

Abstract
Type 1 diabetes (T1D) is a debilitating autoimmune disease that results from T cell-mediated destruction of insulin-producing β cells. Its incidence has increased during the past several decades in developed countries 1, 2, suggesting that changes in the environment (including human microbial environment) may influence disease pathogenesis. The incidence of spontaneous T1D in non-obese diabetic (NOD) mice can be affected by the microbial environment in the animal housing facility3 or by exposure to microbial stimuli, such as injection with mycobacteria or various microbial products 4,5. Here we show that specific-pathogen free (SPF) NOD mice lacking MyD88 protein (an adaptor for multiple innate immune receptors that recognize microbial stimuli) do not develop T1D. The effect is dependent on commensal microbes as germ-free (GF) MyD88-negative NOD mice develop robust diabetes, whereas colonization of these GF NOD.MyD88-negative mice with a defined microbial consortium (representing bacterial phyla normally present in human gut) attenuates T1D. We also find that MyD88-deficiency changes the composition of the distal gut microbiota, and that exposure to the microbiota of SPF NOD.MyD88-negative donors attenuates T1D in GF NOD recipients. Together, these findings indicate that interaction of the intestinal microbes with the innate immune system is a critical epigenetic factor modifying T1D predisposition.
Toll-like receptors (TLRs) are innate pattern recognition receptors 6 involved in host defense7, control over commensal bacteria and the maintenance of tissue integrity8, 9. The role of TLRs involvement in organ-specific autoimmunity is not clear. The MyD88 adaptor protein is used by multiple TLRs (except TLR4 and TLR3, which can or must signal via TRIF, respectively) and other receptors (e.g. interleukin-1 receptor, IL-1R). To test the contributions of these receptors to development of T1D in NOD mice, we examined the effect of MyD88 gene disruption on disease incidence and progression. Two MyD88 knockout (KO) NOD strains were independently derived at the Jackson Laboratory (J) and at Yale University (Y). Both showed a loss of diabetes development during 30 week observation periods when housed under normal specific-pathogen free (SPF) conditions with continuous monitoring for the presence of murine pathogens (Fig. 1). Because multiple TLRs signal through the MyD88 adaptor, follow-up studies were conducted in NOD mice lacking individual TLRs (TLRKO). We found that TLR2 and TLR4 (as well as TLR3, data not shown) were dispensable for development of T1D (or protection from it by complete Freund's adjuvant; Supplemental Fig. 1) when deleted individually, in contrast to the effect of complete protection from diabetes associated with loss of MyD88 (Fig. 1).
Figure 1. MyD88-negative (MyD88KO) mice are completely protected from development of type 1 diabetes.

Incidence of diabetes in two independently derived NOD.MyD88KO and heterozygous NOD.MyD88KO/+ stocks (J-Jackson Laboratory and Y-Yale) (12 back-crosses to NOD). F, females; M, males. n, number of animals per group.
These findings suggested that signaling through receptors which use the MyD88 adaptor is critical for T1D development, and that the autoimmune T cells would likely be affected systemically in NOD.MyD88KO mice. Two types of experiments were performed to examine this hypothesis. First, splenocytes from pre-diabetic MyD88-sufficient and NOD.MyD88KO mice were transferred into immunodeficient NOD.SCID animals. All recipients of control MyD88-sufficient splenocytes (n=5), and 4 out of 5 recipients of NOD.MyD88KO splenocytes became diabetic, arguing against profound systemic tolerance of T cells in NOD.MyD88KO mice. Second, an Elispot analysis of interferon-γ (IFN-γ) production by T cells in response to four peptides known to be recognized by diabetogenic T cells10-13 was performed (Fig. 2). T cells from spleens, mesenteric and pancreatic lymph nodes (MLN and PLN, respectively) were analyzed. Spleens and MLN of NOD.MyD88KO mice contained activated precursors of the diabetogenic T cells, whereas their numbers were clearly reduced in PLN (Fig. 2a). Since individual mice vary in their responses to different peptides14, the overall reactivity to all four peptides is shown in Fig. 2a (see Supplemental Fig. 2 for primary data). The responses to a prevalent diabetes-associated peptide recognized by the 8.3 CD8+ T cell clone 12, 15, were found to be attenuated in the PLN of NOD.MyD88KO mice in a statistically significant manner (Fig. 2b). In addition, adoptively transferred carboxy-fluorescein succinimidyl ester (CFSE)-labeled naïve CD4+ T cells from mice carrying the diabetogenic T cell receptor BDC2.5 proliferated in the PLN but not in other (mesenteric or skin-draining) lymph nodes of the NOD mice, however, their proliferation was clearly attenuated in the PLN of MyD88 KO mice (Fig. 2c).
Figure 2. MyD88 deficiency leads to local tolerance to pancreatic antigens.
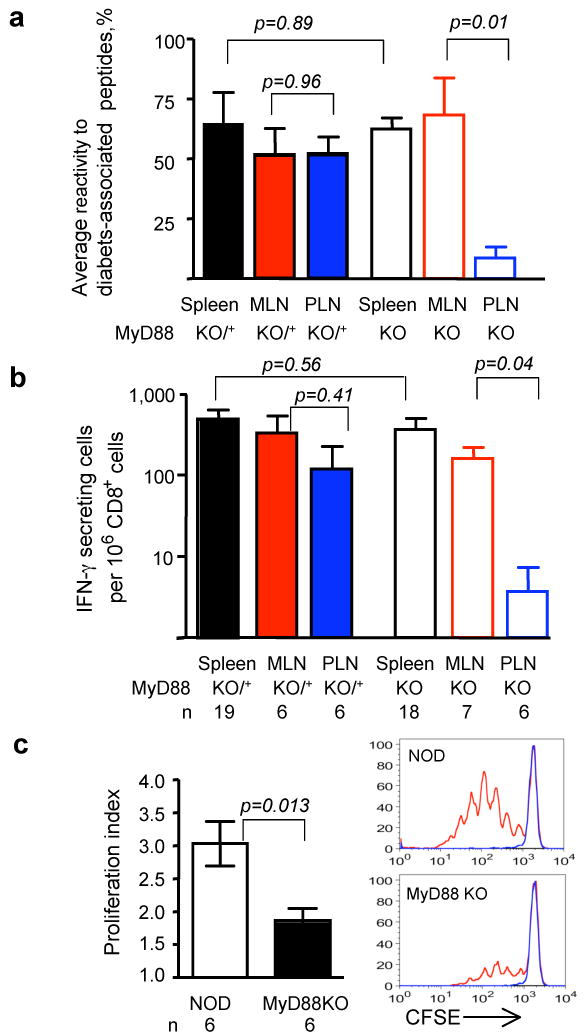
- The overall reactivity of T cells from secondary lymphoid organs of MyD88-sufficient (solid bars) and MyD88-negative (open bars) NOD mice was calculated as a mean of the percentages of mice in the given group reacting to individual diabetes-associated peptides (see Supplemental Fig. 2c).
- The frequency of CD8+ T cells producing IFN-γ in response to peptide recognized by diabetogenic clone 8.3 was determined in the spleens, MLN and PLN of MyD88-sufficient (solid bars) and MyD88-negative (open bars) NOD mice. Error bars – s.e.m.
- Proliferation of CFSE-labeled BDC2.5 CD4+ T cells, depleted of CD25+ T cells, in the PLNs of SPF NOD and NOD.MyD88KO mice assayed on day 3 after i.v. injection. Representative CFSE dilution profiles are shown for BDC2.5+-gated cells in PLN (red) and control skin-draining lymph nodes (blue).
p values in all experiments were determined using unpaired Student's t test. Error bars – s.e.m. n, number of animals per group.
Since the anti-diabetogenic effect of MyD88 deficiency was localized to PLN, it became clear that there was no systemic suppression of immune activation in SPF NOD.MyD88KO mice, making initial conclusions about the requirement of MyD88 signaling for initiation of T1D an oversimplification.
PLN drain both the pancreas and the intestine, and they are an important compartment where islet-specific T cells are activated16, 17. Because MyD88 signaling could be critical for the interactions of the host with the gut microbiota, we explored the hypothesis that the T1D resistance of NOD.MyD88KO could be driven by their intestinal microbiota. First, we treated SPF NOD.MyD88KO mice with a broad-spectrum antibiotic (Sulfatrim) throughout their lifetime. Antibiotic-treated NOD.MyD88KO animals developed T1D at higher rates compared to antibiotic-free NOD.MyD88KO mice (Fig. 3a versus Fig. 1), although they did not quite reach the incidence observed in control NOD.MyD88KO/+ mice. Thus, to fully eliminate any residual microbes, we re-derived NOD and NOD.MyD88 KO animals as germ-free (GF). T1D development in our GF MyD88-sufficient NOD mice was similar to previously reported18 and not dramatically different from mice raised in our SPF facilities (Fig. 3b; Supplemental Figs. 3 and 4). Thus, initiation of autoimmunity is genetically programmed and not affected by the presence of microbiota in immunocompetent SPF NOD animals in high-health-standard facilities.
Figure 3. MyD88-negative NOD mice are protected from diabetes by the gut microbiota.

- Diabetes incidence in SPF NOD.MyD88KO (J) females and control heterozygous NOD.MyD88KO/+ littermates treated with a broad-spectrum antibiotic from birth.
- Diabetes incidence in GF NOD.MyD88KO and control MyD88KO/+ mice. Incidence is shown for male mice; 100% of female NOD.MyD88KO and NOD.MyD88KO/+ female GF mice became diabetic (Supplemental Fig.1).
- Diabetes incidence in gnotobiotic male NOD.MyD88KO and control NOD.MyD88KO/+ mice colonized with a consortium of 6 bacterial strains (ASF 361,519,356,492,502, and 500; see Supplemental results for descriptions). The incidence of diabetes in gnotobiotic NOD.MyD88KO mice was significantly different from the incidence in GF NOD.MyD88KO animals (p<0.001), and in gnotobiotic NOD.MyD88KO/+ mice (p<0.05) (Kaplan Meier test).
- Histological scores of islet destruction in SPF, GF and ASF-colonized NOD.MyD88KO/+ and NOD.MyD88KO mice. Mice in all groups were males, except for the SPF NOD.MyD88KO group, which included both genders.
Most importantly, in contrast to our finding that NOD.MyD88KO mice raised under SPF conditions did not develop T1D, GF NOD.MyD88KO mice robustly developed diabetes (Fig. 3b and Supplemental Fig. 3). This finding clearly shows that neither IL-1R nor MyD88-dependent TLRs are required for activation of an anti-islet T cell response [similar to MyD88KO, autoimmune regulator (Aire)-deficient mice19]. The efficient T cell priming observed in GF NOD.MyD88KO mice does not confirm a previous report that suggested that TLR2 signaling, which is MyD88-dependent, is required for T cell priming in NOD mice20.
To directly show that T1D development in GF NOD.MyD88 KO mice was indeed a consequence of the lack of a microbiota, adult GF NOD.MyD88KO/+ mice were colonized with a consortium of bacterial species contained in the Altered Schaedler Flora (ASF)21 and intercrossed. After introducing the ASF cocktail, PCR assays revealed that 6 of the 8 species colonized the animals (based on sampling of cecal contents, Supplemental results and Supplemental Fig. 5). The ASF-colonized NOD.MyD88KO mice exhibited a significant reduction in the incidence of diabetes (Fig. 3c): only 34% of males became diabetic at 30 weeks of age as compared to >80% of GF NOD.MyD88KO males (Fig. 3b), whereas 70% of ASF-colonized NOD.MyD88KO/+ males developed diabetes with kinetics similar to disease development in SPF NOD.MyD88KO/+ mice (Fig. 3c versus Fig. 1).
A histological analysis of pancreata from SPF, GF and ASF-colonized NOD.MyD88KO mice was performed to compare the effects of the microbiota on T cell-mediated destruction of the islets of Langerhans. The islets of SPF NOD.MyD88KO mice were less infiltrated compared to islets of SPF NOD.MyD88KO/+ mice. Moreover, GF NOD.MyD88KO mice had considerably increased islet infiltration, which was moderated by the introduction of the ASF (Fig. 3d, histological grading shown in Supplemental Fig. 6). Although, the overall effect of ASF on diabetes development was not fully penetrant, these results suggest that bacterial lineages normally present in the gut can modify T1D progression.
We found that MyD88 signaling is critical for development of T1D, and postulated that it is needed for control over component(s) of the microbiota that otherwise (in MyD88KO mice) can protect against development of T1D. To test how MyD88-dependent innate immunity shapes the composition of the gut microbiota, we used a culture-independent, 16S rRNA gene sequence-based approach to characterize the microbial communities of SPF NOD.MyD88KO/+ and NOD.MyD88KO littermates. Because MyD88 deficiency affects T1D development at the early stages (Supplemental Fig. 7), experimental female mice were housed individually and killed at 8 weeks of age. DNA was isolated from their cecal contents and bacterial 16S rRNA genes were amplified by PCR, and the resulting full-length amplicons sequenced (n=36 mice; total of 7,223 16S rRNA gene sequences; average of 201 sequences/animal; average sequence length of 1,310 nucleotides).
In concordance with previous findings in mice and humans22,23,24 two bacterial divisions (phyla) - the Firmicutes (F) and the Bacteroidetes (B) dominated the distal gut (cecal) microbiota of mice from all groups (80.7% and 16.9% of all sequences, respectively). The remainder of the microbiota was composed of divisions commonly encountered at lower abundance in the mouse and human gut: Verrucomicrobia, Proteobacteria, Actinobacteria and the candidate phylum TM725. Furthermore, close relatives of ASF strains were detected in SPF NOD mice.
Analysis of the cecal microbiota of NOD.MyD88KO/+ versus NOD.MyD88KO mice showed significant differences. Antibiotic-free NOD.MyD88KO mice had, on average, a significantly lower F/B ratio when compared to all other groups (one-tailed t-test t=-2.31, p=0.013) (Fig. 4a). Antibiotic treatment of animals eliminated the statistically significant difference in F/B ratio (Fig. 4a). A change of F/B ratio may be important by itself because it can influence the efficiency of processing of otherwise indigestible complex polysaccharides in the diet 26,27. Diet-related changes in diabetes in GF NOD mice have been observed 28; these effects could be related to the components of the diet per se or attributed to the presence of microbial products in the feed.
Figure 4. MyD88 deficiency leads to specific changes in the composition of intestinal microbiota.

- Ratio of Firmicutes to Bacteroidetes in the cecal microbiota of NOD.MyD88KO/+ and NOD.MyD88KO mice who were or were not treated with Sulfatrim. Mean values per mouse ± s.e.m. Untreated NOD.MyD88KO mice had, on average, a significantly lower F/B ratio than all other mice combined (one-tailed t-test, t=-2.31, p=0.013). When comparing post-hoc the effect of sulfatrim in MyD88KO mice only, no significant difference in F/B ratios was observed (t= 0.85, p=0.20).
- Abundance of members of three different bacterial families in the cecal contents of NOD.MyD88KO/+ and NOD.MyD88KO mice (not treated with Sulfatrim). Mean values ± s.e.m. Each of the three families is enriched in NOD.MyD88KO mice (Lactobacilliaceae t=-1.54, p=0.07, Porphyromonadaceae t=2.1, p=0.03, Rikenellaceae t=2.74, p=0.007; one-tailed t-test).
- Clustering of mouse cecal bacterial communities using the unweighted UniFrac metric n=7,223 sequences. Diversity, not abundance of bacteria was taken into consideration. Top panel: Sulfatrim-treated litters. Bottom panel: Untreated litters. Line colors indicate families. Each label is a mouse: red labels are NOD.MyD88KO/+ (H); black labels are NOD.Myd88KO (K). The number is the common mother, S and N designate exposure to Sulfatrim or no Sulfatrim, respectively.
- Histological examination of the pancreata of 8 wk old males from SPF NOD, GF NOD, as well as GF NOD and GF NOD.MyD88KO exposed from birth to microbiota of an SPF NOD.MyD88KO female. The percentages (mean ±s.e.m.) of affected islets with no infiltration (open bars), periinsulitis (blue bars) and true infiltration (combined grades II and III) are shown. p values were obtained using unpaired Student's t test. n, number of animals per group.
To further characterize the changes in microbiota imposed by MyD88 deficiency, 16S rRNA genes were classified taxonomically to the family level (Ribosomal Database Project Classifier29). The proportion of sequences in each family was determined for individual mice, averaged and compared across treatments. The representation of three bacterial families were increased significantly in the microbiota of antibiotic-free SPF NOD.MyD88KO mice compared to the SPF NOD animals: the Lactobacillaceae (Firmicutes), Rikenellaceae and Porphoromadaceae (both Bacteroidetes) (Fig. 4b). Interestingly, the VSL3 probiotic mix, containing four species of Lactobacillaceae affects diabetes30 in NOD mice.
Gut microbial communities are known to be inherited from the mother22: this was also the case in these experiments, where clustering of 16S rRNA genes was strongly influenced by shared mothers (Fig. 4c).
To show that the changes in the intestinal microbiota of MyD88KO animals were responsible for attenuation of T1D development, the newborn progeny of GF NOD mice were exposed to SPF NOD.MyD88 KO females and allowed to mature to 8 weeks of age, after which time their pancreata were removed and analyzed histologically (Fig. 4d). Islet infiltration was significantly reduced in GF NOD animals exposed to microbiota from SPF MyD88 KO mouse compared to GF NOD mice [increased percentage of intact islets (p=0.001) and reduced percentage of infiltrated islets (p=0.0006)].
Although the precise mechanism of induction of local tolerance by the microbiota remains to be elucidated (see Supplemental Fig. 8), the finding that the normal intestinal microbiota can alleviate progression of autoimmune diabetes in a MyD88-independent manner provides a different perspective about disease pathogenesis. Knowledge-based use of live microbial lineages, or microbial products could present new therapeutic options for T1D in the future.
Methods Summary
Mice
B6 mice carrying MyD88 and TLR mutations were backcrossed 10-12 times to NOD/LtJ males and intercrossed to produce KO and heterozygous animals. GF animals were re-derived from NOD/LtJ females impregnated by NOD.MyD88KO males and kept GF at Taconic Farms, Germantown, NY, the University of Chicago and Washington University (St. Loius, MO). ASF was introduced to GF NOD.MyD88KO mice by adding cecal contents from donor mice to sterile drinking water. Wild-type GF mice were colonized with microbiota from SPF NOD.MyD88KO animals by co-housing GF NOD females and newborn progeny with SPF MyD88.KO females.
Histopathology of diabetes
Damage to the islets was scored in a blinded fashion, and graded as follows: 0 –no visible infiltration; I – periinsulitis; II - insulitis with <50% and III – with >50% islet infiltration (Supplemental Fig. 6). At least 100 islets in each group of 5 to 12 animals were scored. In microbiota transfer experiments 20 sections per pancreas cut at 40μm intervals (≥10 islets/section) were examined and scored (combining grades II and III).
Elispot analysis
6×105 splenocytes alone, or 2×105 lymph node cells mixed with 4×105 irradiated splenocytes from B6.NOD-(D17Mit21-D17Mit10)/LtJ (B6.g7) mice, per well of 96-well plates pretreated with anti-IFN-γ antibodies were incubated overnight with listed peptides. Lymphocytes were stained with antibodies against CD4 and CD8 to calculate the frequency of peptide-specific cells per 106 CD4+ or CD8+ T cells.
Sequence and phylogenetic analysis
16S rRNA gene sequences were edited and assembled into consensus sequences using PHRED and PHRAP, aided by XplorSeq23, and bases with a PHRAP quality score of < 20 were trimmed. Sequences were aligned using the Nast online alignment tool (http://greengenes.lbl.gov/cgi-bin/nph-index.cgi), and checked for chimeras using the online Greengenes server (http://greengenes.lbl.gov/cgi-bin/nph-bel3_interface.cgi) with a window size of 300 and using the nast-aligned sequences31.
Supplementary Material
Supplementary Information accompanies the paper on www.nature.com/nature.
Acknowledgments
The authors are thankful to Ann Putnam, Tennessee Park, Desiree Schumann, Maria Prokhorovich, Wei Du, David O'Donnell, Maria Karlsson, and Sabrina Wagoner for their help with experiments, and Sarah Dryden Perkins and Marcelo Garcia for assistance with sequence analysis. This work was supported by the ADA grant 1-05-RA-142 to L.W., JDRF grant 19-2006-1075 to L.W.; NIH grants R37 AI46643 and P30 DK63720, JDRF 4-2005-1168 grant to JAB; NIH grants DK30292 and DK70977, and a W.M. Keck Foundation award to JIG; NIH grant DK063452 to AVC, and JDRF grants 2005-204 and 2007-353 to AVC, and by NIH/NIDDK Digestive Disease Research Core Center grant DK42086.
Footnotes
Full Methods and any associated references are available in the online version of the paper at www.nature.com/nature
Author Contributions L.W. designed and supervised experiments at Yale University; R.E.L. performed analysis of 16S rRNA sequences of the gut microbiota; P.Yu.V. analyzed T1D development in GF and microbiota-colonized mice; P.B.S. performed Elispot analysis; L.A. and A.C.S established and characterized mutant mouse strains at The Jackson Laboratory and at The University of Chicago; C.H., F.S.W. and L.W. characterized mutant mouse strains at Yale University; G.L.Z. was involved in performance of Treg-based assays; J.A.B. designed and supervised the T cell assays; J.I.G. helped with design and interpretation of gut microbial ecology studies and oversaw the microbiota transfer experiments; A.V.C. conceived and designed the project, wrote the manuscript with substantial critical contributions from L.W., R.E.L., F.S.W., J.A.B, and J.I.G.
Author Information 16S rRNA sequences obtained from microbiota of SPF NOD and NOD.MyD88KO mice treated and not-treated with antibiotic were deposited in GenBank under accession numbers EU450891-EU458113. Reprints and permissions information is available at www.nature.com/reprints.
References
- 1.Karvonen M, Tuomilehto J, Libman I, LaPorte R. A review of the recent epidemiological data on the worldwide incidence of type 1 (insulin-dependent) diabetes mellitus. World Health Organization DIAMOND Project Group. Diabetologia. 1993;36:883–92. doi: 10.1007/BF02374468. [DOI] [PubMed] [Google Scholar]
- 2.Patterson CC, Dahlquist G, Soltesz G, Green A. Is childhood-onset type I diabetes a wealth-related disease? An ecological analysis of European incidence rates. Diabetologia. 2001;44(3):B9–16. doi: 10.1007/pl00002961. [DOI] [PubMed] [Google Scholar]
- 3.Pozzilli P, Signore A, Williams AJ, Beales PE. NOD mouse colonies around the world--recent facts and figures. Immunol Today. 1993;14:193–6. doi: 10.1016/0167-5699(93)90160-M. [DOI] [PubMed] [Google Scholar]
- 4.McInerney MF, Pek SB, Thomas DW. Prevention of insulitis and diabetes onset by treatment with complete Freund's adjuvant in NOD mice. Diabetes. 1991;40:715–25. doi: 10.2337/diab.40.6.715. [DOI] [PubMed] [Google Scholar]
- 5.Sadelain MW, Qin HY, Lauzon J, Singh B. Prevention of type I diabetes in NOD mice by adjuvant immunotherapy. Diabetes. 1990;39:583–9. doi: 10.2337/diab.39.5.583. [DOI] [PubMed] [Google Scholar]
- 6.Janeway CA., Jr Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol. 1989;54(Pt 1):1–13. doi: 10.1101/sqb.1989.054.01.003. [DOI] [PubMed] [Google Scholar]
- 7.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 8.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–41. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 9.Strober W. Epithelial cells pay a Toll for protection. Nat Med. 2004;10:898–900. doi: 10.1038/nm0904-898. [DOI] [PubMed] [Google Scholar]
- 10.Wong FS, et al. Identification of an MHC class I-restricted autoantigen in type 1 diabetes by screening an organ-specific cDNA library. Nat Med. 1999;5:1026–31. doi: 10.1038/12465. [DOI] [PubMed] [Google Scholar]
- 11.Graser RT, et al. Identification of a CD8 T cell that can independently mediate autoimmune diabetes development in the complete absence of CD4 T cell helper functions. J Immunol. 2000;164:3913–8. doi: 10.4049/jimmunol.164.7.3913. [DOI] [PubMed] [Google Scholar]
- 12.Amrani A, et al. Perforin-independent beta-cell destruction by diabetogenic CD8(+) T lymphocytes in transgenic nonobese diabetic mice. J Clin Invest. 1999;103:1201–9. doi: 10.1172/JCI6266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haskins K, McDuffie M. Acceleration of diabetes in young NOD mice with a CD4+ islet-specific T cell clone. Science. 1990;249:1433–6. doi: 10.1126/science.2205920. [DOI] [PubMed] [Google Scholar]
- 14.Lieberman SM, et al. Individual nonobese diabetic mice exhibit unique patterns of CD8+ T cell reactivity to three islet antigens, including the newly identified widely expressed dystrophia myotonica kinase. J Immunol. 2004;173:6727–34. doi: 10.4049/jimmunol.173.11.6727. [DOI] [PubMed] [Google Scholar]
- 15.Lieberman SM, et al. Identification of the beta cell antigen targeted by a prevalent population of pathogenic CD8+ T cells in autoimmune diabetes. Proc Natl Acad Sci U S A. 2003;100:8384–8. doi: 10.1073/pnas.0932778100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoglund P, et al. Initiation of autoimmune diabetes by developmentally regulated presentation of islet cell antigens in the pancreatic lymph nodes. J Exp Med. 1999;189:331–9. doi: 10.1084/jem.189.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Turley SJ, Lee JW, Dutton-Swain N, Mathis D, Benoist C. Endocrine self and gut non-self intersect in the pancreatic lymph nodes. Proc Natl Acad Sci U S A. 2005;102:17729–33. doi: 10.1073/pnas.0509006102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suzuki T, et al. Diabetogenic Effects of Lymphocyte Transfusion of the NOD or NOD Nude mouse. In: Rygaard J, B N, Graem N, Spang-Thomsen M, editors. Immune-deficient Animals in Biomedical Research. Karger; Basel: 1985. [Google Scholar]
- 19.Gray DH, Gavanescu I, Benoist C, Mathis D. Danger-free autoimmune disease in Aire-deficient mice. Proc Natl Acad Sci U S A. 2007;104:18193–8. doi: 10.1073/pnas.0709160104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim HS, et al. Toll-like Receptor 2 Senses beta-Cell Death and Contributes to the Initiation of Autoimmune Diabetes. Immunity. 2007;27:321–33. doi: 10.1016/j.immuni.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 21.Dewhirst FE, et al. Phylogeny of the defined murine microbiota: altered Schaedler flora. Appl Environ Microbiol. 1999;65:3287–92. doi: 10.1128/aem.65.8.3287-3292.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ley RE, et al. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A. 2005;102:11070–5. doi: 10.1073/pnas.0504978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444:1022–3. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- 24.Rawls JF, Mahowald MA, Ley RE, Gordon JI. Reciprocal gut microbiota transplants from zebrafish and mice to germ-free recipients reveal host habitat selection. Cell. 2006;127:423–33. doi: 10.1016/j.cell.2006.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ley RE, Peterson DA, Gordon JI. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell. 2006;124:837–48. doi: 10.1016/j.cell.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 26.Turnbaugh PJ, et al. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–31. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 27.Turnbaugh DP, Backhed F, Fulton L, Gordon JI. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe. 2008;3:213–23. doi: 10.1016/j.chom.2008.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Funda PJ, Fundova P, Harrison LC. Proc 13th Int Congr Immunol. 16. MS-11.4. Brazilian Society for Immunology; Rio de Janeiro: 2007. Microflora-dependency of selected diabetes-preventive diets: germ-free and ex-germ-free monocolonized NOD mice as models for studying environmental factors in type 1 diabetes. [Google Scholar]
- 29.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73:5261–7. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Calcinaro F, et al. Oral probiotic administration induces interleukin-10 production and prevents spontaneous autoimmune diabetes in the non-obese diabetic mouse. Diabetologia. 2005;48:1565–75. doi: 10.1007/s00125-005-1831-2. [DOI] [PubMed] [Google Scholar]
- 31.Huber T, Faulkner G, Hugenholtz P. Bellerophon: a program to detect chimeric sequences in multiple sequence alignments. Bioinformatics. 2004;20:2317–9. doi: 10.1093/bioinformatics/bth226. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information accompanies the paper on www.nature.com/nature.