

Abstract
Accumulating evidence supports the idea that two known phosphatidylinositol 3′-Kinase (PI3K) downstream proteins, Fra-1 and Survivin, are potential targets for cancer therapy. Increased expression of Fra-1, a Fos family member of the transcription factor activator protein-1 (AP-1), has been implicated in both the maintenance and progression of the transformed state of several cancer cells. In addition, high Survivin expression in tumors correlates with more aggressive behavior, lower response to chemotherapeutic drugs, and shortened survival time. Previously, we reported that in malignant mesothelioma cells with increased PI3K activity, small molecule inhibitors of the (PI3K)/AKT pathway acted cooperatively with the amphibian ribonuclease chemotherapeutic drug ranpirnase to inhibit cell growth. Because the thiazolidinedione (TZD) anti-diabetic drug, rosiglitazone, targets the PI3K/AKT pathway, we investigated the effect of the combination of these two drugs in cell survival in several cancer cell lines. We show here that the combination of ranpirnase and rosiglitazone synergistically decreases cell viability and increases cell apoptosis in several cancer cell lines. Cell killing is associated with decreased Fra-1 and Survivin expression and knock-down of Fra-1 increases cell killing by ranpirnase in a dose-dependent manner, but not by rosiglitazone. The drug combination does not have a synergistic effect on killing in Fra-1 knock-down cells, demonstrating that Fra-1 modulation accounts in part for the synergism. The novel drug combination of ranpirnase and rosiglitazone is a promising combination to treat cancers with increased PI3K-dependent Fra-1 expression or Survivin.
Keywords: ranpirnase, rosiglitazone, Fra-1, Survivin, apoptosis
INTRODUCTION
Fra-1 and Survivin are key signaling proteins in the development and progression of various cancers. Fos family member Fra-1 accumulates in transcription factor activator protein-1 (AP-1) complexes during the malignant progression of several tumors, and has been implicated in both the maintenance and progression of the transformed state. Fra-1’s causal role in cellular transformation has been documented in several systems, including esophagus (1), breast (2, 3), and thyroid (4, 5). Ectopic expression of Fra-1 in vitro increases the cell motility and metastatic behavior of mammary adenocarcinoma cells (6) and lung epithelial cells (7). Lastly, Fra-1 is induced by lung carcinogens, such as cigarette smoke and asbestos (7). Fra-1 is a predominant component of the AP-1 complex in asbestos-induced mesothelioma and proliferating rat mesothelioma cells, while overexpression of a dominant negative Fra-1 mutant inhibits the growth of these cells in soft agar (8). Survivin is an IAP protein abundantly expressed in fetal tissues (9) and neoplasms (10), but undetectable in most normal, terminally differentiated adult tissues (10). High Survivin expression by tumors correlates with more aggressive behavior, decreased response to chemotherapeutic agents, and shortened survival times, as compared with Survivin-negative cancers (reviewed in (11)). These findings support Fra-1 and Survivin as potential targets for cancer therapy.
Here, we present a novel drug combination (ranpirnase and rosiglitazone) capable of down-regulating both Fra-1 and Survivin in several cancer cell lines. The anticancer effect of ranpirnase (reviewed in (12)) has been documented both in vitro (13–19) and in vivo (14, 16, 18, 20). This amphibian ribonuclease drug holds promise as a chemotherapeutic tool thanks to its low toxicity to normal cells and effectiveness against cancer cells. Previously, we reported that in malignant mesothelioma cells with increased kinase activity levels of AKT, both LY294002 and wortmannin (two inhibitors of the phosphatidylinositol 3′-Kinase (PI3K)/AKT pathway) act cooperatively with ranpirnase to inhibit cell growth (19). Because the anti-diabetic thiazolidinedione (TZD), peroxisome proliferator-activated receptor gamma (PPARγ) agonist, rosiglitazone down regulates the PI3K/AKT pathway, and because TZDs are potentially useful in treating several cancers (21, 22) through both PPARγ-dependent and PPARγ-independent mechanisms, we investigated the combination of the TZD rosiglitazone and ranpirnase. Specifically, we tested the combination of ranpirnase and rosiglitazone in several cancer cell lines, showing that two PI3K downstream targets, Fra-1 (23) and Survivin (24), are down-regulated by the combination. Furthermore, we demonstrated a synergistic, apoptotic effect of ranpirnase and rosiglitazone in some cancer cell lines directly associated with the expression of Fra-1. This drug combination could be an important chemotherapeutic alternative in some cancers.
MATERIAL AND METHODS
Cell lines
The breast cancer cell lines MDA-MB-231 (231), T47D and MCF7; the ovarian cancer cell lines SKOV-3 and CAOV-3; the prostate cancer cell lines PC3, DU-145 and LNCaP; the lung carcinoma cell lines NCI-H292 (H292) and N1792 (all from ATCC, Manassas, Virginia, USA); and the mesothelioma cell lines MP5 and MP2 (kindly donated by Dr. Harvey Pass, New York University) were maintained in frozen stocks. Cells were incubated at 37°C in 5% CO2 until approximately 80–90% confluency in DMEM medium (GIBCO BRL, NY) containing 5% fetal bovine serum (FBS) and 1 g/L of glucose. Cells were then starved O/N in DMEM containing 0.5% FBS before treatments with ranpirnase, rosiglitazone alone or in combination for 2 or 6 days, and then collected. When indicated, media were changed again after treatment to DMEM/F12 containing 0.5% FBS and insulin 1μM to activate the PI3K pathway.
Small molecule inhibitors and chemicals
Stock solution of the PI3K’s small molecular inhibitor LY294002 was diluted in dimethyl sulfoxide (DMSO) and used at effective nontoxic concentrations (20μM) (25) (Calbiochem, La Jolla, CA). Ranpirnase (Onconase, kindly provided by Dr. Kuslima Shogen, AlfaCell Corporation, Bloomfield, NJ), was used at three concentrations (0.1, 1, and 10 μg/ml medium), and was prepared as aliquots in medium from a lyophilized stock solution subsequently frozen at −20°C. Rosiglitazone (Cayman Chemical, Ann Arbor, MI) was dissolved in DMSO and used at concentrations of 10 and 20 μM (<IC50 for all cells tested). GW9662 (Cayman Chemical), an irreversible PPARγ antagonist, was prepared in the same manner, and used at a concentration of 2 μM. All untreated control cells received DMSO in medium.
MTS assay for cell viability
Assays were performed on 96-well microtiter plates after plating of 7.7×104 cells/well. Cells were then cultured for 24 h in complete medium before changing to medium containing 0.5% FBS with ranpirnase at 0.1, 1 or 10 μg/ml, rosiglitazone at 10 or 20 μM, their six combinations, or DMSO-containing medium (solvent control). Cell viability was measured by the colorimetric MTS Assay, CellTiter 96 Aqueous One Solution Cell Proliferation Assay (Promega), per the manufacturer’s recommendations. MTS is a tetrazolium salt that undergoes a color change caused by its bioreduction of MTS into a water-soluble formazan. The conversion of MTS into the aqueous-soluble formazan is accomplished by dehydrogenase enzymes found in active mitochondria, with reaction occurring only in living cells. The quantity of formazan product measured by the amount of 490-nm light absorbance is directly proportional to the number of living cells in culture. Briefly, 20μl of MTS reagent was added per well, and plates incubated at 37°C for 2–3 h. Finally, the absorbance of each well was read at 490 nm and 650nm. ΔOD from these two wavelengths were reported as the corrected viability. Fold changes were calculated with respect to the control as a measure of cell viability.
Flow cytometry
Near confluent cells were maintained in complete medium containing 0.5% FCS overnight before addition of ranpirnase at 1 μg/ml, rosiglitazone 20 μM, their combination, or DMSO-containing medium (solvent control). At 48 h, medium was removed and adherent cells harvested by trypsinization. Combined cells were resuspended at 106/ml in staining solution (50 μg/ml propidium iodide, 0.1% Triton X-100, and 32 μg/ml RNAse A) in phosphate-buffered saline and incubated for 30 min at 37°C before analysis of 10,000 cells/group/time point in triplicate. The distribution of cells, including cells with a hypodiploid DNA content indicative of apoptosis or necrosis, was determined using a Coulter Epics Elite flow cytometer and appropriate software, as described previously (26). To determine number of apoptotic cells, cells were stained with Annexin V and propidium iodide (PI) in the dark for 15 min and 5,000 events per sample, analyzed by flow cytometry as described above. For staining, cell pellets were suspended in 93 μl of 1x binding buffer [10 mM Hepes/NaOH (pH 7.4), 140 mM NaCl, 2.5 mM CaCl2], 5 μl of PI at a final concentration of 2.5 μg/ml (Sigma), and 2 μl of FITC labeled-Annexin V (BD Bioscience, San Jose, CA). Cells with annexin V-positive staining were scored as apoptotic.
Growth curves
Cells (N = 2–3 plates/group/time point) were plated at approximately 1×103 cells per 6-well plate in complete medium, allowed to attach for 24 h, and then treated with inhibitors at different time points. Cells were removed by trypsinization, and aliquots counted using a hemocytometer to determine total cell number.
Western blot analyses
Near confluent MM cells were washed 3× with cold phosphate-buffered saline (PBS) before centrifugation at 14,000 rpm for 1 min. The pellet was resuspended in lysis buffer [20 mM Tris (pH 7.4), 1% Triton X-100, 10% glycerol, 137 mM NaCl, 2 mM EDTA, 25 mM β-glycerophosphate, 1 mM Na3VO4, 2 mM pyrophosphate, 1 mM PMSF, 10 μg/ml leupeptin, 1 mM DTT, 10 mM NaF, 1% aprotinin], incubated at 4°C for 15 min, and centrifuged at 14,000 rpm for 20 min. The amount of protein in each supernatant was determined using the Bio-Rad protein assay (Bio-Rad, Hercules, CA). Thirty μg protein in sample buffer [62.5 mM Tris-HCl (pH 6.8), 2% sodium dodecyl sulfate (SDS), 10% glycerol, 50 mM dithiothreitol, 0.1% w/v bromophenol blue] was electrophoresed in 10% SDS-polyacrylamide gel, and transferred to nitrocellulose using a semi-dry transfer apparatus (Ellard Instrumentation, Ltd., Seattle, WA). Blots were blocked in buffer [Tris-buffered saline (TBS) containing 5% nonfat dry milk plus 0.1% Tween-20 (Sigma)] for 1 h, washed 3X for 5 min each in TBS/0.1% Tween-20, and incubated at 4°C with an antibody specific to Fra-1 (R-20) at a 1:500 dilution or Survivin (D8) at 1:100 dilution (Santa Cruz Biotechnology, Santa Cruz, CA). Blots were then washed 3x with TBS/0.1% Tween-20 and incubated with a specific peroxidase-conjugated secondary antibody for 1 h. After washing blots 3x in TBS/0.1% Tween-20, protein bands were visualized with the LumiGlo enhanced chemiluminescence detection system (Kirkgaard and Perry Laboratories, Gaithersburg, MD) and quantitated by densitometry (27). Blots were reprobed with an antibody to α-Tubulin (Santa Cruz Biotechnology, Santa Cruz, CA) to validate equal loading between lanes.
Constructs and Transfection Techniques
siFra-1 RNA interference (RNAi) duplexes were constructed from sequence information on mature mRNA extracted from the EST database (www.ncbi.nlm.nih.gov) using the open frame region from the cDNA sequence of exon 2 of the Fra-1 gene. The siRNA pool sequences targeting Fra-1 corresponded to the 107–126, 124–143, 230–249 coding regions relative to the first nucleotide of the start codon. The sequences were BLAST-searched (NCBI database) against EST libraries to ensure the specificity of the siRNA molecule. The siRNA duplexes or a scramble control were transfected into cancer lines using Lipofectamine 2000 (Invitrogen) as recommended by the manufacturer. Cells were incubated with complexes overnight, and the medium was replaced the next day. Cells were allowed to recover for 48 hours before treatments.
SYBR Green Real Time Quantitative PCR (RT-QPCR)
Total RNA (1μg) was reverse-transcribed with random primers using the Promega AMV Reverse Transcriptase kit (Promega, Madison, WI) according to the recommendations of the manufacturer. PCR amplifications were performed using the ABI PRISM 7700 Sequence Detection System (Perkin Elmer Applied Biosystems, Foster, CA). Reactions were performed in a 50 μl reaction mixture that included 25 l SYBR Green JumpStart Taq ReadyMix (Sigma, St Louis, MO), μ distilled H2O, DNA template, and 0.2 μM each primer from QuantiteTect primer assays (Qiagen, Valencia, CA). Amplification was performed by initial denaturation at 94 °C for 2 min, and 40 cycles of denaturation at 95 °C for 15 s, annealing at 60 °C for 1 min, and extension for 1 min at 72 °C. This was followed by a dissociation cycle of 95 °C for 15 s, 60 °C for 15 s, and 95 °C for 15s. Threshold cycles (CT) for both Fra-1 mRNAs and the 18S rRNA control were determined. Original input RNA amounts were calculated using the Comparative CT Method (2−dd^CT) to analyze changes in gene expression in the samples relative to the untreated control sample. Duplicate assays were performed with RNA samples isolated from at least 2 independent experiments. The values obtained from cDNAs and 18S controls provided relative gene expression levels for the gene loci investigated (25).
Immunofluorescence
Dual confocal fluorescence approaches were used to determine if co-localization of Fra-1 was specific to nuclear PCNA-positive cells. Cells, grown on coverslips, were fixed in 100% methanol for 1 h on ice, washed in PBS, and incubated in 0.1% Tween-20 in PBS for 30 min at room temperature (RT). After incubation in blocking solution (2% dry milk, 0.1% Tween-20 in PBS) for 30 min at RT, cells were incubated with a cocktail of primary antibodies [mouse anti-PCNA (Pharmagen; 1:1,000) and rabbit polyclonal anti-Fra-1 antibody (R-20) (Santa Cruz Biotechnology Inc., 1:100) for 1 h at RT. Cells were washed twice for 20 min in blocking solution, and once for 10 min in PBS. PCNA was detected using Alexa Fluor 647-goat-anti-mouse IgG (Molecular Probes), diluted 1:400 in 10 μg/ml BSA/PBS. Fra-1 was detected using Alexa Fluor 568-goat-anti-rabbit IgG (Molecular Probes), diluted 1:200 in 10 μg/ml BSA/PBS. Controls were run using only primary or secondary antibodies. Following a final wash in PBS, sections were counter-stained with SYTOX green (1:1,000 in PBS) (Molecular Probes, Eugene, OR), washed 1X in PBS, and mounted on glass slides using AquaPoly/Mount (Polysciences Inc., Warrington, PA), and examined using confocal scanning laser microscopy. For each sample, confocal images were collected in fluorescence modes, followed by electronic merging of the images.
Statistical analyses
All experiments used multiple replicate determinations (N=2, 3 or 8) per group, per time point. Experiments were performed in duplicate. Results were evaluated by one-way analysis of variance using the Student-Newman-Keuls procedure for adjustment of multiple pairwise comparisons between treatment groups. Differences with P values ≤0.05 were considered statistically significant.
RESULTS
The combination of ranpirnase and rosiglitazone synergistically reduced viability and increased death in several cancer cell lines
To test the hypothesis that the combination of the two drugs is synergistically anti-neoplastic, all 12 cancer cell lines were treated with ranpirnase (1 μg/mL), rosiglitazone (20 μM) or their combination for 48h. Cell viability measured by MTS assay showed (Figure 1) that the combination of ranpirnase and rosiglitazone had a significant synergistic effect on the reduction of cell viability in seven out of the twelve cell lines tested (Figure 1).
Figure 1.

The combination of ranpirnase and rosiglitazone increased cytotoxicity after 48 h of treatment compared to ranpirnase or rosiglitazone alone in seven of twelve human cancer cell lines in vitro as determined by the MTS viability assay test. A. T47D. B. MCF7. C. 231. D. LNCaP. E. DU-145. F. PC3. G. SKOV-3. H. CAOV-3. I. MP5. J. MP2. K. H292. L. H1792. C = medium only control; Onc 1 = ranpirnase 1 μg/mL; Rosi20 = rosiglitazone 20 μM; O+R = both. Bars represent mean fold changes ± SEM of 8 samples per group. Experiments repeated twice. * = P ≤ 0.05 in comparison to control; ! = P ≤ 0.05 in comparison to both single treatments.
The combination of ranpirnase and rosiglitazone increased the proportions of subG0/G1 cells
To prove the hypothesis that ranpirnase increased cell killing cooperatively with rosiglitazone, we determined the effects of ranpirnase (1 μg/ml), rosiglitazone (20 μM), and their combination, on the cell cycle kinetics of the 12 cancer cell lines. As early as 48 hours after treatment, five of the twelve cell lines showed a significant increases (P ≤ .05) in the proportions of cells in subG0/G1 (apoptotic and necrotic cells) (Figure 2). These changes occurred with concomitant decreases in cells in G0/G1 and increases in the numbers of cells in G2/M, suggesting cell cycle arrest (data not shown).
Figure 2.

The treatment of cancer cells with the combination of ranpirnase and rosiglitazone for 48 h increased the proportion of subG0/G1 cells in five out of twelve human cancer cells lines, as determined by flow cytometry using Propidium Iodine. A. T47D. B. MCF7. C. 231. D. LNCaP. E. DU-145. F. PC3. G. SKOV3. H. CAOV-3. I. MP5. J. MP2. K. H292. L. H1792. C = medium only control; Onc 1 = ranpirnase 1 μg/mL; Rosi20 = rosiglitazone 20 μM; O+R = both. Bars represent mean ± SEM of 2 samples per group. Experiments repeated twice. * = P ≤ 0.05 in comparison to control; ! = P ≤ 0.05 in comparison to both single treatments.
The combination of ranpirnase and rosiglitazone triggered a reduction in proliferation and increased apoptosis in several cancer cell lines
To determine the mechanism of cell death from the combination of ranpirnase and rosiglitazone, PC3, H1792 and 231 cell lines were examined using the Annexin V assay to detect apoptotic cell death after 6 days of treatment (Figure 3 B, D, and F). These cell lines were originally selected because of their resistance to the drug combination after 48 hours of treatment in previous tests (Figure 2). Compared to control cells, significantly higher concentrations of Annexin V-positive cells were observed. Longer term treatment showed that even these relatively resistant cell lines underwent apoptosis when exposed to the combination for 6 days. Growth curves (Figure 3 A, C and E) also showed a synergistic decrease in cell growth at six days with the combination of ranpirnase and rosiglitazone.
Figure 3.

Cancer cells resistant to synergistic killing by the combination of ranpirnase and rosiglitazone were tested for cell growth (by cell count) and apoptosis (by count of Annexin V-positive cells by flow cytometry) after 6 days of treatment. A–B. PC3. C–D. H1792. E–F. 231. C = medium only control; Onc 1 = ranpirnase 1 μg/mL; Rosi20 = rosiglitazone 20 μM; O+R = both. Symbols represent mean ± SEM of 2 samples per group. Experiments repeated twice. * = P ≤0.05 in comparison to control; ! = P ≤ 0.05 in comparison to both single treatments.
The combination of ranpirnase and rosiglitazone decreased expression of Fra-1 and Survivin
Fra-1 expression in mesothelioma cell lines is regulated by the ERK1/2 or PI3K pathways in a cell-dependent manner (25). To determine which of these two pathways is affected by the combination, we selected two mesothelioma cell lines. The MP5 line has been previously shown to have a PI3K-dependent Fra-1 expression, while the MP2 line has been shown to have an ERK1/2-dependent Fra-1 expression (25). Ranpirnase and rosiglitazone in combination down-regulated Fra-1 only in the cell line with a PI3K-dependent Fra-1 expression, but not in the other cell line (Figure 4). The use of the PI3K inhibitor LY294002 further confirmed the PI3K effect on Fra-1 expression.
Figure 4.

Western blots show that synergistic down-regulation of Fra-1 by treatment with the combination of ranpirnase (1 μg/mL) and rosiglitazone (20 μM) for 48 h is cell dependent. MP5 with a PI3K-dependent Fra-1 expression, as determined by the use of the small molecule inhibitor LY294002 (20 μM), shows the most down-regulating effect. Down-regulation of Survivin is observed in both cell lines A. MP5 and B. MP2. C = Medium only control. Mean ± SEM of N=2 samples per group. Experiments repeated 2×. *= P ≤ 0.05 in comparison to respective control; != P ≤ 0.05 in comparison to both single treatments (ranpirnase and rosiglitazone).
Survivin, another PI3K-dependent protein, was also down-regulated by the PI3K inhibitor, and by the combination of ranpirnase and rosiglitazone in the MP5 cell line, but only by the drug combination in the MP2 cell line.
A second set of cancer lines (231 and DU-145) was treated for 48 h with ranpirnase, rosiglitazone alone, or in combination, and then washed and incubated for three hours with fresh DMEM/F12 complete media containing 0.5% FBS and 1μM insulin. These conditions were previously shown to result in a peak Fra-1 expression in these cells lines (data not shown). Results under these favorable conditions for Fra-1 expression still showed a synergistic down-regulation of Fra-1 in both cell lines (Figure 5). Down-regulation of Survivin was observed only in the DU-145 cell line.
Figure 5.
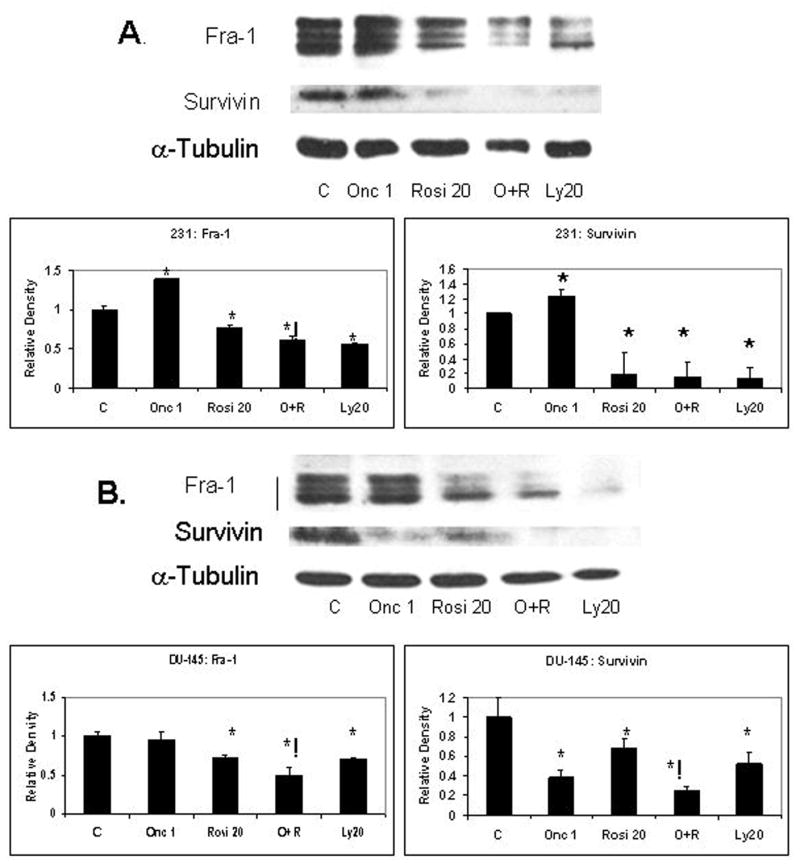
Western blots show that synergistic down-regulation of Fra-1 by treatment with the combination of ranpirnase (1 μg/mL) and rosiglitazone (20 μM) for 48 h and a 3 hour recovery with complete media containing 0.5% FBS and 1μM insulin in A. 231 and B. DU-145 cell lines. The use of the small molecule inhibitor LY294002 (20 μM) shows a PI3K-dependent expression of Fra-1 and Survivin in both cell lines. Synergistic down-regulation of Survivin is observed in DU-145. C = Medium only control. Mean ± SEM of N=2 samples per group. Experiments repeated 2×. *= P ≤ 0.05 in comparison to respective control; ! = P ≤ 0.05 in comparison to both single treatments (ranpirnase and rosiglitazone).
Decreased expression of Fra-1 induced apoptosis
To determine if the down-regulation of Fra-1 accounted for the apoptotic effect produced by the drug combination, all cell lines were transfected with siFra-1 or scramble control and tested for Fra-1 knock-out by RT-QPCR. Only the cells demonstrating >50% reduction in Fra-1 expression were used in this experiment (Figure 6 A). After cell transfection with the RNAi, cells were left for 5 days and tested for apoptosis using the Annexin V assay. As shown in Figure 6B, significantly increased apoptosis was observed in cell lines MP5, 231 and H292. The use of immunofluorescence showed that proliferating cells expressed nuclear Fra-1; dying cells did not (Figure 6B).
Figure 6.

Quantitative RTPCR of the cancer cell lines MP5, PC3, 231 and H292 showed that after transfection with RNAi constructs for Fra-1 (siFra-1) or scramble controls (siC), all cell lines demonstrate Fra-1 knock-down of more than 50% (A). Apoptosis measured as Annexin V-positive cells by flow cytometry (B) shows that Fra-1 knockdown increases apoptosis in three out the four cell lines tested. Immunofluorescence, using PCNA as a proliferation marker (blue), shows that Fra-1 (red) co-localizes with PCNA in the nucleus and that cells undergoing apoptosis (marked by the arrow) have neither PCNA nor Fra-1 expression in the nucleus (C).
Fra-1 expression increased drug resistance to ranpirnase
To directly observe if the knock-down of Fra-1 increased the efficacy of ranpirnase, rosiglitazone, or their combination, cell line H292 (a cell line with high transfection efficiency) was studied. Cells were transfected with siFra-1 or scramble control (siC) and left to rest for 48 h before treatment. Cells were then treated with different concentrations of ranpirnase (0.1, 1 or 10 μg/mL), rosiglitazone (10 or 20 μM), or their combination for 48 h. The PPARγ inhibitor GW9662 (2 μM) was added to observe the influence of PPARγ. Figure 7 shows that ranpirnase reduced viability in a dose-dependent manner. The knock-down of Fra-1 significantly increased the efficacy of ranpirnase alone at all concentrations, but not rosiglitazone alone. The use of the drug combination produced a synergistic effect on the scramble control (siC) transfected cells, but not in Fra-1 knock-down cells. The synergistic effect of rosiglitazone and ranpirnase was not changed by the modulation of PPARγ.
Figure 7.
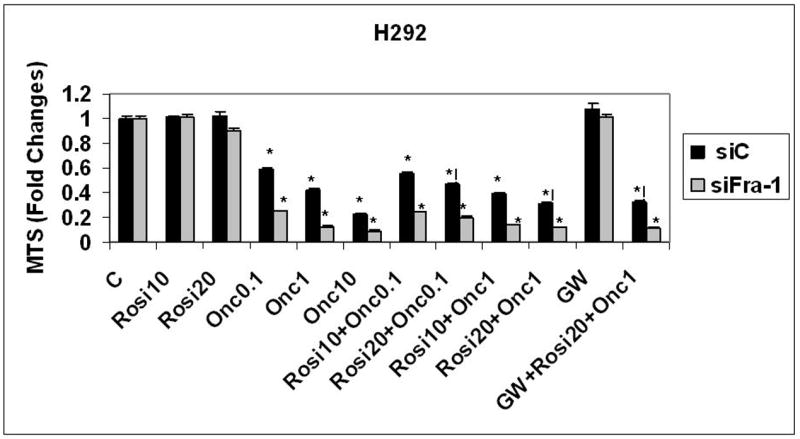
Knock-down of Fra-1 in the H292 lung cancer cell line increases cell killing by ranpirnase (0.1,1, or 10 μg/mL) in a concentration-dependent manner, but not by rosiglitazone (10 or 20 μM). The synergistic effect of ranpirnase (0.1 or 1 μg/mL) and rosiglitazone (10 or 20 μM) for 48 h is observed only in the scramble control (siC) transfected cell lines. The use of the PPARγ antagonist GW9662 (2 μM) did not significantly modify the synergistic effect of ranpirnase and rosiglitazone. C = Medium only control. Mean ± SEM of N=8 samples per group. Experiments repeated 2×. *= P ≤ 0.05 in comparison to respective control; != P ≤ 0.05 in comparison to both single treatments (ranpirnase and rosiglitazone).
DISCUSSION
In human mesothelioma cell lines, we have previously shown that the increased expression of survival pathways, frequently activated in cancer as the PI3K pathway (28), predicts the efficacy of chemotherapeutic drugs such as cisplatin and ranpirnase (19, 29). The data presented here demonstrate that the combination of ranpirnase with rosiglitazone, an anti-diabetic drug targeting the PI3K pathway (30), results in synergistic killing of several cancer cell lines. The mechanism of cell death was determined to involve cell apoptosis. We also investigated whether two recognized PI3K-regulated proteins (Fra-1 (25) and Survivin (24)), key to the development and maintenance of several cancers, were modulated by the drug combination. Furthermore, we investigated the effect of knocking down Fra-1 on the cell killing and apoptosis found with combinations of ranpirnase with rosiglitazone.
Under the experimental conditions of our studies, the combination of ranpirnase and rosiglitazone down-regulates Fra-1 in a cell dependent manner. We have previously shown that the regulation of Fra-1 in mesothelioma could be dependent on the ERK1/ERK2 pathway (8, 25), or on the PI3K pathway (25). The down-regulating effect on Fra-1 produced by the combination of ranpirnase and rosiglitazone suggest that the combination acts through PI3K.
Results presented here also show that the knock-down of Fra-1 can induce apoptosis. This finding could partially explain the killing effect of the drug combination in these cell lines. The knock-down of Fra-1 in the cell line H292 further demonstrates that the synergistic effect of the two drugs is partially related to the modulation of Fra-1. To test whether the synergism between these two drugs was related to rosiglitazone’s ability to activate PPARγ, we used a PPARγ antagonist together with the combination. The synergistic effect was independent of PPARγ.
Clinical trials of ranpirnase alone (Onconase) showed heterogeneity in therapeutic efficacy (19). These differential responses might reflect varying survival signaling mechanisms. Our results suggest that combined therapeutic use of ranpirnase and rosiglitazone may overcome the resistance produced in some cancer cells by the activation of survival pathways and their targets. Further in vivo studies are warranted.
CONCLUSIONS
Antineoplastic drug efficacy depends, in part, on the survival signaling pathways activated in specific tumor cells. The novel drug combination of ranpirnase and rosiglitazone is a promising combination to treat cancers with increased PI3K-dependent Fra-1 expression or Survivin.
Acknowledgments
The authors acknowledge Timothy Hunter, Scott Tighe, Mary Lou Shane, and Meghan Brown from the Vermont Cancer Center (VCC) DNA Analysis Facility at the University of Vermont for performing the Real Time-Quantitative PCR analysis.
This research was supported by National Institutes of Health grants K01 CA104159 (MER-N) and K24 DK068380 (BL).
LIST OF ABBREVIATIONS
- PI3K
Phosphatidylinositol 3′-kinase
- AP-1
Transcription factor, Activator protein-1
- TZD
Thiazolidinedione
- PPARγ
Peroxisome proliferator-activated receptor gamma
- siC
RNA interference scramble control
- siFra-1
RNA interference for Fra-1
- 231
MDA-MB-231 breast cancer cell line
- H292
NCI-H292 lung cancer cell line
Footnotes
COMPETING INTEREST
No conflicts declared.
AUTHORS’ CONTRIBUTIONS
M.E.R-N designed, and performed the research. BL contributed to the analysis and critical reading of the document.
References
- 1.Hu YC, Lam KY, Law S, Wong J, Srivastava G. Identification of Differentially Expressed Genes in Esophageal Squamous Cell Carcinoma (ESCC) by cDNA Expression Array: Overexpression of Fra- 1, Neogenin, Id-1, and CDC25B Genes in ESCC. Clin Cancer Res. 2001;7(8):2213–21. [PubMed] [Google Scholar]
- 2.Chiappetta G, Ferraro A, Botti G, et al. FRA-1 protein overexpression is a feature of hyperplastic and neoplastic breast disorders. BMC Cancer. 2007;7:17. doi: 10.1186/1471-2407-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zajchowski D, Bartholdi M, Gong Y, et al. Identification of gene expression profiles that predict the aggressive behavior of breast cancer cells. Cancer Res. 2001;61:5168–78. [PubMed] [Google Scholar]
- 4.Chiappetta G, Tallini G, De Biasio MC, et al. FRA-1 expression in hyperplastic and neoplastic thyroid diseases. Clin Cancer Res. 2000 Nov;6(11):4300–6. [PubMed] [Google Scholar]
- 5.Casalino L, Bakiri L, Talotta F, et al. Fra-1 promotes growth and survival in RAS-transformed thyroid cells by controlling cyclin A transcription. The EMBO journal. 2007 Apr 4;26(7):1878–90. doi: 10.1038/sj.emboj.7601617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kustikova O, Kramerov D, Grigorian M, et al. Fra-1 induces morphological transformation and increases in vitro invasiveness and motility of epithelioid adenocarcinoma cells. Mol Cell Biol. 1998;18(12):7095–105. doi: 10.1128/mcb.18.12.7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Adiseshaiah P, Lindner DJ, Kalvakolanu DV, Reddy SP. FRA-1 proto-oncogene induces lung epithelial cell invasion and anchorage-independent growth in vitro, but is insufficient to promote tumor growth in vivo. Cancer Res. 2007 Jul 1;67(13):6204–11. doi: 10.1158/0008-5472.CAN-06-4687. [DOI] [PubMed] [Google Scholar]
- 8.Ramos-Nino ME, Timblin CR, Mossman BT. Mesothelial cell transformation requires increased AP-1 binding activity and ERK-dependent Fra-1 expression. Cancer Res. 2002 Nov 1;62(21):6065–9. [PubMed] [Google Scholar]
- 9.Adida C, Crotty PL, McGrath J, Berrebi D, Diebold J, Altieri DC. Developmentally regulated expression of the novel cancer anti-apoptosis gene survivin in human and mouse differentiation. Am J Pathol. 1998 Jan;152(1):43–9. [PMC free article] [PubMed] [Google Scholar]
- 10.Ambrosini G, Adida C, Altieri DC. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med. 1997 Aug;3(8):917–21. doi: 10.1038/nm0897-917. [DOI] [PubMed] [Google Scholar]
- 11.Johnson ME, Howerth EW. Survivin: a bifunctional inhibitor of apoptosis protein. Veterinary pathology. 2004 Nov;41(6):599–607. doi: 10.1354/vp.41-6-599. [DOI] [PubMed] [Google Scholar]
- 12.Ramos-Nino M. Cytotoxic ribonuclease-based cancer therapies. Drugs of the Future. 2007;32(6):517–26. [Google Scholar]
- 13.Halicka HD, Murakami T, Papageorgio CN, et al. Induction of differentiation of leukaemic (HL-60) or prostate cancer (LNCaP, JCA-1) cells potentiates apoptosis triggered by onconase. Cell Prolif. 2000 Dec;33(6):407–17. doi: 10.1046/j.1365-2184.2000.00186.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee I, Lee YH, Mikulski SM, Lee J, Covone K, Shogen K. Tumoricidal effects of onconase on various tumors. J Surg Oncol. 2000 Mar;73(3):164–71. doi: 10.1002/(sici)1096-9098(200003)73:3<164::aid-jso10>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 15.Darzynkiewicz Z, Carter M, Mikulski S, Ardelt W, Shogen K. Cytostatic and cytotoxic effects of Pannon (P-30 Protein), a novel anticancer agent. Cell Tissue Kinet. 1988;21:169–82. doi: 10.1111/j.1365-2184.1988.tb00855.x. [DOI] [PubMed] [Google Scholar]
- 16.Rybak SM, Pearson JW, Fogler WE, et al. Enhancement of vincristine cytotoxicity in drug-resistant cells by simultaneous treatment with onconase, an antitumor ribonuclease. J Natl Cancer Inst. 1996 Jun 5;88(11):747–53. doi: 10.1093/jnci/88.11.747. [DOI] [PubMed] [Google Scholar]
- 17.Juan G, Ardelt B, Li X, et al. G1 arrest of U937 cells by onconase is associated with suppression of cyclin D3 expression, induction of p16INK4A, p21WAF/CIP1 and p27KIP and decreased pRb phosphorylation. Leukemia. 1998;8:1241–8. doi: 10.1038/sj.leu.2401100. [DOI] [PubMed] [Google Scholar]
- 18.Newton DL, Hansen HJ, Mikulski SM, Goldenberg DM, Rybak SM. Potent and specific antitumor effects of an anti-CD22-targeted cytotoxic ribonuclease: potential for the treatment of non-Hodgkin lymphoma. Blood. 2001 Jan 15;97(2):528–35. doi: 10.1182/blood.v97.2.528. [DOI] [PubMed] [Google Scholar]
- 19.Ramos-Nino ME, Vianale G, Sabo-Attwood T, et al. Human mesothelioma cells exhibit tumor cell-specific differences in phosphatidylinositol 3-kinase/AKT activity that predict the efficacy of Onconase. Mol Cancer Ther. 2005 May;4(5):835–42. doi: 10.1158/1535-7163.MCT-04-0243. [DOI] [PubMed] [Google Scholar]
- 20.Mikulski SM, Ardelt W, Shogen K, Bernstein EH, Menduke H. Striking increase of survival of mice bearing M109 Madison carcinoma treated with a novel protein from amphibian embryos. J Natl Cancer Inst. 1990 Jan 17;82(2):151–3. doi: 10.1093/jnci/82.2.151-a. [DOI] [PubMed] [Google Scholar]
- 21.Nunez M, Martin G, Cocca C, et al. Effect of rosiglitazone on N-nitroso-N-methylurea-induced mammary tumors in rat. Anticancer Res. 2006 May-Jun;26(3A):2113–22. [PubMed] [Google Scholar]
- 22.Russu WA. Thiazolidinedione anti-cancer activity: Is inhibition of microtubule assembly implicated? Med Hypotheses. 2007;68(2):343–6. doi: 10.1016/j.mehy.2006.06.054. [DOI] [PubMed] [Google Scholar]
- 23.Ramos-Nino ME, Blumen SR, Sabo-Attwood T, et al. HGF mediates cell proliferation of human mesothelioma cells through a PI3K/MEK5/Fra-1 pathway. Am J Respir Cell Mol Biol. 2008 Feb;38(2):209–17. doi: 10.1165/rcmb.2007-0206OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kawamura K, Fukuda J, Shimizu Y, Kodama H, Tanaka T. Survivin contributes to the anti-apoptotic activities of transforming growth factor alpha in mouse blastocysts through phosphatidylinositol 3′-kinase pathway. Biol Reprod. 2005 Dec;73(6):1094–101. doi: 10.1095/biolreprod.105.042754. [DOI] [PubMed] [Google Scholar]
- 25.Ramos-Nino ME, Blumen SR, Sabo-Attwood T, et al. HGF mediates cell proliferation of human mesothelioma cells through a PI3K/MEK5/Fra-1 pathway. Am J Respir Cell Mol Biol. 2007 Sep 13; doi: 10.1165/rcmb.2007-0206OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.BeruBe K, Quinlan T, Fung H, et al. Apoptosis is observed in mesothelial cells after exposure to crocidolite asbestos. Am J Respir Cell Mol Biol. 1996;15:141–7. doi: 10.1165/ajrcmb.15.1.8679218. [DOI] [PubMed] [Google Scholar]
- 27.Scapoli L, Ramos-Nino ME, Martinelli M, Mossman BT. Src-dependent ERK5 and Src/EGFR-dependent ERK1/2 activation is required for cell proliferation by asbestos. Oncogene. 2004 Jan 22;23(3):805–13. doi: 10.1038/sj.onc.1207163. [DOI] [PubMed] [Google Scholar]
- 28.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002 Jul;2(7):489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 29.Altomare DA, You H, Xiao GH, et al. Human and mouse mesotheliomas exhibit elevated AKT/PKB activity, which can be targeted pharmacologically to inhibit tumor cell growth. Oncogene. 2005 May 16;24:6080–9. doi: 10.1038/sj.onc.1208744. [DOI] [PubMed] [Google Scholar]
- 30.Han S, Roman J. Rosiglitazone suppresses human lung carcinoma cell growth through PPARgamma-dependent and PPARgamma-independent signal pathways. Mol Cancer Ther. 2006 Feb;5(2):430–7. doi: 10.1158/1535-7163.MCT-05-0347. [DOI] [PubMed] [Google Scholar]