

Abstract
Molecules that target microtubules have an important role in the treatment of cancer. A new class of inhibitors of tubulin polymerization based on the 2-(3,4,5-trimethoxybenzoyl)-2-dimethylamino-benzo[b]furan molecular skeleton was synthesized and evaluated for antiproliferative activity, inhibition of tubulin polymerization, and cell cycle effects. The most promising compound in this series was 2-(3,4,5-trimethoxybenzoyl)-3-dimethylamino-6-methoxy-benzo[b]furan, which inhibits cancer cell growth at nanomolar concentrations and interacts strongly with tubulin by binding to the colchicine site.
Keywords: Microtubules, Tubulin polymerization, Colchicine binding site, Combretastatin A-4, Bioisosteric replacement
1. Introduction
Compounds that are able to interfere with the microtubule-tubulin equilibrium in cells are useful in the treatment of human diseases.1 The success of tubulin polymerization inhibitors as anticancer agents has stimulated significant interest in the identification of new compounds that may be more potent or more selective in targeted tissues or tumors.
Among the microtubule depolymerizing agents, combretastatin A-4 (CA-4, 1; Chart 1) is one of more studied compounds. CA-4, isolated from the bark of the South African tree Combretum caffrum,2 strongly inhibits the polymerization of tubulin by binding to the colchicine site.3 Because of its simple structure, a wide number of CA-4 analogues have been developed and evaluated in SAR studies.4
Chart 1.

Inhibitors of tubulin polymerization.
We have earlier reported three different series of 2-(3′,4′,5′-trimethoxybenzoyl)-3-amino benzoheterocyclic derivatives with general structure 2–4, characterized by the presence of the benzo[b]thiophene, 1H-indole, and 1-methylindole skeleton, respectively.5,6 These compounds strongly inhibited tumor cell growth and tubulin polymerization by binding to the colchicine site of tubulin and caused G2/M phase arrest of the cell cycle. The C-6 methoxy group plays an essential role for the inhibition of tubulin polymerization within these series of molecules (compounds 2a–4a).
As a part of our search for novel tubulin polymerization inhibitors, we synthesized and evaluated the biological properties of a new series of benzo[b]furan derivatives with general structure 5. We based this synthesis on the bioisosteric replacement by furan of either the thiophene or pyrrole moieties that characterized the benzo[b]thiophene (2) and indole derivatives 3 and 4. Since the 3,4,5-trimethoxybenzoyl substituent was demonstrated to be essential for the bioactivity of derivatives 2–4, we maintained this substituent at the 2-position of the benzo[b]furan skeleton throughout the present investigation, and we examined the effects of various substituents at the 3-position. This furnished three different small series of compounds.
The first series (derivatives 5a–e) was characterized by the presence of a 3-amino group in the benzo[b]furan skeleton and either no substituent (5a) or a methoxy group at each of the four possible positions on the benzene ring (compounds 5b–e).
In the second series (compounds 5f–k), the 3-amino moiety was replaced with a dimethylamino (5f) or acetamido group (5k), plus a methoxy group at each of the four positions of the benzene ring of the benzofuran moiety combined with the 3-dimethylamino group (5g–j) These compounds allowed us to determine whether the 3-amino substituent could restrict the conformation of the adjacent trimethoxybenzoyl moiety through an intramolecular hydrogen bond with the carbonyl oxygen.
In the third series (5l–n), we examined haloacetyl amides at position 3 of the benzo[b]furan moiety. We were stimulated to do this by previous studies in which the electrophilic nature of the haloacetyl moiety has been used in anticancer drug design.7,8 The alkylation of sulfhydryl groups of tubulin by iodoacetamide9 and the disruption of microtubules by p-bromophenacyl bromide10 have been reported.
We should note that previous studies have yielded a limited series of tubulin inhibitors with the benzo[b]furan molecular skeleton as the core structure. These compounds have general structure 6, which incorporates the 3-(3,4,5-trimethoxybenzoyl)-6-methoxybenzo[b]furan ring system.11 The 6-methoxy substituent is important for maximal activity and appears to correspond to the 4-methoxy group in the B-ring of CA-4.4
2. Chemistry
Derivatives 5a–n were synthesized as shown in Scheme 1. Refluxing 2-hydroxyaldehydes 7a–e and nitroethane in glacial acetic acid in the presence of sodium acetate furnished the corresponding nitriles 8a–e in good yields.12 The subsequent reaction of 8a–e with 2-bromo-1-(3,4,5-trimethoxyphenyl)ethanone5 and potassium carbonate in refluxing acetone yielded the cyclized 3-amino benzo[b]furan derivatives 5a–e, which were transformed into the corresponding 3-dimethylamino compounds 5f–j by treatment with methyl iodide in DMF.
Scheme 1.

Reagents and conditions: (a) C2H5NO2, NaOAc; AcOH; (b) 2-bromo-1-(3,4,5-trimethoxyphenyl)ethanone, K2CO3, (CH3)2CO, reflux, 18 h, (c) Me1, NaH, DMF; (d) CH3COCl, pyridine, CH2Cl2, rt; (e) ClCH2COCl, pyridine, CH2Cl2, rt; (f) BrCH2COBr, pyridine, CH2Cl2, rt; (g) Nal, CH3CON(CH3)2, rt.
Acetylating or haloacetylating the 3-amino group of 5d with acetyl chloride, chloroacetyl chloride, and bromoacetyl chloride in the presence of pyridine provided the acetamide derivatives 5k–m, respectively. The iodoacetyl derivative 5n was prepared from the bromoacetyl derivative 5m by an exchange reaction using sodium iodide in N,N-dimethylacetamide.
3. Biological results and discussion
Table 1 summarizes the growth inhibitory effects of benzo[b]furan derivatives 5a–n against murine leukemia (L1210), murine mammary carcinoma (FM3A), and human T-lymphoblastoid (Molt/4 and CEM) cells, with CA-4 (1) and 2a–4a as reference compounds.
Table 1.
In vitro inhibitory effects of compounds 2–4a, 5a–n, and CA-4 (1) on the proliferation of murine leukemia (L1210), murine mammary carcinoma (FM3A), and human T-lymphocyte (Molt/4 and CEM) cells
| Compound | IC50a (nM) |
|||
|---|---|---|---|---|
| L1210 | FM3A | Molt4/C8 | CEM | |
| 5a | >10,000 | >10,000 | >10,000 | >10,000 |
| 5b | 7500 ± 900 | 7900 ± 2100 | 1800 ± 100 | 6700 ± 100 |
| 5c | >10,000 | >10,000 | >10,000 | >10,000 |
| 5d | 430 ± 40 | 280 ± 160 | 140 ± 20 | 87 ± 22 |
| 5e | >10,000 | >10,000 | 4400 ± 1400 | 7700 ± 400 |
| 5f | >10,000 | 8800 ± 1300 | 2700 ± 500 | >10,000 |
| 5g | >10,000 | >10,000 | >10,000 | >10,000 |
| 5h | 1400 ± 110 | 1200 ± 40 | 470 ± 22 | 1700 ± 100 |
| 5i | 65 ± 5 | 59 ± 5 | 48 ± 4 | 78 ± 3 |
| 5j | 1100 ± 100 | 1200 ± 900 | 670 ± 380 | 1400 ± 80 |
| 5k | 94 ± 7 | 100 ± 0.00 | 62 ± 4.2 | 78 ± 3 |
| 5l | 1100 ± 90 | 1100 ± 90 | 310 ± 28 | 460 ± 33 |
| 5m | 480 ± 15 | 560 ± 12 | 240 ± 19 | 210 ± 11 |
| 5n | 170 ± 30 | 260 ± 60 | 86 ± 5 | 120 ± 0.0 |
| 2a | 39 ± 16 | 46 ± 12 | 10 ± 7 | 7.7 ± 2.9 |
| 3a | 970 ± 24 | 1600 ± 30 | 630 ± 10 | 320 ± 90 |
| 4a | 69 ± 37 | 97 ± 3 | 57 ± 7 | 71 ± 5 |
| CA-4 (1) | 2.8 ± 1.1 | 42 ± 6 | 16 ± 1.4 | 1.9 ± 1.6 |
IC50, compound concentration required to inhibit tumor cell proliferation by 50%. Data are expressed as the mean ± SE from the dose–response curves of at least three independent experiments.
Compound 5i was the most potent compound identified in this study, inhibiting the growth of L1210, FM3A, Molt/4, and CEM cancer cell lines with IC50-values of 65, 59, 48, and 78 nM, respectively. CA-4 (1) and, to a lesser extent, 2a were more potent as antiproliferative agents than 5i with these four cell lines.
The results presented in Table 1 show that the location of the methoxy group on the benzene portion of the benzo[b]furan moiety plays a critical role in inhibition of cell growth, and the most favorable position for the substituent was at C-6, as was observed previously with the thiophene and indole series.5,6 In the series of 3-amino benzo[b]furan derivatives 5b–e, the 6-methoxy derivative 5d had the greatest antiproliferative activity, with IC50-values ranging from 87 to 430 nM against the four cell lines. The 4-methoxy analogue 5b was also over 10-fold less active than 5d in the four cell lines. From the point of view of SAR, the oxygen biostere (5d) did not enhance activity relative to sulfur atom (2a), but did relative to the nitrogen atom (3a).
With the exception of the C-4 methoxy derivative 5g, the methoxy substituent at C-5, C-6, or C-7 (5h–j, respectively) in the 3-dimethylamino series increased antiproliferative activity relative to both the unsubstituted 5f and their parent 3-amino counterparts 5c–e. This suggests that it would be worthwhile introducing dimethylamino modification at the 3-amino group into the thiophene, indole, and methylindole series described previously.5,6
The antiproliferative activity of 5i was superior to that of 1H-indole derivative 3a, comparable to that of 1-methylindole 4a, and slightly less than that of benzo[b]thiophene 2a. Therefore, even though the good activity of 5i required a further modification of the 3-amino group, we conclude that there is validity to the idea of bioisosteric equivalence between benzo[b]furan, 1-methylindole, and benzo[b]thiophene in this class of compounds. Moreover, comparing the structures of 5i with those of 6ab, we conclude that the 3-dimethylamino group of 5i is analogous to the right-hand substituted phenyl ring of 6ab (see Chart 1).
Changing the substituent at C-3 from the dimethylamino group (5i) to an acetamido moiety (5k) caused only a minimal reduction in antiproliferative activities. Consequently, 5k, like 5i, was superior in its antiproliferative properties to the amine, 5d.
Compound 5k was also more potent than 3-haloacetamido derivatives 5l–n against all four cell lines. Of note in haloacetamido series, increasing the size of the halogen atom resulted in increased antiproliferative activity, with the order being I (5n) > Br (5m)>Cl (5k). This order is also inversely related to the electron-withdrawing properties of the halide atoms.
To confirm that the antiproliferative activities of these compounds, like those of the benzothiophene and indole series,5,6 were related to an interaction with the microtubule system, 5d, 5i, and 5k–n were evaluated for their inhibitory effects on tubulin polymerization and on the binding of [3 H]colchicine to tubulin (Table 2).13,14 Comparing benzo-fused heterocyclic compounds 2a–4a and 5i, those containing either a sulfur (2a) or a nitrogen (3a and 4a) in the benzoheterocyclic ring are less effective than oxygen analogue 5i as an inhibitor of tubulin polymerization and [3 H] colchicine binding.
Table 2.
Inhibition of tubulin polymerization and colchicine binding by compounds 2–4a, 5d, 5i, 5k–n, and CA-4
| Compound | Tubulin assemblya IC50 ± SD (μM) |
Colchicine bindingb % ± SD |
|---|---|---|
| 5d | 1.1 ± 0.1 | 73 ± 5 |
| 5i | 0.90 ± 0.0 | 76 ± 5 |
| 5k | 1.6 ± 0.0 | 51 ± 7 |
| 5l | 2.0 ± 0.1 | 47 ± 5 |
| 5m | 2.3 ± 0.5 | 49 ± 2 |
| 5n | 1.8 ± 0.1 | 54 ± 1 |
| 2a | 1.3 ± 0.1 | 60 ± 0 |
| 3a | >40 | nd |
| 4a | 4.6 ± 0.1 | 38 ± 5 |
| CA-4 (1) | 1.2 ± 0.1 | 86 ± 3 |
Inhibition of tubulin polymerization. Tubulin was at 10 μM.
Inhibition of [3H]colchicine binding. Tubulin, colchicine, and tested compound were at 1, 5, and 1 μM, respectively. nd, not determined.
The order of inhibitory action on tubulin assembly was 5i > 5d > 5k > 5n > 5l > 5m, which was consistent with the results of the antiproliferative assays, except that 5d was more potent than 5k. The most potent compound in this series was compound 5i, with an IC50 value of 0.9 μM. This is in agreement with 5i being the compound with the greatest antiproliferative activity. Compounds 5d and 5i were as active as CA-4 as inhibitors of tubulin assembly, although both compounds were less active in their effects on cell growth.
In the colchicine binding studies, compounds 5d and 5i potently inhibited the binding of [3H]colchicine to tubulin, since 73% and 76% inhibition, respectively, occurred with these agents at 1 μM with colchicine at 5 μM. These derivatives were slightly less potent than CA-4, which in these experiments inhibited colchicine binding by 86%, but 5i was twofold more potent than its 1-methylindole counterpart 4a.
Because molecules exhibiting effects on tubulin assembly should cause the alteration of cell cycle parameters with preferential G2-M blockade, flow cytometry analysis was performed to determine the effect of the most active compounds on K562 (human chronic myelogenous leukemia) cells. Cells were cultured for 24 h in the presence of each compound at the IC50 value determined for 24 h of growth (5d = 120 nM, 5i = 68 nM, 5k = 85 nM, 5l = 400 nM, 5m = 212 nM, 5n = 115 nM). Analysis of sub-G0-G1 (apoptotic peak), G0-G1, S, and G2-M peaks revealed that the studied compounds caused somewhat different effects on cell cycle distribution (Fig. 1). While all six compounds caused an increase in the proportion of cells in the G2-M peak, compounds 5d and 5l also caused cells to accumulate in late S phase. In contrast, compound 5n caused the greatest percentage increase in the percentage of cells in apoptotic cells (subG0-G1 peak), as well as substantial proportion of cells in S phase.
Fig. 1.

Effects of compounds 5d (b), 5i (c), 5k (d), 5l (e), 5m (f), and 5n (g) on DNA content/cell following treatment of K562 cells for 24 h. The cells were cultured without compound (Control, a) or with compound used at the concentration leading to 50% cell growth inhibition after 24 h of treatment. Cell cycle distribution was analyzed by the standard propidium iodide procedure as described in Section 5. Sub-G0-G1 (A), G0-G1, S, and G2-M cells are indicated in the control panel.
We performed a series of molecular modeling studies on the series of compounds reported here. In particular, we investigated the use of three different docking software packages, Plants,15 the Surflex module implemented in Sybyl16 and MOE,17 in identifying possible binding conformations for this family of compounds. We also wanted to determine whether any combination of algorithm/scoring function would generate a good correlation between a calculated biological activity and the corresponding experimental value for future use as a predictive model. The results obtained with the different software packages were also rescored using the scoring functions implemented in the CScore module of Sybyl.16
As noted previously, no correlation between the output of the scoring functions and the experimental data was observed.18 However, a correlation was observed between the tubulin polymerization assay results and the calculated RMSD value of the trimethoxyphenyl group of the compounds reported here and the corresponding ring in the DAMA-colchicine co-crystallized with tubulin.19 The best correlation was observed with the MOE docking program (r = 0.8), and the pose with the lowest RMSD value for compound 5i is shown in Figure 2.
Fig. 2.
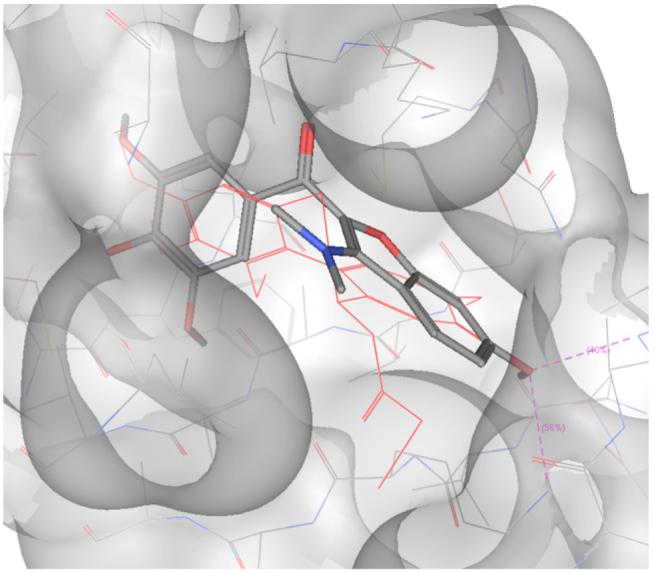
Docking pose of 5i. DAMA-colchicine is represented in red.
From the docking results, it is possible to observe how the methoxy group of the benzofuran ring of 5i can establish two hydrogen bonds with Lys352 and Val181 of β-tubulin. This could explain the difference in activity between 5i and the structurally similar compounds 5f, 5g, 5h, and 5j, which were not able to establish the same interactions, given the different position of the methoxy group on the benzofuran ring. Furthermore, compounds 5g, 5h, and 5j were not docked successfully in the colchicine site by any of the three software packages used.
4. Conclusions
In conclusion, the synthesis and biological evaluation of a new class of synthetic antitubulin compounds based on the 2-(3′,4′,5′-trimethoxybenzoyl) benzo[b]furan skeleton is described. The results showed that compounds 5d and 5i, with a methoxy substitutent at the C-6 position of the benzofuran ring, exhibited the best antiproliferative activity in the 3-amino and 3-dimethylamino benzo[b]furan series, respectively, while shifting the methoxy group in both series to the C-3, C-5, or C-7 position resulted in major losses in activity.
The dimethyl substitution on the amino group at the 3-position of the benzo[b]furan core usually enhanced antiproliferative activity, with the dimethylamino derivatives 5f and 5h–j being more potent than their amino counterparts 5a and 5c–e. In the series of 3-haloacetamido derivatives 5l–n, the order of antiproliferative activity was I > Br > Cl, but the greatest activity was observed with the unsubstituted acetamido derivative 5k. Compound 5k was almost as active as 5i, the most potent antiproliferative agent in the series.
The IC50-values for 5i in the cell lines examined ranged from 48 to 75 nM. The antiproliferative activity of 5i was equivalent to that of the previously reported 1-methylindole derivative 4a and slightly less than that of benzo[b]thiophene 2a. Compound 5i strongly inhibited both the polymerization of tubulin and the binding of [3H]colchicine to tubulin, suggesting that 5i bound to tubulin at a site overlapping the colchicine site. The antimitotic activity of 5i was demonstrated by flow cytometric analysis that showed that 5i had cellular effects typical of agents that bind to tubulin, causing accumulation of cells in G2-M. We also were readily able to model 5i into the colchicine site, with excellent overlap with the colchicinoid bound in X-ray crystal structure.
5. Experimental
5.1. Chemistry
5.1.1. Materials and methods
2-Hydroxybenzaldehyde (7a), 2-hydroxy-6-methoxybenzaldehyde (7b), 2-hydroxy-5-methoxybenzaldehyde (7c), 2-hydroxy-4-methoxybenzaldehyde (7d), 2-hydroxy-3-methoxybenzaldehyde (7e), and 2-hydroxybenzonitrile (8a) are commercially available and were used as received.
1H NMR spectra were recorded on a Bruker AC 200 spectrometer. Chemical shifts (δ) are given in ppm upfield from tetramethylsilane as internal standard, and the spectra were recorded in appropriate deuterated solvents, as indicated. Melting points (mp) were determined on a Buchi–Tottoli apparatus and are uncorrected. All products reported showed 1H NMR spectra in agreement with the assigned structures. Elemental analyses were conducted by the Microanalytical Laboratory of the Chemistry Department of the University of Ferrara. All reactions were carried out under an inert atmosphere of dry nitrogen, unless otherwise described. Standard syringe techniques were applied for transferring dry solvents. Reaction courses and product mixtures were routinely monitored by TLC on silica gel (precoated F254 Merck plates) and visualized with aqueous KMnO4. Flash chromatography was performed using 230–400 mesh silica gel and the indicated solvent system. Organic solutions were dried over anhydrous Na2SO4. Calcium chloride was used in the distillation of DMF, and the distilled solvent was stored over molecular sieves (3 Å).
5.1.2. General procedure A for the synthesis of 2-(3′,4′,5′-trimethoxybenzoyl)-3-amino benzo[b]furan (5a–e)
To a solution of 7a–e (1 mmol) in dry acetone (15 mL) was added 2-bromo-1-(3,4,5-trimethoxyphenyl)-ethanone (289 mg, 1 mmol) and anhydrous potassium carbonate (276 mg, 2 mmol) while stirring, and the reaction mixture was refluxed for 18 h. After cooling, the solvent was evaporated, and the residue was dissolved in a mixture of dichloromethane (15 mL) and water (5 mL). The organic layer was washed with brine, dried and evaporated to obtain a residue, which was purified by flash column chromatography. Then the final solid product was recrystallized from petroleum ether.
5.1.2.1. (3-Aminobenzofuran-2-yl)(3,4,5-trimethoxyphenyl)-methanone (5a)
Following general procedure A, the crude residue purified by flash chromatography using ethyl acetate/petroleum ether 4:6 (v:v) as eluent furnished 5a as a yellow solid (89% yield); mp 134–136 °C. 1H NMR (CDCl3) δ: 3.95 (s, 3H), 3.97 (s, 6H), 6.04 (br s, 2H), 7.28 (t, J = 8.4 Hz, 1H), 7.43 (d, J = 8.4 Hz, 1H), 7.53 (t, J = 8.4 Hz, 1H), 7.60 (s, 2H), 7.64 (d, J = 8.4 Hz, 1H). Anal. Calcd for C18H17NO5: C, 66.05; H, 5.23; N, 4.28. Found: C, 65.95; H, 5.11; N, 4.17.
5.1.2.2. (3-Amino-4-methoxybenzofuran-2-yl)(3,4,5-trimethoxyphenyl)methanone(5b)
Following general procedure A, the crude residue purified by flash chromatography using ethyl acetate/petroleum ether 4:6 (v:v) as eluent furnished 5b as a yellow solid (78% yield); mp 177–179 °C. 1H NMR (CDCl3) δ: 3.91 (s, 3H), 3.94 (s, 3H), 3.97 (s, 6H), 6.52 (br s, 2H), 6.55 (d, J = 7.8 Hz, 1H), 6.98 (d, J = 7.8 Hz, 1H), 7.42 (t, J = 7.8, 1H), 7.57 (s, 2H). Anal. Calcd for C19H19NO6: C, 63.86; H, 5.35; N, 3.92. Found: C, 63.67; H, 5.21; N, 3.78.
5.1.2.3. (3-Amino-5-methoxybenzofuran-2-yl)(3,4,5-trimethoxyphenyl)methanone (5c)
Following general procedure A, the crude residue purified by flash chromatography using ethyl acetate/petroleum ether 4:6 (v:v) as eluent furnished 5c as a yellow solid (76% yield); mp 125–127 °C. 1HNMR (CDCl3) δ: 3.88 (s, 3H), 3.93 (s, 3H), 3.94 (s, 6H), 6.99 (s, 1H), 7.13 (d, J = 8.8 Hz, 1H), 7.26 (br s, 2H), 7.32 (d, J = 8.8 Hz, 1H), 7.58 (s, 2H). Anal. Calcd for C19H19NO6: C, 63.86; H, 5.35; N, 3.92. Found: C, 63.72; H, 5.14; N, 3.80.
5.1.2.4. (3-Amino-6-methoxybenzofuran-2-yl)(3,4,5-trimethoxyphenyl)methanone(5d)
Following general procedure A, the crude residue purified by flash chromatography using ethyl acetate/petroleum ether 4:6 (v:v) as eluent furnished 5d as a yellow solid (77% yield); mp 167–169 °C. 1H NMR (CDCl3) δ: 3.89 (s, 3H), 3.94 (s, 3H), 3.97 (s, 6H), 6.87 (br s, 2H), 6.91 (d, J = 8.2 Hz, 1H), 7.48 (d, J = 8.2 Hz, 1H), 7.52 (s, 1H), 7.54 (s, 2H). Anal. Calcd for C19H19NO6: C,63.86;H, 5.35; N, 3.92. Found: C, 63.77; H, 5.22; N, 3.74.
5.1.2.5. (3-Amino-7-methoxybenzofuran-2-yl)(3,4,5-trimethoxyphenyl)methanone(5e)
Following general procedure A, the crude residue purified by flash chromatography using ethyl acetate/petroleum ether 4:6 (v:v) as eluent furnished 2e as a yellow solid (87% yield); mp 171–173 °C. 1H NMR (CDCl3) δ: 3.93 (s, 3H), 3.97 (s, 6H), 3.99 (s, 3H), 6.84 (br s, 2H), 6.97 (d, J = 6.8 Hz, 1H), 7.21 (m, 2H), 7.66 (s, 2H). Anal. Calcd for C19H19NO6: C, 63.86; H, 5.35; N, 3.92. Found: C, 63.59; H, 5.18; N, 3.80.
5.1.3. General procedure B for the synthesis of 2-(3,4,5-trimethoxybenzoyl)-3-dimethylamino-benzo[b]furans (5f–j)
Sodium hydride (60% oil dispersion, 48 mg, 1 mmol) was carefully added to an ice-cooled solution of CH3I (93 μL, 1.5 mmol) and 5a–e (0.56 mmol) in 2 mL of anhydrous DMF. The reaction vessel was sealed and the mixture was stirred at 40 °C for 48 h. After this period, CH3I (124 μL, 2 mmol) was added, and after 72 h the reaction mixture was diluted with cold water (1 mL) and extracted with CH2Cl2 (3 × 5 mL). The combined organic extracts were washed with water (2 mL) and brine, dried and concentrated in vacuo. The resulting residue was purified by flash chromatography using a mixture of ethyl acetate/petroleum ether as eluent. The final solid product was recrystallized from petroleum ether.
5.1.3.1. (3-(Dimethylamino)benzofuran-2-yl)(3,4,5-trimethoxyphenyl)methanone (5f)
Following general procedure B, the crude residue purified by flash chromatography using ethyl acetate/petroleum ether 3:7 (v:v) as eluent furnished 5f as a yellow oil (46% yield). 1H NMR (CDCl3) δ: 3.32 (s, 6H), 3.93 (s, 3H), 3.96 (s, 6H), 7.30 (t, J = 8.2 Hz, 1H), 7.44 (d, J = 8.2 Hz, 1H), 7.56 (t, J = 8.2 Hz, 1H), 7.64 (s, 2H), 7.68 (d, J = 8.2 Hz, 1H). Anal. (C20H21NO5): C, H, N. Anal. (C19H19NO6): C, H, N. Anal. Calcd for C20H21NO5: C, 67.45; H, 5.96; N, 3.94. Found: C, 67.45; H, 5.82; N, 3.83.
5.1.3.2. (3-(Dimethylamino)-4-methoxybenzofuran-2-yl) (3,4,5-trimethoxyphenyl)methanone (5g)
Following general procedure B, the crude residue purified by flash chromatography using ethyl acetate/petroleum ether 3:7 (v:v) as eluent furnished 5g as a yellow solid (43% yield); mp 148–150 °C. 1H NMR (CDCl3) δ: 3.52 (s, 6H), 3.92 (s, 3H), 3.96 (s, 6H), 3.98 (s, 3H), 6.52 (d, J = 7.8 Hz, 1H), 7.00 (d, J = 7.8 Hz, 1H), 7.40 (t, J = 7.8, 1H), 7.62 (s, 2H). Anal. Calcd for C21H23NO6: C, 65.44; H, 6.02; N, 3.63. Found: C, 65.29; H, 5.88; N, 3.51.
5.1.3.3. (3-(Dimethylamino)-5-methoxybenzofuran-2-yl) (3,4,5-trimethoxyphenyl)methanone (5h)
Following general procedure B, the crude residue purified by flash chromatography using ethyl acetate/petroleum ether 3:7 (v:v) as eluent furnished 5h as a yellow oil (44% yield). 1H NMR (CDCl3) δ: 3.42 (s, 6H), 3.86 (s, 3H), 3.90 (s, 3H), 3.93 (s, 6H), 7.00 (s, 1H), 7.12 (d, J = 8.4 Hz, 1H), 7.34 (d, J = 8.4 Hz, 1H), 7.60 (s, 2H). Anal. Calcd for C21H23NO6: C, 65.44; H, 6.02; N, 3.63. Found: C, 65.33; H, 5.91; N, 3.53.
5.1.3.4. (3-(Dimethylamino)-6-methoxybenzofuran-2-yl) (3,4,5-trimethoxyphenyl)methanone (5i)
Following general procedure B, the crude residue purified by flash chromatography using ethyl acetate/petroleum ether 3:7 (v:v) as eluent furnished 5i as a yellow oil (42% yield). 1H NMR (CDCl3) δ: 3.38 (s, 6H), 3.88 (s, 3H), 3.94 (s, 3H), 3.96 (s, 6H), 6.92 (d, J = 8.0 Hz, 1H), 7.18 (d, J = 8.0 Hz, 1H), 7.54 (s, 1H), 7.68 (s, 2H). Anal. Calcd for C21H23NO6: C, 65.44; H, 6.02; N, 3.63. Found: C, 65.31; H, 5.88; N, 3.48.
5.1.3.5. (3-(Dimethylamino)-7-methoxybenzofuran-2-yl) (3,4,5-trimethoxyphenyl)methanone (5j)
Following general procedure B, the crude residue purified by flash chromatography using ethyl acetate/petroleum ether 3:7 (v:v) as eluent furnished 5j as a yellow solid (93% yield); mp 148–150 °C. 1H NMR (CDCl3) δ: 3.50 (s, 6H), 3.90 (s, 3H), 3.94 (s, 6H), 3.98 (s, 3H), 6.94 (d, J = 6.8 Hz, 1H), 7.18 (m, 2H), 7.62 (s, 2H). Anal. Calcd for C21H23NO6: C, 65.44; H, 6.02; N, 3.63. Found: C, 65.34; H, 5.92; N, 3.52.
5.1.4. Synthesis of (3-acetylamino-6-methoxybenzofuran-2-yl)-(3,4,5-trimethoxyphenyl)methanone (5k)
To a solution of 5d (1 mmol, 358 mg) and pyridine (3 mmol, 242 μL) in dry dichloromethane (5 mL), acetyl chloride (3 mmol, 212 μL) was added at 0 °C. The reaction mixture was stirred for 2 h at room temperature, diluted with dichloromethane (5 mL), washed with water (4 mL), dried over Na2SO4, and concentrated in vacuo. The crude residue purified by flash chromatography using ethyl acetate/petroleum ether 4:6 (v:v) as eluent furnished 5k as a yellow solid (90% yield) after recrystallization from petroleum ether; mp 172–173 °C. 1H NMR (CDCl3) δ: 2.17 (s, 3H), 3.91 (s, 3H), 3.94 (s, 3H), 3.97 (s, 6H), 6.88 (s, 1H), 6.90 (d, J = 9.2 Hz, 1H), 7.53 (s, 2H), 8.47 (d, J = 9.2 Hz, 1H), 10.9 (s, 1H). Anal. (C21H21NO7): C, H, N. Anal. Calcd for C21H21NO7: C, 63.15; H, 5.30; N, 3.51. Found: C, 63.02; H, 5.18; N, 3.38.
5.1.5. Synthesis of (3-chloroacetylamino-6-methoxybenzofuran-2-yl)(3,4,5-trimethoxyphenyl)methanone (5l)
To a solution of 5d (1 mmol, 358 mg) and pyridine (3 mmol, 242 μL) in dry dichloromethane (5 mL), chloroacetyl chloride (3 mmol, 239 μL) was added at 0 °C. The reaction mixture was stirred for 1 h at room temperature, diluted with dichloromethane (10 mL), washed with water (5 mL), dried over Na2SO4, and concentrated in vacuo. The crude residue purified by flash chromatography using ethyl acetate/petroleum ether 3:7 (v:v) as eluent furnished 5l as a green solid (>95% yield) after recrystallization from petroleum ether; mp 123–124 °C. 1H NMR (CDCl3) δ: 3.91 (s, 3H), 3.97 (s, 9H), 4.31 (s, 2H), 6.91 (s, 1H), 6.95 (d, J = 9.2 Hz, 1H), 7.55 (s, 2H), 8.47 (d, J = 9.2 Hz, 1H), 11.7 (s, 1H). Anal. Calcd for C21H20ClNO7: C, 58.14; H, 4.65; N, 3.23. Found: C, 58.01; H, 4.48; N, 3.11.
5.1.6. Synthesis of (3-bromoacetylamino-6-methoxybenzofuran-2-yl)(3,4,5-trimethoxyphenyl)methanone (5m)
To a solution of 5d (1 mmol, 358 mg) and pyridine (3 mmol, 242 μL) in dry dichloromethane (5 mL), bromoacetyl chloride (3 mmol, 250 μL) was added at 0 °C. After 3 h at the same temperature, the reaction mixture was diluted with dichloromethane (5 mL), washed with water (5 mL), dried over Na2SO4, and concentrated in vacuo. The crude residue purified by flash chromatography using ethyl acetate/petroleum ether 3:7 (v:v) as eluent furnished 5 m as a green solid (78% yield) after recrystallization from petroleum ether; mp 99–100 °C. 1H NMR (CDCl3) δ: 3.89 (s, 3H), 3.92 (s, 6H), 3.97 (s, 3H), 4.13 (s, 2H), 6.91 (s, 1H), 6.94 (d, J = 9.2 Hz, 1H), 7.54 (s, 2H), 8.45 (d, J = 9.2 Hz, 1H), 11.6 (s, 1H). Anal. Calcd for C21H20BrNO7: C, 52.73; H, 4.21; N, 2.93. Found: C, 52.62; H, 4.04; N, 2.78.
5.1.7. Synthesis of (3-iodoacetylamino-6-methoxybenzofuran-2-yl)(3,4,5-trimethoxyphenyl)methanone (5n)
A mixture of 5m (1 mmol, 478 mg) and NaI (10 mmol, 1.5 g) in N,N-dimethylacetamide (5 mL) was stirred at room temperature for 18 h. N,N-dimethylacetamide was evaporated under reduced pressure, followed by addition of dichloromethane (15 mL) and a solution of Na2S2O3 (10%, 5 mL). The organic layer was washed with water (5 mL), brine (5 mL), and dried over Na2SO4. After removal of the solvent under reduced pressure, the crude residue purified by flash chromatography using ethyl acetate/petroleum ether 4:6 (v:v) as eluent furnished 5n as a yellow solid (58% yield) after recrystallization from petroleum ether; mp 140–141 °C. 1H NMR (CDCl3) δ: 3.72 (s, 2H), 3.90 (s, 3H), 3.97 (s, 6H), 3. 98 (s, 3H), 6.89 (s, 1H), 6.92 (d, J = 8.8 Hz, 1H), 7.54 (s, 2H), 8.45 (d, J = 8.8 Hz, 1H), 11.5 (s, 1H). Anal. Calcd for C21H20INO7: C, 48.02; H, 3.84; N, 2.67. Found: C, 47.88; H, 3.74; N, 2.56.
5.2. Cell growth inhibitory activity
Murine leukemia L1210, murine mammary carcinoma FM3A, and human T-lymphocyte Molt 4 and CEM cells were suspended at 300,000–500,000 cells/mL of culture medium, and 100 μL of a cell suspension was added to 100 μL of an appropriate dilution of the test compounds in wells of 96-well microtiter plates. After incubation at 37 °C for two (L1210 and FM3A) or three (Molt 4 and CEM) days, cell number was determined using a Coulter counter. The IC50 value was defined as the compound concentration required to inhibit cell proliferation by 50%.
5.3. Effects on tubulin polymerization and on colchicine binding to tubulin
Bovine brain tubulin was purified as described previously.20 To evaluate the effect of the compounds on tubulin assembly in vitro,13 varying concentrations were preincubated with 10 μM tubulin in glutamate buffer at 30 °C and then cooled to 0 °C. After addition of GTP, the mixtures were transferred to 0 °C cuvettes in a recording spectrophotometer and warmed to 30 °C, and the assembly of tubulin was observed turbidimetrically. The IC50 value was defined as the compound concentration that inhibited the extent of assembly by 50% after a 20 min incubation. The ability of the test compounds to inhibit colchicine binding to tubulin was measured as described,14 except that the reaction mixtures contained 1 μM tubulin, 5 μM [3H]colchicine, and 1 μM test compound.
5.4. Flow cytometric analysis of cell cycle distribution
The effects of the most active compounds of the series on cell cycle distribution were studied on K562 cells (myeloblastic leukemia) by flow cytometric analysis after staining with propidium iodide. Cells were exposed for 24 h to each compound used at a concentration corresponding to the IC50 determined after a 24 h incubation. After treatment, the cells were washed once in ice-cold PBS and resuspended at 1 × 106 per mL in a hypotonic fluoro-chrome solution containing propidium iodide (Sigma) at 50 μg/mL in 0.1% sodium citrate plus 0.03% (v/v) nonidet P-40 (Sigma). After a 30 min incubation, the fluorescence of each sample was analyzed as single-parameter frequency histogram, using a FAC-Scan flow cytometer (Becton–Dickinson, San Jose, CA). The distribution of cells in the cell cycle was analyzed with the ModFit LT3 program (Verity Software House, Inc.).
5.5. Molecular modeling
All molecular modeling studies were performed on a MacPro dual 2.66 GHz Xeon running Ubuntu 7. The tubulin structure was downloaded from the PDB (http://www.rcsb.org/–PDB code: 1SA0).19 Hydrogen atoms were added to the protein using Molecular Operating Environment (MOE) 2006.0817 and minimized using the MMFF94x forcefield until a RMSD gradient of 0.05 kcal mol−1 Å−1 was reached. The docking simulations with MOE were performed using the Alpha Triangle placement method and the London dG scoring method. Plants15 was used from the graphical interface included in ZODIAC21 with the default settings. The Surflex module included in Sybyl 7.316 was used with the default settings with a ligand-based (DAMA-colchicine) protomodel. The results obtained with each software package were then rescored using the CScore module in Sybyl 7.3. The RMSD of the trimethoxyphenyl moiety for each of the results obtained was calculated in comparison with ring A of the colchicine analogue.22
References and notes
- 1.(a) Kiselyov A, Bulakin KV. Anti-Cancer Agents Med. Chem. 2007;7:189. doi: 10.2174/187152007780058650. [DOI] [PubMed] [Google Scholar]; (b) Nagle A, Hur W, Gray NS. Curr. Drug Targets. 2006;7:305. doi: 10.2174/138945006776054933. [DOI] [PubMed] [Google Scholar]; (c) Pasquier E, Andrè N, Braguer D. Curr. Cancer Drug Targets. 2007;7:566. doi: 10.2174/156800907781662266. [DOI] [PubMed] [Google Scholar]
- 2.(a) Pettit GR, Singh SB, Hamel E, Lin CM, Alberts DS, Garcia-Kendall D. Experentia. 1989;45:209. doi: 10.1007/BF01954881. [DOI] [PubMed] [Google Scholar]; (b) Chaplin DJ, Horsman MR, Siemann DW. Curr. Opin. Invest. Drugs. 2006;7:522. [PubMed] [Google Scholar]
- 3.Lin CM, Ho HH, Pettit GR, Hamel E. Biochemistry. 1989;28:6984. doi: 10.1021/bi00443a031. [DOI] [PubMed] [Google Scholar]
- 4.Tron GC, Pirali T, Sorba G, Pagliai F, Busacca S, Genazzani AA. J. Med. Chem. 2006;49:3033. doi: 10.1021/jm0512903. [DOI] [PubMed] [Google Scholar]
- 5.Romagnoli R, Baraldi PG, Carrion MD, Lopez Cara C, Preti D, Fruttarolo F, Pavani MG, Tabrizi MA, Tolomeo M, Grimaudo S, Di Antonella C, Balzarini J, Hadfield JA, Brancale A, Hamel E. J. Med. Chem. 2007;50:2273. doi: 10.1021/jm070050f. [DOI] [PubMed] [Google Scholar]
- 6.Romagnoli R, Baraldi PG, Sarkar T, Carrion MD, Lopez-Cara C, Cruz-Lopez O, Preti D, Tabrizi MA, Tolomeo M, Grimaudo S, Di Cristina A, Zonta N, Balzarini J, Brancale A, Hsieh HP, Hamel E. J. Med. Chem. 2008;51:1464. doi: 10.1021/jm7011547. [DOI] [PubMed] [Google Scholar]
- 7.Baker B, Devan BP. J. Am. Chem. Soc. 1989;111:2700. [Google Scholar]
- 8.(a) Jiang J-D, Roboz J, Weisz I, Deng L, Ma L, Holland JF, Bekesi JG. Anti-Cancer Drug Des. 1998;13:735. [PubMed] [Google Scholar]; (b) Song D-Q, Wang Y, Wu L-Z, Yang P, Wang Y-M, Gao L-M, Li Y, Qu J-R, Wang Y-H, Li Y-H, Du NN, Han Y-X, Zhang Z-P, Jiang JD. J. Med. Chem. 2008;51:3094. doi: 10.1021/jm070890u. [DOI] [PubMed] [Google Scholar]
- 9.Luduena RF, Roach MC. Pharmacol. Ther. 1991;49:133. doi: 10.1016/0163-7258(91)90027-j. [DOI] [PubMed] [Google Scholar]
- 10.Hargreaves AJ, Glazier AP, Flaskos J, Mullins FH, McLean WG. Biochem. Pharmacol. 1994;47:1137. doi: 10.1016/0006-2952(94)90384-0. [DOI] [PubMed] [Google Scholar]
- 11.Flynn BL, Hamel E, Jung MK. J. Med. Chem. 2002;45:2670. doi: 10.1021/jm020077t. [DOI] [PubMed] [Google Scholar]
- 12.For the synthesis of 8b–e, see the procedure reported in the article: Karmarkar SN, Kelkar SL, Wadia MS. Synthesis. 1985:510. For the characterization of the 2-hydroxy-6-methoxybenzonitrile (8b) see: Hwu JR, Wong FF, Huang J-J, Tsay S-C. J. Org. Chem. 1997;62:4097.For the characterization of the 2-hydroxy-5-methoxybenzonitrile (8c) and 2-hydroxy-4-methoxybenzonitrile (8d) see: Adachi M, Sugasawa T. Synth. Commun. 1990;20:71.For the characterization of the 2-hydroxy-3-methoxybenzonitrile (8e) see: Dewan SK, Singh R, Kumar A. Synth. Commun. 2004;34:2025.
- 13.Hamel E. Cell Biochem. Biophys. 2003;38:1. doi: 10.1385/CBB:38:1:1. [DOI] [PubMed] [Google Scholar]
- 14.Verdier-Pinard P, Lai J-Y, Yoo H-D, Yu J, Marquez B, Nagle DG, Nambu M, White JD, Falck JR, Gerwick WH, Day BW, Hamel E. Mol. Pharmacol. 1998;53:62. doi: 10.1124/mol.53.1.62. [DOI] [PubMed] [Google Scholar]
- 15.Korb O, Stützle T, Exner TE. Swarm Intell. 2007;1:115. [Google Scholar]
- 16.Tripos SYBYL 7.3. Tripos Inc.; 1699 South Hanley Road, St. Louis, MO 63144, USA: http://www.tripos.com. [Google Scholar]
- 17.Molecular Operating Environment (MOE 2006.08) Chemical Computing Group, Inc.; Montreal, Que., Canada: http://www.chemcomp.com. [Google Scholar]
- 18.De Martino G, Edler MC, La Regina G, Coluccia A, Barbera MC, Barrow D, Nicholson RI, Chiosis G, Brancale A, Hamel E, Artico M, Silvestri R. J. Med. Chem. 2006;49:947. doi: 10.1021/jm050809s. [DOI] [PubMed] [Google Scholar]
- 19.Ravelli RBG, Gigant B, Curmi PA, Jourdain I, Lachkar S, Sobel A, Knossow M. Nature. 2004;428:198. doi: 10.1038/nature02393. [DOI] [PubMed] [Google Scholar]
- 20.Hamel E, Lin CM. Biochemistry. 1984;23:4173. doi: 10.1021/bi00313a026. [DOI] [PubMed] [Google Scholar]
- 21.Zodiac 0.3.5b. http://www.zeden.org.
- 22.Code ‘fragment_rmsd.svl’ obtained from SLV Exchange website. Chemical Computing Group, Inc.; Montreal, Canada: http://svl.chemcomp.com. [Google Scholar]