

Abstract
There is a need to develop improved methods for directing and maintaining the differentiation of human mesenchymal stem cells (hMSC) for regenerative medicine. Here, we present a method for embedding cells in defined protein microenvironments for the directed osteogenic differentiation of hMSC. Composite matrices of collagen I and agarose were produced by emulsification and simultaneous polymerization in the presence of hMSC to produce 30–150 μm diameter hydrogel “beads.” The proliferation, morphology, osteogenic gene expression, and calcium deposition of hMSC in bead environments were compared to other two- and three-dimensional culture environments over 14–21 days in culture. Cells embedded within 40% collagen beads exhibited equivalent proliferation rates to those in gel disks, but showed upregulation of bone sialoprotein and increased calcium deposition over 2D controls. Osteocalcin gene expression was not changed in 3D beads and disks, while collagen type I gene expression was downregulated relative to cells in 2D culture. The hydrogel bead format allows controlled cell differentiation and is a cell delivery vehicle that may also enhance vascular invasion and host incorporation. Our results indicate that the application of such beads can be used to promote the osteogenic phenotype in hMSC, which is an important step toward using them in bone repair applications.
Keywords: mesenchymal stem cell, osteogenic differentiation, collagen I, hydrogels, defined microenvironment
INTRODUCTION
Bone is a dynamic and highly vascularized tissue with tremendous capacity to remodel and heal without leaving a scar.1 With age and disease, however, bone can lose its natural capability to respond to injury and clinical intervention is required to heal larger defects. The need for regenerative bone therapies has driven the field of bone tissue engineering over the past 10 years, yet during this time, no satisfactory bone substitute has been developed.2 A major limitation of bone substitutes currently used is that they do not self-renew, because they are acellular, and thus have a limited useful lifespan.
The current clinical standard for bone grafting is an autogenous graft harvested from the iliac crest. The graft materials include osteogenic cells and growth factors in an appropriate extracellular matrix to promote healing at the site of defect.3 In some patients, such as diabetics, smokers, and those with osteoporosis, tissue of sufficient quality is not available for grafting.2 Therefore, it remains a clinical challenge to stimulate bone formation in applications where healing is delayed or halted. Particularly in areas where host cell infiltration is prevented or there is a lack of vascular supply, the rapid generation of quality bone is needed to prevent morbidity.
In such applications a cell-based therapy could achieve a more robust healing response. The ideal engineered cellular bone graft would need to exhibit the following features: (1) the presence of osteogenic cells to populate the new bone, (2) an appropriate extracellular matrix to provide an osteoconductive scaffold, (3) osteoinductive growth factors to provide signals to the resident cells, and (4) an adequate blood supply to support cell growth and function.4 The development of cell-biomaterial grafting systems is reported in several recent studies, including the cotransplantation of MSC with hydroxyapatite and tricalcium phosphate5 and the use of osteogenically induced MSC seeded on hydroxyapatite/chitin scaffolds promoting bone formation in vivo mouse and rabbit models respectively.6 Human embryonic stem cell derived osteogenic cells have also been used to produce in vivo bone formation on a poly(D,L-lactic-co-glycolic acid)/hydroxyapatite composite scaffold when transplanted in a mouse model.7 A variety of materials have been introduced as potential bone substitutes and some have begun to incorporate growth factors to enhance regeneration. Still, the development of a mechanically sound and reliable cell-based grafting system has yet to see broad success in clinical application.
Human mesenchymal stem cells (hMSC) are a known osteogenic cell source.8 Their ability to differentiate along the osteogenic lineage has been demonstrated in a variety of systems both in vitro and in vivo. In vitro, osteogenic differentiation of hMSC is often induced by culture in the presence of ascorbic acid-2-phosphate, β-glycerophosphate, and the synthetic glucocorticoid dexamethasone. However, in some studies this method has been reported to suppress bone formation and promote cellular toxicity when used in vivo.9,10 In vivo, most efforts to encourage bone formation by hMSC have used osteoinductive scaffolds, with controlled growth factor release. A main limiting factor in using hMSC therapeutically, however, is inadequate control over the induction and maintenance of their differentiation. The ability to provide potent and consistent cues in the immediate cellular niche will be critical to the success of future stem cell therapies.
A number of physiological extracellular matrix proteins have been used as scaffolds for bone tissue engineering, including collagen I,11 fibrin,12 and hyaluronan.13 There have been many approaches to the incorporation of cells into bone grafts including the use of processed scaffolds for the seeding of cells and hydrogels. Protein hydrogels that directly encapsulate cells within a three-dimensional (3D) microenvironment have many advantages. Solubilized collagen I can be mixed with living cells during polymer gelation to directly embed cells in a fibrillar collagen matrix. Collagen I is a major component of the organic phase in bone and hMSC grow and differentiate in reconstituted collagen I hydrogels.14 Adhesion to collagen I via the α2β1 integrin is sufficient to induce osteogenic differentiation of hMSC even in the absence of exogenous soluble stimuli in two-dimensional (2D) culture.15
We previously have reported on a system to embed cells in 3D hydrogel microbeads consisting of collagen I and agarose16 and now have applied this system to the study and direction of hMSC differentiation. In the present study, we examined the osteogenic potential of hMSC within such microenvironments in the absence of soluble osteogenic supplements. We have quantified cellular function within defined collagen/agarose beads in terms of the ability to proliferate, express key bone markers and calcify the matrix. Our work has established the potential of these defined microenvironments for hMSC culture, expansion, and differentiation, with the overall goal of developing a system to deliver predifferentiated hMSC to sites of bone injury in order to enhance repair.
MATERIALS AND METHODS
hMSC were purchased from Lonza Group Ltd. (Switzerland). hMSC tissue culture media (DMEM) was purchased from Mediatech (Cellgro, Herndon VA) and penicillin G-streptomycin sulfate fungizone (FPS) from Hyclone (Fisher Scientific, Fair Lawn, NJ). Fetal bovine serum (FBS) was purchased from Gemini Bio-Products (Woodland, CA). Trypsin-EDTA and agarose was obtained from Sigma Chemical Co. (St. Louis, MO). Purified bovine collagen I was purchased from MP Biomedicals (Solon, OH). The Trizol reagent for RNA isolation was purchased from Invitrogen (Carlsbad, CA) and QuantiTect® SYBR® Green One Step RT-PCR Kit from Qiagen (Valencia, CA). Primers were ordered from Integrated DNA Technologies (Coralville, IA). Alizarin Red S was purchased from Sigma Chemical Co (St. Louis, MO). Hoechst 33258 dye was purchased from Molecular Probes (Eugene, OR) and Proteinase K from Promega (Madison, WI). Unless otherwise specified, the other standard reagents were obtained from Fisher Scientific (Fair Lawn, NJ).
Cell Culture
Cryopreserved hMSC were grown according to manufacturer’s instructions. hMSC were cultured in Dulbecco’s Modification of Eagle’s Medium 1× (DMEM) supplemented with 10% FBS and fungizone/penicillin/streptomycin (FPS) [10,000 units/mL]. Medium was changed every 3 days and cultures were incubated at 37°C in a humidified atmosphere containing 95% air and 5% CO2. Cells were detached using trypsin-EDTA and passaged into fresh culture flasks upon reaching confluence. hMSC were used between passages 4 and 6. In preparation for incorporation in 3D constructs cells were washed with PBS, detached with trypsin-EDTA, collected, and counted using a Beckman Coulter Counter.
Bead Preparation
Beads were prepared as previously described16 and were cultured for up to 21 days. The bead production system is shown schematically in Figure 1. Cells were counted and resuspended at a concentration of 1.0 × 106 cells/mL in a mixture of 5× DMEM, FBS, 0.1N NaOH, 4.0 mg/mL collagen I, and 2.0% agarose (these components were added sequentially in the order listed). The collagen solution was added immediately after the addition of NaOH to neutralize the solution and prevent cellular shock. As demonstrated in our previous work,16 a variety of compositions can be formulated using this system. In the present work, we made 0, 12, 25, and 40% wt/wt collagen/agarose beads to determine the effect of composition on bead recovery in the process. Subsequent experiments used only 0 and 40% wt/wt collagen/agarose beads, because this composition exhibited the most significant cellular response in our previous studies.
Figure 1.

Schematic of the process of bead preparation and phase contrast image of resulting 40% type I collagen beads and disk. Cells were directly embedded within a three-dimensional composite hydrogel microenvironment through an emulsification-based encapsulation process. Cells were combined with liquid ECM components (collagen Type I and agarose) and the cell-ECM suspension was emulsified by continuous stirring in a warm PDMS bath. Matrix gelation was then initiated by cooling the bath and gelled beads were collected through successive PBS washes, and subsequently were cultured under normal cell culture conditions. Corresponding disk constructs were made by molding gels in a 6-well plate. Large inset shows a phase contrast image of 40% wt/wt collagen/agarose beads (acellular). Bead diameter ranged from ~30 to 150 μM. Smaller inset shows a 40% wt/wt collagen/agarose disk immediately after gelation.
The emulsification vessel consisted of a bath of PDMS fluid kept at 37°C. A stirrer motor fitted with a double-bladed impeller was used to stir the bath and create a complex flow pattern that encouraged break up of liquid droplets. The suspension of living cells in ECM solution was rapidly injected into the warm, continuously stirred PDMS phase. Total emulsification time was 6 min, at which time a secondary bath containing ice water was fitted around the PDMS bath to achieve rapid cooling and gelation of the emulsified droplets into beads. The system was cooled for 30 min with continuous stirring. This allowed for gelation of the spherical bead phase, while preventing reagglomeration of the cell/ECM mixture. All material from the emulsification vessel was then transferred to centrifuge tubes along with an equal volume of sterile PBS, and this mixture was spun at 1000 rpm for 5 min. The formed cell/ECM beads preferentially partitioned into the aqueous phase and allowed the PDMS to be aspirated away. Two similar wash steps with PBS followed and the washed beads were transferred to culture dishes and kept in culture medium.
3D Gel and Coated Plate Preparation
Three dimensional gels and two-dimensional coated plates were prepared similarly to the beads. Gels were made containing hMSC in 40% collagen/agarose and were prepared as described for bead preparation, except instead of injection into the emulsification vessel the cell/ECM mixture was injected directly into a 6-well plate. Gels were kept at 4°C for 3 min and then transferred to 37°C and incubated for 30 min to allow complete gelation prior to the addition of culture medium. After 24 h, the gels were released from the edge of the well to allow for compaction. Cellular compaction was kept constant at 1.0 × 106 cells per mL/ECM.
Plates were coated with the 40% collagen/agarose ECM mixture without cells and allowed to gel. One milliliter of ECM mixture was injected into a 6-well plate to coat the bottom and placed in 4°C for 3 min and then moved to 37°C and incubated for 30 min. Cells were then washed with PBS, detached using trypsin-EDTA and plated on top of the coated wells. Cells were plated at density of 1 × 105 cells/well and cultured at 37°C.
Second Harmonic Generation (SHG) and Scanning Electron Microscopy (SEM)
Beads were imaged using a Zeiss LSM 510 two photon confocal microscope (Zeiss, Thornwood, NY). Samples were excited by a two photon laser at 820 nm and emissions were collected at 480 nm. Images were taken at a 65× magnification and 3D stacks were collected. SHG imaging of pure agarose constructs showed no signal. Microstructure imaging was performed at day 3 and day 7, because these time points represent the early and later stages of gel compaction in this hydrogel system. Our aim was to observe how the collagen in the bead matrix was distributed and subsequently actively remodeled by the cellular component.
Visualization of scaffold microarchitecture was completed with a LEO 1550 VP Field Emission SEM (SEM, Zeiss, Thornwood NY). Disks were fixed in 4% glutaraldehyde for 1 h, washed with PBS and dehydrated by freeze drying for SEM preparation. Samples were sputter coated with gold before imaging.
RNA Isolation
RNA was isolated using the TRIzol Reagent (Invitrogen, Carlsbad, CA) after 14 days in culture. This time point was chosen based upon previous studies that examined the osteogenic differentiation of hMSC on 2D rigid collagen I substrates. Constructs were homogenized in the TRIzol reagent using the TissueRuptor power homogenizer. Isolation was performed as per manufacturer’s instructions and total isolated RNA was dissolved in RNase/DNase free water and stored at −20°C. Total RNA was quantified using a NanoDrop spectrophotometer (NanoDrop Technologies, Wilmington, DE).
Quantitative Real Time RT-PCR
Quantitative reverse transcription RT-PCR was performed using the LightCycler® 480 Real-Time PCR System (Roche, Pleasonton, CA) and the QuantiTect® SYBR® Green RT-PCR Kit with HotStar Taq DNA Polymerase. 1× QuantiTect SYBR Green, 0.5 μM Primer F and Primer R, 0.5 μL/reaction QuantiTect RT Mix were combined with sample RNA to yield a 20 μL reaction volume. Primers used for amplification of differentiation marker genes were designed using OligoPerfect™ (Invitrogen, Carlsbad, CA) and purchased through IDT Technologies (Coralville, IA). Primers used in this study are found in Table I. RT-PCR was performed according to the following protocol defined by the manufacturer: RT 20 min at 50, 20°C/s ramp, PCR activation 15 min at 95°C, 20°C/s ramp, 35–55 cycles of [denaturation 15 s at 94°C, 20/s ramp, annealing 20–30 s at 50–60°C, 20°C/s ramp, extension 30 s 72°C, 2°C/s ramp]. The markers of cell phenotype used were collagen type I (COL1), bone sialoprotein (BSP), and bone GLA protein (BGLAP, also known as osteocalcin). All samples were loaded in duplicate and normalized to total RNA content and to the performance of the housekeeping gene, GAPDH. Fold differences in gene expression were relative to hMSC cultured on tissue culture plastic (TCP) and calculated using the ΔΔCt method.17
TABLE I.
Real Time RT-PCR Primers
| Gene/NCBI Designation | Primer Sequences |
|---|---|
| Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) | Forward 5′-CGACCACTTTGTCAAGCTCA-3′,
Reverse 5′-AGGGGTCTACATGGCAACTG-3′ |
| Bone sialoprotein (BSP) | Forward 5′-CTGCTTCCTCACTCCAGGAC-3′,
Reverse 5′-GATTGCTTCCTCTGGCAGTC-3′ |
| Osteocalcin (BGLAP) | Forward 5′-GACTGTGACGAGTTGGCTGA-3′,
Reverse 5′-CTGGAGAGGAGCAGAACTGG-3′ |
| Collagen I (COL I) | Forward 5′-GACGTCCTGGTGAAGTTGGT-3′,
Reverse 5′-ACCAGGGAAGCCTCTCTCTC-3′ |
Calcium Deposition Assay—Alizarin Red
The Alizarin Red assay used here was adapted from a published quantitative dye extraction protocol.18 At day 21, samples were washed twice with PBS and fixed in 10% formaldehyde for 15 min at room temperature. Samples were then washed twice with excess dH2O and incubated in the presence of 40 mM Alizarin Red Solution (pH = 4.1) for 20 min at room temperature with shaking. Samples were washed four times with excess dH2O with shaking for 5 min per wash. Pelleted samples were mixed with 800 μL of 10% acetic acid, homogenized, and shaken for 1.5 h at room temperature. Homogenized samples were loaded in a 96-well plate and quantified using densitometry. Acellular controls for 3D samples were processed simultaneously and subtracted as background from samples. All measurements were normalized to volume and total cell number as determined by a DNA assay.
DNA Assay
Cell number was assessed at different time points to assess cell recovery (day 0), to characterize cell proliferation (day 15), and to normalize calcium assays (day 21). To extract the cells from the three-dimensional constructs and two-dimensional collagen/agarose coated samples, constructs were freeze dried and digested in Proteinase K at 55°C for 16–20 h, to break apart the matrix and allow for cellular release. DNA content was analyzed using the Hoechst 33258 dye and samples were diluted and loaded into a 96-well plate in duplicate. Following the addition of the DNA binding, dye samples were incubated protected from light for 15 min. Fluorescence was quantified using a fluorescence microplate reader (BioTek, Winooksi, VT) at an excitation wavelength of 350 nm and an emission wavelength of 460 nm. The DNA content of each 3D construct was converted to cell number based on cell and DNA standards.
Statistical Analysis
All experiments were repeated a minimum of three times, and the representing data are presented as mean ± SEM. Statistical analyses were performed using Student’s unpaired t test, and a p-value < 0.05 was considered significant.
RESULTS
To determine the efficiency of our encapsulation process, we quantified cell number immediately following the production of both 0 and 40 wt/wt % collagen/agarose beads. Cell number was quantified using a DNA binding assay, and the fraction of encapsulated cells was calculated based on an initial cell number of 3 million cells per bead batch. As shown in Figure 2(A), ~1.1–1.4 million cells were recovered for both bead types, representing a cell encapsulation efficiency of ~40–45%. To determine whether cell loss was associated with loss of matrix material or whether cells were lost independently, we assessed the mass recovery of collagen/agarose matrix in beads as compared to disk constructs (which exhibit no mass loss). As shown in Figure 2(B), recovery of matrix ranged from 44–53%, similar to the cell recovery level, with the degree of recovery increasing with increasing collagen content in the matrix (p < 0.05).
Figure 2.

Recovery of both cellular and matrix material was ~40–50% following encapsulation. Panel A: hMSC encapsulated in 0 and 40 wt/wt % beads were assayed for total cell number. Results are presented as a percent recovered of original (3 × 106 cells). There was no statistical difference between the two 3D samples (p = 0.7, n = 5). Panel B: 0, 12, 25, and 40 wt/wt % collagen/agarose beads were made and immediately assayed for total recovered mass. Results are presented as a percent recovered of total material used, as measured by equivalent disk constructs. *Statistical significance p < 0.05, n = 3, relative to 0% bead mass recovery.
SEM and two-photon confocal microscopy using SHG were used to image the bead matrix. The use of both these techniques allowed for the imaging of both the surface and interior of the 3D constructs to provide a more complete view of the microstructure of the hydrogel beads. Figure 3 shows SEM micrographs of the surface of 0 and 40% composite matrices. Pure agarose matrix (panel A of Figure 3) exhibited a granular appearance with no fibrillar structure. Gels containing 40 wt % collagen (panel B of Figure 3) showed a prominent fibrillar collagen network that was surrounded by an agarose phase. SHG imaging of hydrogel matrices at day 3 and day 7 in culture is shown in Figure 4. Only the collagen fibrils are evident in these images, as agarose does not produce an SHG signal. It can be seen that the fibrillar collagen network was consolidated over time in culture, producing a more dense mesh of collagen fibers.
Figure 3.

Scanning Electron Microscopy (SEM) reveals distinct microarchitecture between pure agarose and collagen/agarose composites. Panel A: Pure agarose gel. Panel B: 40 wt/wt % collagen/agarose gel. Scale bar represents 1 μM.
Figure 4.

Second harmonic generation (SHG) imaging reveals changing collagen type I fibril networks over time. Panels A and B: 25 wt/wt % collagen/agarose hydrogel after culture with hMSC for 3 (A) and 7 (B) days. Panels C and D: 40 wt/wt % collagen/agarose hydrogel after culture with hMSC for 3 (C) and 7 (D) days. Scale bar represents 20 μM.
Figure 5 shows the degree of hMSC proliferation on and in 40% collagen/agarose gels. hMSC were cultured either on top of gel substrates, in gel disks, or in gel beads for 15 days, at which time cell number was determined by DNA binding assay. Because hMSC number decreased due to cell loss during the encapsulation process (see Figure 2, above), the degree of proliferation was based on the corrected cell number at day 0. hMSC cultured in three dimensions exhibited approximately threefold proliferation after 15 days in culture and there was no statistical significance between disk and bead geometries. In contrast, hMSC grown on top of an acellular 40 wt/wt % collagen/agarose gel exhibited a 15-fold increase in cell number over 15 days.
Figure 5.
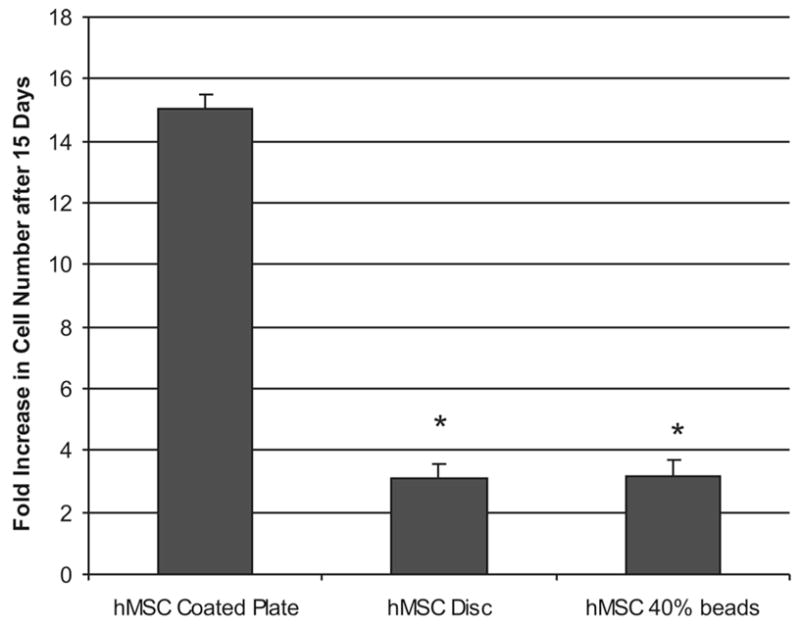
hMSC proliferate in collagen/agarose 3D microenvironments, but to a lower degree than in monolayer culture. Fold increase is represented here as change in total cell number from day 0 after 15 days in culture. *Statistical significance p < 0.05, n = 3, relative to the 2D collagen/agarose-coated plate control. Proliferation in 3D disks is not significantly different from beads.
To better characterize cellular function within these defined microenvironments and assess the effect of a three-dimensional culture on the process of osteogenic differentiation, real-time RT-PCR was used to detect the relative expression of genes important in bone formation. BSP and osteocalcin gene expression was assessed because these proteins are known to be involved in matrix mineralization. Collagen I (COLI) gene expression was also assessed since it is a primary constituent of bone and of the hydrogel matrices being examined. hMSC were cultured for 2 weeks on TCP, on 40% collagen/agarose substrates, in 40% collagen/agarose disks, or in 40% collagen/agarose beads. RT-PCR results were normalized to GAPDH and are expressed in Figure 6 as a fold increase over TCP. hMSC cultured in both 3D collagen microenvironments (disk and bead) exhibited statistically significant increases in BSP gene expression (p < 0.05) relative to TCP, showing ~5.5- and 3.5-fold increases respectively. In contrast, expression of bone GLA protein (BGLAP, also known as osteocalcin), was not statistically altered in 3D matrices, and was slightly downregulated on 2D hydrogel substrates. Expression of COLI was markedly reduced in all hydrogel treatments, relative to TCP.
Figure 6.
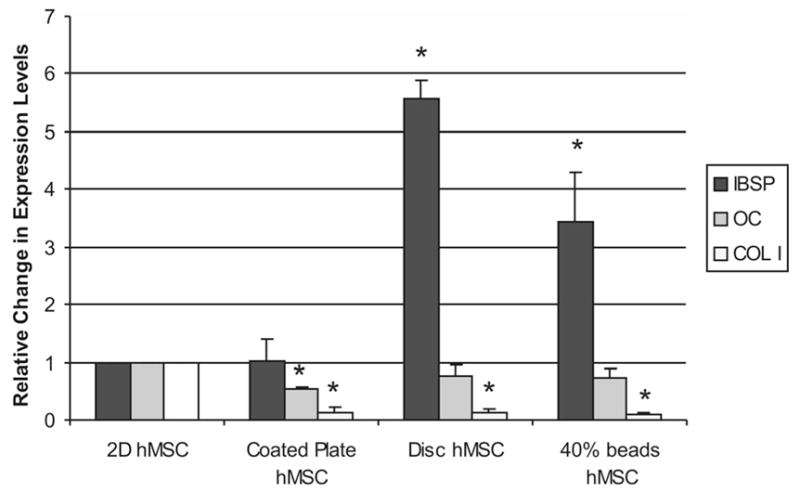
3D culture upregulates the expression of bone sialoprotein, does not affect osteocalcin and downregulates collagen I production over 14 days in culture. Fold change is relative to hMSC grown on tissue culture plastic (TCP) and normalized to the housekeeping gene GAPDH. *Statistical significance p < 0.05, n = 3 relative to 2D hMSC plated on TCP.
A hallmark of hMSC osteogenic differentiation is the mineralization of the protein matrix surrounding the cells. To assess the extent of extracellular calcium deposition we used a modified Alizarin Red staining assay that allowed for the semiquantification of matrix calcification and direct comparison of 2D and 3D samples (Figure 7). After 21 days in culture, hMSC grown in 40% collagen/agarose disks showed an ~4-fold greater degree of calcium deposition than cells grown on a 2D collagen/agarose substrate (p < 0.05). hMSC in pure agarose beads deposited calcium to the same degree as those on the 2D substrate, but cells in 40% collagen/agarose beads exhibited ~3-fold increased deposition.
Figure 7.

3D culture promotes calcium deposition into the extracellular matrix over 21 days in culture. Extracellular calcium deposition is detected using an adapted Alizarin Red assay and quantified using densitometry. Samples are normalized to total cell number. *Statistical significance p < 0.05, n = 3 relative to 2D hMSC on collagen/agarose hydrogel.
DISCUSSION
In the work presented here, we have further characterized a previously established procedure for creating protein-based 3D microenvironments for the culture of hMSC. In our earlier studies, we established control of bead size and composition varying both the emulsification parameters as well as collagen I content. We also were able to establish the effect of collagen concentration on altered hMSC morphology as a result of integrin-mediated adhesion events between the hMSC and collagen I fibers.16 Such adhesion to matrix is associated with the osteogenic differentiation of hMSC in 2D culture.15 In this study, we continued the characterization of the emulsification process itself as well as the osteogenic potential of hMSC encapsulated within 3D collagen/agarose microenvironments.
Matrix-based directed differentiation of hMSC has potential advantages over other techniques currently used in bone tissue engineering. Collagen-based materials have been used as a matrix in a variety of applications, including cartilage and bone.19–21 Cells can be homogenously entrapped and distributed directly within collagen hydrogels, whereas approaches using preformed collagen scaffolds generally necessitate seeding of cells onto only the surface of the constructs.19,21,22 The microbead format of our system has the potential to be used as a minimally invasive technique for delivering cells already encased in a matrix. In addition, packed beds of beads maintain void spaces between beads and these void spaces may promote the ingrowth of a vascular supply, which in turn may enhance graft viability and function.
The injection of slurries of microbeads that contain encapsulated, predifferentiated hMSC offers the potential to fill defects in bone and promote more rapid and appropriate healing. In particular, this method is suited to confined defects that can be conformally filled with beads, but which will not be required to bear load until healing has progressed. Example applications where this type of cell therapy may have utility are in implant fixation, spinal fusions, and the treatment of avascular necrosis. For applications that require immediate load bearing, more mechanically robust, synthetic scaffolds will be more likely to have utility, and there are a number of approaches that have targeted these applications.
The use of synthetic polymers for tissue engineering generally requires that the material be functionalized for cell adhesion, and these materials may not degrade consistently or benignly within the host.23–27 In addition to the structural role of collagen I in scaffold development, cellular adhesion to this matrix protein elicits cellular responses without the use of soluble stimuli or scaffold functionalization. The use of such natural matrix components allows for cell-mediated degradation of the construct to allow for further incorporation via secreted enzymes.27–31 In our current system, the polysaccharide agarose provides structural support to the hydrogel system and is used to facilitate harvesting of formed beads from the emulsification vessel. Agarose has been shown to be well integrated with host tissue in a rat dermal model; agarose gel was degraded and newly formed collagen fibers were visible in the implants with no noticeable granuloma formation.32 The need for a structural filler (e.g., alginate) in the production of collagen beads has been also reported in other bead formulation protocols.33,34 However, the ability of hMSC to interact with the collagen I fibers does not appear to be inhibited by the presence of agarose as a filler material, and our studies have shown that the collagen component of the beads has clear effects on the function of the embedded cells.
The method we have developed for making collagen-agarose beads can reliably produce beads of desired composition, size, and size distribution. However, the results of the current study show that cell and matrix recovery is only in the range of 40–50%. Importantly, it should be noted that the bead batches produced in this study used only 3.0 mL of cell/ECM material. It is likely that a large portion of the lost material is a “fixed loss” due to adherence of the formed beads to the emulsification impeller and apparatus, but that this loss will not scale with batch size, so that larger batches will exhibit larger percent recoveries. Improved material recovery is a goal of future process optimization.
Cells embedded inside 3D collagen I gels tend to exhibit lower proliferation rates, compared to cells grown as monolayers in 2D.22,35 We observed a similar effect with hMSC in the 3D collagen/agarose bead system. hMSC in 40% collagen-agarose beads and disks did proliferate, exhibiting about a 3-fold increase in cell number over 15 days in culture. However, this proliferation rate was much lower than hMSC grown on top of the same material, which increased in number ~15-fold over the same time. Decreased proliferation in 3D hydrogels also has been observed in other studies using MSC.36 It is not clear why a 3D environment leads to decreased cell proliferation, but this effect may be due to steric hindrance caused by the matrix or altered signaling that causes MSC to exit the cell cycle. It is encouraging that hMSC were able to proliferate in collagen/agarose beads, though at a lower rate than in monolayer culture. Lower proliferation rates may in fact be more conducive to allowing controlled differentiation of populations of hMSC.
Osteogenic differentiation of hMSC in 3D scaffolds is often induced through the use of potent growth factors27,37,38 and/or other biochemical stimulants.39 A 3D matrix microstructure can also promote multilineage differentiation of hMSC.40 In the present study, we demonstrated significant upregulation of BSP gene expression as a result of 3D culture in collagen/agarose matrices, without the use of exogenous growth factors or soluble biochemicals. Osteocalcin gene expression was not affected by 3D culture. Collagen I was markedly downregulated, most likely because the collagen I receptor α1β1 integrin, which is expressed by hMSC, provides negative feedback on collagen synthesis.41 Our results also showed increased calcium deposition by hMSC in 3D matrices, as compared to 2D cultures. Calcium deposition was higher in disk constructs, which may reflect the enhanced diffusion of material out of the beads. This would be a positive feature of the beads, since it would allow diffusion of mineral to the matrix surrounding the beads in a bone healing situation, thereby enhancing interaction at the host/graft interface.
A continuing challenge in implementing improved cellular grafting techniques is the inability to maintain cell viability and phenotype. The encapsulation of hMSC in a defined protein hydrogel microenvironment offers the possibility of directing osteoblastic differentiation without the use of exogenous biochemical stimulation. Use of the bead format can facilitate delivery of the cells directly into a site of bone injury, and the surrounding matrix may serve as a buffer to allow hMSC to maintain both their viability and differentiation potential while being exposed to the damaged environment of the defect. The potential applications of this type of encapsulation system also extend to cell-based diagnostics and therapeutic protein production via biotechnology, both cases where the control of cell function is important. Future studies will include improvements to the bead production process, the incorporation of other matrix materials (e.g., fibronectin, vitronectin) to enhance the osteogenic response of encapsulated hMSC, and further characterization of hMSC phenotype within varying bead microenvironments.
Acknowledgments
Contract grant sponsor: National Institute of Arthritis and Musculoskeletal and Skin Diseases; Contract grant number: R01-AR053231
References
- 1.Sommerfeldt DW, Rubin CT. Biology of bone and how it orchestrates the form and function of the skeleton. Eur Spine J. 2001;10(Suppl 2):S86–S95. doi: 10.1007/s005860100283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Salgado AJ, Coutinho OP, Reis RL. Bone tissue engineering: State of the art and future trends. Macromol Biosci. 2004;4:743–765. doi: 10.1002/mabi.200400026. [DOI] [PubMed] [Google Scholar]
- 3.Orban JM, Marra KG, Hollinger JO. Composition options for tissue-engineered bone. Tissue Eng. 2002;8:529–539. doi: 10.1089/107632702760240454. [DOI] [PubMed] [Google Scholar]
- 4.Goldberg VM. Selection of bone grafts for revision total hip arthroplasty. Clin Orthop Relat Res. 2000;381:68–76. doi: 10.1097/00003086-200012000-00008. [DOI] [PubMed] [Google Scholar]
- 5.Shi S, Bartold PM, Miura M, Seo BM, Robey PG, Gronthos S. The efficacy of mesenchymal stem cells to regenerate and repair dental structures. Orthod Craniofac Res. 2005;8:191–199. doi: 10.1111/j.1601-6343.2005.00331.x. [DOI] [PubMed] [Google Scholar]
- 6.Ge Z, Baguenard S, Lim LY, Wee A, Khor E. Hydroxyapatitechitin materials as potential tissue engineered bone substitutes. Biomaterials. 2004;25:1049–1058. doi: 10.1016/s0142-9612(03)00612-4. [DOI] [PubMed] [Google Scholar]
- 7.Kim S, Kim SS, Lee SH, Eun AS, Gwak SJ, Song JH, Kim BS, Chung HM. In vivo bone formation from human embryonic stem cell-derived osteogenic cells in poly(D,L-lactic-co-glycolic acid)/hydroxyapatite composite scaffolds. Biomaterials. 2007 doi: 10.1016/j.biomaterials.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 8.Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S, Marshak DR. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284:143–147. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- 9.Bruder SP, Jaiswal N, Ricalton NS, Mosca JD, Kraus KH, Kadiyala S. Mesenchymal stem cells in osteobiology and applied bone regeneration. Clin Orthop Relat Res. 1998;355(Suppl):S247–S256. doi: 10.1097/00003086-199810001-00025. [DOI] [PubMed] [Google Scholar]
- 10.Ng PC, Lam CW, Wong GW, Lee CH, Cheng PS, Fok TF, Chan IH, Wong E, Cheung K, Lee SY. Changes in markers of bone metabolism during dexamethasone treatment for chronic lung disease in preterm infants. Arch Dis Child Fetal Neonatal Ed. 2002;86:F49–F54. doi: 10.1136/fn.86.1.F49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiao Y, Qian H, Young WG, Bartold PM. Tissue engineering for bone regeneration using differentiated alveolar bone cells in collagen scaffolds. Tissue Eng. 2003;9:1167–1177. doi: 10.1089/10763270360728071. [DOI] [PubMed] [Google Scholar]
- 12.Perka C, Schultz O, Spitzer RS, Lindenhayn K, Burmester GR, Sittinger M. Segmental bone repair by tissue-engineered periosteal cell transplants with bioresorbable fleece and fibrin scaffolds in rabbits. Biomaterials. 2000;21:1145–1153. doi: 10.1016/s0142-9612(99)00280-x. [DOI] [PubMed] [Google Scholar]
- 13.Solchaga LA, Dennis JE, Goldberg VM, Caplan AI. Hyaluronic acid-based polymers as cell carriers for tissue-engineered repair of bone and cartilage. J Orthop Res. 1999;17:205–213. doi: 10.1002/jor.1100170209. [DOI] [PubMed] [Google Scholar]
- 14.Xiao G, Wang D, Benson MD, Karsenty G, Franceschi RT. Role of the α2-integrin in osteoblast-specific gene expression and activation of the Osf2 transcription factor. J Biol Chem. 1998;273:32988–32994. doi: 10.1074/jbc.273.49.32988. [DOI] [PubMed] [Google Scholar]
- 15.Salasznyk RM, Williams WA, Boskey A, Batorsky A, Plopper GE. Adhesion to vitronectin and collagen I promotes osteogenic differentiation of human mesenchymal stem cells. J Biomed Biotechnol. 2004;2004:24–34. doi: 10.1155/S1110724304306017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Batorsky A, Liao J, Lund AW, Plopper GE, Stegemann JP. Encapsulation of adult human mesenchymal stem cells within collagen-agarose microenvironments. Biotechnol Bioeng. 2005;92:492–500. doi: 10.1002/bit.20614. [DOI] [PubMed] [Google Scholar]
- 17.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 18.Gregory CA, Gunn WG, Peister A, Prockop DJ. An Alizarin red-based assay of mineralization by adherent cells in culture: Comparison with cetylpyridinium chloride extraction. Anal Biochem. 2004;329:77–84. doi: 10.1016/j.ab.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 19.Kakudo N, Shimotsuma A, Miyake S, Kushida S, Kusumoto K. Bone tissue engineering using human adipose-derived stem cells and honeycomb collagen scaffold. J Biomed Mater Res A. 2007;84A:191–197. doi: 10.1002/jbm.a.31311. [DOI] [PubMed] [Google Scholar]
- 20.Malicev E, Radosavljevic D, Velikonja NK. Fibrin gel improved the spatial uniformity and phenotype of human chondrocytes seeded on collagen scaffolds. Biotechnol Bioeng. 2007;96:364–370. doi: 10.1002/bit.21038. [DOI] [PubMed] [Google Scholar]
- 21.Vunjak-Novakovic G, Meinel L, Altman G, Kaplan D. Bioreactor cultivation of osteochondral grafts. Orthod Craniofac Res. 2005;8:209–218. doi: 10.1111/j.1601-6343.2005.00334.x. [DOI] [PubMed] [Google Scholar]
- 22.Brannvall K, Bergman K, Wallenquist U, Svahn S, Bowden T, Hilborn J, Forsberg-Nilsson K. Enhanced neuronal differentiation in a three-dimensional collagen-hyaluronan matrix. J Neurosci Res. 2007;85:2138–2146. doi: 10.1002/jnr.21358. [DOI] [PubMed] [Google Scholar]
- 23.Na K, Kim SW, Sun BK, Woo DG, Yang HN, Chung HM, Park KH. Osteogenic differentiation of rabbit mesenchymal stem cells in thermo-reversible hydrogel constructs containing hydroxyapatite and bone morphogenic protein-2 (BMP-2) Biomaterials. 2007;28:2631–2637. doi: 10.1016/j.biomaterials.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 24.Kim G, Okumura M, Bosnakovski D, Ishiguro T, Park CH, Kadosawa T, Fujinaga T. Effects of ascorbic acid on proliferation and biological properties of bovine chondrocytes in alginate beads. Jpn J Vet Res. 2003;51:83–94. [PubMed] [Google Scholar]
- 25.Nuttelman CR, Mortisen DJ, Henry SM, Anseth KS. Attachment of fibronectin to poly(vinyl alcohol) hydrogels promotes NIH3T3 cell adhesion, proliferation, and migration. J Biomed Mater Res. 2001;57:217–223. doi: 10.1002/1097-4636(200111)57:2<217::aid-jbm1161>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 26.Chiu JB, Liu C, Hsiao BS, Chu B, Hadjiargyrou M. Functionalization of poly(L-lactide) nanofibrous scaffolds with bioactive collagen molecules. J Biomed Mater Res A. 2007;83A:1117–1127. doi: 10.1002/jbm.a.31279. [DOI] [PubMed] [Google Scholar]
- 27.Pratt AB, Weber FE, Schmoekel HG, Muller R, Hubbell JA. Synthetic extracellular matrices for in situ tissue engineering. Biotechnol Bioeng. 2004;86:27–36. doi: 10.1002/bit.10897. [DOI] [PubMed] [Google Scholar]
- 28.Noth U, Rackwitz L, Heymer A, Weber M, Baumann B, Steinert A, Schutze N, Jakob F, Eulert J. Chondrogenic differentiation of human mesenchymal stem cells in collagen type I hydrogels. J Biomed Mater Res A. 2007;83:626–635. doi: 10.1002/jbm.a.31254. [DOI] [PubMed] [Google Scholar]
- 29.Sivakumar P, Czirok A, Rongish BJ, Divakara VP, Wang YP, Dallas SL. New insights into extracellular matrix assembly and reorganization from dynamic imaging of extracellular matrix proteins in living osteoblasts. J Cell Sci. 2006;119:1350–1360. doi: 10.1242/jcs.02830. [DOI] [PubMed] [Google Scholar]
- 30.Ehrbar M, Djonov VG, Schnell C, Tschanz SA, Martiny-Baron G, Schenk U, Wood J, Burri PH, Hubbell JA, Zisch AH. Cell-demanded liberation of VEGF121 from fibrin implants induces local and controlled blood vessel growth. Circ Res. 2004;94:1124–1132. doi: 10.1161/01.RES.0000126411.29641.08. [DOI] [PubMed] [Google Scholar]
- 31.Ehrbar M, Rizzi SC, Hlushchuk R, Djonov V, Zisch AH, Hubbell JA, Weber FE, Lutolf MP. Enzymatic formation of modular cell-instructive fibrin analogs for tissue engineering. Biomaterials. 2007;28:3856–3866. doi: 10.1016/j.biomaterials.2007.03.027. [DOI] [PubMed] [Google Scholar]
- 32.Fernandez-Cossio S, Leon-Mateos A, Sampedro FG, Oreja MT. Biocompatibility of agarose gel as a dermal filler: Histologic evaluation of subcutaneous implants. Plast Reconstr Surg. 2007;120:1161–1169. doi: 10.1097/01.prs.0000279475.99934.71. [DOI] [PubMed] [Google Scholar]
- 33.Bosnakovski D, Mizuno M, Kim G, Takagi S, Okumura M, Fujinaga T. Chondrogenic differentiation of bovine bone marrow mesenchymal stem cells (MSCs) in different hydrogels: Influence of collagen type II extracellular matrix on MSC chondrogenesis. Biotechnol Bioeng. 2006;93:1152–1163. doi: 10.1002/bit.20828. [DOI] [PubMed] [Google Scholar]
- 34.Chiu CT, Chang WC, Wang YJ. Microspheres of collagen/β-TCP with an open network fibrillar structure strengthened by chitosan. Artif Cells Blood Substit Immobil Biotechnol. 2007;35:309–317. doi: 10.1080/10731190701378626. [DOI] [PubMed] [Google Scholar]
- 35.Hong H, McCullough CM, Stegemann JP. The role of ERK signaling in protein hydrogel remodeling by vascular smooth muscle cells. Biomaterials. 2007;28:3824–3833. doi: 10.1016/j.biomaterials.2007.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Markusen JF, Mason C, Hull DA, Town MA, Tabor AB, Clements M, Boshoff CH, Dunnill P. Behavior of adult human mesenchymal stem cells entrapped in alginate-GRGDY beads. Tissue Eng. 2006;12:821–830. doi: 10.1089/ten.2006.12.821. [DOI] [PubMed] [Google Scholar]
- 37.Heckmann L, Fiedler J, Mattes T, Dauner M, Brenner RE. Interactive effects of growth factors and 3D-scaffolds on multipotent mesenchymal stromal cells. Biotechnol Appl Biochem. 2007 doi: 10.1042/BA20070071. [DOI] [PubMed] [Google Scholar]
- 38.Hosseinkhani H, Hosseinkhani M, Khademhosseini A, Kobayashi H. Bone regeneration through controlled release of bone morphogenetic protein-2 from 3-D tissue engineered nano-scaffold. J Contr Release. 2007;117:380–386. doi: 10.1016/j.jconrel.2006.11.018. [DOI] [PubMed] [Google Scholar]
- 39.Hosseinkhani H, Hosseinkhani M, Gabrielson NP, Pack DW, Khademhosseini A, Kobayashi H. DNA nanoparticles encapsulated in 3D tissue-engineered scaffolds enhance osteogenic differentiation of mesenchymal stem cells. J Biomed Mater Res A. 2007 doi: 10.1002/jbm.a.31327. [DOI] [PubMed] [Google Scholar]
- 40.Li WJ, Tuli R, Huang X, Laquerriere P, Tuan RS. Multilineage differentiation of human mesenchymal stem cells in a three-dimensional nanofibrous scaffold. Biomaterials. 2005;26:5158–5166. doi: 10.1016/j.biomaterials.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 41.Gardner HA. Integrin signaling in fibrosis and scleroderma. Curr Rheumatol Rep. 1999;1:28–33. doi: 10.1007/s11926-999-0021-5. [DOI] [PubMed] [Google Scholar]