Google Scholar]
- Tietze F. Enzymatic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal Biochem. 1969;27:502–522. doi: 10.1016/0003-2697(69)90064-5. [DOI] [PubMed] [Google Scholar]
- Wang S, Chen H, Sheen L, Lii C. Methionine and cysteine affect glutathione level, glutathione-related enzyme activities and the expression of glutathione s-transferase isozymes in rat hepatocytes. J Nutr. 1997;127:2135–2141. doi: 10.1093/jn/127.11.2135. [DOI] [PubMed] [Google Scholar]