

Abstract
Curcuminoid extract and piperine are being evaluated for beneficial effects in Alzheimer’s disease, among other intractable disorders. Consequently, we studied the potential for herb-drug interactions involving cytochrome P450 (CYP), UDP-glucuronosyltransferase (UGT), and sulfotransferase (SULT) enzymes. The curcuminoid extract inhibited SULT > CYP2C19 > CYP2B6 > UGT > CYP2C9 > CYP3A activities with IC50 values ranging from 0.99 ± 0.04 to 25.3 ± 1.3 μM, while CYP2D6, CYP1A2, and CYP2E1 activities were less affected (IC50 values >60 μM). Inhibition of CYP3A activity by curcuminoid extract was consistent with competitive inhibition (Ki = 11.0 ± 1.3 μM), while inhibition of both CYP2C9 and CYP2C19 activities were consistent with mixed competitive-noncompetitive inhibition (10.6 ± 1.1 μM and 7.8 ± 0.9 μM, respectively). Piperine was a relatively selective noncompetitive inhibitor of CYP3A (IC50 5.5 ± 0.7 μM, Ki = 5.4 ± 0.3 μM) with less effect on other enzymes evaluated (IC50 >29 μM). Curcuminoid extract and piperine inhibited recombinant CYP3A4 much more potently (by >5-fold) than CYP3A5. Pure synthetic curcuminoids (curcumin, demethoxycurcumin, and bisdemethoxycurcumin) were also evaluated for their effects on CYP3A, CYP2C9, UGT, and SULT activities. All three curcuminoids had similar effects on CYP3A, UGT, and SULT activity, but demethoxycurcumin (IC50 = 8.8 ± 1.2 μM) was more active against CYP2C9 than either curcumin or bisdemethoxycurcumin (IC50 >50 μM). Based on these data and expected tissue concentrations of inhibitors, we predict that an orally administered curcuminoid/piperine combination is most likely to inhibit CYP3A, CYP2C9, UGT, and SULT metabolism within the intestinal mucosa.
Introduction
Curcuminoids are polyphenolic compounds found in high concentrations in turmeric and are responsible for giving this food spice its distinctive yellow color. There are three principal curcuminoids in extracts of turmeric, including curcumin, demethoxycurcumin, and bisdemethoxycurcumin (Fig. 1). Extracts containing curcuminoids are available over-the-counter as herbal supplements and currently are being evaluated in various clinical trials for the treatment of colon and pancreatic cancers, multiple myeloma, Alzheimer’s disease, and ulcerative colitis (Cruz-Correa et al., 2006; Hanai et al., 2006)1. Curcuminoids and especially bisdemethoxycurcumin have been shown to improve immune deficiency in cells from Alzheimer’s disease patients (Fiala et al., 2007).
Figure 1.

Structures of curcuminoids studied.
Typically, high doses (2–8 g per day) of curcuminoids are being administered to patients in clinical trials because of their limited bioavailability. Another approach to increase systemic curcuminoid concentrations is the use of combination formulations of curcuminoids and piperine, specifically to enhance curcuminoid bioavailability. Piperine, found in the extract of black pepper, enhances the relative oral bioavailability of curcuminoids by as much as 20-fold in healthy human volunteers (Shoba et al., 1998). Piperine also enhances the bioavailability of several other compounds including phenytoin, coenzyme Q10, theophylline, and propranolol (Bano et al., 1991; Badmaev et al., 2000; Pattanaik et al., 2006). Several mechanisms have been postulated for piperine’s bioavailability-enhancing effect including the formation of apolar complexes with other compounds, inhibition of efflux transport, and inhibition of gut metabolism (Atal et al., 1985; Khajuria et al., 1998; Bhardwaj et al., 2002).
Although health benefits may be derived from the use of curcuminoids and piperine, these compounds have the potential to interact with co-administered drugs (i.e., drug-herb interaction) through inhibition of drug metabolism mediated by UDP-glucuronosyltransferase (UGT), sulfotransferase (SULT), and cytochrome P450 (CYP) enzymes. Curcuminoids have been shown to inhibit glucuronidation of mycophenolic acid, bilirubin, capsaicin, and 1-naphthol in LS180 and Caco-2 cells (both are human colon cancer cell lines) and human UGT1A1-transfected COS-1 cells (Basu et al., 2003; Basu et al., 2004; Basu et al., 2004; Naganuma et al., 2006). Effects on glucuronidation by human tissues have not yet been evaluated. Sulfation of 4-nitrophenol was also potently inhibited by curcuminoids in human liver cytosol with IC50 values ranging from 14 to 380 nM (Eaton et al., 1996; Vietri et al., 2003). In rat liver microsomes, curcuminoids inhibited CYP1A1, 1A2, and 2B1 metabolism with IC50 values ranging from 4.2 to 20 μM (Thapliyal and Maru, 2001). Most recently, in vivo studies of curcuminoids given to rats showed elevated plasma levels of co-administered midazolam, a marker substrate for CYP3A metabolism (Zhang et al., 2007). However, it is not known whether curcuminoids can inhibit any human CYP.
Piperine inhibits CYP3A-mediated verapamil oxidation in human liver microsomes with IC50 values ranging from 36 to 77 μM (Bhardwaj et al., 2002). As yet, effects on other human CYPs have not been reported. Piperine also inhibited glucuronidation of (−)-epigallocatechin-3-gallate (EGCG) in mice resulting in increased EGCG bioavailability (Lambert et al., 2004). In the same study, 20 μM piperine was found to inhibit the formation of EGCG-3′-glucuronide in human HT-29 colon adenocarcinoma cells by ~50%. Other rodent studies have shown that piperine can inhibit both CYP and UGT activities in vivo (Atal et al., 1985; Reen and Singh, 1991). However, effects of piperine on sulfation or glucuronidation by human tissues have not been reported.
In conclusion, preliminary evidence indicates that curcuminoids and piperine have the potential to inhibit human CYP, UGT and SULT enzymes, primarily based on studies in rodent models. Given the potential for more widespread use of curcuminoids and piperine, particularly if the results of ongoing clinical trials show promise, coupled with the likelihood that these herbal medications would be given with traditional medications, it is important to determine whether curcuminoids and/or piperine can alter the disposition and effects of co-administered drugs in human patients. As a first step in this process, we report a comprehensive evaluation of the inhibitory effects of these compounds on CYP, UGT, and SULT enzymes using recombinant human CYPs, human liver fractions, and human intestinal cell lines. Results from these in vitro studies will be used to design herb-drug interaction studies in human volunteers using enzyme selective probe drugs. Because studies to date have used turmeric extracts exclusively that include multiple curcuminoids, we also synthesized the 3 main curcuminoids (curcumin, demethoxycurcumin, and bisdemethoxycurcumin) and used these to determine whether the observed inhibition by turmeric extract is dependent on a particular curcuminoid.
Materials and Methods
Materials
Curcuma longa (turmeric) extract (catalog #C1386; referred to here as “curcuminoid extract”) and piperine (catalog #P49007, 97% purity) were purchased from Sigma-Aldrich (St. Louis, MO). Curcuminoid extract is listed in the Sigma-Aldrich catalog as “curcumin” because it contains mainly curcumin (~70%) with smaller amounts of demethoxycurcumin and bisdemethoxycurcumin (exact amounts were determined experimentally below). However, note that for simplicity the molar concentrations of the curcuminoid extract given below were calculated based on the molecular weight of curcumin and assumes 100% purity. Unless otherwise noted, most other reagents were also purchased from Sigma-Aldrich. Bupropion and 4-hydroxybupropion (Glaxo-Wellcome, Research Triangle Park, NC); chlorzoxazone and 6-hydroxychlorzoxazone (R. W. Johnson Pharmaceutical Research Institute, Spring House, PA); triazolam, 1-hydroxytriazolam, and 4-hydroxytriazolam (Upjohn, Kalamazoo, MI); acetaminophen, acetaminophen glucuronide, and acetaminophen sulfate (McNeil Consumer Products Co., Fort Washington, PA); and flurbiprofen and hydroxyflurbiprofen (gift of Dr. Timothy Tracy) were provided as indicated by their respective manufacturers. Pronethalol was provided by ICI Limited (Macclesfield, UK). Recombinant CYP3A4 and CYP3A5 Supersomes (without added cytochrome b5) were purchased from BD Gentest (Woburn, MA). LS180 cells were purchased from American Type Culture Collection (Manassas, VA) and maintained in MEM with 2 mM L-glutamine and Earle’s salts and supplemented with 0.1 mM non-essential amino acids, 1.0mM sodium pyruvate, 50 U/mL penicillin, 50 μg/mL streptomycin, and 10% fetal bovine serum (all purchased from Invitrogen, Carlsbad, CA).
Synthesis of pure curcuminoids
The curcumin derivatives, 4a-b were synthesized as outlined in Scheme 1 based on methods previously published (Nurfina et al., 1997; Kim and Yang, 2004). Acetylacetone was treated with boric anhydride to give the boron complex 1. Condensation of the aldehydes 2a-b with the boron complex 1 in the presence of n-butylamine followed by acid decomposition of the boron complex 3 afforded the curcumin derivatives 4a-b. Demethoxycurcumin 4c was synthesized in a similar fashion using an equimolar mixture of vanillin and 4-hydroxybenzaldehyde (2c). The reaction afforded a mixture of the three curcuminoids; the demethoxycurcumin component was the major product.
Scheme 1. Scheme for the synthesis of pure curcuminoids.

i) Boric anhydride; ii) benzaldehydes 2a-c, tributylborate, EtOAc; 100°C; iii) n-BuNH2, 85°C; iv) 0.4N HCl, 60°C.
General
All reactions were run under an argon atmosphere with magnetic stirring using oven-dried glassware unless otherwise indicated. Air- and moisture-sensitive liquids were transferred via syringe through rubber septa. Silica gel (60 Å, 40–63 m mesh) from Silicycle (Quebec City, Quebec, Canada) was used for column chromatography. Ethyl acetate was dried by filtration through neutral alumina and was stored over activated 4 Å molecular sieves under Argon prior to use. All other solvents and reagents were used as received. 1H NMR was recorded at 300.0 MHz with a Varian Mercury 300 instrument (Palo Alto, CA). Chemical shifts were reported in ppm (δ) relative to CDCl3 at 7.26 ppm. NMR spectra wereμ recorded in CDCl3 unless stated otherwise. ESI-MS data were obtained using a Hitachi model M-8000 mass spectrometer (San Jose, CA).
5-Hydroxy-1,7-bis-(4-hydroxy-phenyl)-hepta-1,4,6-trien-3-one (bisdemethoxycurcumin), 4a
A mixture of acetylacetone (2 mL, 19.5 mmol) and boric anhydride (1 g, 12.8 mmol) was stirred at room temperature for 2h under an atmosphere of argon. 4-Hydroxybenzaldehyde (9.52 g, 78 mmol) was dissolved in dry ethyl acetate (150 mL) in a round bottom flask fixed with a condenser under an atmosphere of argon. Tributyl borate (21 mL, 78 mmol) was added to the solution via a syringe. The stirred mixture was heated to 100°C for one hour and the mixture turned into an orange solution. The boron complex was added to this mixture and the reaction mixture was stirred at 100°C for an additional hour. The mixture was cooled to 85°C and butylamine (7.7 mL, 78 mmol) was added in 1.9 mL portions every 5 minutes and the mixture turned into a deep red solution after addition of the first portion. The solution became brown after complete addition of butylamine. The mixture was stirred at 100°C for an additional 30 min, and then cooled to 50°C. Hydrochloric acid (0.4 N, 60 mL) was added and the mixture was stirred for another 30 min. The layers were separated and the organic material was successively washed with water and brine. The solution was dried over Na2SO4, filtered and concentrated to dryness. The crude product was subject to flash chromatography (hexane/EtOAc, 2:1, v:v) to afford 0.98 g, 16% yield of the desired product as an orange powder. Rf = 0.15; MP = 199.9 °C; ESI-MS: m/z 309 (MH)+, 331 (MNa)+, 307 (MH)−; 1H NMR δ 7.5 (d, J = 15.6 Hz, 2H, Ph-CH-), 7.29 (m, 4H, Ph), 6.62–6.78 (m, 4H, Ph), 6.37 (d, J = 15.6 Hz, 2H, -CH-), 5.7 (s, 1H, -CO-CH-CO-).
5-Hydroxy-1,7-bis-(4-hydroxy-3-methoxy-phenyl)-hepta-1,4,6-trien-3-one (curcumin), 4b was synthesized according to the general procedure described for compound 4a. The crude product was purified by flash chromatography (EtOAc/hexane, 1:1, v:v) to give 22 % yield of the desired product as an orange powder. Rf = 0.35; ESI-MS: m/z 369 (MH+), 367 (MH−); 1H NMR δ 7.41 (d, J = 15.6 Hz, 2H, Ph-CH-), 6.92 (m, 4H, Ph), 6.72 (m, 2H, Ph), 6.34 (d, J = 15.6 Hz, 2H, -CH-), 5.69 (s, 1H, -CO-CH-CO-), 3.77 (s, 6H, CH3).
1-(4-Hydroxy-3-methoxy-phenyl)-7-(4-hydroxy-phenyl)-hepta-1,6-diene-3,5-dione (demethoxycurcumin), 4c was synthesized according to the general procedure described for compound 4a. An equimolar mixture of vanillin (0.5 g, 3.3 mmol) and 4-hydroxybenzaldehyde (0.4 g, 3.3 mmol)d was employed for the synthesis of compound 4c. Flash chromatography using MeOH/CH2Cl2 (4:96, v:v, Rf = 0.21) gave separation from the other curcumin (curcumin and bisdemethoxycurcumin) derivatives formed in the reaction. A second chromatography using hexane/EtOAc (2:1, v:v, Rf = 0.17) afforded the pure demethoxycurcumin 4c as a yellow powder (17 mg). ESI-MS m/z 339 (MH+), 337 (MH−); 1H NMR (CD3OD) δ7.59 (m, 2H), 7.49 (d, J = 8.1 Hz, 2H), 7.22 (m, 1H), 7.11 (m, 1H), 6.82 (m, 3H), 6.61 (d, J = 8.1 Hz, 2H), 5.96 (s, 1H), 3.92 (s, 3H).
Assay of curcuminoid extract for curcuminoid content
The curcuminoid extract was initially prepared by completely dissolving 11 mg of powder in 20 mL of pure acetonitrile. This was prepared further for HPLC analysis by diluting 400 μL of this solution with 600 μL 10 mM ammonium acetate, pH 4.2, and 20 μL of 500 μM phenacetin (internal standard) in methanol. The absolute and relative concentrations of individual curcuminoids in this solution were then determined by reference to synthetic curcuminoids prepared for HPLC in a similar fashion. Details of the HPLC method used to separate and quantitate each of the curcuminoids are given in Table 1.
Table 1.
Details of analytical methods.
| Assay | Mobile Phase | Gradient Profile | Detection Method | Retention Times |
|---|---|---|---|---|
| Curcuminoids | 10 mM ammonium acetate (pH 4.2) | 40% CH3CN (0–20 min) → 75% CH3CN (25 min) →40% CH3CN (26–28 min) | Curcuminoids: 425 nm Phenacetin: 254 nm | Curcumin: 25.2 min Demethoxy: 24.6 min Bisdemethoxy: 23.6 min Phenacetin (IS): 3.6 min |
| Triazolam | 10 mM phosphate buffer (pH 7.0) | 25% CH3CN/ 15% CH3OH (0–17 min) → 32% CH3CN/ 20% CH3OH (18 min) → 45% CH3CN/ 20% CH3OH (25 min) → 25% CH3CN/ 15% CH3OH (27–32 min) | 220 nm | 1′-OH-triazolam: 15.2 min 4-OH-triazolam:16.7 min Diazepam (IS): 26.5 min |
| Flurbiprofen | 20 mM phosphate buffer (pH 2.2) | 35% CH3CN (0 min) → 50% CH3CN (5 min) → 75% CH3CN (15 min) → 35%CH3CN (17–20 min) | 4′-OH-flurbiprofen: ex 260 nm, em 320 nm Phenacetin: 220 nm | 4′-OH-flurbiprofen: 8.9 min Phenacetin (IS): 6.5 min |
| S-mephenytoin | 0.5% formic acid | 10% CH3OH (0 min) → 80% CH3OH (4 min) → 10% CH3OH (4.1–12 min) | 4-OH-mephenytoin (MS-MS): 235.5 (width 5) → 150.2 Diazepam (MS): 285.3 | 4-OH-mephenytoin: 3.7 min Diazepam (IS): 5.4 min |
| Dextromethorphan | 20 mM phosphate buffer (pH 2.2) | 20% CH3CN (0–5 min) → 60% CH3CN(14 min) →20% CH3CN (15–18 min) | ex 280 nm, em 310 nm | Dextrorphan: 7.8 min Pronethalol (IS): 12.1 min |
| Phenacetin | 0.5% formic acid | 0% CH3CN (0 min) → 50% CH3CN (8 min) → 0% CH3CN (8.1–12 min) | APAP (+, MS-MS): 152.1 (1.0 width) → 110.0 Sulfaphenazole (+): 315.3 | APAP: 2.9 min Sulfaphenazole (IS): 6.6 min |
| Chlorzoxazone | 20 mM phosphate buffer (pH 2.2) | 18% CH3CN (0 min) → 50% CH3CN (15 min) → 18% CH3CN (16–20 min) | 295 nm | 6-OH-chlorzoxazone: 8.8 min Phenacetin (IS): 12.8 min |
| Bupropion | 0.02% TFA | 5% CH3CN (0 min) → 60% CH3CN (5 min)→5% CH3CN (5.2–10 min) | Hydroxybupropion (+, MS- MS): 256.0 (2.0 width) → 238.0 Trazodone (+): 372.6 | Hydroxybupropion: 2.1 min Trazodone (IS): 4.1 min |
| Acetaminophen (+ Mode) | 10 mM Ammonium Acetate (pH 4.2) | 0% CH3CN (0–4 min) → 50% CH3CN(14 min) →0% CH3CN (14.1–23 min) | APAP-glucuronide (MS-MS): 345.2 (10 width) → 152.2 Sulfaphenazole: 315.3 | APAP-glucuronide: 1.4 min Sulfaphenazole (IS): 10.7 min |
| Acetaminophen (− Mode) | 10 mM Ammonium Acetate (pH 4.2) | 0% CH3CN (0 min) → 50% CH3CN (10 min) → 0% (10.1–15 min) | APAP-sulfate (MS-MS): 230.2 (10 width) → 150.2 Sulfaphenazole: 313.3 | APAP-sulfate: 2.9 min Sulfaphenazole (IS): 7.1 min |
IS: internal standard, ex: excitation; em: emission
Acetaminophen UGT and SULT activities in LS180 cells
LS180 cells were seeded into 24-well plates at 175,000 cells per well. The cells were incubated in 10 mM acetaminophen alone (including DSMO vehicle at 0.5% final concentration) or combined with varying concentrations of curcuminoids (0.19 μM-200 μM) or piperine (0.05–50 μM). After 24 hr, cells and media were collected, and protein precipitated by adding an equal volume (1 mL) of a 5% acetic acid/95% acetonitrile solution that included sulfaphenazole (200 μM) as an internal standard. After mixing and then centrifuging at 13,000 g for 5 min, the supernatant was evaporated in a vacuum oven set at 40°C, and then re-suspended in mobile phase for LC-MS analysis (see below). Cell viability was also measured using a trypan blue exclusion assay to determine possible treatment effects.
Human liver tissue fraction preparation
Microsomes were prepared by ultracentrifugation as described in detail previously (von Moltke et al., 1993). Human liver tissue was acquired from the International Institute for the Advancement of Medicine (Exton, PA), the Liver Tissue Procurement and Distribution System, University of Minnesota (Minneapolis, MN), or the National Disease Research Interchange (Philadelphia, PA). All livers were either intended for transplantation but failed to tissue match or were normal tissue adjacent to surgical biopsies. Microsomal pellets were suspended in 100 mM potassium phosphate buffer containing 20% glycerol and stored at −80°C until use. Liver quality was assessed by systematically comparing enzyme activity results for individual livers to the median activity value for all livers (n=54 total). Livers that consistently showed low activities (<30% of median) for all activities (over 10 different CYP and UGT activities) were excluded from use in these studies. Donor characteristics of each of the livers have been reported previously (Hesse et al., 2004). Liver microsomes from 50 different individuals were pooled for UGT activity assays. Liver microsomes prepared individually from three individual donors with known high activities (from previous work) for the respective CYP were chosen as follows: CYP3A (LV41, LV49, LV50); CYP2C9 (LV8, LV13, LV14); CYP2D6 (LV4, LV10, LV15); CYP2C19 (LV8, LV12, LV14); CYP1A2 (LV8, LV28, LV29); CYP2E1 (LV10, LV13, LV14); and CYP2B6 (LV39, LV43, LV50).
Pooled cytosol-enriched fractions were prepared from four livers (LV39, LV41, LV48, and LV49) by differential centrifugation using a protocol as previously described with minor modifications (Wang et al., 2004). Briefly, liver tissue was diced with a clean razor on ice and immediately added to 4 volumes of ice-cold 1.15% KCl dissolved in 50 mM potassium phosphate buffer (pH 7.4). The tissue was homogenized using a glass-Teflon homogenizer, centrifuged at 11,000 g for 15 min at 4°C, and the resulting supernatant centrifuged at 110,000 g for 1 hr. The high speed supernatant (cytosol enriched fraction) was assayed for protein content (bicinchoninic method), diluted to 15 mg/mL in 50 mM phosphate buffer (pH 7.4) including 5% glycerol, and stored at −80°C until use.
CYP activities
Incubation conditions for human liver microsomes and recombinant CYP3A4 and CYP3A5 were similar to those used previously unless otherwise noted (von Moltke et al., 2002; Patki et al., 2003). Briefly, for IC50 determinations, triazolam (250 μM, CYP3A), flurbiprofen (5 μM, CYP2C9), dextromethorphan (25 μM, CYP2D6), phenacetin (100 μM, CYP1A2), chlorzoxazone (50 μM, CYP2E1), S-mephenytoin (25 μM, CYP2C19), or bupropion (50 μM, CYP2B6) dissolved in methanol were added to a series of incubation tubes and the solvent removed by vacuum. For Ki determinations, triazolam concentrations ranged from 50 to 1000 μM, flurbiprofen concentrations ranged from 1 to 20 μM, and for S-mephenytoin concentrations ranged from 5 to 100 μM. Various concentrations of curcuminoids (0.19–200 μM) or piperine (0.05–50 μM) dissolved in DMSO (0.25% final concentration for CYP3A, CYP2D6, CYP2B6, and CYP1A2 assays), acetone (0.25% final concentration for CYP2C9 and CYP2C19 assays), or methanol (1% final concentration for CYP2E1 assay) were added directly to the incubation mixture. All dose responses for determining IC50 values included 12 concentrations including 0 μM except for time-dependent inhibition experiments which contained 6 concentrations surrounding the expected IC50 value. Incubation mixtures contain 50 mM phosphate buffer, 5 mM MgCl2, 0.5 mM NADP+, and an isocitrate/isocitric dehydrogenase regenerating system. Reactions were initiated with 0.25 mg/mL microsomal protein or 0.125 mg/mL recombinant protein and incubated at 37°C. All CYP activity experiments were performed as independent experiments with HLM from three different individuals as described in the “Human liver tissue fraction preparation” section. Reactions were terminated with addition of 80 μL (200 μL for CYP3A assay, acetonitrile only) acetonitrile containing 5% acetic acid and cooled on ice at 20 min (CYP3A, CYP2C9, CYP2D6, CYP2B6, CYP1A2, or CYP2E1) or 60 min (CYP2C19) after initiation of the reaction. Internal standard was added, the mixture centrifuged (13,000 g for 5 min), and the supernatant was either transferred directly to a sample vial (CYP3A, CYP2C9, and CYP2D6) or dried-down by vacuum oven and re-dissolved in mobile phase (CYP2B6, CYP1A2, CYP2E1, or CYP2C19) for HPLC-UV/FL or LC-MS analysis (described below).
Acetaminophen glucuronidation activities
Glucuronidation activities were measured by a method similar to that used previously unless otherwise noted (Court et al., 2001). Briefly, incubation buffer containing 50 mM phosphate buffer, pH 7.5, with 5 mM MgCl2, 20 mM UDPGA, and 0.05 mg/mL alamethicin was added to 10 mM acetaminophen that had been previously evaporated by vacuum. Vehicle alone (0.5% DMSO final concentration) or various concentrations of curcuminoids (0.19–200 μM) or piperine (0.05–50 μM) were added directly to the incubation mixture. Microsomes were added for a final concentration of 0.5 mg/mL with a final assay volume of 100 μL. Incubation time was 3 hr at 37°C with the reaction terminated by the addition of 50 μL of 5% acetic acid in acetonitrile with 200 μM sulfaphenazole as the internal standard. Under these conditions, product formation was linear with respect to time and protein concentration for up to 4 hr and up to 1 mg/ml protein, respectively. Samples were mixed and centrifuged at 13,000 g for 5 min. The resulting supernatant was removed, evaporated by vacuum, and reconstituted with 100 μL of water for LC-MS analysis (see below).
Acetaminophen sulfotransferase activities
Acetaminophen sulfation was assayed according to the method described by Wang et al. (2004) with minor changes. Briefly, incubation buffer containing 50 mM phosphate buffer (pH 7.5) and 100 μM PAPS was added to 10 mM acetaminophen that had been previously evaporated in a vacuum oven. Vehicle alone (0.5% DMSO final concentration) or various concentrations of curcuminoids (0.19 μM-200 μM) were added directly to the incubation mixture. Human liver cytosol was added (final protein concentration of 0.5 or 2 mg/mL) with a final incubation volume of 200 μL. The sulfation reaction was allowed to proceed for 3 hr at 37°C, and the reaction was terminated by the addition of 300 μL of 5% acetic acid in acetonitrile with 200 μM sulfaphenazole as the internal standard. Under these conditions, product formation was linear with respect to time and protein concentration for up to 6 hr and up to 4 mg/ml protein, respectively. Samples were mixed and then centrifuged at 13,000 g for 5 min. The resulting supernatant was removed, evaporated by vacuum, and reconstituted with 100 μL of mobile phase for LC-MS analysis.
Analysis of samples
All HPLC-UV/FL measurements were performed with an HPLC system (Model 1100, Agilent, Palto Alto, CA) using a 250 × 4.6-mm C18 column (Synergi Hydro-RP, Phenomenex, Torrance, CA) serially connected to a UV absorbance diode array detector and a fluorescence detector. LC-MS was done with an HPLC instrument (Surveyor, Thermofinnigan, Somerset, NJ) using a 150 × 3 mm column (Synergi Fusion-RP, Phenomenex) connected serially to an ion-trap mass detector (LCQ Deca XP Max, Thermofinnigan) with either electrospray ionization source (most assays) or atmospheric pressure chemical ionization source (S-mephenytoin hydroxylation assay). Details for each method are given in Table 1. Flow rates were set at 1 mL/min for all HPLC-UV/FL measurements and at 400 μL/min for S-mephenytoin and bupropion LC-MS measurements and at 500 μL/min for phenacetin and acetaminophen (APAP) LC-MS measurements. In addition, positive ions were monitored in all LC-MS methods except acetaminophen sulfate and its internal standard sulfaphenazole, which required negative ion monitoring.
Data analysis
IC50 values (inhibitor concentration resulting in 50% decrease in enzyme activity) and CC50 values (compound concentration resulting in 50% decrease in cell viability) were calculated using Prism Software (San Diego, CA). Ki values were also determined for selected activities by nonlinear regression analysis of untransformed data to the mixed competitive-noncompetitive, noncompetitive, or competitive inhibition models using SigmaPlot (San Jose, CA). Goodness of fit critieria (R2 and Akaike’s Information Criterion) were used to determine the inhibition model that best described the data. Results are presented as the mean of triplicate determinations ± standard error unless described otherwise.
Results
Curcumin, demethoxycurcumin, and bisdemethoxycurcumin content of the curcuminoid extract
HPLC-UV analysis of the curcuminoid extract showed the content of individual curcuminoids to be 66%, 23%, and 11% for curcumin, demethoxycurcumin, and bisdemethoxycurcumin, respectively, with these curcuminoids representing essentially 100% of the powder by weight. This was in close agreement with the manufacturer’s product data sheet that listed curcumin content of 65 to 70% with the major impurities being demethoxycurcumin and bisdemethoxycurcumin.
Inhibition of CYP activities in HLMs by curcuminoids and piperine
We first tested the inhibitory effects of the curcuminoid extract on CYP-specific functional activities for CYP3A (triazolam hydroxylation), CYP2C9 (flurbiprofen hydroxylation), CYP2C19 (S-mephenytoin hydroxylation), CYP1A2 (phenacetin de-ethylation), CYP2D6 (dextromethorphan demethylation), CYP2E1 (chlorzoxazone hydroxylation), and CYP2B6 (bupropion hydroxylation) in human liver microsomes. Moderate to potent inhibition (IC50 < 50 μM) was observed with the curcuminoid extract for most CYP isoforms with a relative order of inhibitory potency of CYP2C19 > CYP2B6 > CYP2C9 > CYP3A (Table 2). Relatively weak inhibition (IC50 > 60 μM) was observed for CYP2D6 and CYP1A2, while less than 20% inhibition was observed for CYP2E1 activity at the highest concentration evaluated (200 μM). Inhibition of CYP3A by the curcuminoid extract was best described by the competitive inhibition model with a Ki value of 11.0 ± 1.3 μM for triazolam 1′-hydroxylation. In contrast, inhibition by the curcuminoid extract of both CYP2C9 and CYP2C19 were best described by mixed competitive-noncompetitive inhibition models (α values of 2.5 for CYP2C9 and 2.8 for CYP2C19) with Ki values of 10.6 μ 1.1 μM and 7.8 ± 0.9 μM, respectively.
Table 2. IC50 values for inhibition by curcuminoid extract and piperine of CYP selective activities measured in human liver microsomes.
Functional activities evaluated included triazolam 1′-hydroxylation (CYP3A), flurbiprofen hydroxylation (CYP2C9), S-mephenytoin hydroxylation (CYP2C19), phenacetin de-ethylation (CYP1A2), dextromethorphan demethylation (CYP2D6), chlorzoxazone hydroxylation (CYP2E1), and bupropion hydroxylation (CYP2B6). IC50 values were generated by nonlinear regression curve fitting using Prism software. Also shown are IC50 values for control inhibitors that are known to be relatively specific for each of the CYP isoforms evaluated measured in the same set of human liver microsomes. Shown are the mean ± standard errors of triplicate determinations using HLM from three different individuals.
| Isozyme | IC50 Curcuminoid Extract (μM) | IC50 Piperine (μM) | IC50 Control Inhibitor (μM) | Control Inhibitor |
|---|---|---|---|---|
| CYP3A | 25.3 ± 1.3 | 5.5 ± 0.7 | 0.10 ± 0.04 | Ketoconazole |
| CYP2C9 | 13.5 ± 1.4 | 40.7 ± 4.1 | 0.44 ± 0.03 | Sulfaphenazole |
| CYP2D6 | 63.6 ± 4.8 | >50 | 0.13 ± 0.01 | Quinidine |
| CYP2C19 | 7.4 ± 1.2 | >50 | 6.5 ± 1.3 | Omeprazole |
| CYP1A2 | 95.4 ± 17.1 | 29.8 ± 3.6 | 0.10 ± 0.02 | α-Naphthoflavone |
| CYP2E1 | >200 | >50 | 5.8 ± 0.8 | Diethyldithiocarbamate |
| CYP2B6 | 9.4 ± 1.9 | >50 | 5.0 ± 0.5 | thio-TEPA |
In contrast, piperine inhibited only 3 of the 7 CYP activities evaluated to any significant extent (IC50 < 50 μM), with highest potency observed for the CYP3A activity, that showed over 5 times lower IC50 values than the CYP1A2 and CYP2C9 activities (Table 2). Less than 50% inhibition was observed for the remaining CYP activities at the highest piperine concentration evaluated (50 μM). Inhibition of CYP3A by piperine was best described by the non-competitive inhibition model with a Ki value of 5.4 ± 0.3 μM for triazolam 1′-hydroxylation
Inhibition of recombinant CYP3A4 and CYP3A5 by curcuminoids and piperine
Because both piperine and curcuminoid extract inhibited the metabolism of triazolam in human liver microsomes, and triazolam is metabolized by both of the major human adult CYP3A isoforms (CYP3A4 and CYP3A5), we further evaluated the effect of curcuminoid extract and piperine using recombinant enzymes (rCYP3A4 and rCYP3A5). We found that rCYP3A4 was much more sensitive to inhibition than rCYP3A5 with the curcuminoid extract displaying IC50 values of 20.3 ± 0.7 μM and >200 μM for triazolam 1′-hydroxylation by rCYP3A4 and rCYP3A5, respectively (Fig. 2A). Similarly, piperine had IC50 values of 8.0 ± 1.8 μM and >50 μM for triazolam 1′-hydroxylation by CYP3A4 and CYP3A5, respectively (Fig. 2B).
Figure 2. Concentration-dependent inhibition by curcuminoid extract (A) and piperine (B) of triazolam 1′-hydroxylation by recombinant CYP3A4 and CYP3A5.
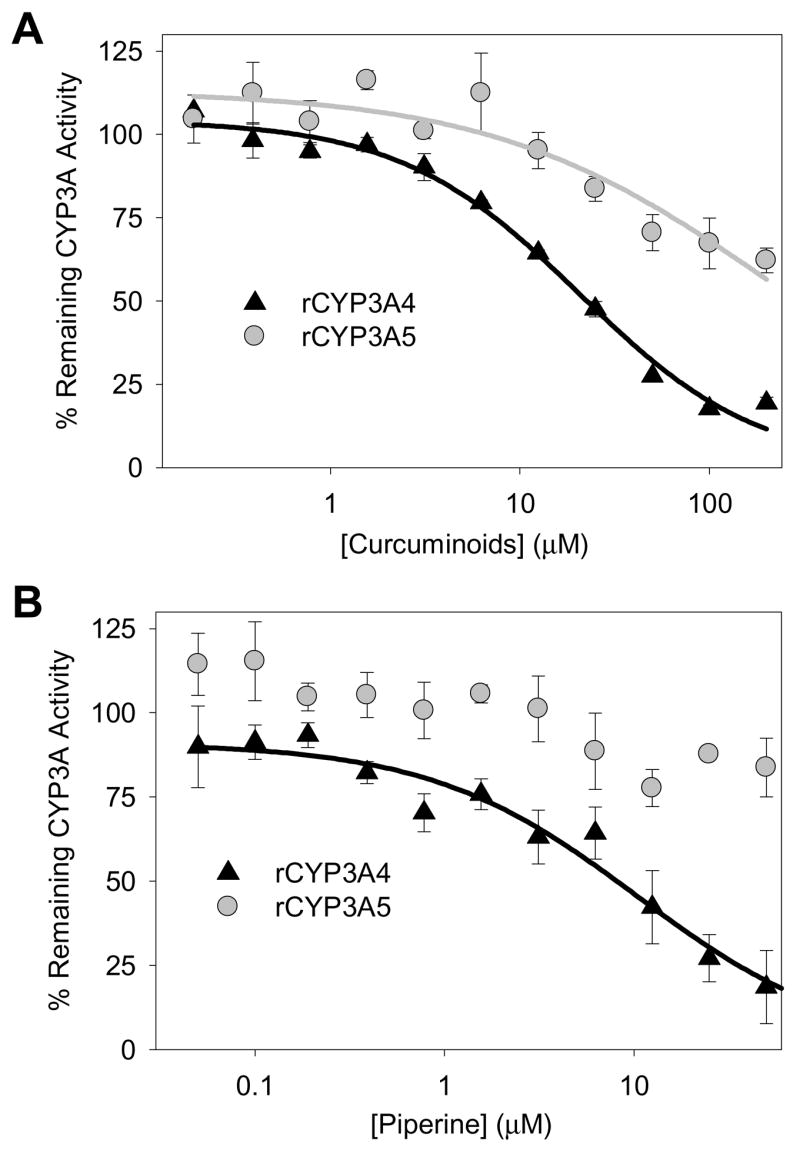
Methods section provides detailed assay information. Triazolam 4-hydroxylation data (provided by the same assay) was essentially the same. Data points correspond to the mean ± standard error of triplicate determinations in a single experiment.
Inhibition of CYP3A and CYP2C9 activities in HLMs by synthetic curcuminoids
The ihibition of CYP3A and CYP2C9 activities was further evaluated using pure synthetic curcumin, demethoxycurcumin, and bisdemethoxycurcumin. Individual synthetic curcuminoids as well as a mixture of the 3 compounds (referred to as the “curcuminoid mixture”) were studied. The curcuminoid mixture contained pure curcumin, demethoxycurcumin, and bisdemethoxycurcumin in proportions similar to that found in the curcuminoid extract (70:20:10 by weight, respectively). The effect on CYP3A activities were similar for the individual curcuminoids in comparison with the curcuminoid mixture, and also the curcuminoid extract with IC50 values ranging from 32.1 to 58.0 μM (Table 3). In contrast, there were substantial differences in IC50 values for inhibition of CYP2C9 activities for the different curcuminoid preparations in that demethoxycurcumin showed potent inhibition with an IC50 value of 8.8 ± 1.2 μM, while both curcumin and bisdemethoxycurcumin showed only minimal inhibition (61 and 72%, respectively) at the highest concentration evaluated (i.e., 50 μM) (Table 3). The curcuminoid mixture had an intermediate IC50 value for inhibition of CYP2C9 activity of 36.3 ± 3.4 μM.
Table 3. IC50 values for inhibition by a curcuminoid extract, curcumin, demethoxycurcumin, bisdemethoxycurcumin, and a curcuminoid mixture of CYP3A, CYP2C9, and UGT activities in human liver microsomes (HLM) or intact LS180 cells and SULT activity in human liver cytosol (HLC) or intact LS180 cells.
Functional activities evaluated included triazolam 1′-hydroxylation (CYP3A), flurbiprofen hydroxylation (CYP2C9), acetaminophen glucuronidation (UGT), and acetaminophen sulfation (SULT). IC50 values from experiments determining the effect of 20 minute preincubation on the extent of inhibition of CYP3A-mediated triazolam 1-hydroxylation by curcuminoids are also shown. Shown are the mean ± standard errors of triplicate determinations.
| Enzyme | Model System | IC50 Curcuminoid Extract (μM) | IC50 Pure Curcumin (μM) | IC50 Pure Demethoxycurcumin (μM) | IC50 Pure Bis- demethoxycurcumin (μM) | IC50 Curcuminoid Mixture (μM) |
|---|---|---|---|---|---|---|
| CYP3A | HLM(No Preincubation) | 32.1 ± 0.6 | 58.0 ± 3.5 | 43.5 ± 3.8 | 41.7 ± 3.5 | 36.4 ± 3.8 |
| CYP3A | HLM(Preincubation) | 24.1 ± 2.0 | 47.1 ± 2.0 | 31.5 ± 0.5 | 35.3 ± 2.7 | ND |
| CYP2C9 | HLM | 13.5 ± 1.4 | >50 | 8.8 ± 1.2 | >50 | 36.3 ± 3.4 |
| SULT | HLC | 0.99 ± 0.04 | 5.9 ± 0.4 | 1.70 ± 0.06 | 0.95 ± 0.02 | 1.36 ± 0.06 |
| SULT | LS180 cells | 5.2 ± 0.6 | 2.6 ± 0.4 | 12.5 ± 0.4 | 5.6 ± 0.8 | 9.6 ± 1.4 |
| UGT | HLM | 133.5 ± 17.9 | ND | ND | ND | ND |
| UGT | LS180 cells | 12.1 ± 0.4 | 2.2 ± 0.1 | 13.9 ±2.3 | 9.8 ± 1.4 | 12.2 ± 1.8 |
ND: not determined
Effect of preincubation on inhibition by curcuminoid extract and piperine
The effect of a 20 min preincubation (i.e., inhibitor incubated for 20 minutes with microsomes, MgCl2, and NADPH regenerating system prior to substrate addition) was evaluated for the curcuminoid extract (CYP3A, 2C9 and 2B6 activities) and piperine (CYP3A activity only) over a range of inhibitor concentrations. As shown in Fig. 3A, preincubation enhanced the effect of the curcuminoid extract on CYP3A activity at inhibitor concentrations over 25 μM. In contrast, there was no enhancement of inhibition of CYP2C9 or CYP2B6 activities by preincubation of the curcuminoid mixture or inhibition of CYP3A activities by piperine (Figs. 3B-D). The effect of time and inhibitor concentration on CYP3A inhibition by curcuminoid extract were further explored at a curcuminoid extract concentration of 12.5 μM and 50 μM with preincubation for 0, 20, 40, and 60 min (Fig. 4). Again, there was no effect of preincubation on the degree of inhibition of CYP3A activity with the 12.5 μM curcuminoid extract concentration, while 50 μM resulted in enhanced inhibition following 20, 40, and 60 min preincubation compared with control samples that were not preincubated.
Figure 3. Effect of 20 minute preincubation (with inhibitor and NADPH regeneration system) on the extent of inhibition of CYP3A-mediated triazolam 1′-hydroxylation by curcuminoid extract (A) and piperine (B), of CYP2C9-mediated flurbiprofen hydroxylation by curcuminoid extract (C), and of CYP2B6-mediated bupropion hydroxylation by curcuminoid extract (D) measured in human liver microsomes.

Assay details are provided in the text. Data points correspond to the mean ± standard error of triplicate determinations using HLM from three different individuals.
Figure 4. Effect of duration of preincubation (0, 20, 40, and 60 min with inhibitor and NADPH regeneration system) on the extent of inhibition of CYP3A-mediated triazolam 1′-and 4-hydroxylation by 12.5μM (A) and 50μM (B) curcuminoid extract in human liver microsomes.

Assay details are provided in the text. Column heights correspond to the mean ± standard error of triplicate determinations using HLM from three different individuals.
Time-dependent inhibition of CYP3A activity by curcuminoids was further evaluated using synthetic curcumin, demethoxycurcumin, and bisdemethoxycurcumin. As shown in Figs. 5A-C, small leftward shifts in IC50 curves were observed for all three compounds following 20 min preincubation, although the effect was more pronounced for curcumin (58.0 ± 2.0 μM vs. 47.1 ± 2.0 μM with preincubation) and demethoxycurcumin (43.5 ± 3.8 μM vs. 31.5 ± 0.5 μM with preincubation) compared with bisdemethoxycurcumin (41.8 ± 3.5 μM vs. 35.3 ± 2.7 μM with preincubation). However, for all three compounds, the relative shift (less than 30% decrease in IC50) was notably less than that observed for troleandomycin, a well-characterized mechanism-based CYP3A inhibitor, that showed about a 60% decrease in IC50 with a 20 min preincubation (1.89 ± 0.13 μM vs. 0.59 ± 0.03 μM with preincubation, Fig. 5D).
Figure 5. Effect of 20 minute preincubation (with inhibitor and NADPH regeneration system) on the extent of inhibition of CYP3A-mediated triazolam 1′-hydroxylation by curcumin (A), demethoxycurcumin (B), bisdemethoxycurcumin (C), and troleandomycin (D, control CYP3A inhibitor) in human liver microsomes.

Assay details are provided in the text. Data points correspond to the mean ± standard error of triplicate determinations using HLM from three different individuals.
Inhibition of acetaminophen glucuronidation by curcuminoids, piperine, and calphostin-C
Inhibition of acetaminophen glucuronidation by the curcuminoid mixture was evaluated using human liver microsomes, intact LS180 cells, and LS180 cell homogenates. In preliminary experiments, intact LS180 colon cells were treated with 50 μM curcuminoid extract and acetaminophen glucuronidation activities were measured in washed cell homogenates collected for up to 24 hr after treatment. As shown in Fig. 6 there was time-dependent inhibition with a maximum effect (86% decrease in glucuronidation activity) observed at 1 hr after treatment and complete restoration of baseline activity by 24 hr. The restoration of glucuronidation activity was closely paralleled by a decrease in curcuminoid concentration detected in the media by HPLC over the same 24 hr time period (Fig. 6). Concentrations of each individual curcuminoid decreased at similar rates (data not shown).
Figure 6. Time course of inhibition of acetaminophen glucuronidation in cultured LS180 cells treated with 50 μM curcuminoid extract.
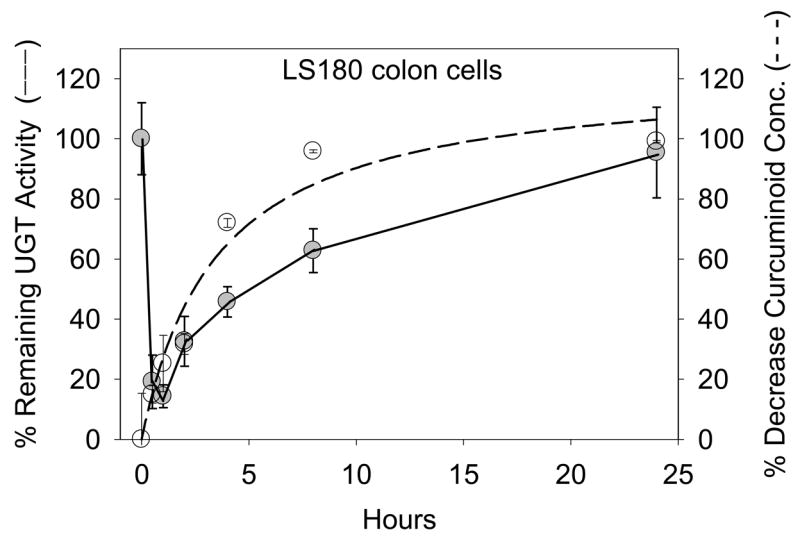
LS180 cells were collected at various times after 50μM curcuminoid treatment, washed, homogenized, and incubated with 10mM acetaminophen for 3hrs in the presence of UDPGA, MgCl2, and alamethicin. Percent UGT activity (−) was calculated compared to DMSO (0.25%) treated controls. Curcuminoid concentration (sum of curcumin, demethoxycurcumin, and bisdemethoxycurcumin) in the media was also monitored over the same time span and is shown as a percent decrease from the initial concentration (- - -). Data points correspond to the mean ± standard error of triplicate determinations in a single experiment.
The effect of curcuminoid extract concentration on acetaminophen glucuronidation by intact LS180 cells was evaluated by incubating cells for 24 hr in the presence of acetaminophen with increasing curcuminoid extract concentrations. This treatment duration was determined as the minimum necessary to allow accurate measurement of decreases in acetaminophen metabolite formation by cells using the LC-MS assay technique. Results were compared to effects on acetaminophen glucuronidation by LS180 cell homogenates and human liver microsomes. As shown in Fig. 7A, the curcuminoid extract showed the greatest inhibition potency (lowest IC50 values) in intact LS180 cells (12.1 ± 0.4 μM) compared with LS180 cell homogenates (60.9 ± 12.2 μM) and human liver microsomes (133.5 ± 17.9 μM). Evaluation of cell toxicity effects by trypan blue exclusion assay showed that the concentration of curcuminoid extract that resulted in a 50% decrease in cell viability (CC50) was 77 ± 5 μM in the presence of 10 mM acetaminophen, and 94 ± 6 μM in the absence of acetaminophen, which was more than 6 times the IC50 of the curcuminoid extract in intact cells.
Figure 7. Concentration-dependent inhibition of acetaminophen glucuronidation by curcuminoid extract (A) and calphostin-C (B) in human liver microsomes and LS180 cells and of acetaminophen sulfation by curcuminoid extract (C) in human liver cytosol and LS180 cells.
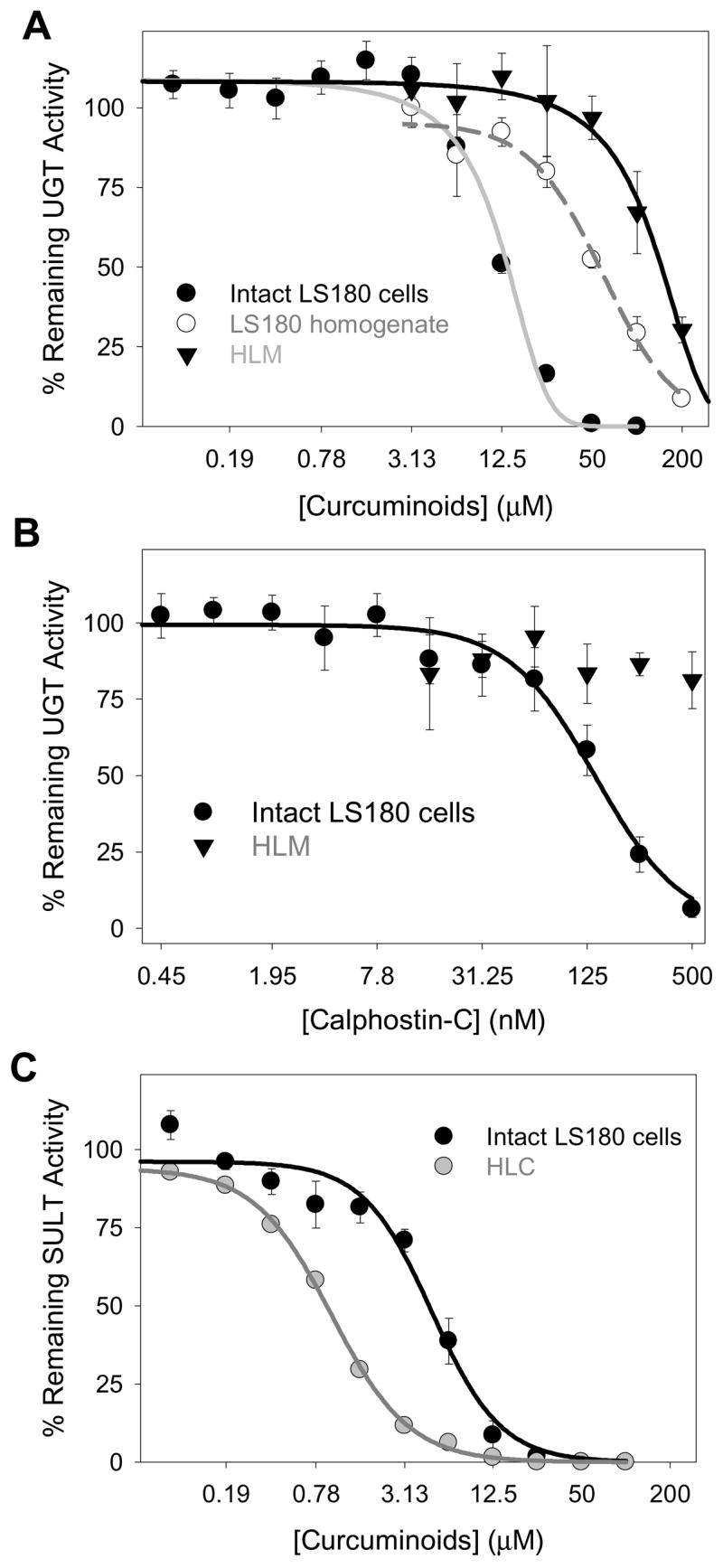
In panel A, inhibition of acetaminophen glucuronidation by curcuminoid extract was compared between intact LS180 cells, homogenates of (washed and untreated) LS180 cells, and human liver microsomes. In panel B, the effect of a selective PKC inhibitor, calphostin-C, on acetaminophen glucuronidation was evaluated using both intact LS180 cells and human liver microsomes. In panel C, inhibition of acetaminophen sulfation by curcuminoid extract was compared between intact LS180 cells and human liver cytosol. Methods section provides detailed assay information. Data points correspond to the mean ± standard error of at least three determinations. LS180 experiments were repeated on three different days.
Inhibition of acetaminophen glucuronidation by intact LS180 cells was also determined using the individual synthetic curcuminoids and a mixture of these compounds. As shown in Table 3, the effect of the individual synthetic curcuminoids on glucuronidation was similar to that of the curcuminoid mixture and curcuminoid extract (IC50 values ranged from 9.8 to 12.2 μM) except for curcumin, which showed a much lower IC50 value of 2.2 ± 0.1 μM.
In contrast to the curcuminoids, piperine had no effect on acetaminophen glucuronidation by LS180 cells (intact cells or homogenates) or by human liver microsomes at concentrations up to 50 μM piperine.
In order to further explore the mechanism of curcuminoid inhibition of acetaminophen glucuronidation in LS180 cells, calphostin-C, a selective PKC inhibitor, was evaluated both in human liver microsomes and intact LS180 cells. Calphostin-C was a potent inhibitor of acetaminophen glucuronidation in the intact LS180 cells with an IC50 of 140 ± 30 nM, but had much less of an effect on glucuronidation in HLM with essentially no inhibition observed at 500 nM (Fig. 7B).
Inhibition of acetaminophen sulfation by curcuminoids and piperine
The effects of the curcuminoid extract and piperine on acetaminophen sulfation were evaluated using intact LS180 cells and human liver cytosol. In contrast to the effects on glucuronidation where greater inhibition was observed in intact cells versus liver fractions, curcuminoid extract resulted in more than 4-fold greater inhibition of acetaminophen sulfation in liver fractions compared with intact LS180 cells with IC50 values of 5.2 ± 0.6 μM and 0.99 ± 0.04 μM, respectively (Fig. 7C). Evaluation of effects of individual synthetic curcuminoids using human liver cytosol showed substantial inhibition of sulfation with all compounds (IC50 <10 μM), although somewhat less with curcumin (IC50 = 5.9 ± 0.4 μM) compared with the other curcuminoids, the synthetic curcuminoid mixture, and the curcuminoid extract (IC50 values less than 2 μM) (Table 3). This trend was reversed with intact LS180 cells (Table 3), in that the IC50 value for curcumin was somewhat lower (2.6 ± 0.4 μM) compared with the other synthetic curcuminoids, curcuminoid mixture and curcuminoid extract (5.6 to 9.6 μM). Also noteworthy was that (except for curcumin) IC50 values for inhibition of acetaminophen sulfation by curcuminoids were consistently lower (i.e., by about 80%) for human liver cytosol compared with intact LS180 cells (Table 3).
Similar to the previously observed lack of effect on acetaminophen glucuronidation, piperine at concentrations of up to 50 μM showed no inhibition of acetaminophen sulfation in either human liver cytosol or intact LS180 cells.
Effect of piperine on inhibition of acetaminophen glucuronidation and sulfation by curcuminoid extract
IC50 values for inhibition of acetaminophen sulfation and glucuronidation by curcuminoid extract in intact LS180 cells were unchanged by the addition of 50 μM piperine to the culture medium. IC50 values for curcuminoid extract treatment alone for acetaminophen glucuronidation and sulfation were 10.0 ± 0.7 μM and 4.3 ± 0.5 μM (mean ± standard error of triplicate determinations from one experiment), respectively, while IC50 values for curcuminoid extract treatment plus 50 μM piperine were 11.0 ± 1.2 μM and 4.1 ± 1.0 μM, respectively.
Discussion
The most significant and novel finding of this study is that curcuminoid extract is a moderately potent inhibitor of a substantial number of the major human drug metabolizing CYP enzymes. The rank order of potency of CYP inhibition by curcuminoid extract was CYP2C19 > CYP2B6 > CYP2C9 > CYP3A while the remaining CYPs were less susceptible to inhibition (IC50 > 60 μM). In contrast, piperine was a relatively selective inhibitor of CYP3A. These results confirm and extend upon a previous observation that piperine is a relatively potent inhibitor of CYP3A-mediated verapamil metabolism (Bhardwaj et al., 2002). Both curcuminoid extract and piperine were also shown to inhibit CYP3A4 much more potently (i.e., 5–10 fold) than CYP3A5. This differential inhibition (CYP3A4≫CYP3A5) has been shown for larger sets of compounds, although there is currently no known explanation (Patki et al., 2003; Soars et al., 2006; O’Donnell et al., 2007)
Inhibition of microsomal CYP3A activity by curcuminoids was both time-dependent (at substrate concentrations over 25 μM) and competitive. The observation of time dependency of inhibition at high curcuminoid concentrations but not at low concentrations is likely complex and might involve low affinity enzymatic production of curcuminoid metabolites with increased CYP3A inhibition potency or inhibitory reactive oxygen species. Although it is also possible that the curcuminoid extract might contain low amounts of another CYP3A inhibitor, this was ruled out by showing similar time-dependent inhibition by pure curcumin and demethoxycurcumin. It should be noted that the extent of the time-dependent inhibition potency shift was much less than that observed for the mechanism-based inhibitor troleandomycin as well as other known mechanism-based inhibitors (lopinavir, ritonavir, and amprenavir) tested in the same experimental system (von Moltke et al., 2000; Weemhoff et al., 2003). Preincubation with curcuminoid extract did not enhance inhibition of CYP2C9 and CYP2B6 activities. In fact, inhibition of CYP2B6 decreased with preincubation, which is consistent with consumptive depletion of the curcuminoid(s) to non-inhibitory metabolite(s) of CYP2B6.
Unlike CYP3A which was equally inhibited by all 3 curcuminoids, CYP2C9 was selectively inhibited by demethoxycurcumin. This difference may reflect the more restrictive substrate preference of CYP2C9 compared with CYP3A enzymes. We also found that the IC50 of the curcuminoid mixture was more than 2-fold higher compared with the curcuminoid extract. This might be explained by the presence of additional highly potent CYP2C9 inhibitor(s) in the curcuminoid extract (and not in the synthetic curcuminoids), although such compounds were not detected by the analytical methods utilized to determine curcuminoid purity.
Curcuminoids were also found to inhibit both UGT- and SULT-mediated metabolism. We found time-dependent inhibition of acetaminophen glucuronidation in intact LS180 cells with nearly 90% inhibition measured at 1hr (Fig. 6). This is similar to what has been reported previously for other UGT substrates (Basu et al., 2003; Basu et al., 2004; Basu et al., 2004). Loss of inhibition with culture time probably resulted from metabolism of curcuminoids by the cells because there was a parallel decrease in curcuminoids remaining in the media. Interestingly, the curcuminoid mixture more potently inhibited acetaminophen glucuronidation in intact LS180 cells compared to both the LS180 cell lysates and HLM (Fig. 7A). This result has several potential explanations including nonspecific cellular toxicity effects of curcuminoids, enhanced intracellular inhibitor concentration, or a specific but indirect effect on glucuronidation through inhibition of enzymes required for UGT phosphorylation as shown by Basu et al. (2003). Non-specific cell toxicity effects are not likely because trypan blue exclusion experiments showed that toxic effects only occurred at curcuminoid concentrations that greatly exceeded concentrations causing UGT inhibition. Accumulation of curcuminoids within the intact cells is also not likely since acetaminophen sulfation (measured simultaneously) was much less potently inhibited in intact cells as compared with cell fractions. A more likely explanation for differential inhibition potency between intact cell and cell fraction experiments is inhibition of UGT phosphorylation enzymes (probably protein kinase C) that are required for enzyme function. Calphostin, a selective protein kinase C inhibitor, inhibited acetaminophen glucuronidation by intact LS180 cells, but not by cell fractions. Evaluation of effects of the pure curcuminoids on LS180 cell glucuronidation indicated that curcumin is a more potent inhibitor than demethoxycurcumin or bisdemethoxycurcumin. While this might indicate that curcumin has more activity against PKC compared with the other curcuminoids, curcumin is more lipophilic than the other curcuminoids, so this effect might be the result of higher cell permeability of curcumin.
Of the activities evaluated, acetaminophen sulfation by HLC was most potently inhibited by curcuminoid extract with an IC50 value of 0.99 μM. Similar inhibition potency of curcuminoid extract has also been reported for sulfation of 4-nitrophenol by HLC with an IC50 value of 0.38 μM, although IC50 values as low as 14 nM have been reported for other substrates and tissues (Eaton et al., 1996; Vietri et al., 2003). Differences in inhibition potencies between the three pure curcuminoids were also observed with curcumin being the least potent inhibitor of HLC acetaminophen sulfation compared with the other curcuminoids, the pure mixture and the extract. In contrast, pure curcumin was somewhat more potent compared with other curcuminoids in inhibiting acetaminophen sulfation by intact LS180 cells perhaps reflecting differences in the SULT enzymes expressed in HLC and LS180 cells, or alternatively, differences in cellular permeability of the different curcuminoids.
Acetaminophen sulfation and glucuronidation activities were not affected by piperine at concentrations up to 50 μM. However, in rodents and human HT-29 colon adenocarcinoma cells, piperine has been shown to inhibit glucuronidation of EGCG and 3-OH-benzo(a)pyrene at concentrations as low as 8 μM (Atal et al., 1985; Lambert et al., 2004). There are several explanations for this difference including differences in UGT enzymes mediating EGCG and 3-OH-benzo(a)pyrene glucuronidation and the relatively high concentration of acetaminophen used (i.e., 10 mM, corresponding to predicted intestinal concentrations), which may have minimized competitive inhibition by piperine. We also performed a preliminary experiment to assess the effect of piperine on the inhibition of acetaminophen glucuronidation and sulfation by curcuminoid extract in LS180 cells. We observed no additional inhibition suggesting that at least for this model system, piperine does not enhance the potency of curcuminoid inhibition such as by enhancing curcuminoid cell penetration.
Pharmacokinetic studies in cancer patients and healthy volunteers have shown that at doses of 8 g per day of pure curcumin (99.3%), 3.6 g of curcuminoids, or piperine (20 mg) co-administered with 2 g of curcumin, the highest mean maximum plasma concentration (Cmax) was 1.77 ± 1.87 μM (Shoba et al., 1998; Cheng et al., 2001; Sharma et al., 2004). Because of low systemic availability, the most likely site for a metabolic interaction of curcuminoids with other drugs is in the intestine. We would predict that curcuminoids’ concentration in the proximal intestine lumen following a single dose of 3000 mg with a 1000 mL stomach volume will be approximately 10 mM, which is significantly higher than many of the IC50 values determined in this study. Piperine plasma levels have not been reported; however, we also expect some effects of piperine on drug metabolism in the intestine (approximately 80 μM concentration from a 20 mg dose and 1000 mL stomach volume) and perhaps also on the liver.
Also of importance in predicting possible intestinal interactions between curcuminoids and drug metabolizing enzymes is the relative intestinal expression of the intestinal enzymes. Based on studies quantifying CYP protein, the CYP3A subfamily enzymes and to a lesser extent CYP2C9 are the predominant CYP isozymes expressed in the intestine (Paine et al., 2006). Although multiple UGT and SULT isozymes have been identified in the intestinal mucosa by mRNA, protein, and activity assays, it is not yet clear which isozymes predominate, and to what extent intestinal glucuronidation and sulfation of drugs can affect drug bioavailability (Her et al., 1996; Gregory et al., 2004; Wang et al., 2006).
One issue relevant to predicting in vivo drug-herb interactions from in vitro data is non specific binding of inhibitor to microsomes, which if significant can lead to underestimation of inhibition potency. Indeed, there is some evidence for binding of curcuminoids to serum albumin (Barik et al., 2003). Although the experiments presented here utilized relatively low microsomal protein concentrations (0.5 mg protein/mL or less), which should have minimized such an effect, the extent of microsomal binding, of curcuminoids should be evaluated in future studies.
In conclusion, these data indicate that administration of a curcuminoid extract-piperine combination at doses routinely used in clinical studies could reach concentrations likely to inhibit CYP3A, CYP2C9, UGT, and SULT enzymes particularly in the intestine. Consequently, controlled drug interaction studies in human subjects are warranted to investigate the effect of curcuminoid and piperine combination on drugs metabolized by these enzymes.
Acknowledgments
We thank David J. Greenblatt and Richard I. Shader for their critical review of the manuscript and helpful suggestions.
This publication was made possible by Grant Number F31AT003973 from the National Center for Complementary and Alternative Medicine (NCCAM), National Institutes of Health (Bethesda, MD) to L.P.V. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NCCAM, or the National Institutes of Health. Other support was also provided by grants R01GM061834 and R21GM074369 from the National Institute of General Medical Sciences (NIGMS), National Institutes of Health (Bethesda, MD) to M.H.C. and a grant from the Alzheimer’s Drug Discovery Foundation (ADDF) (www.alzdiscovery.org.) to J.R.C. The ADDF catalyzes and funds drug discovery and drug development for Alzheimer’s disease and cognitive aging.
Nonstandard abbreviations
- UGT
UDP-glucuronosyltransferase
- UDPGA
UDP-glucuronic acid
- HLMs
human liver microsomes
- HLC
human liver cytosol
- APAP
acetaminophen
- CYP
cytochrome P450
- SULT
sulfotransferase
Footnotes
National meetings where parts of this work were originally presented: Volak LP, Bekaii-Saab TS, Greenblatt DJ, and Court MH (2006) Curcumin, a component of curry spice, inhibits metabolism of acetaminophen (Tylenol®) in LS180 colon cells: potential role of PKC in modulating acetaminophen glucuronidation. Drug Metab Rev 38 (Supplement 2):164.
Volak LP, Greenblatt DJ, and Court MH (2007) Curcumin, a component of turmeric, and piperine, a component of black pepper, inhibit human cytochrome P450 (CYP) enzymes in vitro. J Clin Pharmacol 47:1183-1211.
Information about curcuminoid clinical trials was also obtained on 7/3/07 from www.clinicaltrials.gov by searching for the keyword curcumin. Specific ClinicalTrials.gov identifiers for trials mentioned in text are NCT00094445, NCT00113841, and NCT00099710.
Reprint requests should be addressed to: Michael H. Court, Tufts University School of Medicine, Department of Pharmacology and Experimental Therapeutics, 136 Harrison Ave., Boston, MA 02111, Michael.Court@tufts.edu
References
- Atal CK, Dubey RK, Singh J. Biochemical basis of enhanced drug bioavailability by piperine: evidence that piperine is a potent inhibitor of drug metabolism. J Pharmacol Exp Ther. 1985;232:258–262. [PubMed] [Google Scholar]
- Badmaev V, Majeed M, Prakash L. Piperine derived from black pepper increases the plasma levels of coenzyme Q10 following oral supplementation. J Nutr Biochem. 2000;11:109–113. doi: 10.1016/s0955-2863(99)00074-1. [DOI] [PubMed] [Google Scholar]
- Bano G, Raina RK, Zutshi U, Bedi KL, Johri RK, Sharma SC. Effect of piperine on bioavailability and pharmacokinetics of propranolol and theophylline in healthy volunteers. Eur J Clin Pharmacol. 1991;41:615–617. doi: 10.1007/BF00314996. [DOI] [PubMed] [Google Scholar]
- Barik A, Priyadarsini KI, Mohan H. Photophysical studies on binding of curcumin to bovine serum albumins. Photochem Photobiol. 2003;77:597–603. doi: 10.1562/0031-8655(2003)077<0597:psoboc>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Basu NK, Ciotti M, Hwang MS, Kole L, Mitra PS, Cho JW, Owens IS. Differential and special properties of the major human UGT1-encoded gastrointestinal UDP-glucuronosyltransferases enhance potential to control chemical uptake. J Biol Chem. 2004;279:1429–1441. doi: 10.1074/jbc.M306439200. [DOI] [PubMed] [Google Scholar]
- Basu NK, Kole L, Kubota S, Owens IS. Human UDP-glucuronosyltransferases show atypical metabolism of mycophenolic acid and inhibition by curcumin. Drug Metab Dispos. 2004;32:768–773. doi: 10.1124/dmd.32.7.768. [DOI] [PubMed] [Google Scholar]
- Basu NK, Kole L, Owens IS. Evidence for phosphorylation requirement for human bilirubin UDP-glucuronosyltransferase (UGT1A1) activity. Biochem Biophys Res Commun. 2003;303:98–104. doi: 10.1016/s0006-291x(03)00241-9. [DOI] [PubMed] [Google Scholar]
- Bhardwaj RK, Glaeser H, Becquemont L, Klotz U, Gupta SK, Fromm MF. Piperine, a major constituent of black pepper, inhibits human P-glycoprotein and CYP3A4. J Pharmacol Exp Ther. 2002;302:645–650. doi: 10.1124/jpet.102.034728. [DOI] [PubMed] [Google Scholar]
- Bjornsson TD, Callaghan JT, Einolf HJ, Fischer V, Gan L, Grimm S, Kao J, King SP, Miwa G, Ni L, Kumar G, McLeod J, Obach SR, Roberts S, Roe A, Shah A, Snikeris F, Sullivan JT, Tweedie D, Vega JM, Walsh J, Wrighton SA. The conduct of in vitro and in vivo drug-drug interaction studies: a PhRMA perspective. J Clin Pharmacol. 2003;43:443–469. [PubMed] [Google Scholar]
- Cheng AL, Hsu CH, Lin JK, Hsu MM, Ho YF, Shen TS, Ko JY, Lin JT, Lin BR, Ming-Shiang W, Yu HS, Jee SH, Chen GS, Chen TM, Chen CA, Lai MK, Pu YS, Pan MH, Wang YJ, Tsai CC, Hsieh CY. Phase I clinical trial of curcumin, a chemopreventive agent, in patients with high-risk or pre-malignant lesions. Anticancer Res. 2001;21:2895–2900. [PubMed] [Google Scholar]
- Court MH, Duan SX, von Moltke LL, Greenblatt DJ, Patten CJ, Miners JO, Mackenzie PI. Interindividual variability in acetaminophen glucuronidation by human liver microsomes: identification of relevant acetaminophen UDP-glucuronosyltransferase isoforms. J Pharmacol Exp Ther. 2001;299:998–1006. [PubMed] [Google Scholar]
- Cruz-Correa M, Shoskes DA, Sanchez P, Zhao R, Hylind LM, Wexner SD, Giardiello FM. Combination treatment with curcumin and quercetin of adenomas in familial adenomatous polyposis. Clin Gastroenterol Hepatol. 2006;4:1035–1038. doi: 10.1016/j.cgh.2006.03.020. [DOI] [PubMed] [Google Scholar]
- Eaton EA, Walle UK, Lewis AJ, Hudson T, Wilson AA, Walle T. Flavonoids, potent inhibitors of the human P-form phenolsulfotransferase. Potential role in drug metabolism and chemoprevention. Drug Metab Dispos. 1996;24:232–237. [PubMed] [Google Scholar]
- Fiala M, Liu PT, Espinosa-Jeffrey A, Rosenthal MJ, Bernard G, Ringman JM, Sayre J, Zhang L, Zaghi J, Dejbakhsh S, Chiang B, Hui J, Mahanian M, Baghaee A, Hong P, Cashman J. Innate immunity and transcription of MGAT-III and Toll-like receptors in Alzheimer’s disease patients are improved by bisdemethoxycurcumin. Proc Natl Acad Sci U S A. 2007;104:12849–12854. doi: 10.1073/pnas.0701267104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory PA, Lewinsky RH, Gardner-Stephen DA, Mackenzie PI. Regulation of UDP glucuronosyltransferases in the gastrointestinal tract. Toxicol Appl Pharmacol. 2004;199:354–363. doi: 10.1016/j.taap.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Hanai H, Iida T, Takeuchi K, Watanabe F, Maruyama Y, Andoh A, Tsujikawa T, Fujiyama Y, Mitsuyama K, Sata M, Yamada M, Iwaoka Y, Kanke K, Hiraishi H, Hirayama K, Arai H, Yoshii S, Uchijima M, Nagata T, Koide Y. Curcumin maintenance therapy for ulcerative colitis: randomized, multicenter, double-blind, placebo-controlled trial. Clin Gastroenterol Hepatol. 2006;4:1502–1506. doi: 10.1016/j.cgh.2006.08.008. [DOI] [PubMed] [Google Scholar]
- Her C, Szumlanski C, Aksoy IA, Weinshilboum RM. Human jejunal estrogen sulfotransferase and dehydroepiandrosterone sulfotransferase: immunochemical characterization of individual variation. Drug Metab Dispos. 1996;24:1328–1335. [PubMed] [Google Scholar]
- Hesse LM, He P, Krishnaswamy S, Hao Q, Hogan K, von Moltke LL, Greenblatt DJ, Court MH. Pharmacogenetic determinants of interindividual variability in bupropion hydroxylation by cytochrome P450 2B6 in human liver microsomes. Pharmacogenetics. 2004;14:225–238. doi: 10.1097/00008571-200404000-00002. [DOI] [PubMed] [Google Scholar]
- Khajuria A, Zutshi U, Bedi KL. Permeability characteristics of piperine on oral absorption--an active alkaloid from peppers and a bioavailability enhancer. Indian J Exp Biol. 1998;36:46–50. [PubMed] [Google Scholar]
- Kim H, Yang C. Synthetic curcumin derivatives inhibit Jun-Fos-DNA complex formation. Bull Korean Chem Soc. 2004;25:1769–1774. [Google Scholar]
- Lambert JD, Hong J, Kim DH, Mishin VM, Yang CS. Piperine enhances the bioavailability of the tea polyphenol (-)-epigallocatechin-3-gallate in mice. J Nutr. 2004;134:1948–1952. doi: 10.1093/jn/134.8.1948. [DOI] [PubMed] [Google Scholar]
- Naganuma M, Saruwatari A, Okamura S, Tamura H. Turmeric and curcumin modulate the conjugation of 1-naphthol in Caco-2 cells. Biol Pharm Bull. 2006;29:1476–1479. doi: 10.1248/bpb.29.1476. [DOI] [PubMed] [Google Scholar]
- Nurfina AN, Reksohadiprodjo MS, Timmerman H, Jenie UA, Sugiyanto D, van der Goot H. Synthesis of some symmetrical curcumin derivatives and their antiinflammatory activity. Eur J Med Chem. 1997;32:321–328. [Google Scholar]
- O’Donnell CJ, Grime K, Courtney P, Slee D, Riley RJ. The development of a cocktail CYP2B6, CYP2C8, and CYP3A5 inhibition assay and a preliminary assessment of utility in a drug discovery setting. Drug Metab Dispos. 2007;35:381–385. doi: 10.1124/dmd.106.012344. [DOI] [PubMed] [Google Scholar]
- Paine MF, Hart HL, Ludington SS, Haining RL, Rettie AE, Zeldin DC. The human intestinal cytochrome P450 “pie”. Drug Metab Dispos. 2006;34:880–886. doi: 10.1124/dmd.105.008672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patki KC, von Moltke LL, Greenblatt DJ. In vitro metabolism of midazolam, triazolam, nifedipine, and testosterone by human liver microsomes and recombinant cytochromes p450: role of cyp3a4 and cyp3a5. Drug Metab Dispos. 2003;31:938–944. doi: 10.1124/dmd.31.7.938. [DOI] [PubMed] [Google Scholar]
- Pattanaik S, Hota D, Prabhakar S, Kharbanda P, Pandhi P. Effect of piperine on the steady-state pharmacokinetics of phenytoin in patients with epilepsy. Phytother Res. 2006;20:683–686. doi: 10.1002/ptr.1937. [DOI] [PubMed] [Google Scholar]
- Reen RK, Singh J. In vitro and in vivo inhibition of pulmonary cytochrome P450 activities by piperine, a major ingredient of piper species. Indian J Exp Biol. 1991;29:568–573. [PubMed] [Google Scholar]
- Sharma RA, Euden SA, Platton SL, Cooke DN, Shafayat A, Hewitt HR, Marczylo TH, Morgan B, Hemingway D, Plummer SM, Pirmohamed M, Gescher AJ, Steward WP. Phase I clinical trial of oral curcumin: biomarkers of systemic activity and compliance. Clin Cancer Res. 2004;10:6847–6854. doi: 10.1158/1078-0432.CCR-04-0744. [DOI] [PubMed] [Google Scholar]
- Shoba G, Joy D, Joseph T, Majeed M, Rajendran R, Srinivas PS. Influence of piperine on the pharmacokinetics of curcumin in animals and human volunteers. Planta Med. 1998;64:353–356. doi: 10.1055/s-2006-957450. [DOI] [PubMed] [Google Scholar]
- Soars MG, Grime K, Riley RJ. Comparative analysis of substrate and inhibitor interactions with CYP3A4 and CYP3A5. Xenobiotica. 2006;36:287–299. doi: 10.1080/00498250500446208. [DOI] [PubMed] [Google Scholar]
- Thapliyal R, Maru GB. Inhibition of cytochrome P450 isozymes by curcumins in vitro and in vivo. Food Chem Toxicol. 2001;39:541–547. doi: 10.1016/s0278-6915(00)00165-4. [DOI] [PubMed] [Google Scholar]
- Vietri M, Pietrabissa A, Mosca F, Spisni R, Pacifici GM. Curcumin is a potent inhibitor of phenol sulfotransferase (SULT1A1) in human liver and extrahepatic tissues. Xenobiotica. 2003;33:357–363. doi: 10.1080/0049825031000065197. [DOI] [PubMed] [Google Scholar]
- von Moltke LL, Durol AL, Duan SX, Greenblatt DJ. Potent mechanism-based inhibition of human CYP3A in vitro by amprenavir and ritonavir: comparison with ketoconazole. Eur J Clin Pharmacol. 2000;56:259–261. doi: 10.1007/s002280000125. [DOI] [PubMed] [Google Scholar]
- von Moltke LL, Greenblatt DJ, Harmatz JS, Shader RI. Alprazolam metabolism in vitro: studies of human, monkey, mouse, and rat liver microsomes. Pharmacology. 1993;47:268–276. doi: 10.1159/000139107. [DOI] [PubMed] [Google Scholar]
- von Moltke LL, Weemhoff JL, Perloff MD, Hesse LM, Harmatz JS, Roth-Schechter BF, Greenblatt DJ. Effect of zolpidem on human cytochrome P450 activity, and on transport mediated by P-glycoprotein. Biopharm Drug Dispos. 2002;23:361–367. doi: 10.1002/bdd.329. [DOI] [PubMed] [Google Scholar]
- Wang LQ, Falany CN, James MO. Triclosan as a substrate and inhibitor of 3′-phosphoadenosine 5′-phosphosulfate-sulfotransferase and UDP-glucuronosyl transferase in human liver fractions. Drug Metab Dispos. 2004;32:1162–1169. doi: 10.1124/dmd.104.000273. [DOI] [PubMed] [Google Scholar]
- Wang M, Ebmeier CC, Olin JR, Anderson RJ. Sulfation of tibolone metabolites by human postmenopausal liver and small intestinal sulfotransferases (SULTs) Steroids. 2006;71:343–351. doi: 10.1016/j.steroids.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Weemhoff JL, von Moltke LL, Richert C, Hesse LM, Harmatz JS, Greenblatt DJ. Apparent mechanism-based inhibition of human CYP3A in-vitro by lopinavir. J Pharm Pharmacol. 2003;55:381–386. doi: 10.1211/002235702739. [DOI] [PubMed] [Google Scholar]
- Zhang W, Tan TM, Lim LY. Impact of curcumin-induced changes in P-glycoprotein and CYP3A expression on the pharmacokinetics of peroral celiprolol and midazolam in rats. Drug Metab Dispos. 2007;35:110–115. doi: 10.1124/dmd.106.011072. [DOI] [PubMed] [Google Scholar]