

Abstract
Although agents that inhibit DNA synthesis are widely used in the treatment of cancer, the optimal method for combining such agents and the mechanism of their synergy is poorly understood. The present study examined the effects of combining gemcitabine and SN-38 (the active metabolite of irinotecan), two S phase-selective agents that individually have broad antitumor activity, in human cancer cells in vitro. Colony forming assays revealed that simultaneous treatment of Ovcar-5 ovarian cancer cells or BxPC-3 pancreatic cancer cells with gemcitabine and SN-38 resulted in antagonistic effects. In contrast, sequential treatment with the two agents in either order resulted in synergistic antiproliferative effects, although the mechanism of synergy varied with the sequence. In particular, SN-38 arrested cells in S phase, enhanced the accumulation of gemcitabine metabolites and diminished checkpoint kinase 1, thereby sensitizing cells in the SN-38 → gemcitabine sequence. Gemcitabine treatment followed by removal allowed prolonged progression through S phase, contributing to synergy of the gemcitabine → SN-38 sequence. Collectively, these results suggest that S phase selective agents might exhibit more cytotoxicity when administered sequentially rather than simultaneously.
Gemcitabine (2′,2′-difluoro 2′-deoxycytidine), a pyrimidine-based antimetabolite, is active against cancers of the pancreas, lung, breast and ovary as well as some lymphomas (Ryan et al., 2006). According to current understanding, this agent is taken into target cells mainly by the equilibrative nucleoside transporter hENT1 (Damaraju et al., 2003) and sequentially phosphorylated to the 5’-mono-, di- and tri-phosphate derivatives (Plunkett et al., 1995; Ryan et al., 2006). The antiproliferative and cytotoxic effects of gemcitabine have been attributed to two major factors, the ability of gemcitabine diphosphate to inhibit ribonucleotide reductase, thereby depleting deoxyribonucleotide triphosphates required for DNA synthesis, and incorporation of gemcitabine into DNA, where it stalls advancing replication forks one base pair beyond the site of incorporation (Plunkett et al., 1995; Ryan et al., 2006). In addition, it has been suggested that upon incorporation into DNA gemcitabine acts as a topo I poison, stabilizing covalent DNA-topo I complexes that then contribute to DNA damage and cytotoxicity (Pourquier et al., 2002).
Over the past decade there has been considerable interest in combining gemcitabine with a variety of agents that have different mechanisms of action, including doxorubicin, cisplatin, paclitaxel, docetaxel, capecitabine, vinorelbine or ionizing radiation. Particularly pertinent to the present study have been previous attempts to combine gemcitabine with irinotecan, a semisynthetic derivative of camptothecin that is approved for the treatment of colorectal cancer and also is active against pancreatic cancer, non-small cell lung cancer, and breast cancer (Sparreboom and Zamboni, 2006). Irinotecan is a prodrug that is hydrolyzed by carboxylesterases in vivo to SN-38 (7-ethyl-10-hydroxycamptothecin), a classical topo I poison that is thought to kill susceptible cells by stabilizing covalent topo I-DNA complexes, thereby creating an opportunity for the formation of DNA double-strand breaks and other lethal lesions when replication forks collide with the stabilized ternary complexes (Hsiang et al., 1989; Pommier, 2006).
Previous studies have demonstrated that gemcitabine diminishes DNA replication by not only depleting deoxyribonucleotide triphosphates (Plunkett et al., 1995), but also causing the sequential activation of the kinases ATR and Chk1, phosphorylation and degradation of the Cdc25a phosphatase, and resulting inability to activate cyclin-dependent kinases required for replication (Karnitz et al., 2005; Shi et al., 2001). Additional studies have demonstrated that SN-38 triggers the same replication checkpoint (Cliby et al., 2002; Flatten et al., 2005). On the other hand, it also appears that gemcitabine (Huang and Plunkett, 1995) and camptothecin analogues (Holm et al., 1989; Hsiang et al., 1989) require ongoing DNA synthesis to kill target cells. These observations raise the concern that gemcitabine and topo I poisons might be antagonistic when administered simultaneously, as one agent might inhibit the DNA replication required for killing by the other.
The earliest studies examining the gemcitabine/irinotecan combination reported enhanced antiproliferative effects in MCF-7 breast cancer and SCOG small cell lung cancer cells treated with these agents simultaneously in vitro (Bahadori et al., 1999). Although subsequent phase I (Kakolyris et al., 2002; Rocha Lima et al., 1999) and phase II trials of the combination (Rocha Lima et al., 2002) appeared promising, two phase III trials comparing the gemcitabine/irinotecan combination to gemcitabine alone in patients with advanced pancreatic cancer showed no increase in time to progression or survival with the combination (Rocha Lima et al., 2004; Stathopoulos et al., 2006). Given the overlapping spectrum of activity of these agents and the tolerability of the combination, these were disappointing results. Nonetheless, there is continuing interest in combining this pair of agents, as indicated by recent phase II trials of gemcitabine and irinotecan in patients with relapsed non-small cell lung cancer (Kosmas et al., 2007; Rocha-Lima et al., 2007), small cell lung cancer (Akerley et al., 2007; Ohyanagi et al., 2008) and pancreatic and biliary cancer (Sun et al., 2007). Importantly, all of these trials, including the negative phase III trials, were performed by administering gemcitabine and irinotecan simultaneously.
In the present study we have reexamined the effect of administering gemcitabine and SN-38 to solid tumor cell lines in vitro. As predicted, the effects were antagonistic when the two agents were administered simultaneously. On the other hand, the antiproliferative effects of the two agents were synergistic, particularly at high drug concentrations, when the agents were administered sequentially in either sequence. Further experiments examined the mechanisms of this sequence-dependent synergy.
METHODS
Materials
Gemcitabine was obtained from Jack Kovach (Stony Brook, NY). SN-38 was a kind gift from L.P. McGovern (Pfizer, Kalamazoo, MI). Reagents were purchased from the following companies: Opti-MEM medium and Lipofectamine 2000 from Invitrogen (Carlsbad, CA); PI, Tween 20 and BSA from Sigma (St. Louis, MO); and RNase A from Worthington Biochemical (Lakewood, NJ). Antibodies to the following antigens were purchased from the indicated suppliers: phospho-Ser345-Chk1, Cell Signaling Technology (Beverly, MA); β-actin and Chk1, Santa Cruz Biotechnology (Santa Cruz, CA); and BrdU, Becton-Dickinson (Mountain View, CA). Murine monoclonal antibodies that recognize topo I, heat shock protein 90 and lamin A were kind gifts from Y.-C. Cheng (Yale University, New Haven, CT), David Toft (Mayo Clinic, Rochester, MN) and Frank McKeon (Harvard Medical School, Boston, MA), respectively.
Cell culture
BxPC-3 pancreatic cancer cells and Ovcar-5 ovarian cancer cells (from American Type Culture Collection, Manassas, VA and the National Cancer Institute, Rockville, MD, respectively) were cultured in RPMI-10% (v/v) FCS containing 100 units/ml penicillin G, 100 μg/ml streptomycin, and 2 mM glutamine (medium A). After subconfluent monolayers were trypsinized, aliquots containing 500 Ovcar-5 cells or 750 BxPC-3 cells were plated in multiple 60-mm dishes containing 3 ml of medium A and incubated for 12-16 h at 37 °C to allow cells to attach. Graded concentrations of drugs or equivalent volumes of diluent (0.1% DMSO) were then added to triplicate plates. After a 24-h incubation, plates were washed twice in serum-free RPMI 1640 and incubated in drug-free medium A for an additional 7 days. The resulting colonies were stained with Coomassie blue and counted. Diluent-treated control plates typically contained 150-200 colonies.
To examine the effect of sequential drug exposure, cells that had been allowed to adhere for 12-16 h were exposed to diluent or graded concentrations of SN-38 in 3 ml medium A for 24 h, washed twice with serum-free RPMI 1640, exposed to diluent or graded concentrations of gemcitabine in 3 ml medium A for 24 h, washed twice in serum-free RPMI 1640, and incubated in drug-free medium A for 7-8 days to allow colonies to form. Exposure to the reverse sequence was performed in a similar fashion.
Analysis of combined drug effects
Concentration-effect curves were initially generated for each agent to estimate its IC50 for the cell line under study. In subsequent experiments, cells were treated with serial dilutions of each drug individually and with both drugs simultaneously at a fixed ratio of doses that typically corresponded to 3/8, 1/2, 5/8, 3/4, 7/8, 1, and 1-1/2 times the individual IC50s. Fractional survival (f) was calculated by dividing the number of colonies in drug-treated plates by the number of colonies in control plates. Data were subsequently analyzed by the median effect method (Chou and Talalay, 1984) using Calcusyn software (Biosoft, Cambridge, UK).
For each level of cytotoxicity (f = 0.95, 0.90, 0.85, … 0.05), the CI was calculated according to the assumption that the effects of the agents are mutually exclusive, an assumption supported by the mechanistic experiments described below as well as theoretic considerations (Berenbaum, 1989). In this method, which is equivalent to isobologram analysis (Berenbaum, 1989), synergy is indicated by CI <1, additivity by CI = 1, and antagonism by CI >1 (Chou and Talalay, 1984).
Cell cycle analysis
Logarithmically proliferating Ovcar-5 cells were incubated with one or both drugs simultaneously or sequentially as indicated in the text, washed with drug-free RPMI 1640, released by trypsinization, and sedimented at 200 × g for 10 min. After a wash with ice-cold PBS, cells were fixed at 4 °C in 50% (v/v) ethanol, digested with RNase A, stained with PI, and subjected to flow microfluorimetry as described previously (Meng et al., 2003). After 30,000 events per sample were collected, data were analyzed using ModFit software (Verity Software, Topsham, ME).
SN-38 accumulation and cell size determination
To assess the effect of gemcitabine or SN-38 on SN-38 accumulation, log phase Ovcar-5 cells were incubated for 24 h with diluent, 50 nM gemcitabine, 20 nM SN-38 or both drugs simultaneously. At the completion of the incubation, cells were released by trypsinization, sedimented at 200 × g for 6 min, washed twice with serum free-RPMI 1640, and resuspended in RPMI 1640 containing 10% FCS. Half of each sample was treated with 10 μM SN-38 (added from a 10 mM stock in DMSO) and the other half was treated with an equivalent amount of DMSO. After a 30-min incubation at 37 °C, each sample was analyzed on a Becton Dickinsson LSRII flow cytometer using an excitation wavelength of 355 nm and an emission wavelength of 510 ± 20 nm. Following acquisition of 30,000 events per sample, the relative amount of SN-38 in the cells was calculated by subtracting the peak height of the aliquot treated with DMSO alone from the peak height of the sample treated with SN-38.
To assess cell volume, images of cells examined using a Zeiss Axiovert 200M microscope were captured with a Zeiss Axiocam high resolution digital camera controlled with Zeiss KS400 software, which was used to measure the diameters of 80-100 individual cells from each treatment (DMSO, 50 nM gemcitabine, 20 nM SN-38, or gemcitabine + SN-38 for 24 h). From the individual diameters, volumes of the cells were derived and used in calculations to determine SN-38 whole cell uptake.
Gemcitabine di- and tri-phosphate accumulation
To assess the effect of SN-38 on gemcitabine di-and tri-phosphate accumulation, 5 × 106 Ovcar-5 cells were incubated for 24 h with DMSO, 50 nM gemcitabine, 20 nM SN-38, or both drugs simultaneously. At the completion of the incubation, cells were released by trypsinization, sedimented at 200 × g for 6 min, washed twice with ice cold serum-free RPMI 1640, and counted. Aliquots containing 5 × 106 cells for each treatment were resuspended in RPMI 1640 containing 10% FCS. After samples were treated with 10 μM gemcitabine for 4 h at 37 °C, nucleotides were extracted using a modification of a previously described method (Ruiz van Haperen et al., 1994). All steps were performed at 4 °C. In brief, cells were sedimented at 850 × g for 10 min, washed with PBS, resuspended in 135 μl PBS supplemented with 15 μl of internal standard (araCTP), and lysed by vigorous agitation for 1 min after addition of trichloroacetic acid to a final concentration of 10% (w/v). Following a 10-min incubation, insoluble material was sedimented at 850 × g for 10 min. The supernatant was mixed with 400 μl of 1:4 (v/v) trioctylamine:trichlorotrifluoroethane (prepared fresh daily) and sedimented at 10000 × g for 1 min. The upper aqueous layer was transferred to a fresh tube and stored at -20 °C until analyzed by high performance liquid chromatography.
Gemcitabine di- and triphosphate were separated from each other and from endogenous nucleotides by strong anion exchange (SAX) chromatography using a ZirChrom SAX column and a gradient elution with 3 buffers: (A) 10 mM K2HPO4 (pH 6.8), 100 mM NaCl, 0.01 mg/ml sodium azide; (B) 40 mM K2HPO4 (pH 6.8), 400 mM NaCl, 0.01 mg/ml sodium azide and (C) 100 mM [NH4]2HPO4 (pH 6.85), 0.01 mg/ml sodium azide. Separation was accomplished on a column heated to 65 °C using a gradient from 100% A and to 18.6% A/62% B/19.4% C at 70 min. Analytes were detected at 280 nm. Sensitivity, precision and accuracy as well as metabolite stability were similar to parameters previously reported (Ruiz van Haperen et al., 1994).
Immunoblotting
Following treatment with drug or diluent as indicated in the figure legends, cells were washed three times with ice-cold RPMI 1640 medium containing 10 mM HEPES (pH 7.4 at 4°C) and solubilized by addition of 6 M guanidine hydrochloride containing 250 mM Tris-HCl (pH 8.5 at 20°C), 10 mM EDTA, 1% (v/v) 2-mercaptoethanol, and 1 mM freshly added phenylmethylsulfonyl fluoride. After preparation for electrophoresis as described previously (Kaufmann et al., 1997), aliquots containing 50 μg of protein (determined by the bicinchoninic acid method of Smith et al., 1985) were separated on SDS-polyacrylamide gels containing 5-15% (w/v) acrylamide gradients, electrophoretically transferred to nitrocellulose, and probed with immunological reagents as described (Kaufmann, 2001). Alternatively, cell lysates prepared from siRNA-transfected cells were subjected to electrophoresis and probed by immunoblotting as described (Arlander et al., 2003).
siRNA transfections
On day 1, Ovcar-5 cells (6-8 × 105) were plated in 35-mm tissue culture dishes and incubated overnight. On day 2, after cells were washed twice with Opti-MEM medium, 2 ml of Opti-MEM were added to each plate. Four hundred nmol Chk1 siRNA (Arlander et al., 2003; Flatten et al., 2005) or, as a control, luciferase siRNA (Dharmacon, Lafayette, CO) were complexed with 10 μl of Lipofectamine 2000 in 0.5 ml of Opti-MEM for 20 min. Following addition of the lipid-siRNA complexes to the cells, the cultures were incubated for 4-7 h before addition of 1 ml of Opti-MEM containing 35% FCS. The transfections were repeated on day 3. On day 4, the cultures were trypsinized and replated in 100-mm tissue culture dishes containing medium A. On day 5, cells were washed, harvested for immunoblotting or exposed to drugs as described above.
BrdU staining
BrdU incorporation into DNA was assessed as previously described (Cliby et al., 2002). Briefly, cells were pretreated with gemcitabine, SN-38 or both for 12 or 24 h. After incubation with 20 μM BrdU for 30 min, cells were trypsinized, centrifuged at 200 × g, washed in ice-cold PBS, and fixed in 66% (v/v) ethanol at -20 °C. After rehydration with PBS, samples were incubated with 0.04% (w/v) pepsin in 0.1 N HCl for 30 min at 20°C in the dark, washed in PBS containing 0.5% (w/v) Tween 20 and 0.5% (w/v) bovine serum albumin (PBS-TB), incubated in 2 N HCl for 30 min at 37 °C, neutralized with 0.1 M sodium borate, and washed again in PBS-TB. All further steps were performed in the dark at 20-22 °C unless otherwise indicated. Samples were incubated with anti-BrdU antibody in PBS-TB for >1 h, washed in PBS-TB, treated with fluorescein-conjugated goat anti-mouse IgG in PBS-TB for 30 min, washed in PBS-TB, incubated for 20 min at 37 °C in PBS-TB supplemented with 20 μg/ml PI and 0.1 mg/ml RNase A, and subjected to flow cytometry. After collection of 20-30,000 events per sample, staining was analyzed using Becton Dickinson CellQuest software (San Jose, CA). Relative levels of BrdU incorporation were calculated as the ratio of the mean fluorescence intensity of drug-treated samples to mean fluorescence intensity of diluent treated samples.
Statistics
Unless otherwise indicated, clonogenic experiments were repeated until at least three independent experiments yielded correlation coefficients R > 0.9 for all three median effect lines. Results of multiple experiments were summarized by indicating the mean ± s.d. of the CI at the indicated level of colony inhibition. Error bars in various figures likewise indicate mean ± s.d. of at least three independent experiments unless otherwise indicated.
RESULTS
Simultaneous administration of SN-38 and gemcitabine produces antagonism
To assess the effect of combining SN-38 and gemcitabine in vitro, we exposed human cancer cell lines to increasing concentrations of each agent individually and in combination. Ovcar-5 and BxPC-3, two human carcinoma lines that, like the majority of human cancers, have mutant p53 and a defective G1/S checkpoint, were utilized for these studies. When Ovcar-5 cells were exposed to one or both agents for 24 h, washed and allowed to form colonies, simultaneous exposure to both agents diminished colony formation more than exposure to gemcitabine alone, but not more than SN-38 alone (Fig. 1A). Analysis by the median effect method, a widely used mathematical approach to assessing the effects of combining two or more agents (Berenbaum, 1989; Chou and Talalay, 1984), yielded a CI value that was consistently >1 (Fig. 1A, right panel), indicating antagonism.
Figure 1. Effects of simultaneous gemcitabine and SN-38 exposure on clonogenic survival.
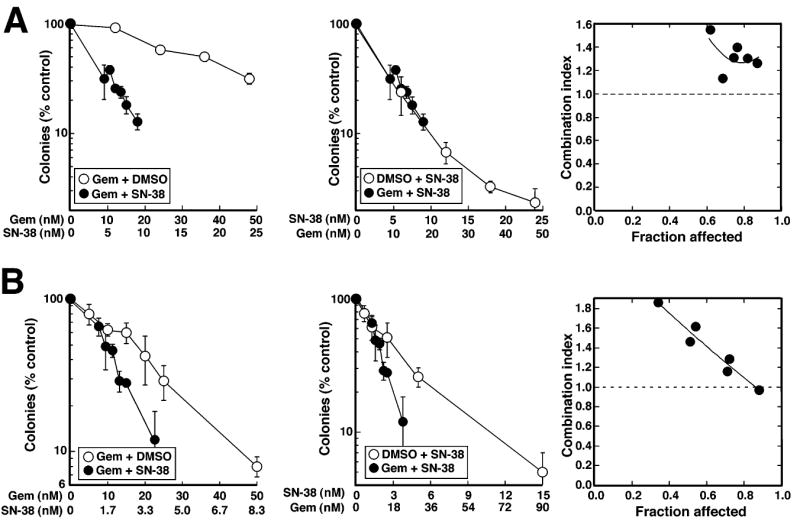
A,Ovcar-5 cells were treated for 24 h with gemcitabine alone, SN-38 alone, or both drugs simultaneously at a fixed molar ratio of 2:1. At the end of the incubation, cells were washed and incubated in drug-free medium until colonies formed. Data from the same experiment are plotted as a function of either SN-38 or gemcitabine concentration. Error bars, ± s.d. from triplicate aliquots. Right panel indicates CI as a function of drug effects. Circles indicate CI calculated from data in left and middle panels under the assumption that effects are mutually exclusive; and line represents second-order regression line calculated from data points. Synergy is indicated by CI < 1, whereas antagonism is indicated by CI > 1. B, BxPC-3 cells were treated for 24 h with the indicated concentrations of gemcitabine alone, SN-38 alone or gemcitabine + SN-38 simultaneously at a 6:1 ratio for 24 h and analyzed as described in panel A. Results in A and B are each representative of 3 independent experiments.
To rule out the possibility that this lack of synergism was unique to Ovcar-5 cells, BxPC-3 pancreatic adenocarcinoma cells were subjected to the same analysis. Simultaneously administered gemcitabine and SN-38 again failed to synergize (Fig. 1B).
Effects of gemcitabine and SN-38 on drug accumulation
To rule out the possibility that an unanticipated effect of one agent on the uptake and/or activation of the other might be responsible for this lack of synergism, we measured accumulation of SN-38 and the active phosphorylated metabolites of gemcitabine. To evaluate SN-38 accumulation, a flow cytometry-based assay for cellular SN-38 content was developed and characterized (Fig. S1). Cellular uptake was linear over the 0.5 – 10 μM SN-38 range (Fig. S1B), providing attomole sensitivity in single cells. When this assay was utilized to assess SN-38 accumulation after treatment for 24 h with 50 nM gemcitabine, mean cellular SN-38 fluorescence was 140 ± 10% (mean ± s.d., n = 3 independent experiments) of diluent treated cells (Fig. 2A). A similar increase in SN-38 content was observed after pretreatment with 20 nM SN-38 or both drugs simultaneously for 24 h (Fig. 2A). Further analysis demonstrated that the volume of the drug treated cells also increased to ~150% of diluent-treated cells (Fig. 2B), possibly reflecting the cell cycle arrest described below. Taking this increased cell volume into account, the SN-38 concentration was essentially unaltered in cells treated with gemcitabine or the combination, ruling out diminished SN-38 accumulation as a cause for the antagonism.
Figure 2. Effects of drug treatment on SN-38 and gemcitabine cellular accumulation.

A, effects of drug treatments on mean SN-38 accumulation using the method characterized in Fig. S1. Cells were pretreated with the indicated agent for 24 h, released by trypsinization, washed, and incubated in 10 μM SN-38 for 20 min before analysis of cell-associated SN-38 fluorescence by flow microfluorimetry. Error bars, ± s.d. from 3 independent experiments. B, mean cell volumes as a function of drug treatment. Cells were treated with the indicated agent for 24 h, released by trypsinization, washed, resuspended in medium A, and photographed to measure cell diameter. Error bars, ± S.E.M. from 80-100 cells subjected to each treatment. C, effect of drug treatment on subsequent formation of gemcitabine di- and triphosphate. Cells were pretreated with the indicated agent for 24 h, released by trypsinization, washed, counted, and incubated in 10 μM gemcitabine for 4 h before analysis of intracellular gemcitabine metabolites by HPLC. Error bars, ± range from 2 independent experiments
Diminished uptake and phosphorylation of gemcitabine also could not be implicated in the lack of synergism. As indicated in Fig. 2C, the accumulation of active gemcitabine metabolites was 2.5-fold higher on a cellular basis after treatment with the combination. After correction for the increase in cell volume (Fig. 2B), this amounted to an ~80% increase in active gemcitabine metabolites after treatment with the combination. Further experiments turned to other potential explanations for the lack of synergism.
Effects of SN-38 and gemcitabine on the S phase checkpoint
While this work was in progress, Zhang et al. reported that hydroxyurea and camptothecin cause proteasome-mediated downregulation of Chk1 in A549 cells (Zhang et al., 2005), while Morgan et al. reported that gemcitabine and radiation induced accumulation of phospho-Chk1 without any change in total Chk1 (Morgan et al., 2005). To extend these earlier studies, we examined the effects of SN-38 and gemcitabine on levels of phospho-Chk1 and Chk1. As indicated in Fig. 3, SN-38 and gemcitabine as well as the combination induced Chk1 phosphorylation, indicating that the upstream components of the 9-1-1/ATR signaling pathway are intact. Moreover, SN-38 and gemcitabine induced Chk1 downregulation; and this effect was greater with the combination (cf. lanes 12-17 vs. 6-11). As the drug concentrations increased, the constant phospho-Chk1 signal in the face of diminished total Chk1 content suggested an increase in the percentage of Chk1 that is phosphorylated. All of these changes occurred with little alteration in levels of the SN-38 target topo I except at the highest SN-38 concentration (Fig. 3A). Similar results were obtained in BxPC-3 cells as well.1
Figure 3. Gemcitabine and SN-38 induce Chk-1 phosphorylation and degradation.

Log-phase Ovcar-5 cells were treated for 24 h with diluent (0.1% DMSO, lanes 1 and 6), gemcitabine at 12.5, 25, 50 or 100 nM (lanes 2-5, respectively), or SN-38 at 2.5, 5, 10, 20 and 40 nM in the absence (lanes 7-11) or presence (lanes 12-17) of 50 nM gemcitabine. At the end of the incubation, whole cell lysates were prepared, subjected to SDS-PAGE, transferred to nitrocellulose, and probed with reagents that recognize phospho-Ser345-Chk1, total Chk1, topo I or, as control, β-actin. Because of the limited number of lanes on our electrophoresis apparatus, lanes 1-5 and 6-17 represent two different membranes from the same experiment that were probed simultaneously.
To further assess the effects of these agents, cells were incubated with each drug or the combination for 12 or 24 h, pulsed with BrdU for 30 min, and immediately stained for BrdU incorporation into DNA. Results of this analysis are illustrated in Fig. 4A and summarized in Fig. 4B. Treatment with gemcitabine resulted in diminished BrdU incorporation throughout S phase (Fig. 4A). Treatment with SN-38 for 12 h resulted in a marked decrease in BrdU incorporation predominantly in late S phase with accumulation of cells earlier in S phase (Fig. 4A) as previously reported for topotecan (Cliby et al., 2002). Finally, simultaneous treatment with both agents resulted in a pattern similar to gemcitabine alone.
Figure 4. Effects of gemcitabine, SN-38 and the combination on DNA synthesis and cell cycle distribution of OVCAR-5 cells.
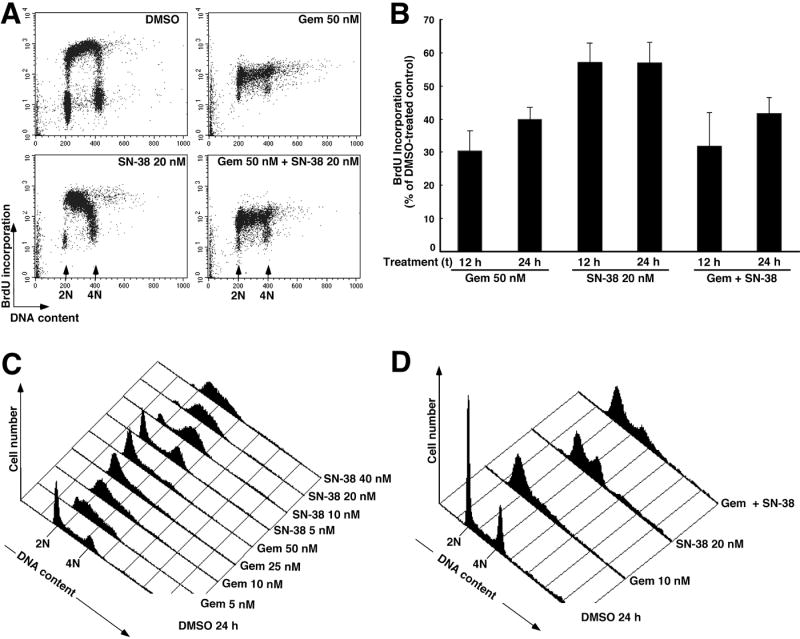
A, BrdU incorporation assessed during a 30 min incubation that started 12 h after addition of diluent, gemcitabine, SN-38 or both drugs simultaneously. B, quantification of BrdU incorporation after the indicated treatments. Error bars, ± S.E.M. from 4-6 independent experiments. C, histograms showing DNA content after treatment with progressively higher concentrations of gemcitabine or SN-38 for 24 h followed by PI staining. D, histograms showing DNA content after treatment with the two drugs individually or simultaneously for 24 h. Panels C and D are representative of 3-6 experiments for each treatment.
Additional experiments examined the cell cycle effects of gemcitabine, SN-38, and both drugs simultaneously. As indicated in Fig. 4C, a 24-h treatment with gemcitabine resulted in a cell cycle arrest that occurred progressively earlier in S phase as the dose was increased, paralleling results reported in ML-1 cells (Shi et al., 2001).2 Low concentrations of SN-38 caused arrest in late S or G2, whereas higher concentrations caused accumulation of the cells progressively earlier in S phase (Fig. 4C). At intermediate SN-38 concentrations (20 nM), the S and G2 populations were sometimes distinct (Fig. 4D) and sometimes continuous (Fig. 4C). When cells were treated with gemcitabine and SN-38 for 24 h simultaneously, the cell cycle distribution was similar to that seen after gemcitabine alone (Fig. 4D). In particular, the marked S phase accumulation seen in cells treated with SN-38 was less evident. Similar results were observed in BxPC-3 cells (Fig. S2), ruling out the possibility that these cell cycle effects were unique to Ovcar5 cells.
Sequential exposure to SN-38 and gemcitabine results in synergistic antiproliferative effects
Results presented in Fig. 4 indicate that SN-38 and gemcitabine both inhibit DNA synthesis and arrest cell cycle progression, providing a potential explanation for lack of synergy when these agents were administered simultaneously (Fig. 1). The downregulation of Chk1 seen after both agents (Fig. 3), coupled with previous reports that Chk1 downregulation sensitizes cells to these drugs (Arlander et al., 2003; Flatten et al., 2005; Zhang et al., 2005), prompted us to examine the effect of administering these agents sequentially. When Ovcar-5 cells were exposed to SN-38 for 24 h, washed, and treated for 24 h with gemcitabine, colony formation was inhibited more than when cells were exposed to either drug alone (Fig. 5A). Analysis by the median effect method indicated that results were essentially additive at lower drug concentrations but synergistic at higher concentrations, with CI values of 1.08 ± 0.14 (mean ± s.d., n = 4 independent experiments) at the IC50 of the combination and 0.74 ± 0.06 at the IC90. When BxPC-3 cells were treated with SN-38 was followed by gemcitabine (Fig. S3A), similar results were observed, with CI values of 0.92 ± 0.18 and 0.38 ± 0.22 (n=3) at the IC50 and IC90, respectively.
Figure 5. Sequence-dependent synergy of gemcitabine and SN-38 in Ovcar-5 cells.

A, Ovcar-5 cells were treated for 24 h with diluent or gemcitabine, washed and treated for another 24 h with diluent or SN-38 using a fixed gemcitabine:SN-38 molar ratio of 10:1. At the end of the incubation, cells were washed and incubated in drug-free medium until colonies formed. Data from the same experiment are plotted as a function of either SN-38 or gemcitabine concentration. Error bars, ± s.d. from triplicate plates. Right panel indicates CI as a function of drug effects. Circles indicate CI calculated from data in left and middle panels under the assumption that effects are mutually exclusive; and line represents second-order regression line calculated from data points. Synergy is indicated by CI < 1, whereas antagonism is indicated by CI > 1. B, Ovcar-5 cells were treated for 24 h with diluent or SN-38, washed and treated for another 24 h with diluent or gemcitabine using a fixed SN-38:gemcitabine ratio of 1:5. Results are representative of 3-5 independent experiments, which are summarized in the text as mean ± s.d. of CI.
Synergy was also observed with the opposite sequence. When Ovcar-5 cells were treated for 24 h with gemcitabine, washed, and exposed to SN-38 for an additional 24 h, colony formation again was diminished more than with either agent alone (Fig. 5B). While the CI was 0.99 ± 0.07 (n=5) at the IC50 of the combination, it was 0.79 ± 0.12 at the IC90, again suggesting synergy at higher drug concentrations (Fig. 5B, right panel). Likewise, the results were synergistic when BxPC-3 cells were exposed to gemcitabine followed by SN-38 (Fig. S3B), with CI values of 0.7 ± 0.1 and 0.31 ± 0.07 (n=3) at the IC50 and IC90, respectively. Subsequent experiments were undertaken to better understand how, at higher concentrations, exposure to one agent could sensitize cells to subsequent exposure to the other.
Chk1 downregulation sensitizes Ovcar-5 cells to gemcitabine, facilitating synergy during sequential SN-38 → gemcitabine treatment
The ability of SN-38 to cause Chk1 downregulation (Fig. 3), coupled with the previous observation that Chk1 downregulation enhances gemcitabine cytotoxicity (Arlander et al., 2003; Karnitz et al., 2005), raised the possibility that SN-38-induced Chk1 downregulation might sensitize the cells to gemcitabine. To assess this possibility, Ovcar-5 cells were initially transfected with luciferase (control) or Chk1 siRNA, treated for 24 h with gemcitabine, washed, and incubated for 7 days to allow surviving cells to form colonies. Results of this analysis demonstrated that Chk1 downregulation (Fig. 6A, inset) resulted in increased gemcitabine sensitivity, with a 2-fold decrease in the IC50 (Fig. 6A).
Figure 6. Chk1 siRNA sensitizes cells to gemcitabine.
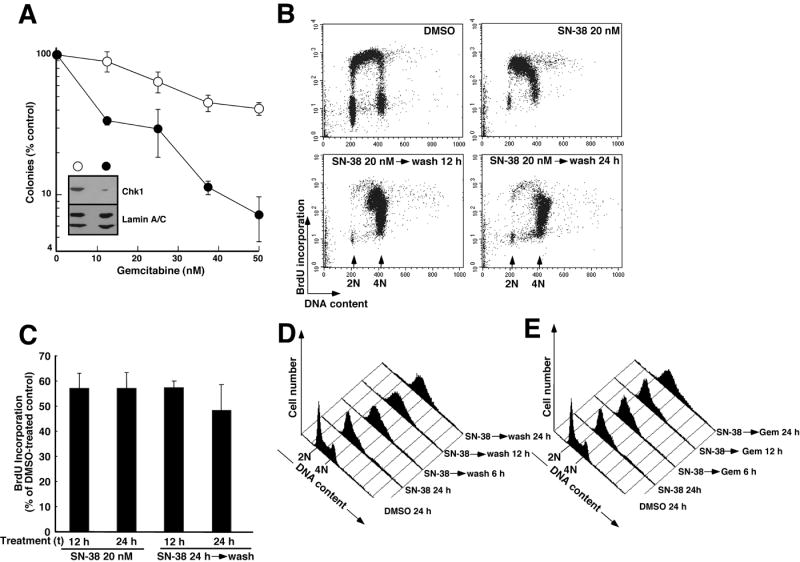
A, Ovcar-5 cells transfected twice with luciferase siRNA (open circles) or Chk1 siRNA (filled circles) were treated for 24 h with diluent (0.1% DMSO) or the indicated concentration of gemcitabine for 24 h, washed and incubated in drug-free medium for 8 d to allow colonies to form. Error bars, ± s.d. from triplicate samples. Inset in A, whole cell lysates were prepared from additional transfected cells at the time of drug treatment, subjected to SDS-PAGE and probed for Chk1. Lamins A and C served as loading controls. B, BrdU incorporation assessed during a 30 min incubation that started 12 h after addition of diluent or 20 nM SN-38. Alternatively, cells were treated for 24 h with SN-38, then washed and incubated in drug-free medium for 12 or 24 h. C, quantification of BrdU incorporation after the treatments shown in panel B. Error bars, ± S.E.M. from 4-6 independent experiments. D, histograms showing DNA content after treatment with 20 nM SN-38 for 24 h. Alternatively, cells were treated with SN-38 for 24 h, washed, and incubated in drug-free medium. Samples were harvested every 3 h after SN-38 removal. For clarity, only histograms obtained 6, 12 and 24 h after SN-38 removal are shown. E, histograms showing time-course of cell cycle progression in cells exposed to 20 nM SN-38 for 24 h, washed and treated with 10 nM gemcitabine for the indicated length of time before PI staining and analysis as indicated in panel D. Panels D and E come from the same experiment and are representative of 4 independent experiments.
To search for additional changes that might contribute to the observed synergy, further experiments examined changes in DNA synthesis and cell cycle distribution when SN-38 was removed from Ovcar-5 cells after a 24-h treatment. This analysis demonstrated little if any increase in DNA synthesis (Figs. 6B and 6C) as cells progressed from S phase into G2 and then arrested (Fig. 6D). Further analysis, however, indicated that cells treated with gemcitabine after SN-38 removal remained arrested in S phase longer and progressed into G2 somewhat more slowly than cells treated with diluent after SN-38 removal (cf. Fig. 6D vs. 6E), raising the possibility that the synchronization in S phase by SN-38 might have predisposed the cells to the effects of gemcitabine. Similar effects were observed in BxPC-3 (Fig. S4A), ruling out the possibility that these effects were limited to Ovcar5 cells. Moreover, results shown in Fig. 2C indicate that SN-38 treatment increases subsequent levels of gemcitabine active metabolites, providing another factor that might contribute to the synergy of the SN-38 → gemcitabine sequence.
Synergistic effects of sequential gemcitabine → SN-38 treatment occur independent of Chk1 downregulation
A parallel series of experiments was performed to examine the mechanistic basis for the synergy observed with the gemcitabine → SN-38 sequence. The ability of gemcitabine to cause Chk1 downregulation (Fig. 3), coupled with the previous observation that Chk1 downregulation enhances cytotoxicity of topo I poisons in HeLa, U2OS and A549 cells (Flatten et al., 2005; Zhang et al., 2005), raised the possibility that gemcitabine-induced Chk1 downregulation might sensitize the cells to SN-38. When Ovcar-5 cells were transfected with Chk1 siRNA, treated for 24 h with SN-38, washed, and incubated for 7 days to allow surviving cells to form colonies, however, no sensitization was observed in any of the six experiments performed (Fig. 7A). Further experiments demonstrated that Chk1 siRNA abrogated the dramatic S phase arrest observed with SN-38 (Fig. 7B), ruling out the possibility that the cell cycle effects of SN-38 were mediated by some other pathway.
Figure 7. Synchronous S phase progression as a potential cause of SN-38 sensitization.
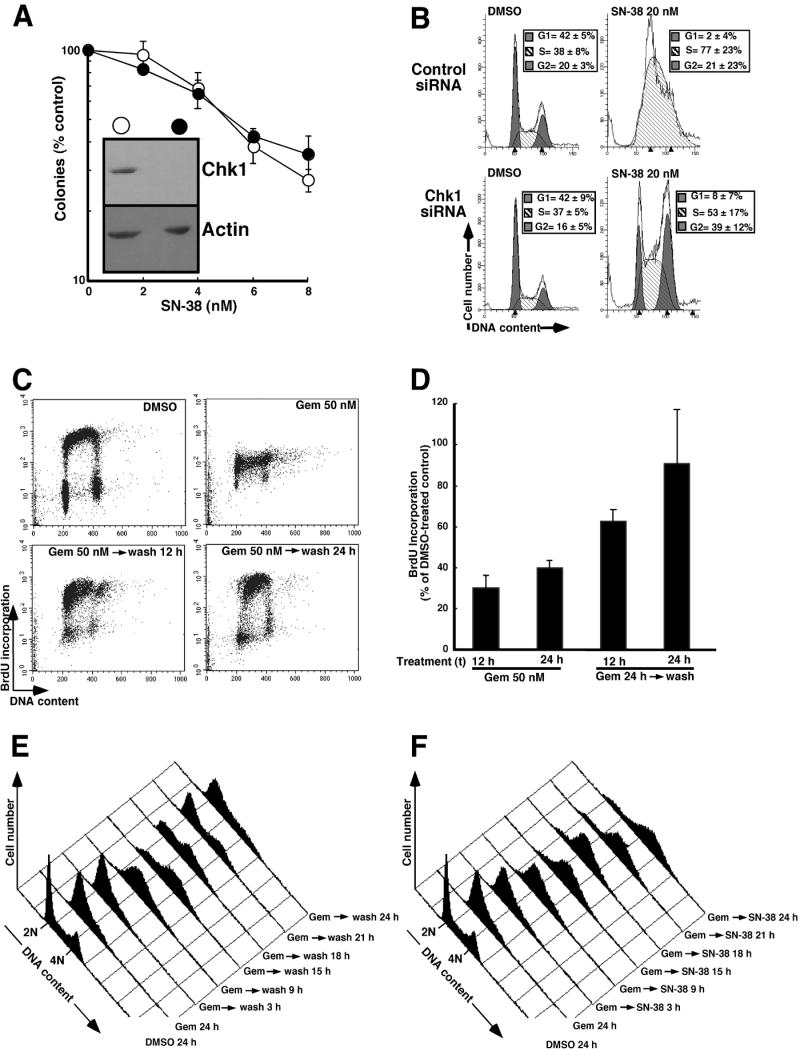
A, After transfection with luciferase siRNA (open circles) or Chk1 siRNA (filled circles) were treated for 24 h with diluent (0.1% DMSO) or the indicated concentration of SN-38 for 24 h, Ovcar-5 cells washed and incubated in drug-free medium for 8 d to allow colonies to form. Error bars, ± s.d. from triplicate plates. B, cell cycle distribution of Ovcar-5 cells transfected as described above and exposed for 24 h to diluent or 20 nM SN-38. Histograms are representative of at least 4 independent experiments, with cell cycle distributions summarized as mean ± s.d of those experiments in the inset of each histogram. C, BrdU incorporation assessed during a 30 min incubation that started 12 h after addition of diluent or 50 nM gemcitabine. Alternatively, cells were treated with 50 nM gemcitabine for 24 h, washed, and incubated in drug-free medium for 12 or 24 h. D, quantification of BrdU incorporation after the treatments shown in panel C. Error bars, ± S.E.M. from 4-6 independent experiments. E, DNA histograms showing cell cycle progression after a 24 h treatment with 50 nM gemcitabine followed by removal of the drug. Samples were taken every 3 h after gemcitabine removal. For clarity, only histograms obtained 3, 9, 15, 18, 21 and 24 h after gemcitabine removal are shown. F, histograms showing time-course of cell cycle progression in cells sequentially exposed to gemcitabine followed by SN-38. After a 24-h treatment with 50 nM gemcitabine, cells were washed and exposed to 20 nM SN-38 for the indicated length of time before PI staining and analysis as indicated in panel E. Panels E and F come from the same experiment and are representative of 3 independent experiments.
To search for other changes that might contribute to the observed synergy, further experiments examined changes in DNA synthesis and cell cycle distribution when gemcitabine was removed after a 24 h treatment. Results of this analysis (Figs. 7C and 7D) demonstrated that gemcitabine removal resulted in increased DNA synthesis (Fig. 7C) and slow progression of cells through S phase during the ensuing 12-15 h (Figs. 7E and 7F), thereby providing a large fraction of cells in the cell cycle phase in which they are ordinarily most sensitive topo I poisons (D’Arpa et al., 1990). Similar results were observed in BxPC-3 cells (Fig. S4B). Collectively, these results suggest that the recovery of DNA synthesis after gemcitabine removal and subsequent synchronous progression through S phase might contribute to the synergy observed with the gemcitabine → SN-38 sequence. In addition, by 24 h after removal of gemcitabine, a substantial fraction of the Ovcar-5 cells treated with SN-38 had progressed to an aneuploid state containing >4n DNA (Fig. 7F), suggesting that this sequence had also impaired the G2/M checkpoint more than either drug alone.
DISCUSSION
In the present study, we have demonstrated that the antiproliferative effects of the topo I poison SN-38 and the antimetabolite gemcitabine are antagonistic when the agents are administered to cell lines simultaneously. This antagonism appears to reflect, at least in part, the ability of each of these agents to diminish the DNA replication on which the other agent relies for its cytotoxicity. In contrast, exposure to the same agents in either sequence results in synergistic antiproliferative effects, particularly at higher drug concentrations. Our further studies indicate that the mechanism of this sequence-dependent synergy varies depending on the sequence. Collectively, these results not only have implications for potential future development of the gemcitabine/irinotecan combination, but also provide new insight into the issue of how best to combine S phase poisons for maximal cytotoxic effects.
When SN-38 and gemcitabine were administered simultaneously, antiproliferative effects of the combination were at best additive and often less than additive. Similar results were observed in the Ovcar-5, BxPC3 and A549 cell lines (Figs. 1A and 1B and data not shown), ruling out the possibility that the antagonistic effects of the two agents were limited to a single cell line. These observations differ from a previous report indicating that the effects of treating breast and small cell lung cancer lines with gemcitabine and irinotecan simultaneously in vitro are synergistic (Bahadori et al., 1999). There are several potential explanations for these divergent conclusions. First, the present experiments utilized therapeutically achievable nanomolar concentrations of SN-38 rather than 10 – 100 μM concentrations of irinotecan, a prodrug that requires esterase-mediated cleavage for activation. Second, we utilized gemcitabine at concentrations below 100 nM, whereas the preceding paper utilized concentrations up to 100 μM. Third, we utilized a long-term assay (colony forming ability) rather than a short-term dye reduction assay to assess viability. Finally, we employed cell types (ovarian and pancreatic cancer) that are known to be responsive to topo I poisons and gemcitabine. Consistent with our results, antagonism has also been reported when U937 human leukemia cells are simultaneously exposed to gemcitabine and SN-38 (Shanks et al., 2005), although the mechanism of this antagonism was not examined.
A number of experiments were performed to assess the mechanism of antagonism observed in the present study. To assess the effect of gemcitabine on SN-38 uptake, we developed a novel flow cytometry-based assay that allowed detection of SN-38 in living cells based on the intrinsic fluorescence of the SN-38 ring structure (Fig. S1). This assay permitted analysis of SN-38 uptake on a cell-by-cell basis for the first time and demonstrated that SN-38 uptake is extremely rapid, being complete within less than a minute (Fig. S1). When this assay was applied to cells that were pretreated with gemcitabine, no decrease in SN-38 uptake was observed (Figs. 2A and 2B). Likewise, SN-38 failed to diminish gemcitabine uptake and phosphorylation (Fig. 2C). Accordingly, diminished drug uptake cannot account for the antagonism observed during simultaneous exposure. Additional experiments also ruled out the possibility that gemcitabine caused topo I downregulation as an explanation for the antagonistic effect of simultaneous treatment (Fig. 3).
Further experiments demonstrated that gemcitabine diminished DNA synthesis (Figs. 4A and 4B), decreasing the very events required for maximal cytotoxic effects of camptothecin analogs (Holm et al., 1989; Hsiang et al., 1989). Likewise, SN-38 inhibited BrdU incorporation (Fig. 4A) and arrested the cells in G2 at low concentrations and S phase at higher concentrations (Fig. 4C), confirming earlier results (Cliby et al., 2002; Nelson and Kastan, 1994) and providing an explanation for the less than additive effects of simultaneously administered gemcitabine. Similar antagonistic antiproliferative effects have been observed when colon cancer cells are treated with SN-38 and 5-fluorouracil simultaneously (Mullany et al., 1998) and are a potential problem with many combinations that include two agents that depend upon ongoing DNA synthesis for their cytotoxicity.
In contrast to simultaneous administration, sequential administration of SN-38 and gemcitabine resulted in greater than additive antiproliferative effects, particularly at high but therapeutically achievable drug concentrations (Figs. 5 and S3). Further experiments suggested a number of factors that might contribute to this synergy.
Three factors potentially contribute to the synergy of the SN-38 → gemcitabine combination. First, SN-38 treatment resulted in a modest increase in gemcitabine active metabolites (Fig. 2C). This might reflect the well established increase in deoxycytidine kinase activity observed in S phase cells (Wan and Mak, 1978). Second, SN-38 synchronized the cells in S phase (Figs. 4C, 6D and S2), a phase of the cell cycle in which gemcitabine is particularly toxic (Huang and Plunkett, 1995), potentially enhancing the cytotoxicity of gemcitabine when it was administered after the camptothecin analog. Finally, SN-38 caused a decrease in Chk1 levels (Fig. 3) consistent with the hydroxyurea- and camptothecin-induced, proteasome-mediated Chk1 downregulation previously reported (Zhang et al., 2005). Further experiments demonstrated that treatment with Chk1 siRNA enhanced the cytotoxicity of gemcitabine in Ovcar-5 cells (Fig. 6A). Importantly, this sensitization was observed when Chk1 was only partially downregulated (inset, Fig. 6A), raising the possibility that Chk1 downregulation, along with enhanced accumulation of gemcitabine metabolites and cell synchronization, might contribute to the synergy of the SN-38 → gemcitabine sequence.
In the same experiments, Chk1 siRNA failed to sensitize Ovcar-5 cells to SN-38 (Fig. 7A). Flow cytometry demonstrated that Chk1 siRNA diminished the SN-38-induced S phase arrest (Fig. 7B), ruling out the possibility that S phase arrest was dependent on some other pathway. These results clearly dissociate the effects of Chk1 on cell cycle progression and cytotoxicity. Ongoing studies are attempting to understand why some cell lines such as HeLa, U2OS and A549 cells are sensitized to topo I poisons by Chk1 siRNA (Flatten et al., 2005; Zhang et al., 2005) and others (Fig. 7A) are not. Nonetheless, these results suggested that gemcitabine-induced Chk1 downregulation (Fig. 3) is not sufficient to explain the synergy of the gemcitabine → SN-38 sequence. Additional analysis demonstrated that gemcitabine induced a late G1/early S phase arrest (Figs. 4C and S2) in association with a marked decrease in DNA synthesis (Figs. 4A and 4B). After gemcitabine removal, DNA synthesis increased (Fig. 7C and 7D) as cells went from the G1/S boundary to G2 (Figs. 7E and S4B), providing a prolonged period of DNA synthesis that afforded the opportunity for cytotoxic interactions between advancing replication forks and SN-38-stabilized topo I cleavage complexes in the gemcitabine → SN-38 sequence (Fig. 7F). In addition, treatment with SN-38 after gemcitabine withdrawal led to increased formation of aneuploid cells (cf. Figs. 7E and 7F at 24 h after diluent or SN-38 addition), likely reflecting a further decrease in Chk1 levels with the combination3 and consequent abrogation of the G2/M checkpoint that might have further enhanced the cytotoxicity of the combination.
During the course of these studies we had the opportunity to compare the effects of gemcitabine and SN-38. Even though both of these agents diminished DNA synthesis (Fig. 4B), there were notable differences. Gemcitabine caused diminished BrdU incorporation throughout S phase, whereas SN-38 caused a more prominent decrease in BrdU incorporation in late S phase (Fig. 4A). Moreover, high doses of gemcitabine induced an arrest at the G1/S border after 24 h of treatment, whereas SN-38 arrested cells in S phase (Figs. 4C and S2). Finally, Chk1 downregulation sensitized Ovcar-5 cells to gemcitabine but not SN-38 (Figs. 6A and 7A). These differences are difficult to reconcile with a previous claim that the ability of gemcitabine to act as a topo I poison upon incorporation into DNA plays a major role in the action of this antimetabolite.
As indicated in the Introduction, the best approach for combining two S phase-selective agents remains an open question. Results of phase III trials demonstrated that simultaneous administration of irinotecan and gemcitabine failed to significantly increase either time to progression or survival in patients with advanced pancreatic cancer compared to gemcitabine alone (Rocha Lima et al., 2004; Stathopoulos et al., 2006). This was a disappointing result because of the single-agent activity of each of these drugs in pancreatic cancer. Nonetheless, because of the broad spectrum of activity of these two agents, there continues to be considerable interest in further study of this combination (Akerley et al., 2007; Kosmas et al., 2007; Rocha-Lima et al., 2007; Saif et al., 2007; Sun et al., 2007). With only one exception (Saif et al., 2007), studies of the combination have involved simultaneous rather than sequential exposure to these agents. Results of the present study suggest that the antiproliferative effects of these agents are synergistic only when they are administered sequentially. Further study is required to see whether these observations can be translated into therapeutic benefit.
Supplementary Material
A, histogram showing cell fluorescence after treatment of Ovcar-5 cells with diluent (DMSO) or 10 μM SN-38. B, increase in mean fluorescence intensity as a function of SN-38 concentration. C, time course of SN-38 uptake into cells assessed by measuring mean fluorescence intensity every 10 sec after addition of 10 μM SN-38 to cells in a temperature controlled 37 °C tube holder.
Histograms show DNA content after treatment with 40 nM gemcitabine, 10 nM SN-38 or both drugs simultaneously for 24 h.
A, BxPC-3 cells were treated for 24 h with diluent or SN-38, washed and treated for another 24 h with diluent or gemcitabine using a fixed molar ratio of 8.33:1 (gemcitabine:SN-38). At the end of the incubation, cells were washed and incubated in drug-free medium until colonies formed. Data from the same experiment are plotted as a function of either SN-38 or gemcitabine concentration. Error bars, ± s.d. from triplicate plates. Right panel indicates CI as a function of drug effects. Circles indicate CI calculated from data in left and middle panels under the assumption that effects are mutually exclusive; and line represents second-order regression line calculated from data points. Synergy is indicated by CI < 1, whereas antagonism is indicated by CI > 1. B, BxPC-3 cells were treated for 24 h with diluent or gemcitabine, washed and treated for another 24 h with diluent or SN-38 at a fixed SN-38:gemcitabine ratio of 1:6.
A, histograms showing DNA content after treatment with 10 nM SN-38 for 24 h followed by fixation and PI staining. Alternatively, cells were treated with 10 nM SN-38 for 24 h, washed, and incubated in drug-free medium or 40 nM gemcitabine for 24 h before fixation. B, histograms showing DNA content after treatment with 40 nM gemcitabine for 24 h. Alternatively, cells were treated with gemcitabine for 24 h, washed, and incubated in drug-free medium or 10 nM SN-38 for 24 h.
Acknowledgments
Kind gifts of reagents from L.P. McGovern, Jack Kovach, David Toft, Frank McKeon and Y-C. Cheng are gratefully acknowledged, as are helpful discussions with Charles Erlichman, Zhenkun Lou and Kriste Lewis; assistance of Phyllis Svingen with some of the clonogenic assays; technical assistance of the Mayo Clinic Flow Cytometry Shared Resource; secretarial assistance of Deb Strauss; and helpful suggestions of the two anonymous reviewers.
Abbreviations used
- BrdU
5-bromo-2’-deoxyuridine
- BSA
bovine serum albumin
- Chk1
checkpoint kinase 1
- CI
combination index
- EDTA
ethylenediaminetetraacetic acid
- FCS
heat-inactivated fetal calf serum
- IC50
concentration that inhibits colony formation by 50%
- PBS
calcium- and magnesium-free Dulbecco’s phosphate buffered saline
- PI
propidium iodide
- SDS
sodium dodecyl sulfate
- SN-38
7-ethyl-10-hydroxycamptothecin
- topo I
topoisomerase I
Footnotes
Supported in part by R01 CA7309.
D.A.L. and S.H.K., unpublished observations.
A similar effect was observed in HCT116 and BxPC-3 cells (M. G-P. and S.H.K., unpublished observations).
M. G-P. and S.H.K., unpublished observations.
References
- Akerley W, McCoy J, Hesketh PJ, Goodwin JW, Bearden JD, Atkins JN, Chansky K, Crowley JJ, Gandara DR. Gemcitabine and irinotecan for patients with untreated extensive stage small cell lung cancer: SWOG 0119. J Thorac Oncol. 2007;2(6):526–530. doi: 10.1097/JTO.0b013e318060d2dc. [DOI] [PubMed] [Google Scholar]
- Arlander SJH, Eapen AK, Vroman BT, McDonald RJ, Toft DO, Karnitz LM. Hsp90 Inhibition Depletes Chk1 and Sensitizes Tumor Cells to Replication Stress. J Biol Chem. 2003;278(52):52572–52577. doi: 10.1074/jbc.M309054200. [DOI] [PubMed] [Google Scholar]
- Bahadori HR, Rocha Lima CM, Green MR, Safa AR. Synergistic effect of gemcitabine and irinotecan (CPT-11) on breast and small cell lung cancer cell lines. Anticancer Res. 1999;19(6B):5423–5428. [PubMed] [Google Scholar]
- Berenbaum MC. What is Synergy? Pharmacol Rev. 1989;41(2):93–141. [PubMed] [Google Scholar]
- Chou T-C, Talalay P. Quantitative Analysis of Dose-Effect Relationships: The Combined Effects of Multiple Drugs or Enzyme Inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- Cliby WA, Lewis KA, Lilly KK, Kaufmann SH. S Phase and G2 Arrests Induced by Topoisomerase I Poisons Are Dependent on ATR Kinase Function. J Biol Chem. 2002;277(2):1599–1606. doi: 10.1074/jbc.M106287200. [DOI] [PubMed] [Google Scholar]
- D’Arpa P, Beardmore C, Liu LF. Involvement of Nucleic Acid Synthesis in Cell Killing Mechanisms of Topoisomerase Poisons. Cancer Res. 1990;50:6919–6924. [PubMed] [Google Scholar]
- Damaraju VL, Damaraju S, Young JD, Baldwin SA, Mackey J, Sawyer MB, Cass CE. Nucleoside anticancer drugs: the role of nucleoside transporters in resistance to cancer chemotherapy. Oncogene. 2003;22(47):7524–7536. doi: 10.1038/sj.onc.1206952. [DOI] [PubMed] [Google Scholar]
- Flatten K, Dai NT, Vroman BT, Loegering D, Erlichman C, Karnitz LM, Kaufmann SH. The Role of Checkpoint Kinase 1 in Sensitivity to Topoisomerase I Poisons. J Biol Chem. 2005;280:14349–14355. doi: 10.1074/jbc.M411890200. [DOI] [PubMed] [Google Scholar]
- Holm C, Covey JM, Kerrigan D, Pommier Y. Differential Requirement of DNA Replication for the Cytotoxicity of DNA Topoisomerase I and II Inhibitors in Chinese Hamster DC3F Cells. Cancer Res. 1989;49(22):6365–6358. [PubMed] [Google Scholar]
- Hsiang Y-H, Lihou MG, Liu LF. Arrest of Replication Forks by Drug-Stabilized Topoisomerase I-DNA Cleavable Complexes as a Mechanism of Cell Killing by Camptothecin. Cancer Res. 1989;49:5077–5082. [PubMed] [Google Scholar]
- Huang P, Plunkett W. Fludarabine- and Gemcitabine-Induced Apoptosis: Incorporation of Analogs into DNA is a Critical Event. Cancer Chemother Pharmacol. 1995;36(3):181–188. doi: 10.1007/BF00685844. [DOI] [PubMed] [Google Scholar]
- Kakolyris SS, Kouroussis C, Koukourakis M, Kalbakis K, Mavroudis D, Vardakis N, Georgoulias V. A dose-escalation study of irinotecan (CPT-11) in combination with gemcitabine in patients with advanced non-small cell lung cancer previously treated with a cisplatin-based front line chemotherapy. Anticancer Res. 2002;22(3):1891–1896. [PubMed] [Google Scholar]
- Karnitz LM, Flatten KS, Wagner JM, Loegering D, Hackbarth JS, Arlander SJ, Vroman BT, Thomas MB, Baek YU, Hopkins KM, Lieberman HB, Chen J, Cliby WA, Kaufmann SH. Gemcitabine-induced activation of checkpoint signaling pathways that affect tumor cell survival. Mol Pharmacol. 2005;68(6):1636–1644. doi: 10.1124/mol.105.012716. [DOI] [PubMed] [Google Scholar]
- Kaufmann SH. Reutilization of Immunoblots After Chemiluminescent Detection. Anal Biochem. 2001;296:283–286. doi: 10.1006/abio.2001.5313. [DOI] [PubMed] [Google Scholar]
- Kaufmann SH, Svingen PA, Gore SD, Armstrong DK, Cheng Y-C, Rowinsky EK. Altered Formation of Topotecan-Stabilized Topoisomerase I-DNA Adducts in Human Leukemia Cells. Blood. 1997;89:2098–2104. [PubMed] [Google Scholar]
- Kosmas C, Tsavaris N, Syrigos K, Koutras A, Tsakonas G, Makatsoris T, Mylonakis N, Karabelis A, Stathopoulos GP, Kalofonos HP. A phase I-II study of bi-weekly gemcitabine and irinotecan as second-line chemotherapy in non-small cell lung cancer after prior taxane + platinum-based regimens. Cancer Chemother Pharmacol. 2007;59(1):51–59. doi: 10.1007/s00280-006-0242-5. [DOI] [PubMed] [Google Scholar]
- Meng X, Chandra J, Loegering D, Van Becelaere K, Kottke TJ, Gore SD, Karp JE, Sebolt-Leopold JS, Kaufmann SH. Central role of FADD in Apoptosis Induction by the Mitogen Activated Activated Protein Kinase Kinase Inhibitor CI1040 (PD184352 in Acute Lymphocytic leukemia Cell Lines in Vitro. J Biol Chem. 2003;278:47236–47339. doi: 10.1074/jbc.M304793200. [DOI] [PubMed] [Google Scholar]
- Morgan MA, Parsels LA, Parsels JD, Mesiwala AK, Maybaum J, Lawrence TS. Role of checkpoint kinase 1 in preventing premature mitosis in response to gemcitabine. Cancer Res. 2005;65(15):6835–6842. doi: 10.1158/0008-5472.CAN-04-2246. [DOI] [PubMed] [Google Scholar]
- Mullany S, Svingen PA, Kaufmann SH, Erlichman C. Effect of Adding the Topoisomerase I Poison 7-Ethyl-10-Hydroxycamptothecin (SN-38) to 5-Fluorouracil and Folinic Acid in HCT-8 Cells: Elevated dTTP Pools and Enhanced Cytotoxicity. Cancer Chemother Pharmacol. 1998;42:391–399. doi: 10.1007/s002800050835. [DOI] [PubMed] [Google Scholar]
- Nelson WG, Kastan MB. DNA Strand Breaks: The DNA Template Alterations that Trigger p53-Dependent DNA Damage Response Pathways. Mol Cell Biol. 1994;14(3):1815–1823. doi: 10.1128/mcb.14.3.1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohyanagi F, Horiike A, Okano Y, Satoh Y, Okumura S, Ishikawa Y, Nakagawa K, Horai T, Nishio M. Phase II trial of gemcitabine and irinotecan in previously treated patients with small-cell lung cancer. Cancer Chemother Pharmacol. 2008;61(3):503–508. doi: 10.1007/s00280-007-0496-6. [DOI] [PubMed] [Google Scholar]
- Plunkett W, Huang P, Xu YZ, Heinemann V, Grunewald R, Gandhi V. Gemcitabine: metabolism, mechanisms of action, and self-potentiation. Semin Oncol. 1995;22(Suppl 11):3–10. [PubMed] [Google Scholar]
- Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer. 2006;6(10):789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- Pourquier P, Gioffre C, Kohlhagen G, Urasaki Y, Goldwasser F, Hertel LW, Yu S, Pon RT, Gmeiner WH, Pommier Y. Gemcitabine (2’,2’-difluoro-2’-deoxycytidine), an antimetabolite that poisons topoisomerase I. Clin Cancer Res. 2002;8(8):2499–2504. [PubMed] [Google Scholar]
- Rocha Lima CM, Green MR, Rotche R, Miller WH, Jr, Jeffrey GM, Cisar LA, Morganti A, Orlando N, Gruia G, Miller LL. Irinotecan plus gemcitabine results in no survival advantage compared with gemcitabine monotherapy in patients with locally advanced or metastatic pancreatic cancer despite increased tumor response rate. J Clin Oncol. 2004;22(18):3776–3783. doi: 10.1200/JCO.2004.12.082. [DOI] [PubMed] [Google Scholar]
- Rocha Lima CM, Savarese D, Bruckner H, Dudek A, Eckardt J, Hainsworth J, Yunus F, Lester E, Miller W, Saville W, Elfring GL, Locker PK, Compton LD, Miller LL, Green MR. Irinotecan plus gemcitabine induces both radiographic and CA 19-9 tumor marker responses in patients with previously untreated advanced pancreatic cancer. J Clin Oncol. 2002;20(5):1182–1191. doi: 10.1200/JCO.2002.20.5.1182. [DOI] [PubMed] [Google Scholar]
- Rocha Lima CMS, Leong S-S, Sherman CA, Perkel JA, Putman-Hair T, Safa AR, Green MR. Phase I Study of CPT-11 and Gemcitabine in Patients with Solid Tumors. Cancer Therapeutics. 1999;2:58–66. [Google Scholar]
- Rocha-Lima C, Herndon J, 2nd, Lee M, Atkins J, Mauer A, Vokes E, Green M. Phase II trial of irinotecan/gemcitabine as second-line therapy for relapsed and refractory small-cell lung cancer: Cancer and Leukemia Group B Study 39902. Ann Oncol. 2007;18(2):331–337. doi: 10.1093/annonc/mdl375. [DOI] [PubMed] [Google Scholar]
- Ruiz van Haperen VW, Veerman G, Boven E, Noordhuis P, Vermorken JB, Peters GJ. Schedule dependence of sensitivity to 2’,2’-difluorodeoxycytidine (Gemcitabine) in relation to accumulation and retention of its triphosphate in solid tumour cell lines and solid tumours. Biochem Pharmacol. 1994;48(7):1327–1339. doi: 10.1016/0006-2952(94)90554-1. [DOI] [PubMed] [Google Scholar]
- Ryan DP, Garcia-Carbonero R, Chabner BA. Cytidine Analogues. In: Chabner BA, Lango DL, editors. Cancer Chemotherapy and Biotherapy. Lippincott Williams & Wilkins; 2006. pp. 183–211. [Google Scholar]
- Saif MW, Sellers S, Li M, Wang W, Cusimano L, Wang H, Zhang R. A phase I study of bi-weekly administration of 24-h gemcitabine followed by 24-h irinotecan in patients with solid tumors. Cancer Chemother Pharmacol. 2007;60(6):871–882. doi: 10.1007/s00280-007-0434-7. [DOI] [PubMed] [Google Scholar]
- Shanks RH, Rizzieri DA, Flowers JL, Colvin OM, Adams DJ. Preclinical evaluation of gemcitabine combination regimens for application in acute myeloid leukemia. Clin Cancer Res. 2005;11(11):4225–4233. doi: 10.1158/1078-0432.CCR-04-2106. [DOI] [PubMed] [Google Scholar]
- Shi Z, Azuma A, Sampath D, Li YX, Huang P, Plunkett W. S-Phase arrest by nucleoside analogues and abrogation of survival without cell cycle progression by 7-hydroxystaurosporine. Cancer Res. 2001;61:1065–1072. [PubMed] [Google Scholar]
- Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Measurement of Protein Using Bicinchoninic Acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- Sparreboom A, Zamboni WC. Topoisomerase I-Targeting Drugs. In: Chabner BA, Longo DL, editors. Cancer Chemotherapy and Biotherapy. Lippincott Williams & Wilkins; 2006. pp. 371–413. [Google Scholar]
- Stathopoulos GP, Syrigos K, Aravantinos G, Polyzos A, Papakotoulas P, Fountzilas G, Potamianou A, Ziras N, Boukovinas J, Varthalitis J, Androulakis N, Kotsakis A, Samonis G, Georgoulias V. A multicenter phase III trial comparing irinotecan-gemcitabine (IG) with gemcitabine (G) monotherapy as first-line treatment in patients with locally advanced or metastatic pancreatic cancer. Br J Cancer. 2006;95(5):587–592. doi: 10.1038/sj.bjc.6603301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Hewitt MR, Theobald MR, Hershock D, Haller DG. A phase 1 study of fixed dose rate gemcitabine and irinotecan in patients with advanced pancreatic and biliary cancer. Cancer. 2007;110(12):2768–2774. doi: 10.1002/cncr.23098. [DOI] [PubMed] [Google Scholar]
- Wan CW, Mak TW. Deoxycytidine kinase and cytosine nucleoside deaminase activities in synchronized cultures of normal rat kidney cells. Cancer Res. 1978;38(9):2768–2772. [PubMed] [Google Scholar]
- Zhang Y-W, Otterness DM, Chiang GG, Xie W, Liu Y-C, Mercurio F, Abraham RT. Genotoxic Stress Targets Human Chk1 for Degradation by the Ubiquitin-Proteasome Pathway. Mol Cell. 2005;19:607–618. doi: 10.1016/j.molcel.2005.07.019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A, histogram showing cell fluorescence after treatment of Ovcar-5 cells with diluent (DMSO) or 10 μM SN-38. B, increase in mean fluorescence intensity as a function of SN-38 concentration. C, time course of SN-38 uptake into cells assessed by measuring mean fluorescence intensity every 10 sec after addition of 10 μM SN-38 to cells in a temperature controlled 37 °C tube holder.
Histograms show DNA content after treatment with 40 nM gemcitabine, 10 nM SN-38 or both drugs simultaneously for 24 h.
A, BxPC-3 cells were treated for 24 h with diluent or SN-38, washed and treated for another 24 h with diluent or gemcitabine using a fixed molar ratio of 8.33:1 (gemcitabine:SN-38). At the end of the incubation, cells were washed and incubated in drug-free medium until colonies formed. Data from the same experiment are plotted as a function of either SN-38 or gemcitabine concentration. Error bars, ± s.d. from triplicate plates. Right panel indicates CI as a function of drug effects. Circles indicate CI calculated from data in left and middle panels under the assumption that effects are mutually exclusive; and line represents second-order regression line calculated from data points. Synergy is indicated by CI < 1, whereas antagonism is indicated by CI > 1. B, BxPC-3 cells were treated for 24 h with diluent or gemcitabine, washed and treated for another 24 h with diluent or SN-38 at a fixed SN-38:gemcitabine ratio of 1:6.
A, histograms showing DNA content after treatment with 10 nM SN-38 for 24 h followed by fixation and PI staining. Alternatively, cells were treated with 10 nM SN-38 for 24 h, washed, and incubated in drug-free medium or 40 nM gemcitabine for 24 h before fixation. B, histograms showing DNA content after treatment with 40 nM gemcitabine for 24 h. Alternatively, cells were treated with gemcitabine for 24 h, washed, and incubated in drug-free medium or 10 nM SN-38 for 24 h.