

Abstract
Objective
Thoracic aortic aneurysms (TAAs) develop by a multifactorial process involving maladaptive signaling pathways which alter the aortic vascular environment. Transforming growth factor-beta (TGF-β) has been implicated in regulating the structure and composition of the extracellular matrix by differential activation of various intracellular signaling pathways. However, whether and to what degree TGF-β signaling contributes to TAA development remains unclear. Accordingly, the hypothesis that alterations in TGF-β signaling occur during aneurysm formation was tested in a murine model of TAA.
Methods
TAAs were surgically induced in mice (C57BL/6J) and aortas were analyzed at predetermined time points (1-, 2-, and 4- wks post-TAA induction). Quantitative real-time PCR (QPCR) was performed to evaluate the expression of 84 relevant TGF-β superfamily genes, and the protein levels of key signaling intermediates were measured by immunoblotting. Results were compared to unoperated reference control mice.
Results
QPCR revealed increased expression of TGF-β superfamily ligands (Gdf -2, -6, -7, Inhba), ligand inhibitors (Bmper, Chrd, Gsc), and transcriptional regulators (Dlx2, Evi1), among other genes (Cdkn2b, Igf1, IL-6). Protein levels of TGF-β receptorII, Smad2, Smad1/5/8, phospho-Smad1/5/8, and Smurf1 were increased from control values post-TAA induction. Both TGF-β receptorI and Smad4 were decreased from control values, while ALK-1 levels remained unchanged.
Conclusions
These alterations in the TGF-β pathway suggest a mechanism by which primary signaling is switched from a TGF-βRI/Smad2-dependent response, to an ALK-1/Smad1/5/8 response, representing a significant change in signaling outcome which may enhance matrix degradation.
Keywords: TGF-β, aneurysm, signal transduction, extracellular matrix, remodeling
Thoracic aortic aneurysms (TAAs) develop through a multifactorial process involving both intracellular and extracellular mechanisms.[1,2] The signaling pathways driving these mechanisms cause maladaptive remodeling of the vascular extracellular matrix (ECM), ultimately leading to an increased propensity for dilatation, dissection, or rupture of the aortic wall.[3,4] ECM remodeling is influenced by mechanisms that balance both matrix deposition and matrix degradation suggesting that within the aneurysmal aorta this balance is disrupted in favor of enhanced proteolysis.[4,5] Previous studies have principally focused on the dysregulation of specific extracellular protease systems, such as the matrix metalloproteinases (MMPs), hence little information exists regarding the upstream signaling pathways that drive aneurysm development.
One upstream signaling protein known to alter the structure and composition of the ECM, and known to play an important role in vascular remodeling is transforming growth factor-beta (TGF-β).[6,7] TGF-β is a member of a superfamily of ligands and receptors that include the TGF-βs, bone morphogenetic proteins (BMPs), and the activins/inhibins. These soluble peptide growth factors are produced by multiple cell types and participate in a wide array of cellular responses including: proliferation, angiogenesis, differentiation, apoptosis, and wound healing.[8,9] TGF-β is probably best known for its role in collagen synthesis related to fibrotic disease.[10] The classical profibrotic TGF-β signaling pathway is initiated upon binding of ligand to the type II TGF-β receptor (TGF-βRII). The type II receptor is autophosphorylated, which then recruits and transphosphorylates a type I receptor (TGF-βRI). The activated type I receptor in turn activates a receptor-Smad (R-Smad). The R-Smad then binds the common co-Smad and translocates to the nucleus. Once in the nucleus, it binds transcriptional co-factors and forms an activated transcriptional complex; this leads to the induction of target genes that favor matrix deposition.[11] The complexity of this signaling pathway is conferred by the presence of over 30 different functional ligands, 5 different type II receptors, 7 different type I receptors, and 8 different Smad proteins.[12,13] Additionally, there are known alternative signaling events that are mediated by type II TGF-β receptors in the absence of type I receptors, type 1 receptors in the absence of R-Smads, and interactions between activated R-Smads and other intracellular signaling proteins independent of the co-Smad.[14,15] Furthermore, the TGF-β signaling response can be modified based on the activation of other intracellular signaling pathways.
Previous studies have implicated altered TGF-β signaling in the pathogenesis of TAAs. For example, in Marfan syndrome type-1, enhanced TGF-β pathway activation was associated with aneurysm formation secondary to the fibrillin-1 mutation; whereas in MFS type-2 and Loeys-Dietz syndrome, mutations in both TGF-β receptors were associated with aneurysm development.[16-18] Additionally, familial thoracic aortic aneurysm and dissection has also been associated with the expression of a mutated TGF-β receptors.[19] Interestingly, in all cases, alterations in TGF-β signaling were associated with aortic pathology leading to degenerative changes within the aortic vascular ECM.
Because of the role that TGF-β is known to play in regulating the structure and composition of the ECM it was hypothesized that alterations in TGF-β signaling occur during aneurysm formation. Thus, using a unique murine model of TAA, the present study had two objectives. The first objective was to assess altered gene transcription in aortic tissues during the early stages of aneurysm development. To concentrate specifically on the role of TGF-β signaling, a commercially available pathway-focused polymerase chain reaction (PCR) gene expression profiling array was used, which provided analysis of 84 established TGF-β/BMP-inducible genes. The second objective was to extend the gene profiling results by analyzing key signaling intermediates in the TGF-β pathway at the protein level. Together, these data provide a framework that begins to define the role of TGF-β signaling in the early stages of aortic dilatation during aneurysm formation.
METHODS
Experimental design
The present study examined a total of 56 C57BL/6J mice (comprised of approximately equal numbers of males (56%) and females (44%)); 3 groups of mice under went TAA induction surgery with terminal points at 1-week (n=12), 2-weeks (n=15), and 4-weeks (n=13) post-TAA induction. The descending thoracic aortas were excised and processed accordingly for histology, gene expression profiling, and immunoblotting studies. Results were compared to age-matched unoperated mice (no TAA induction; n=16) which served as a reference control group. All animals were 8 to 12 weeks of age at the time of the initial surgical procedure. This animal protocol was approved by the Medical University of South Carolina Institutional Animal Care and Use Committee (ARC #2146), and all mice were treated and cared for in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (National research Council, Washington, DC, 1996).
Operative procedure
Murine TAAs were induced as previously described.[20] Mice were anesthetized by nose-cone with 2% isoflurane. The descending thoracic aorta was exposed and a sponge soaked in 0.5M calcium chloride was placed on the periadventitial surface for 15 minutes. The chest was closed in layers and the mice were allowed to recover.
Terminal surgical procedure, aortic harvest
At terminal study, the mice were again anesthetized using 2% isoflurane and the initial incision was reopened. The descending thoracic aorta was carefully excised and either snap-frozen and stored at −80°C for later analysis, or placed into RNALater solution (Ambion, Inc. Austin, TX) at 4°C for 24 hrs for prior to RNA extraction.
Aortic diameter measurements and architecture
Aortic outer diameter measurements were made as previously described.[21] Paraffin embedded formalin-fixed aortic tissue sections (4 µm) were stained with hematoxylin and eosin (H&E) for general architecture, Verhoeff-Van Gieson for elastin fibers, and picrosirius red (PSR) for collagen content.
PCR-based TGF-β/BMP pathway-focused gene expression profiling
Six mice were used for pathway-focused gene expression profiling studies (n=3; 2-wk TAA induced mice vs. n=3; unoperated reference control mice). The tissue specimens were homogenized in buffer RLT (RNeasy Kit) using a rotor-stator type tissue grinder and total RNA was extracted using the RNeasy Fibrous Tissue Mini Kit (Qiagen, Inc., Valencia, CA) according to the manufacturer's instructions including both Proteinase K and DNase digestion steps. The isolated RNA was assessed for quality and quantity using the Experion Automated Electrophoresis System (Bio-Rad Laboratories, Hercules, CA). High-quality RNA (1 µg) was reverse transcribed using the iScript cDNA Synthesis Kit (Bio-Rad Laboratories, Hercules, CA). The remaining total RNA was stored at −80°C and the freshly generated cDNA was immediately used for polymerase chain reaction (PCR)-based gene expression profiling using the TGF-β/BMP Pathway-focused RT2Profiler™ PCR Array (Cat# APMM-035A, SuperArray Bioscience Corp., Fredrick, MD) in combination with the RT2 Real-Time™ SYBR Green/Fluorescein PCR master mix (Cat# PA-011, SuperArray Bioscience Corp., Fredrick, MD). The manufacturer's recommended cycling parameters were used with the Bio-Rad MyIQ Single-Color Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, CA). Negative controls were run to verify the absence of genomic DNA contamination (no reverse transcription control), and the absence of overall DNA contamination in the PCR system and working environment (no template control). Fold change in gene expression was determined using the ΔΔCt method.
Immunoblotting
Resected aortic tissue specimens (1-week (n=12), 2-weeks (n=12), and 4-weeks (n=12); reference controls (n=12)) were homogenized in cold acidic extraction buffer, and the relative abundance of TGF-β pathway components was determined using standard immunoblotting techniques.[22] Briefly, 10 µg of each aortic homogenate was fractionated on a 4-12% Bis-Tris gradient polyacrylamide gel (Invitrogen Corp., Carlsbad, CA) and transferred to nitrocellulose membrane (; Bio-Rad, Hercules, CA). The membranes were incubated with specific antibodies (0.4 µg/ml in 5% non-fat dry milk/PBS) for key components of the TGF-β pathway (TGF-β1, TGF-β2, TGF-β3, TGF-βRII, TGF-βRI, Smad2, pSmad2, Smad 1/5/8, pSmad1/5/8, Smad4, and Smurf1, Cell Signaling Technology Inc., Danvers, MA; and ALK-1, Santa Cruz Biotechnology, Inc, Santa Cruz, CA), and positive immunoreactivity was identified by chemiluminescence detection (Western Lighting Chemiluminescence Reagent Plus; Perkin Elmer, Shelton, CT) following exposure to film (Hyperfilm; GE Healthcare, Piscataway, NJ).
Data analysis
All statistical procedures were carried out using the Stata statistical package (Intercooled Stata v8.2; StataCorp LP, College Station, TX). Change in aortic diameter was expressed as a percentage increase (mean ± SEM) from baseline value in each mouse and significant changes were determined using one-sample mean comparison testing versus a fixed value of 100. Comparisons between groups were made using ANOVA with Tukey's wsd post hoc analysis. In both cases, p<0.05 was considered significant.
Fold change in gene expression was performed using the ΔΔCt method, and expressed as the ratio of 2-wk TAA to the unoperated reference control. Statistical differences in mean Ct values between treatment groups were determined using a two-tailed Student's t-Test; p≤0.05 was considered significant.
Immunoblotting results were quantitated by densitometric image analysis (Gel Pro Analyzer; Media Cybernetics, Silver Spring, MD). Protein abundance was expressed as a percentage increase or decrease as compared to reference controls (mean ± SEM) set at 100%. Statistical differences were determined using one-sample mean comparison test versus a fixed value of 100. Differences between groups were determined using ANOVA with Tukey's wsd post hoc analysis. Values of p<0.05 were considered significant.
RESULTS
Aortic diameter measurements and architecture
Aortic diameter measurements revealed a progressive increase in aortic diameter, consistent with the early stages of TAA formation (Figure 1A). Histologically, at 4-week post-TAA induction flattening of the elastic lamellae and thinning of the aortic wall were evident as early indicators of aneurysm formation (Figure 1B).
Figure 1. Aortic dilatation and architecture.

A. Aortic diameters were measured in each mouse at baseline and terminal surgery (average baseline value = 654.4 µm). Aortic diameter increased progressively at 1-week, 2-weeks, and 4-weeks post-TAA induction; * p<0.05 vs. baseline, # p<0.05 vs. 1-week post-TAA, + p<0.05 vs. 2-week TAA. B. Cross-sections of aorta from an unoperated and 4-week TAA mouse stained with hematoxylin and eosin (H&E), Verhoeff van Geison (VVG) for elastin, and picrosirius red (PSR) for collagen, demonstrating thinning and flattening of the elastic lamellae during TAA development. Scale bar is equal to 50 µm.
PCR-based TGF-β/BMP pathway-focused gene expression profiling
Transcriptional activity was assessed in response to the activation of TGF-β/BMP signaling pathways by quantitative real-time PCR-based gene expression profiling. Expression results were obtained from three unoperated reference control mice and three 2-wk TAA mice (Table 1). Of the 84 TGF-β pathway-focused genes assessed, 22 genes were induced in the 2-wk TAA group by at least 2-fold, and 12 of those genes displayed a significant increase in mean Ct value as compared to the unoperated controls (p≤0.05) (Figure 2). Of the genes induced at least 2-fold, 8 were TGF-β superfamily ligands, 5 were ligand binding proteins/inhibitors, 5 were downstream effector genes, 3 were transcriptional regulators, and 1 was a B-cell lineage marker.
Table 1. Genes induced in aortic tissue harvested from mice at 2-weeks post TAA induction.
Data compiled using GeneCards: encyclopedia for genes, proteins, and diseases. Rebhan, M., Chalifa-Caspi, V., Prilusky, J., Lancet, D.; Weizmann Institute of Science, Bioinformatics Unit and Genome Center (Rehovot, Israel), 1997. (URL: http//www.genecards.org/). Bold text denotes p≤0.05 mean Ct value of 2-week TAA vs. unoperated reference control.
| Gene Symbol | Gene | Function | Fold Induction | p-value | |
|---|---|---|---|---|---|
| TGF-β superfamily Ligands | Bmp2 | Bone morphogenetic Protein 2 | BMP receptor ligand, TGF-β superfamily, induces cartilage and bone formation | 2.36 | 0.10 |
| Gdf1 | Growth & differentiation factor-1 | Ligand for TGF-β superfamily receptors, mediates differentiation | 2.95 | 0.14 | |
| Gdf2 | Growth & differentiation factor-2 (BMP-9) | Ligand for TGF-β superfamily receptors, involved in bone formation | 4.56 | 0.05 | |
| Gdf6 | Growth & differentiation factor-6 | Ligand for TGF-β superfamily receptors, required for bone formation | 8.92 | 0.02 | |
| Gdf7 | Growth & differentiation factor-7 | Ligand for TGF-β superfamily receptors, mediates differentiation | 4.09 | 0.03 | |
| Inhba | Activin beta-A chain | Ligand for TGF-β receptors | 1.66 | 0.05 | |
| Nodal | Nodal, mouse homolog | Ligand for TGF-β receptors, involved in embryonic development | 3.98 | 0.14 | |
| Tgfb2 | Transforming growth factor β2 | Ligand for TGF-β receptors, involved in extracellular matrix formation | 2.04 | 0.06 | |
| Tgfb3 | Transforming growth factor β3 | Ligand for TGF-β receptors, involved in extracellular matrix formation | 2.14 | 0.21 | |
| Ligand Inhibitors | Bmper | BMP binding endothelial regulator | Inhibitor of BMP function | 2.17 | 0.05 |
| Chrd | Chordin precursor | Dorsalizing factor; sequesters BMPs in latent complexes | 3.16 | 0.03 | |
| Gsc | Homeobox protein Goosecoid | Regulates chordin (Chrd) function | 3.90 | 0.02 | |
| Tdgf1 | Teratocarcinoma-derived growth factor-1 (Cripto) | Binds to TGF-β, inhibits TGF-β receptor complex formation | 2.47 | 0.12 | |
| Ltbp2 | Latent TGF-β binding protein 2 | Binds latency associated peptide, similar to Ltbp1; sequesters TGF-β in ECM | 2.94 | 0.13 | |
| Nbl1 | Neuroblastoma suppressor of tumorigenicity 1 | Tumor suppressor gene, cell cycle regulator (G1/S), BMP signaling inhibitor | 2.67 | 0.30 | |
| TGF-β Effector Genes | Cdkn2b | Cyclin-dependent Kinase Inhibitor 2b, p15 | Potent inhibitor of CDK4 and CDK6, mediates cell cycle arrest | 2.16 | 0.05 |
| Col1a1 | Collagen, type I, alpha 1 | Type I collagen, fibrillar collagen | 3.32 | 0.21 | |
| Col1a2 | Collagen, type I, alpha 2 | Type I collagen, fibrillar collagen | 2.92 | 0.19 | |
| Col3a1 | Collagen, type III, alpha 1 | Type III collagen, fibrillar collagen | 4.91 | 0.15 | |
| Trxn. Regs. | Dlx2 | Homeobox protein DLX-2 | Drosophila distal-less homolog, may interact/regulate Smad4 function | 12.86 | 0.03 |
| Evi1 | Ectropic virus integration site-1 protein | Transcriptional co-repressor, involved in embryonic development | 2.55 | 0.02 | |
| Runx1 | Runt-related transcription factor 1 (PEA2-alphaB) | Transcription factor, enhancer for Evi-1, involved in development | 2.30 | 0.09 | |
| Other | Cd79a | B-cell antigen receptor complex, alpha precursor | B-cell lineage marker, associated with IgM-receptor, mouse mb-1 homolog | 3.62 | 0.10 |
| Igf1 | Insulin-like growth factor 1 | Growth factor, induced by TGF-β, can modify TGF-β signaling response | 1.45 | 0.03 | |
| Il6 | Interleukin 6, interferon beta 2 | Cytokine normally repressed by TGF-β, can induce MMP expression | 8.81 | 0.01 |
Figure 2. Results of pathway-focused gene expression profiling.

The TGF-β/BMP signaling pathway was explored by pathway-focused gene expression profiling. Relative gene expression was calculated using the ΔΔCt method and expressed as fold gene induction of 2-week TAA/unoperated control. Results shown for all genes expressed at least 2-fold or genes which displayed a statistically significant change in mean Ct value. * p≤0.05 2-week TAA vs. unoperated control.
Analysis of key TGF-β pathway components
In addition to gene expression profiling, classical TGF-β pathway components were assessed at the protein level by immunoblotting. Of the three TGF-β ligands measured (TGF-β1, TGF-β2, and TGF-β3), only TGF-β3 was detectable and demonstrated a significant decrease from control levels at 2-wks post-TAA induction with no significant change observed at 1-wk or 4-wks (Figure 3). The type-II TGF-β receptor (TGF-βRII) was increased from reference control values at 2-wks and 4-wks post-TAA induction (Figure 4A), while the type I TGF-β receptor (TGF-βRI) was decreased from reference controls at 2-wks and 4-wks (Figure 4B). The abundance of a second type I TGF-β receptor, ALK-1, did not change from control levels over the time course studied (Figure 4C). The TGF-β signaling intermediate Smad2 was elevated from reference control values at all time points post-TAA induction (Figure 5A), while phosphorylated-Smad2 was detectable at very low levels, but failed to reach a significant difference from control at any time point (data not shown). Smad1/5/8 was significantly elevated from reference control at 2-wks and 4-wks post-TAA induction (Figure 5B), and a significant increase in Smad1/5/8 phosphorylation (pSmad1/5/8) was observed at 4-wks (Figure 5C). The co-Smad (Smad4) was significantly decreased from reference control values at all time points post-TAA induction (Figure 6). The TGF-β-induced E3 ubiquitin ligase, Smad ubiquitination regulatory factor-1 (Smurf1), was significantly elevated from reference control values at all time points post-TAA induction (Figure 7).
Figure 3. Analysis of TGF-β3 protein abundance.

TGF-β3 protein levels were decreased from control at 2-weeks post-TAA induction. * p<0.05 vs. 100%
Figure 4. Analysis of TGF-β receptor protein abundance.

A. TGF-βRII abundance was elevated from control at 2-weeks and 4-weeks post-TAA induction. B. TGF-βRI abundance decreased from control at 2-weeks and 4-weeks post-TAA induction. C. ALK-1 abundance did not change over the time course measured. * p<0.05 vs. 100%, # p<0.05 vs. 1-week TAA.
Figure 5. Analysis of R-Smad protein abundance.

A. Smad2 abundance was increased from control at 1-week, 2-weeks, and 4-weeks post-TAA induction. B. Smad1/5/8 was increased from control at 2-weeks and 4-weeks post-TAA induction. C. The phosphorylation of Smad1/5/8 (pSmad1/5/8) was elevated from control at 4-weeks post-TAA induction. * p<0.05 vs. 100%
Figure 6. Analysis of co-Smad protein abundance.

Smad4 decreased from control at 1-week, 2-weeks, and 4-weeks post-TAA induction. * p<0.05 vs. 100%
Figure 7. Analysis of Smurf1 protein abundance.
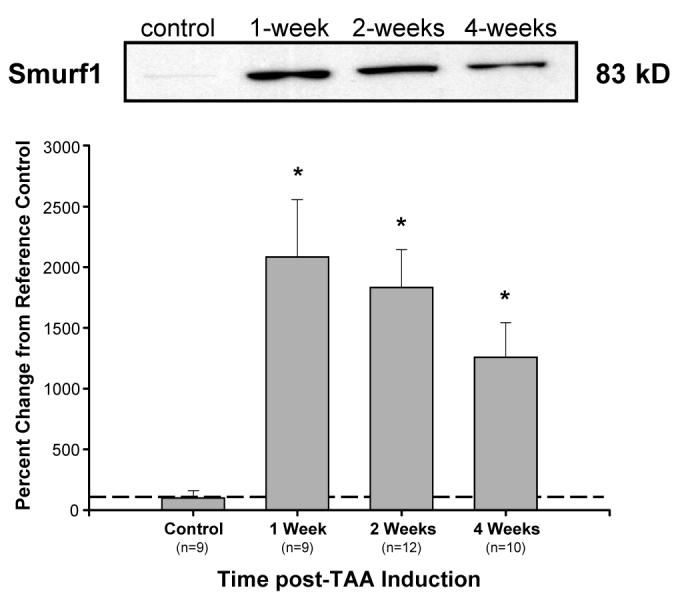
Smurf1 abundance increased from control at 1-week, 2-weeks, and 4-weeks post-TAA induction. * p<0.05 vs. 100%
DISCUSSION
Dysregulation of the TGF-β signaling pathway has been identified as a primary genetic defect in several disorders leading to the development of ascending aortic aneurysms.[16-19] However, paradoxical discoveries implicating both enhanced TGF-β signaling and loss of TGF-β receptor kinase function have obfuscated a clear functional role for TGF-β in TAA formation. In an effort to better define TGF-β signaling in TAA development, the present study examined changes in the TGF-β signaling pathway during the early stages of aortic dilatation in a unique murine model of TAA. Using pathway-focused gene expression profiling and targeted protein analysis, the unique findings from this study revealed transcriptional activation of several established TGF-β/BMP responsive genes that coincided with a down regulation of several key signaling components within the classical TGF-β pathway. Together these data suggest that the termination of Smad2-mediated signaling, determined in-part by the degradation of TGF-βRI, may enhance signaling by ALK-1/TGF-βRII and stimulate matrix degradation leading to TAA development.
To better understand the molecular changes accompanying aneurysm formation, TGF-β/BMP pathway-focused gene expression profiling was performed comparing mRNA isolated from 2-wk TAA specimens to a cohort of unoperated reference controls. Genes that displayed a 2-fold or better change in expression, or genes that displayed a significant difference as determined by t-test were categorized into 5 groups based on previously reported protein function (Table 1.; groups: TGF-β superfamily ligands, ligand inhibitors, TGF-β effector genes, transcriptional regulators, and other).
Several genes encoding for TGF-β superfamily ligands were elevated in the 2-wk TAA tissues, including growth differentiation factor (Gdf) -2, -6, and -7, along with the activin/inhibin β-A chain. These ligands have been implicated in wide array of functions including embryonic development, differentiation, and bone formation, as well as elastin and type I collagen expression.[23,24] Interestingly, the present study suggests that activin (composed of homo-or hetero-dimers of the inhibin βA- or βB- subunits) may be produced during aneurysm formation, consistent with a previous report demonstrating activin production in response to vascular injury and remodeling.[25] In addition, several ligand regulatory proteins also displayed elevated mRNA levels. The BMP binding endothelial regulator (Bmper) was elevated greater than 2-fold and functions to inhibit BMP signaling. Additionally, chordin (Chrd), which binds and sequesters BMPs in latent complexes, and its functional regulator Goosecoid (Gsc) were significantly expressed over control. These data, combined with the elevated expression levels of BMP-9 (Gdf-2), and a 2.36-fold elevation of BMP-2, suggest that activation of the BMP signaling pathway may play a significant role in aortic dilatation and aneurysm formation. In fact, treatment of C2C12 fibroblasts with BMP-2 has previously been shown to result in the potent induction of MMP-13 and concomitant repression of TIMP-1;[26] both of which would serve to enhance TAA formation in the present model system. Together, the regulation of ligand expression and signaling may serve to stimulate other signaling pathways that contribute to aneurysm development or drive a process of differentiation capable of altering the phenotype of resident vascular cells.
Evidence of functional signaling through the classical TGF-β pathway is supported by results showing elevated mRNA levels of several downstream effector genes. Cyclindependent kinase 4 inhibitor/p15 (Cdkn2b) was elevated 2-fold over control and has been implicated in regulating TGF-β-induced cell cycle arrest.[27] Additionally, expression of the type I and type III fibrillar collagens were also elevated, though none of the mean Ct values reached statistical significance (Col1a1, 3.32-fold; Col1a2, 2.92-fold; Col3a1, 4.91-fold). While this provides evidence that the TGF-β signaling pathway is intact, the unexpected lack of induction of other well described TGF-β inducible genes such as plasminogen activator inhibitor type 1 (PAI-1, 1.27-fold decrease) was not observed. This may suggest that signaling through the classical TGF-β pathway (Smad2/3-mediated) has been redirected through an alternate pathway that shares regulation of some genes (e.g. Smad1/5/8).
Several transcriptional regulators also displayed increased mRNA expression in 2-wk TAA specimens. The mouse homolog of the drosophila distal-less homeobox gene (Dlx2) was significantly induced almost 13-fold over reference control values; representing the largest increase in mRNA expression measured in these specimens. Dlx2 has been previously identified as a downstream target of TGF-β and BMP-2 signaling, and other Dlx family members have been shown to directly interact with Smad4 and inhibit transcription of TGF-β superfamily induced genes.[28,29] Thus, the robust induction of Dlx2 mRNA may represent a mechanism to attenuate Smad4-dependent signaling responses during TAA development. In addition, the mRNA induction of the ectropic virus integration site-1 gene (Evi1) and Runt-related transcription factor-1 (Runx1), both function as a transcriptional co-repressors, and may also serve to attenuate TGF-β-induced transcription of specific genes.[9,30,31] Interestingly, Runx1 which was elevated 2.3-fold over reference control values but failed to reach statistical significance, has previously been demonstrated as a repressor of tissue inhibitor of metalloproteinase-1 (TIMP-1) transcription; its function would thus serve to lower TIMP-1 levels and enhance matrix degradation.[30] Previous work by this laboratory has demonstrated that the targeted deletion of TIMP-1 alone is sufficient to accelerate aortic dilatation and TAA formation in this murine model.[32]
Two other genes, insulin-like growth factor 1 (Igf1) and interleukin 6 (IL-6), also displayed elevated expression in the 2-wk TAA specimens. While it has previously been shown that both genes can be induced in response to TGF-β,[33-35] they are both regulators of major signaling pathways that have potential to modify TGF-β signaling responses. Interestingly, IL-6 has been shown enhance TGF-β signaling by increasing receptor segregation into clathrin-containing endosomes that are recycled to the plasma membrane.[36] Furthermore, increased IL-6 protein levels were identified in aortic tissues samples from patients with abdominal aortic aneurysms (AAA), and increased circulating levels of IL-6 were identified in patients AAA and TAA.[37,38] Moreover, studies in other cell types demonstrated that in vitro treatment with IL-6 can enhance cell motility,[39] and induce the expression and secretion of several MMPs including MMP-9.[40,41] This may provide a mechanism to explain MMP-9 induction in endogenous cells associated with early aortic dilatation during murine TAA formation,[21] and may indicate that altered TGF-β signaling in this model is associated with increased matrix degradation.
Together, these transcriptional changes reflect a program that stimulates production of alternate TGF-β superfamily ligands and regulators that could redirect signaling through pathways that lead to enhanced matrix degradation; potentially mediated by the induction of MMP-9/MMP-13 and repression of TIMP-1. Moreover, implications of the inhibition/repression of Smad4-mediated transcription may also indicate that Smad-independent pathways participate in this response.
In order to further define a role for altered TGF-β signaling in aneurysm formation, key signaling intermediates within the classical TGF-β pathway were measured at the protein level. The unique findings revealed an increase in TGF-βRII, while TGF-βRI and Smad4 protein levels were decreased over the time course of the study. This was accompanied by an increase in Smad2, with no change in Smad2 phosphorylation, suggesting that TGF-βRI/Smad2-mediated signaling is inhibited during the early stages of aortic dilatation. Because TGF-β has been shown to induce production of specific E3-ubiquitin ligases (Smurf1, Smurf2), and because TGF-βRI and Smad4 have previously been described as substrates for Smurf1,[42,43] Smurf1 protein abundance was measured in the TAA samples and found to be robustly elevated at all time points post-TAA induction. Thus, the elevated levels of Smurf1 may in part be responsible for the loss of TGF-βRI and Smad4 during TAA formation. Together, this suggests that the loss of classical profibrotic TGF-β signaling may result in an altered balance between matrix deposition and matrix degradation, favoring enhanced matrix degradation within the vascular wall.
In addition to TGF-βRI, is a second type I receptor, ALK-1, is capable of interacting with TGF-βRII and signaling in response to TGF-β.[12,13] Interestingly, ALK-1 levels remained unchanged at all time points post-TAA induction. Thus, the combined effect of decreased TGF-βRI protein levels, unchanged levels of ALK-1, and increased abundance of TGF-βRII, may result in an increase in ALK-1/TGF-βRII heteromeric receptor complexes. Whereas the activation of TGF-βRI results in Smad2/3 phosphorylation, the activation of ALK-1 results in the phosphorylation of Smad1/5/8.[44,45] Accordingly, Smad1/5/8 levels were measured and demonstrated to be elevated at 2-weeks and 4-weeks post TAA induction. Furthermore, Smad1/5/8 phosphorylation was significantly increased at 4-weeks. Thus, these data suggest that the systematic down-regulation of TGF-βRI/Smad2 signaling may give way to increased signaling through ALK-1/Smad1/5/8 and thereby alter the response to TGF-β during TAA development. This represents a significant change in signaling outcome, as ALK-1-mediated responses often occur in opposition to TGF-βRI responses.[46,47] In fact, ALK-1 signaling has been identified as an antagonistic mediator of TGF-βRI signaling.[48] Interestingly, TGF-βRI and ALK-1 play important roles in angiogenesis,[46,49] and defects in these receptor signaling pathways have been associated with vascular pathology.[49,50] Because cell migration and angiogenesis require the production and release of MMPs,[51] it is interesting to speculate that activation of ALK-1 may induce angiogenic factors that contribute to the degradation of the vascular ECM in aneurysm development. Indeed enhanced angiogenesis has been implicated in the development of abdominal aortic aneurysms.[52-54]
Mapping complex signaling pathways by gene expression profiling and immunoblotting is not accomplished without encountering several limitations. First, because the pathway-focused gene expression profiling studies were performed using a limited cohort of mice, experimental variation in the small population could misrepresent significant results. To attempt to circumvent this, data were reported for genes that were expressed greater than 2-fold, or genes that displayed a significant difference in mean Ct value from control. Furthermore, because there are many post-transcriptional and post-translational modifications that regulate protein abundance, alterations in mRNA levels may not necessarily translate to alterations at the protein level. Additional studies will be required to verify that altered gene transcription truly correlates to altered protein production and abundance. Second, care should be taken when comparing the protein levels of two different signaling intermediates analyzed by immunoblotting. All results were expressed as a percent change from reference control, therefore the relative difference in protein abundance between analytes (e.g. TGF-βRI and ALK-1) can not be inferred from the present results. Moreover, many members of the TGF-β superfamily were not measured directly and may significantly contribute to alterations in the vascular ECM during aneurysm formation. Last, each major cell population within the aortic vascular wall (endothelial cells, smooth muscle cells, and fibroblasts), is capable of responding to TGF-β ligands, and may not respond in the same fashion. Spatiotemporal autocrine and paracrine responses may influence the other cell types present in local environment. Hence, future experiments will be required to determine the critical cell types involved and at what point intervention is capable of restoring the homeostatic balance between matrix degradation and matrix deposition.
Nevertheless, the present report demonstrates increased transcriptional activation of several well established TGF-β/BMP-inducible genes which coincides with altered protein levels of key classical TGF-β pathway components. Together these data imply that alterations in TGF-β signaling are associated with aortic dilatation during the early stages of aneurysm development, and suggests a mechanism by which primary signaling is switched from a TGF-βRI/Smad2 mediated response, to an ALK-1/Smad1/5/8 response that is capable of inducing matrix degradation. Additional studies using pathway-specific knockout strategies or specific pharmacologic inhibitors to further delineate the specific roles of each signaling arm are required. Advancing our understanding of TGF-β signaling and the role of this enigmatic pathway during TAA development may therefore identify potential therapeutic targets capable of inhibiting aortic dilatation or even reversing aneurysm disease.
Acknowledgments
This work was supported by NIH/NHLBI R01 HL075488-04, HL059165-07, and Research Institute of the Department of Veterans Affairs Award.
Footnotes
Portions of this work were presented at the American Heart Association Scientific Sessions, Chicago, IL (November 12-15, 2006)
REFERENCES
- 1.Isselbacher EM. Thoracic and abdominal aortic aneurysms. Circulation. 2005;111:816–828. doi: 10.1161/01.CIR.0000154569.08857.7A. [DOI] [PubMed] [Google Scholar]
- 2.Thompson RW. Reflections on the pathogenesis of abdominal aortic aneurysms. Cardiovasc Surg. 2002;10:389–394. doi: 10.1016/s0967-2109(02)00042-x. [DOI] [PubMed] [Google Scholar]
- 3.Liapis CD, Paraskevas KI. The pivotal role of matrix metalloproteinases in the development of human abdominal aortic aneurysms. Vasc Med. 2003;8:267–271. doi: 10.1191/1358863x03vm504ra. [DOI] [PubMed] [Google Scholar]
- 4.Thompson RW, Geraghty PJ, Lee JK. Abdominal aortic aneurysms: basic mechanisms and clinical implications. Curr Probl Surg. 2002;39:110–230. doi: 10.1067/msg.2002.121421. [DOI] [PubMed] [Google Scholar]
- 5.Tamarina NA, McMillan WD, Shively VP, Pearce WH. Expression of matrix metalloproteinases and their inhibitors in aneurysms and normal aorta. Surgery. 1997;122:264–271. doi: 10.1016/s0039-6060(97)90017-9. discussion 271-262. [DOI] [PubMed] [Google Scholar]
- 6.Bobik A. Transforming growth factor-betas and vascular disorders. Arterioscler Thromb Vasc Biol. 2006;26:1712–1720. doi: 10.1161/01.ATV.0000225287.20034.2c. [DOI] [PubMed] [Google Scholar]
- 7.Ghosh AK. Factors involved in the regulation of type I collagen gene expression: implication in fibrosis. Exp Biol Med (Maywood) 2002;227:301–314. doi: 10.1177/153537020222700502. [DOI] [PubMed] [Google Scholar]
- 8.Massague J. TGFbeta signaling: receptors, transducers, and Mad proteins. Cell. 1996;85:947–950. doi: 10.1016/s0092-8674(00)81296-9. [DOI] [PubMed] [Google Scholar]
- 9.Massague J. How cells read TGF-beta signals. Nat Rev Mol Cell Biol. 2000;1:169–178. doi: 10.1038/35043051. [DOI] [PubMed] [Google Scholar]
- 10.Roberts AB, Sporn MB, Assoian RK, Smith JM, Roche NS, Wakefield LM, Heine UI, Liotta LA, Falanga V, Kehrl JH, et al. Transforming growth factor type beta: rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc Natl Acad Sci U S A. 1986;83:4167–4171. doi: 10.1073/pnas.83.12.4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 12.Feng XH, Derynck R. Specificity and Versatility in TGF-beta signaling through Smads. Annu Rev Cell Dev Biol. 2005;21:659–693. doi: 10.1146/annurev.cellbio.21.022404.142018. [DOI] [PubMed] [Google Scholar]
- 13.Moustakas A, Souchelnytskyi S, Heldin CH. Smad regulation in TGF-beta signal transduction. J Cell Sci. 2001;114:4359–4369. doi: 10.1242/jcs.114.24.4359. [DOI] [PubMed] [Google Scholar]
- 14.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 15.Moustakas A, Heldin CH. Non-Smad TGF-beta signals. J Cell Sci. 2005;118:3573–3584. doi: 10.1242/jcs.02554. [DOI] [PubMed] [Google Scholar]
- 16.Hasham SN, Willing MC, Guo DC, Muilenburg A, He R, Tran VT, Scherer SE, Shete SS, Milewicz DM. Mapping a locus for familial thoracic aortic aneurysms and dissections (TAAD2) to 3p24-25. Circulation. 2003;107:3184–3190. doi: 10.1161/01.CIR.0000078634.33124.95. [DOI] [PubMed] [Google Scholar]
- 17.Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, Meyers J, Leitch CC, Katsanis N, Sharifi N, Xu FL, Myers LA, Spevak PJ, Cameron DE, De Backer J, Hellemans J, Chen Y, Davis EC, Webb CL, Kress W, Coucke P, Rifkin DB, De Paepe AM, Dietz HC. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37:275–281. doi: 10.1038/ng1511. [DOI] [PubMed] [Google Scholar]
- 18.Neptune ER, Frischmeyer PA, Arking DE, Myers L, Bunton TE, Gayraud B, Ramirez F, Sakai LY, Dietz HC. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet. 2003;33:407–411. doi: 10.1038/ng1116. [DOI] [PubMed] [Google Scholar]
- 19.Pannu H, Fadulu VT, Chang J, Lafont A, Hasham SN, Sparks E, Giampietro PF, Zaleski C, Estrera AL, Safi HJ, Shete S, Willing MC, Raman CS, Milewicz DM. Mutations in transforming growth factor-beta receptor type II cause familial thoracic aortic aneurysms and dissections. Circulation. 2005;112:513–520. doi: 10.1161/CIRCULATIONAHA.105.537340. [DOI] [PubMed] [Google Scholar]
- 20.Ikonomidis JS, Gibson WC, Gardner J, Sweterlitsch S, Thompson RP, Mukherjee R, Spinale FG. A murine model of thoracic aortic aneurysms. J Surg Res. 2003;115:157–163. doi: 10.1016/s0022-4804(03)00193-8. [DOI] [PubMed] [Google Scholar]
- 21.Jones JA, Barbour JR, Lowry AS, Bouges S, Beck C, McClister DM, Jr., Mukherjee R, Ikonomidis JS. Spatiotemporal expression and localization of matrix metalloproteinase-9 in a murine model of thoracic aortic aneurysm. J Vasc Surg. 2006;44:1314–1321. doi: 10.1016/j.jvs.2006.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spinale FG, Coker ML, Thomas CV, Walker JD, Mukherjee R, Hebbar L. Time-dependent changes in matrix metalloproteinase activity and expression during the progression of congestive heart failure: relation to ventricular and myocyte function. Circ Res. 1998;82:482–495. doi: 10.1161/01.res.82.4.482. [DOI] [PubMed] [Google Scholar]
- 23.Francis-West PH, Parish J, Lee K, Archer CW. BMP/GDF-signalling interactions during synovial joint development. Cell Tissue Res. 1999;296:111–119. doi: 10.1007/s004410051272. [DOI] [PubMed] [Google Scholar]
- 24.Wolfman NM, Hattersley G, Cox K, Celeste AJ, Nelson R, Yamaji N, Dube JL, DiBlasio-Smith E, Nove J, Song JJ, Wozney JM, Rosen V. Ectopic induction of tendon and ligament in rats by growth and differentiation factors 5, 6, and 7, members of the TGF-beta gene family. J Clin Invest. 1997;100:321–330. doi: 10.1172/JCI119537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Molloy CJ, Taylor DS, Pawlowski JE. Novel cardiovascular actions of the activins. J Endocrinol. 1999;161:179–185. doi: 10.1677/joe.0.1610179. [DOI] [PubMed] [Google Scholar]
- 26.Nakashima A, Tamura M. Regulation of matrix metalloproteinase-13 and tissue inhibitor of matrix metalloproteinase-1 gene expression by WNT3A and bone morphogenetic protein-2 in osteoblastic differentiation. Front Biosci. 2006;11:1667–1678. doi: 10.2741/1912. [DOI] [PubMed] [Google Scholar]
- 27.Hannon GJ, Beach D. p15INK4B is a potential effector of TGF-beta-induced cell cycle arrest. Nature. 1994;371:257–261. doi: 10.1038/371257a0. [DOI] [PubMed] [Google Scholar]
- 28.Chiba S, Takeshita K, Imai Y, Kumano K, Kurokawa M, Masuda S, Shimizu K, Nakamura S, Ruddle FH, Hirai H. Homeoprotein DLX-1 interacts with Smad4 and blocks a signaling pathway from activin A in hematopoietic cells. Proc Natl Acad Sci U S A. 2003;100:15577–15582. doi: 10.1073/pnas.2536757100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu SC, Harris MA, Rubenstein JL, Mundy GR, Harris SE. Bone morphogenetic protein-2 (BMP-2) signaling to the Col2alpha1 gene in chondroblasts requires the homeobox gene Dlx-2. DNA Cell Biol. 2001;20:359–365. doi: 10.1089/10445490152122479. [DOI] [PubMed] [Google Scholar]
- 30.Bertrand-Philippe M, Ruddell RG, Arthur MJ, Thomas J, Mungalsingh N, Mann DA. Regulation of tissue inhibitor of metalloproteinase 1 gene transcription by RUNX1 and RUNX2. J Biol Chem. 2004;279:24530–24539. doi: 10.1074/jbc.M311804200. [DOI] [PubMed] [Google Scholar]
- 31.Kurokawa M, Mitani K, Irie K, Matsuyama T, Takahashi T, Chiba S, Yazaki Y, Matsumoto K, Hirai H. The oncoprotein Evi-1 represses TGF-beta signalling by inhibiting Smad3. Nature. 1998;394:92–96. doi: 10.1038/27945. [DOI] [PubMed] [Google Scholar]
- 32.Ikonomidis JS, Gibson WC, Butler JE, McClister DM, Sweterlitsch SE, Thompson RP, Mukherjee R, Spinale FG. Effects of deletion of the tissue inhibitor of matrix metalloproteinases-1 gene on the progression of murine thoracic aortic aneurysms. Circulation. 2004;110:II268–273. doi: 10.1161/01.CIR.0000138384.68947.20. [DOI] [PubMed] [Google Scholar]
- 33.Bloor CA, Knight RA, Kedia RK, Spiteri MA, Allen JT. Differential mRNA expression of insulin-like growth factor-1 splice variants in patients with idiopathic pulmonary fibrosis and pulmonary sarcoidosis. Am J Respir Crit Care Med. 2001;164:265–272. doi: 10.1164/ajrccm.164.2.2003114. [DOI] [PubMed] [Google Scholar]
- 34.Okazaki R, Durham SK, Riggs BL, Conover CA. Transforming growth factor-beta and forskolin increase all classes of insulin-like growth factor-I transcripts in normal human osteoblast-like cells. Biochem Biophys Res Commun. 1995;207:963–970. doi: 10.1006/bbrc.1995.1279. [DOI] [PubMed] [Google Scholar]
- 35.Park JI, Lee MG, Cho K, Park BJ, Chae KS, Byun DS, Ryu BK, Park YK, Chi SG. Transforming growth factor-beta1 activates interleukin-6 expression in prostate cancer cells through the synergistic collaboration of the Smad2, p38-NF-kappaB, JNK, and Ras signaling pathways. Oncogene. 2003;22:4314–4332. doi: 10.1038/sj.onc.1206478. [DOI] [PubMed] [Google Scholar]
- 36.Zhang XL, Topley N, Ito T, Phillips A. Interleukin-6 regulation of transforming growth factor (TGF)-beta receptor compartmentalization and turnover enhances TGF-beta1 signaling. J Biol Chem. 2005;280:12239–12245. doi: 10.1074/jbc.M413284200. [DOI] [PubMed] [Google Scholar]
- 37.Dawson J, Cockerill GW, Choke E, Belli AM, Loftus I, Thompson MM. Aortic aneurysms secrete interleukin-6 into the circulation. J Vasc Surg. 2007;45:350–356. doi: 10.1016/j.jvs.2006.09.049. [DOI] [PubMed] [Google Scholar]
- 38.Middleton RK, Lloyd GM, Bown MJ, Cooper NJ, London NJ, Sayers RD. The proinflammatory and chemotactic cytokine microenvironment of the abdominal aortic aneurysm wall: a protein array study. J Vasc Surg. 2007;45:574–580. doi: 10.1016/j.jvs.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 39.Sehgal PB, Tamm I. Interleukin-6 enhances motility of breast carcinoma cells. Exs. 1991;59:178–193. doi: 10.1007/978-3-0348-7494-6_12. [DOI] [PubMed] [Google Scholar]
- 40.Kusano K, Miyaura C, Inada M, Tamura T, Ito A, Nagase H, Kamoi K, Suda T. Regulation of matrix metalloproteinases (MMP-2, -3, -9, and -13) by interleukin-1 and interleukin-6 in mouse calvaria: association of MMP induction with bone resorption. Endocrinology. 1998;139:1338–1345. doi: 10.1210/endo.139.3.5818. [DOI] [PubMed] [Google Scholar]
- 41.Lin T, Zhang W, Fan Y, Mulholland M. Interleukin-1beta and interleukin-6 stimulate matrix metalloproteinase-9 secretion in cultured myenteric glia. J Surg Res. 2007;137:38–45. doi: 10.1016/j.jss.2006.05.043. [DOI] [PubMed] [Google Scholar]
- 42.Ebisawa T, Fukuchi M, Murakami G, Chiba T, Tanaka K, Imamura T, Miyazono K. Smurf1 interacts with transforming growth factor-beta type I receptor through Smad7 and induces receptor degradation. J Biol Chem. 2001;276:12477–12480. doi: 10.1074/jbc.C100008200. [DOI] [PubMed] [Google Scholar]
- 43.Moren A, Imamura T, Miyazono K, Heldin CH, Moustakas A. Degradation of the tumor suppressor Smad4 by WW and HECT domain ubiquitin ligases. J Biol Chem. 2005;280:22115–22123. doi: 10.1074/jbc.M414027200. [DOI] [PubMed] [Google Scholar]
- 44.Chen YG, Massague J. Smad1 recognition and activation by the ALK1 group of transforming growth factor-beta family receptors. J Biol Chem. 1999;274:3672–3677. doi: 10.1074/jbc.274.6.3672. [DOI] [PubMed] [Google Scholar]
- 45.Oh SP, Seki T, Goss KA, Imamura T, Yi Y, Donahoe PK, Li L, Miyazono K, ten Dijke P, Kim S, Li E. Activin receptor-like kinase 1 modulates transforming growth factor-beta 1 signaling in the regulation of angiogenesis. Proc Natl Acad Sci U S A. 2000;97:2626–2631. doi: 10.1073/pnas.97.6.2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goumans MJ, Lebrin F, Valdimarsdottir G. Controlling the angiogenic switch: a balance between two distinct TGF-b receptor signaling pathways. Trends Cardiovasc Med. 2003;13:301–307. doi: 10.1016/s1050-1738(03)00142-7. [DOI] [PubMed] [Google Scholar]
- 47.Goumans MJ, Valdimarsdottir G, Itoh S, Lebrin F, Larsson J, Mummery C, Karlsson S, ten Dijke P. Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFbeta/ALK5 signaling. Mol Cell. 2003;12:817–828. doi: 10.1016/s1097-2765(03)00386-1. [DOI] [PubMed] [Google Scholar]
- 48.Goumans MJ, Valdimarsdottir G, Itoh S, Rosendahl A, Sideras P, ten Dijke P. Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. Embo J. 2002;21:1743–1753. doi: 10.1093/emboj/21.7.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bertolino P, Deckers M, Lebrin F, ten Dijke P. Transforming growth factor-beta signal transduction in angiogenesis and vascular disorders. Chest. 2005;128:585S–590S. doi: 10.1378/chest.128.6_suppl.585S. [DOI] [PubMed] [Google Scholar]
- 50.Fernandez LA, Sanz-Rodriguez F, Blanco FJ, Bernabeu C, Botella LM. Hereditary hemorrhagic telangiectasia, a vascular dysplasia affecting the TGF-beta signaling pathway. Clin Med Res. 2006;4:66–78. doi: 10.3121/cmr.4.1.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rundhaug JE. Matrix metalloproteinases and angiogenesis. J Cell Mol Med. 2005;9:267–285. doi: 10.1111/j.1582-4934.2005.tb00355.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Choke E, Cockerill GW, Dawson J, Wilson RW, Jones A, Loftus IM, Thompson MM. Increased angiogenesis at the site of abdominal aortic aneurysm rupture. Ann N Y Acad Sci. 2006;1085:315–319. doi: 10.1196/annals.1383.007. [DOI] [PubMed] [Google Scholar]
- 53.Miwa K, Nakashima H, Aoki M, Miyake T, Kawasaki T, Iwai M, Oishi M, Kataoka K, Ohgi S, Ogihara T, Kaneda Y, Morishita R. Inhibition of ets, an essential transcription factor for angiogenesis, to prevent the development of abdominal aortic aneurysm in a rat model. Gene Ther. 2005;12:1109–1118. doi: 10.1038/sj.gt.3302496. [DOI] [PubMed] [Google Scholar]
- 54.Paik DC, Fu C, Bhattacharya J, Tilson MD. Ongoing angiogenesis in blood vessels of the abdominal aortic aneurysm. Exp Mol Med. 2004;36:524–533. doi: 10.1038/emm.2004.67. [DOI] [PubMed] [Google Scholar]