

Abstract
Because damage to sympathetic nerve terminals occurs in a variety of diseases, we tested the hypothesis that nerve terminal damage per se is sufficient to impair ganglionic neurotransmission in vivo. First, we measured the effect of nerve terminal damage produced by the sympathetic nerve terminal toxin 6-hydroxydopamine (6-OHDA) on ganglionic levels of several neurotrophins thought to promote neurotransmission. 6-OHDA-induced nerve terminal damage did not decrease the expression of neurotrophin-4 or brain-derived neurotrophic factor mRNA in the celiac ganglia but did decrease the ganglionic content of both nerve growth factor protein (nadir = -63%) and the mRNA of the alpha-3 subunit of the nicotinic cholinergic receptor (nadir = -49%), a subunit required for neurotransmission. Next, we tested whether this degree of receptor deficiency was sufficient to impair activation of celiac ganglia neurons. Impaired fos mRNA responses to nicotine administration in the celiac ganglia of 6-OHDA-pretreated rats correlated temporally with suppressed expression of functional nicotinic receptors. We verified by Fos protein immunohistochemistry that this ganglionic impairment was specific to principal ganglionic neurons. Last, we tested whether centrally initiated ganglionic neurotransmission is also impaired following nerve terminal damage. The principal neurons in rat celiac ganglia were reflexively activated by 2-deoxy-glucose-induced glucopenia, and the Fos response in the celiac ganglia was markedly inhibited by pretreatment with 6-OHDA. We conclude that sympathetic nerve terminal damage per se is sufficient to impair ganglionic neurotransmission in vivo and that decreased nicotinic receptor production is a likely mediator.
Keywords: 6-hydroxydopamine, neurotransmission, nerve growth factor, nicotinic acetylcholine receptor, fos mRNA, Fos protein
Studies of the effects of damage to sympathetic nerve fibers on their cell bodies have traditionally relied on the extreme experimental maneuver of complete axotomy. Such studies have proven valuable both for demonstrating the proof of principle that axonal damage can produce cell body dysfunction and for exploring the mechanisms involved. It is possible to extrapolate from these earlier studies that a less severe neural insult, such as damage restricted to nerve terminals, would also impair ganglionic function, but this hypothesis has not been directly tested. Furthermore, cell body dysfunction induced by axotomy has no influence on neurotransmitter release from nerve terminals because by definition axotomy severs all connection between the cell body and its terminals. In contrast, impaired cell body function following incomplete nerve terminal damage can have functional consequences by impairing neurotransmitter release from spared terminals. Thus, we thought it important to determine the effect of a less extreme and more disease-relevant sympathetic nerve fiber injury, nerve terminal damage, on ganglionic neurotransmission.
Indeed, several diseases result in peripheral nerve damage restricted to sympathetic nerve terminals. Chaga’s disease is characterized by loss of cardiac sympathetic nerve terminals (Camargos et al., [2000]), as are myocardial infarction (Mathes et al., [1971]; Kozlovskis et al., [1986]) and the more chronic syndrome of diabetic autonomic neuropathy (Langer et al., [1995]; Schmid et al., [1999]). Postural tachycardic syndrome is associated with sympathetic denervation of the legs but not the arms (Jacob et al., [2000]), and some reports suggest loss of sympathetic nerve terminals in the salivary glands of patients with Sjögren’s disease (Konttinen et al., [1992]). Finally, we have shown a marked loss of sympathetic nerve terminals selective to the pancreatic islet early in autoimmune diabetes (Mei et al., [2002], [2006a]; Mundinger et al., [2003]).
This islet-specific nerve terminal loss is associated with increased galanin expression in sympathetic neuronal cell bodies in celiac ganglia (Mei et al., [2006b]), which project axons to the pancreas (Quinson et al., [2001]). Similarly, myocardial infarction increases galanin expression in the corresponding stellate ganglion (Habecker et al., [2005]). Although the function of damage-induced galanin expression is unknown, such ganglionic neuropeptide expression has been used simply as a marker of neuronal responses to axonal damage, and we hypothesize that it may also be a marker of impaired function. For example, axotomy-induced galanin expression in superior cervical ganglia (Hyatt-Sachs et al., [1996]; Zigmond, [1997]; Landry et al., [2000]) is associated with impaired ganglionic neurotransmission (Purves [1975]; Purves et al., [1988]). However, axotomy has shown to produce much greater neuropeptide expression in this ganglion than that induced by nerve terminal damage (Hyatt-Sachs et al., [1996]). Therefore, the question of whether damage to sympathetic nerve terminals per se can impair ganglionic neurotransmission remains open. If so, the diseases detailed above may have an additional, unrecognized defect in the sympathetic pathway.
It should also be emphasized that although axotomy-induced impairment of ganglionic neurotransmission is an accepted concept, the actual assessment of neurotransmission has been based on in vitro measurement: reduced excitatory postsynaptic potentials to electrical stimulation in explanted ganglia (Purves, [1975]; Purves et al., [1988]). Although it may be assumed that axotomy would impair physiologically induced ganglionic neurotransmission in vivo, this assumption has not been directly tested. Therefore, in the current study we thought it important to employ an in vivo index of ganglionic neurotransmission.
We also sought to determine the mechanism by which damage to sympathetic nerve terminals per se might impair ganglionic neurotransmission. Potential mechanisms of impaired ganglionic neurotransmission include (1) reduced synaptic contact between preganglionic nerve terminals and the dendrites of principal ganglionic cells, (2) reduced preganglionic release of acetylcholine neurotransmitter, and (3) dysfunction of the nicotinic receptors on the dendrites of ganglionic neurons. Because neurotrophins are thought to regulate these three components of ganglionic neurotransmission (Purves et al., [1988]; Causing et al., [1997]; Zhou et al., [1998]; Roosen et al., [2001]), reductions in neurotrophins might ultimately cause any of the dysfunctions we observed. Neurotrophin-4 (NT-4; Roosen et al., [2001]) and brain-derived neurotrophic factor (BDNF; Causing et al., [1997]) are produced in sympathetic ganglia, as judged by measurable ganglionic mRNA, and both neurotrophins are thought to have their trophic effects on the incoming preganglionic nerves. For example, both NT-4- and BDNF-knockout mice have reduced preganglionic nerve terminals and axons (Causing et al., [1997]; Roosen et al., [2001]), and NT-4-deficient mice have reduced preganglionic cell bodies as well (Roosen et al., [2001]). Therefore, NT-4 or BDNF deficiency could impair ganglionic neurotransmission by reducing both the number of synapses and the amount of neurotransmitter released in the ganglia. However, this mechanism of ganglionic impairment remains speculative because neither NT-4 nor BDNF deficiency has been demonstrated following axon or terminal damage.
Although NT-4 and BNDF are synthesized in ganglia, neurotrophin 3 (NT-3) and nerve growth factor (NGF) are target tissue-derived neurotrophins that are retrogradely transported to ganglionic neurons after binding to terminal trk-C and trk-A receptors, respectively. There is a decrease in the number of principal neurons in cultured ganglia of embryonic NT-3-knockout mice (Wyatt et al., [1997]). Furthermore, because apoptosis is also increased (Wyatt et al., [1997]), it has been suggested that neuronal survival is codependent on NT-3 and NGF, at least in neonatal development. However, ganglionic NT-3 protein content in rats drops below detectable limits at about 6 weeks of age (Zhang and Rush, [2001]). Consequently, NT-3′s role in the maintenance of either neuronal number or neuronal function in adult rats is limited, and looking for decreases in ganglionic NT-3 protein in adult animals after axotomy or nerve terminal damage is not currently feasible.
In contrast, a decrease in the ganglionic concentration of the prototypic sympathetic neurotrophin nerve growth factor has been observed after axotomy (Zhou et al., [1994]), postganglionic nerve terminal damage (Korsching and Thoenen, [1985]; Schmidt et al., [2000]), and colchicine treatment (Korsching and Thoenen, [1985]). NGF is produced and secreted constitutively by sympathetically innervated tissues (Korsching and Thoenen, [1983a], [1985]; Wetmore and Olson, [1995]; Schmid et al., [1999]) and is taken up by trk-A receptors on postganglionic sympathetic nerve terminals (Gatzinsky et al., [2001]), and the complex is retrogradely transported to neuronal cell bodies residing in sympathetic ganglia (Korsching and Thoenen, [1983b]). The decrease in NGF following both axotomy and axonal crush is accompanied by impaired ganglionic neurotransmission in vitro, as demonstrated by decreased excitatory postsynaptic potentials in postganglionic axons (Purves, [1975]). Impaired neurotransmission is restricted to the axotomized neuron, as adjacent ganglionic neurons with intact axons retain their function (De Castro et al., [1995]). Importantly, this ganglionic impairment induced by axonal damage is partially reversed by NGF treatment (Nja and Purves, [1978]). Thus, it is likely that impaired ganglionic neurotransmission following axotomy is at least partly caused by a deficiency in NGF in ganglionic neurons; perhaps, the same is true following nerve terminal damage.
Ganglionic NGF deficiency is associated with two specific postsynaptic defects that have the potential to impair ganglionic neurotransmission. First, dendritic arborization in sympathetic ganglia is reduced following axotomy (Yawo, [1987]; Purves et al., [1988]). This effect is reproduced by treatment with antibodies against NGF (Ruit et al., [1990]), and more importantly, NGF treatment markedly increases the surface area of dendrites (Purves et al., [1988]; Snider, [1988]). Second, ganglionic mRNA transcripts (Zhou et al., [1998]) and the protein content (Yeh et al., [2001]; Zhou et al., [2001]) of several subunits of the nicotinic acetylcholine receptor, the classical mediator of ganglionic neurotransmission, are decreased following axotomy. Oddly, administration of NGF antiserum does not decrease nicotinic receptor subunit expression (Zhou et al., [1998]). However, adding NGF to the culture medium of explanted superior cervical ganglia partially prevents a decrease in the level of alpha-3 subunit of the nicotinic acetylcholine receptor (A3-NR), a subunit required for neurotransmission (Yeh et al., [2001]). Therefore, decreased NGF accumulation in sympathetic ganglia neurons following postganglionic axotomy or nerve terminal damage may impair ganglionic neurotransmission by reducing the dendritic surface area of the neuronal cell body or possibly by decreasing the number of functional nicotinic receptors on those withdrawn dendrites.
The goal of the present study was to test the hypothesis that nerve terminal damage per se is sufficient to impair ganglionic neurotransmission in vivo. First, we investigated the effect of sympathetic nerve terminal damage on several potential mediators of impaired ganglionic neurotransmission. We measured the time course of changes in celiac ganglia neurotrophins (NGF, NT-4, and BDNF) in response to nerve terminal damage induced by the sympathetic nerve terminal toxin 6-hydroxydopamine. Based on our NGF findings, we also measured the time course of changes in A3-NR mRNA in celiac ganglia following nerve terminal destruction. Next, we performed three separate investigations of ganglionic function to determine if A3-NR deficiencies produced by nerve terminal damage were associated with impaired ganglionic neurotransmission in vivo. To do so, we chemically activated nicotinic receptors and measured celiac ganglia fos mRNA and Fos protein responses, expecting impaired responses in rats pretreated with 6-hydroxydopamine. We also activated celiac ganglia neurons reflexively by central glucopenia, again expecting impaired responses in rats with nerve terminal damage. Our results show that sympathetic nerve terminal damage can indeed impair ganglionic neurotransmission in vivo and that decreased nicotinic acetylcholine receptor production is one likely mediator. These data imply that diseases characterized by sympathetic nerve terminal damage may also have an unrecognized defect in ganglionic neurotransmission that contributes to the impairment of the sympathetic pathway.
MATERIALS AND METHODS
Animals and Pretreatments
All experiments were performed on male Wistar rats (280-350 g; Simonsen Labs, Gilroy, CA) housed in groups under a 12-hr light/12-hr dark cycle. Rats had ad libitum access to pelleted chow, and only those included in the glucopenia studies were subjected to an overnight fast to minimize individual variability in basal plasma glucose levels. Water was available at all times.
6-Hydroxydopamine (6-OHDA; Sigma, Kansas City, MO) was administered to several groups of rats to selectively destroy sympathetic nerve terminals: five groups for the time course study of nerve growth factor (NGF; n = 4-6 rats per group); six groups for the time course study of the alpha-3 subunit of the nicotinic acetylcholine receptor (A3-NR), neurotrophin 4 (NT-4) and brain-derived neurotrophic factor (BDNF) expression (n = 3-5 rats per group); six groups for the study of chronic fos expression (n = 4 rats per group); two groups for the study of impaired acute fos mRNA response to nicotine (n = 6-8 rats per group); and one group each for the study of impaired Fos protein responses to nicotine and 2-DG (n = 4-6 rats per group).
6-OHDA was dissolved in saline and ascorbic acid (10%) immediately before intraperitoneal administration to rats (100 mg/kg), and care was taken to protect the solution from light in order to prevent degradation. This dose/route of 6-OHDA destroys approximately 90% of peripheral sympathetic nerve terminals (Mei et al., [2001]). Successful absorption of 6-OHDA was verified by the appearance of a ruffled coat and lethargy 15 min after injection and by weight loss 24 hr after injection. Acute, terminal studies were performed 1-9 days later. Neurally intact control rats received a vehicle pretreatment of saline and ascorbic acid (10%, 2 mL/kg, ip).
All rats included in these studies were certified as healthy by the veterinary medical officer and exhibited normal grooming and feeding behavior on the day of study. Research involving animals was conducted in an AAALAC-accredited facility, and all protocols were approved by the Institutional Animal Care and Use Committee of the Seattle VA Puget Sound Health Care System.
Experimental Design
First, we sought to determine the effect of sympathetic nerve terminal destruction on the ganglionic content of NGF protein and the ganglionic expression of NT-4, BDNF, and A3-NR, factors involved either directly or indirectly in ganglionic neurotransmission. Between 1 and 9 days following 6-OHDA pretreatment, rats were anesthetized with isofluorane, and a midline laparotomy was performed to harvest the celiac ganglion. A second incision was performed at the level of the bifurcation of the right carotid artery to harvest the superior cervical ganglion, which was analyzed for BDNF only. Celiac ganglia for NGF protein content measurement were snap-frozen on dry ice and stored at -80°C until assayed by ELISA (see below). Ganglia for A3-NR, NT-4, and BDNF mRNA measurement were placed in RNA-later (76104; Qiagen, Valencia, CA), placed in a refrigerator overnight, and then frozen at -80°C until assayed by RT-PCR (see below).
We compared the temporal changes in A3-NR expression following sympathetic nerve terminal destruction to the impaired ganglionic activation in response to a nicotine injection. However, before we could use the index of fos mRNA in the celiac ganglia to quantify successful neurotransmission in response to nicotine stimulation, we first needed to perform two control studies. In the first control study, we determined the time of peak fos mRNA expression after acute nicotine (2mg/kg sc, in 0.5 mL/kg); rats were sacrificed for celiac ganglia harvest either 15, 30, 45, or 60 min after nicotine injection. Successful nicotine absorption was verified by an immediate and pronounced Straubtail reaction. Ganglia were frozen in RNA-later for analysis of their fos mRNA. In the second control study, we determined the chronic effect of 6-OHDA alone on fos expression, independent of acute effects of nicotine stimulation. We treated rats with 6-OHDA (100 mg/kg, ip) and sacrificed them for celiac ganglia harvest either 1, 2, 3, 5, 7, or 9 days later. Again, ganglia were frozen in RNA-later and assayed for fos mRNA.
To identify the cell type responsible for chronic fos expression due to 6-OHDA alone (see the second control study, above), we harvested celiac ganglia 5 days after 6-OHDA; however, this time the ganglia were fixed and analyzed by immunohistochemical staining for Fos protein (described below).
Guided by the results of these two control studies, we sought to determine if the decrease in A3-NR mRNA induced by nerve terminal destruction was associated temporally with impaired ganglionic neurotransmission. Nicotine (2 mg/kg sc, in 0.5 mL/kg) was administered to neurally intact, vehicle-treated control rats and to rats that had nerve terminal damage caused by pretreatment with 6-OHDA, either 2 or 5 days previously; these 6-OHDA-pretreated rats were separate from those studied for effects on NGF content and A3-NR and NT-4 expression. Rats were anesthetized 30min after nicotine administration to capture the maximum increase in fos mRNA, as determined in the first control study above. Acute fos response to nicotine (fos) was calculated as the total fos mRNA 30 min after nicotine stimulation minus the chronic effect of 6-OHDA pretreatment to elevate fos mRNA in satellite cells, independent of nicotine.
To verify that the impaired fos mRNA response to nicotine following 6-OHDA was specific to principal ganglionic neurons, we repeated the nicotine study 5 days after 6-OHDA, this time using immunohistochemistry to localize and quantify successful neurotransmission in principal ganglia neurons. Five days after pretreatment with either saline or 6-OHDA, overnight fasted rats received a subcutaneous injection of nicotine (2 mg/kg in 0.5 mL/kg). After waiting 120 min for optimal production and nuclear translocation of Fos protein (>Koistinaho, [1991]), animals were briefly anesthetized with isofluorane (4% induction, 2% maintenance in O2), and the celiac ganglia were fixed in situ, as described previously (Mei et al., [2001]). Fixed ganglia were harvested and placed in 25% sucrose (0.01M PBS, pH 7.4) for overnight dehydration. The next day, the tissue was embedded in mounting medium (Tissue-Tek, Miles Inc., Elkhart, IN), frozen on dry ice, and stored at -80°C until it was sectioned and stained for nuclear Fos protein (see below).
We have previously demonstrated that central glucopenia, induced by 2-deoxyglucose (2-DG), reflexively activates pancreatic (Havel et al., [1988]) and hepatic (Mundinger et al., [1997]) sympathetic nerves, whose cell bodies reside primarily in the celiac ganglia (Quinson et al., [2001]). Therefore, 2-DG was used to physiologically activate celiac ganglia neurons (Mei et al., [2001]), and we tested for impairment of that activation following sympathetic nerve terminal destruction. Five days after pretreatment with either saline or 6-OHDA, overnight fasted rats received an intraperitoneal injection of 2-DG (200 mg/kg). After waiting 120 min, each animal was anesthetized, and a midline laparotomy was performed to expose the inferior vena cava. Vena caval blood for epinephrine determination was drawn on a mixture (20μL/mL blood) of EGTA (0.09 mg/mL) and glutathione (0.06 mg/mL). Samples were immediately placed on ice and centrifuged (3,000 rpm, 20 min, 3°C), and the plasma was frozen (-80°C) until assay. Immediately after blood sampling, celiac ganglia were fixed in situ, harvested, dehydrated, and stored, as described above. Later, they were sectioned and stained for nuclear Fos protein.
To demonstrate that ganglionic Fos responses to intraperitoneal 2-DG were due to central glucopenia and resultant activation of preganglionic nerves of the celiac ganglia, two groups of rats were given injections into the third cerebral ventricle via chronic catheter (Sipols et al., [1995]). On the day of the study, overnight fasted rats received either 2-DG or L-glucose, a nonmetabolizable isomer of glucose, in the third ventricle (50μmol in 10μL). The 10μL was given over 6 min to minimize acute fluctuations in cerebral ventricular pressure. One hundred and twenty minutes after injections, rats were anesthetized and laparotomized, a vena caval blood sample was taken, and ganglia were fixed in situ, as described above. Celiac ganglia were then harvested and stained for Fos protein, and blood was assayed for determination of plasma epinephrine level.
Tissue and Plasma Analysis
The NGF peptide content in whole ganglia was determined by ELISA. The kit (Immunoassay System #G7630, Promega, Madison, WI) uses a modification of the high-affinity two-site immunoassay originally described by Korsching and Thoenen ([1983b]). Peptide extractions and the assay were performed separately on each individual ganglion, without pooling.
The expression of mRNA for NT-4, BDNF, A3-NR, and fos in sympathetic ganglia was determined by RT-PCR. Individual celiac or superior cervical ganglia were homogenized-frozen in 600 L of RLT buffer (RNeasy Protect Mini Kit 74124; Qiagen, Valencia, CA). The tissue lysate was then centrifuged, and the supernatant was collected and applied to an RNeasy minicolumn (Qiagen) to purify the isolated RNA. RNA extraction was performed separately on each individual ganglion, without pooling. Total RNA from each ganglion was frozen at -80°C for later RT-PCR.
To obtain cDNA for NT-4, A3-NR, BDNF, or fos, total RNA in the samples was reverse-transcribed into single-strand cDNA using an Applied Biosystems (Foster City, CA) High Capacity cDNA Archive Kit (product 4322171), as we have done previously (Mei et al., [2006b]). Quantitative RT-PCR was performed using an ABI Prism 7000 sequence detection system (Applied Biosystems) and the Taqman MGB probe Assay on Demand for A3-NR, BDNF, and fos geneexpression (Applied Biosystems Rn00583820_ml, Rn00560868_ml, and Rn02396739_ml, respectively); the rat NT-4 mix was designed using Primer Express Software (Applied Biosystems). Taqman rodent glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Applied Biosystems 4308313) was used as an endogenous control to ensure that 6-OHDA or nicotine treatment affected only NT-4, BDNF, A3-NR, and fos mRNA expression. The PCR reaction was performed at 50°C for 2 min and then at 95°C for 10 min, followed by 40 cycles at 95°C for 15 sec and 60°C for 1 min. In an attempt to reliably measure BDNF mRNA, these particular runs were extended to 50 cycles. The cycle threshold (CT) for NT-4, BDNF, A3-NR, or fos and their controls was determined using Taqman SDS software. NT-4, BDNF, A3-NR, or fos mRNA was expressed relative to GAPDH as ΔCT for each ganglion to correct for any differences in either size or extraction/purification/transcription efficiency between ganglia. The difference in NT-4, BDNF, A3-NR, or fos expression between experimental and control groups was calculated as ΔΔCT. The change in NT-4, BDNF, A3-NR, or fos expression (relative to the control) in 6-OHDA rats was calculated as 2ΔΔCT using a method previously described in detail (Livak and Schmittgen, [2001]).
Because the NT-4 primer/probe was designed specifically for these studies, we tested its amplification efficiency. We found that the efficiency of the NT-4 amplification was approximately half that of GAPDH; therefore, we corrected all NT-4 CT values, as directed by the producers of the primer/probe (Applied Biosystems). Although this correction factor had a large effect on both absolute NT-4 CT and ΔCT values in a sample, it had only a minor effect on both the ΔΔCT value and the calculated NT-4 expression relative to the control.
Immunohistochemistry was performed to quantify nuclear Fos protein responses to ganglionic stimulation by nicotine, 2-DG ip, or 2-DG 3cv. We have previously described in detail the method of identifying and manually quantifying Fos-positive nuclei of principal ganglionic cells in the celiac ganglia (Mei et al., [2001]).
Statistics
In the two studies in which only two groups were compared, nuclear Fos and epinephrine responses to 2-DG and nicotine in 6-OHDA-pretreated rats were compared to those of their saline-pretreated controls using a two-sample t test. In all other studies, experimental values were compared to controls using a one-way analysis of variance to test for an overall effect and using a post hoc Dunnett’s test to determine significance between control and experimental groups. All data are expressed as means ± SEMs.
RESULTS
Nerve Terminal Damage Decreases Nerve Growth Factor Content But Not Neurotrophin-4 or Brain-Derived Neurotrophic Factor Expression in Celiac Ganglia
To investigate the effect of nerve terminal damage on mediators of ganglionic neurotransmission, we measured the effect of the sympathetic nerve terminal toxin 6-hydroxydopamine (6-OHDA) on the ganglionic content of nerve growth factor (NGF) and expression of neurotrophin-4 (NT-4) and brain-derived neurotrophic factor (BDNF). These neurotrophins are thought to have post- and presynaptic trophic effects, respectively. NGF is produced in and secreted by sympathetically innervated tissue. This prototypic neurotrophin then binds the trk-A receptor on sympathetic nerve terminals, and the bound NGF/trk-A complex is retrogradely transported to principal ganglionic cell bodies in sympathetic ganglia. We therefore expected nerve terminal damage to decrease ganglionic NGF protein content. Indeed, 6-OHDA decreased the NGF content in the celiac ganglia (P < 0.001 overall; Fig. 1): NGF content in the celiac ganglion of neurally intact, vehicle-treated rats was 286 ± 25 pg/ganglion (n = 10), and this level was reduced to 136 ± 22 pg/ganglion 1 day after treatment with 6-OHDA (n = 6, P < 0.01 versus neurally intact). A nadir was reached 2 days after 6-OHDA treatment (105 ± 11 pg/ganglion, n = 4, P < 0.01), and significant reduction in NGF content was still present 8 days after nerve terminal destruction (n = 5, P < 0.01). Thus, sympathetic nerve terminal destruction deprives the corresponding neuronal cell bodies in the celiac ganglia of a trophic factor thought to maintain ganglionic neurotransmission.
Figure 1.

Nerve growth factor (NGF) protein content in the celiac ganglion (CG) was reduced following treatment with 6-hydroxy-dopamine (6-OHDA). Each data point represents a separate group of rats. In all figures, data are expressed as means ± SEMs.
Although NGF is transported to neuronal cell bodies in peripheral sympathetic ganglia, NT-4 is produced there (Roosen et al., [2001]). We therefore measured NT-4 mRNA, not protein, in the celiac ganglia, both in the presence and the absence of nerve terminal destruction. To our surprise, 6-OHDA did not decrease NT-4 expression in the celiac ganglia. In fact, NT-4 expression was elevated in the celiac ganglia following 6-OHDA (P < 0.05 overall). One day after 6-OHDA, NT-4 expression was increased 4-to 5-fold (P < 0.05 versus vehicle treatment; Fig. 2), and it was also elevated 9 days after treatment with 6-OHDA (P < 0.05). Thus, if nerve terminal damage impairs ganglionic neurotransmission, ganglionic NT-4 deficiency is unlikely to be a mediator.
Figure 2.

Neurotrophin-4 (NT-4) mRNA content in the CG was not reduced following treatment with 6-OHDA.
Because knocking out BDNF in mice produces effects similar to knocking out NT-4, we measured BDNF mRNA in celiac ganglia, both in the presence and the absence of nerve terminal destruction. To our surprise, BDNF mRNA expression was not consistently measurable in celiac ganglia from control animals, even after extending the RT-PCR amplification to 50 cycles: only 19% of the samples from rats pretreated with vehicle yielded a value above background. In celiac ganglia from animals treated with 6-OHDA, however, BDNF mRNA was more consistently measurable (100% and 75% above background in rats pretreated with 6-OHDA 7 and 9 days earlier, respectively), suggesting that BDNF expression probably increased, but certainly did not decrease, following nerve terminal damage. Using the superior cervical ganglion as a positive control (data not shown), we demonstrated (1) the ability to detect BDNF mRNA in peripheral sympathetic ganglia, (2) that BDNF expression in superior cervical ganglia did not decrease following treatment with 6-OHDA, and (3) that there was 50-to 100-fold less BDNF mRNA than NT-4 mRNA in control superior cervical ganglia. With very low levels of celiac ganglion BDNF mRNA and no hint of a decrease in the ganglionic content of either BDNF or NT-4 mRNA after 6-OHDA, trk-B-mediated impairment in preganglionic innervation is unlikely to be produced by nerve terminal damage.
Nerve Terminal Damage Decreases Nicotinic Receptor Expression in Celiac Ganglia
Next, we sought to determine if the reduction of NGF content in the celiac ganglia was associated with a decrease in functional nicotinic acetylcholine receptors, the classical mediators of neurotransmission in sympathetic ganglia. First, we measured the effect of 6-OHDA on the ganglionic expression of the alpha-3 subunit of the nicotinic acetylcholine receptor (A3-NR) because this subunit is essential for neurotransmission in sympathetic ganglia (Xu et al., [1999]); later, we measured the response of the ganglionic neuron to direct activation of these receptors with nicotine. A3-NR expression in the celiac ganglia was reduced following nerve terminal destruction (P < 0.001 overall; Fig. 3), but it was not significant until the third day after treatment with 6-OHDA (-49%, P < 0.01 versus vehicle treatment). Thereafter, A3-NR subunit expression remained low up to 7 days after 6-OHDA (P < 0.01) and tended to be low at 9 days. It is likely that the decrease in the A3-NR message results in a decrease in A3-NR protein, a critical protein for neurotransmission. If so, there is a rationale for testing for impaired ganglionic neurotransmission following 6-OHDA.
Figure 3.

Alpha-3 subunit of nicotinic acetylcholine receptor (A3-NR) mRNA content in the CG was reduced following treatment with 6-OHDA.
fos Expression in Celiac Ganglia: Acute Stimulation by Nicotine and Chronic Stimulation by 6-OHDA
To directly test whether the observed deficiency in A3-NR expression following 6-OHDA was sufficient to impair ganglionic neurotransmission, we directly activated those receptors with subcutaneous injections of nicotine and measured the fos mRNA response in the celiac ganglia. However, we first had to determine the optimal time after nicotine administration to harvest celiac ganglia for RT-PCR analysis. We did so by measuring fos mRNA every 15 min after subcutaneous nicotine administration for 1 hr. Fos mRNA in the celiac ganglia increased 15 min after subcutaneous nicotine (n = 6, P < 0.01 versus saline treatment; Fig. 4A), peaked at 30 min (P < 0.01), and then returned toward baseline levels after 45 and 60 min. Therefore, in all subsequent studies, we harvested celiac ganglia 30 min after nicotine administration.
Figure 4.


A: Time course of acute stimulation of fos mRNA in the CG by nicotine. B: Time course of chronic stimulation of fos mRNA in the CG by 6-OHDA.
Second, we needed to determine the chronic effect of 6-OHDA treatment per se on fos mRNA expression, independent of acute nicotine stimulation. We therefore measured fos mRNA in rats treated only with 6-OHDA. 6-OHDA, in the absence of nicotine, chronically stimulated fos mRNA in the celiac ganglia (P < 0.01 overall, Fig. 4B): fos mRNA increased 1 day after nerve terminal destruction (9.51-fold ± 4.00-fold over saline treatment, n = 4, P < 0.05), remained elevated for 5 days, and tended to recover thereafter. Of note, fos mRNA expression was elevated 8.92-fold ± 2.66-fold and 9.13-fold ± 2.41-fold over control 2 and 5 days after 6-OHDA, respectively. This chronically elevated fos mRNA was accounted for when calculating fos responses to acute nicotine stimulation in 6-OHDA-pretreated rats (see below).
To determine the cell type responsible for the increased fos mRNA following 6-OHDA, we stained celiac ganglia sections for Fos protein in rats with and without nerve terminal damage. Rats treated with 6-OHDA 5 days previously had Fos-positive staining in small cells adjacent to the large principal ganglia cells; presumably, these small cells were satellite cells. Rats pretreated with vehicle had no such nonneuronal Fos staining (Fig. 5). Both saline-and 6-OHDA-pretreated rats lacked positive Fos protein staining in principal ganglia cells. Therefore, the chronic effect of 6-OHDA alone to increase fos mRNA in rat celiac ganglia is likely a result of increased fos expression restricted to nonprincipal ganglia cells.
Figure 5.
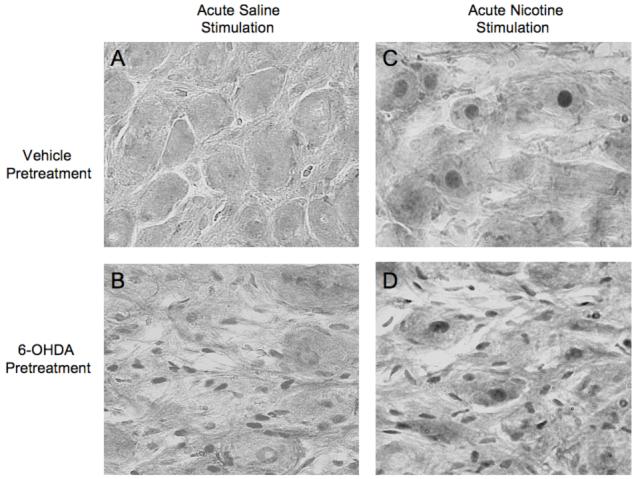
A: Vehicle pretreatment does not produce a chronic Fos response in either presumed satellite cells or principal neurons of the CG. B: 6-OHDA pretreatment produces a chronic Fos response restricted to the nuclei of presumed satellite cells of the CG, with no Fos observed in the nuclei of principal ganglion neurons. C: Acute nicotine stimulation induces Fos staining restricted to the nuclei of principal ganglia neurons, with no Fos staining in presumed satellite cells. D: In rats pretreated with 6-OHDA and given nicotine acutely, Fos is in the nuclei of both presumed satellite cells and principal neurons.
Nerve Terminal Damage Impairs Celiac Ganglia fos mRNA and Fos Protein Responses to Nicotine
To determine if nerve terminal damage impairs ganglionic neurotransmission, a process that requires functional nicotinic acetylcholine receptors, we calculated celiac ganglia fos mRNA responses to nicotine stimulation in rats with and without prior treatment with 6-OHDA. Furthermore, to temporally relate impaired receptor activation to decreases of A3-NR expression, we performed these nicotine studies both 2 and 5 days after 6-OHDA, when there is normal and deficient A3-NR expression in the celiac ganglia, respectively (see Fig. 3). In neurally intact rats, nicotine stimulation produced a 30.16-fold ± 3.43-fold increase in fos mRNA expression over saline-stimulated controls (n = 6; Fig. 6). Because these rats were not pretreated with 6-OHDA, this increase was solely a result of acute stimulation by nicotine. Two days after nerve terminal destruction with 6-OHDA, when A3-NR expression in the celiac ganglia was not reduced (see Fig. 3), fos mRNA expression after nicotine stimulation were increased 42.26-fold ± 4.68-fold over saline controls. Subtracting the chronic effects of 6-OHDA per se on satellite cell fos mRNA (8.92-fold, 2 days after 6-OHDA), the change in fos mRNA because of acute nicotine stimulation of principal ganglia cells (fos) was 33.34-fold ± 4.68-fold greater (n = 8; Fig. 6), a response not significantly different from the 30.16-fold ± 3.43-fold increased response observed in neurally intact rats. Therefore, the fos response in celiac ganglia to nicotine stimulation was not impaired during after 6-OHDA when A3-NR expression was normal. However, 5 days after nerve terminal destruction, when A3-NR expression in the celiac ganglia was reduced, fos mRNA expression after nicotine stimulation was 26.82-fold ± 1.88-fold greater than that of the saline controls. Subtracting the chronic effects of 6-OHDA on satellite cells (9.13-fold, 5 days after 6-OHDA), the fos mRNA response of principal ganglia cells to acute nicotine stimulation was 17.69-fold ± 1.88-fold greater, a marked impairment compared with the 30.16-fold ± 3.43-fold greater response in neurally intact rats, (P < 0.01; Fig. 6). Thus, activation of celiac ganglia neurons by nicotine was impaired only when ganglionic A3-NR expression was suppressed, suggesting that A3-NR deficiency contributes to impaired ganglionic neurotransmission seen after nerve terminal destruction.
Figure 6.

Acute fos mRNA response to nicotine in the CG (Δfos) was impaired by 6-OHDA pretreatment 5 days, but not 2 days, earlier. Δfos to nicotine per se in 6-OHDA pretreated rats was calculated by subtracting the chronic effect of 6-OHDA on fos (see Fig, 4B) from the total fos seen 30 min after nicotine stimulation. See the Results section for further details of the calculation (*significantly different from saline pretreatment).
To verify that the impaired fos mRNA response to nicotine following 6-OHDA was specific to principal ganglionic neurons, we repeated the nicotine study 5 days after 6-OHDA, this time counting Fos-positive nuclei in principal ganglia neurons using immunohistochemistry. In animals pretreated with vehicle 5 days previously, the acute Fos response to nicotine stimulation was 5.21 ± 1.50 Fos-positive nuclei/mm2 (n = 5, Fig. 7). As expected, the acute Fos response to nicotine stimulation was significantly reduced in rats pretreated 5 days earlier with 6-OHDA (0.96 ± 0.55 Fos-positive nuclei/mm2, P < 0.05, n = 6; Fig. 7). Thus, in two separate experiments using two separate indices of principal ganglia activation by nicotine, activation of principal ganglia neurons in the celiac ganglia was impaired 5 days after nerve terminal damage, a time when A3-NR was suppressed.
Figure 7.

Acute Fos protein response to nicotine in the nuclei of principal neurons of the CG was impaired by 6-OHDA pretreatment 5 days earlier (*significantly different from saline pretreatment).
Nerve Terminal Damage Impairs the Celiac Ganglia Fos Response to Neuroglucopenia
To determine if physiologically stimulated neurotransmission would also be impaired following nerve terminal damage, we induced moderate neuroglucopenia and measured the Fos responses in principal neurons of the celiac ganglia in rats with and without 6-OHDA treatment 5 days previously. Rats pretreated with vehicle that received an acute intraperitoneal injection of the glucopenic agent 2-deoxy-D-glucose (2-DG) had 9.71 ± 2.43 Fos-positive neurons/mm2 (n = 5; Fig. 8), confirming the moderate activation of celiac ganglia neurons during glucopenic stress we have shown previously (Mei et al., [2001]). In rats whose nerve terminals were damaged by pretreatment 5 days earlier with 6-OHDA, the celiac ganglia Fos response to 2-DG was reduced to only 2.34 ± 1.08 Fos-positive neurons/mm2 (n = 7, P < 0.005 versus vehicle pretreatment; Fig. 8). Thus, the ability of principal ganglia neurons to activate during physiologic stimulation was markedly impaired following postganglionic nerve terminal damage.
Figure 8.

Acute Fos protein response to 2-deoxy-glucose (2-DG) in principal neurons of the CG was impaired by 6-OHDA pretreatment 5 days earlier (*significantly different from saline pretreatment).
We assumed that this impaired ganglionic activation was a result of decreased responsiveness of the ganglionic cell bodies. To rule out the alternative, namely, that 6-OHDA treatment had suppressed sympathetic outflow from the brain, we measured epinephrine levels in peripheral plasma during neuroglucopenia. Epinephrine levels in vehicle pretreated rats was 2,108 ± 476 pg/mL following 2-DG, indicative of reflexive activation of the sympatho-adrenal system during neuroglucopenia (Havel et al., [1988]; Cryer, [1993]; Veneman et al., [1994]; Mundinger et al., [1997]). As expected, the epinephrine response to 2-DG was not impaired in rats pretreated with 6-OHDA (2,427 ± 610 pg/mL, P = NS vs. vehicle pretreatment), suggesting normal sympathetic outflow from the brain to the abdomen in animals with peripheral sympathetic nerve terminal damage.
To verify that activation of celiac ganglia neurons during intraperitoneal 2-DG was initiated by central rather than peripheral ganglionic glucopenia, we measured Fos expression in the celiac ganglia in response to intracerebral ventricular administration of 2-DG. 2-DG given in the third ventricle of neurally intact rats produced 7.41 ± 0.96 Fos-positive neurons/mm2 in the celiac ganglion (n = 8), a level similar to that achieved during our intraperitoneal 2-DG administration. In contrast, an equiosmolar dose of L-glucose given in the third ventricle produced no significant activation (0.16 ± 0.16 Fos-positive neurons/mm2, n = 6, P < 0.001 versus 2-DG 3cv). Likewise, plasma epinephrine levels during third-ventricular 2-DG administration (1,740 ± 974 pg/mL) tended to be greater than during L-glucose (547 ± 248 pg/mL). Thus, central 2-DG administration increases sympathetic outflow to and results in the activation of principal neurons in the celiac ganglia. It is likely that intraperitoneal 2-DG does the same.
DISCUSSION
Both nerve terminal damage and axotomy increase neuropeptide expression in the nerve cell body, but the effect of nerve terminal damage is smaller. We therefore asked if nerve terminal damage would be sufficient to produce impairment of ganglionic neurotransmission similar to that produced by axotomy. The present study has demonstrated that damage to sympathetic nerve terminals is indeed sufficient to impair ganglionic neurotransmission. Furthermore, we found a decrease in the expression and function of ganglionic nicotinic receptors that suggested a likely mechanism for this cell body dysfunction. These findings imply that diseases characterized by partial loss of sympathetic nerve terminals (Mathes et al., [1971]; Kozlovskis et al., [1986]; Konttinen et al., [1992]; Langer et al., [1995]; Schmid et al., [1999]; Camargos et al., [2000]; Jacob et al., [2000]; Mei et al., [2002], [2006a]) may have a larger defect in the overall sympathetic pathway than that estimated by the degree of peripheral nerve terminal loss alone.
To investigate the effect of nerve terminal damage on mediators of ganglionic function, we looked for 6-hydroxydopamine (6-OHDA)-induced deficiencies in ganglionic neurotrophins. One such neurotrophin is neurotrophin-4 (NT-4), which is secreted constitutively (Hibbert et al., [2003]) by sympathetic ganglionic neurons into the synaptic cleft, is bound by -trk-B receptors (Ip et al., [1993]) on preganglionic nerve terminals, and is then retrogradely transported to preganglionic cell bodies located in the intermediolateral horn of the spinal cord. There, NT-4 supports not only spinal neuron survival but also their function by maintaining the connectivity of preganglionic nerve terminals with the dendrites of sympathetic ganglia neurons. We therefore reasoned that NT-4 deficiency could impair ganglionic neurotransmission by reducing both the number of synapses and the amount of neurotransmitter released by the preganglionic nerve terminals in the ganglia. However, to our knowledge, there are no published studies demonstrating decreased ganglionic NT-4 expression following nerve terminal damage. We found that NT-4 mRNA in the celiac ganglia was modestly elevated not decreased following 6-OHDA. Thus, if nerve terminal damage does decrease preganglionic innervation, it is not because of NT-4 insufficiency.
It could also be posited that there is a similar preganglionic role for brain-derived neurotrophic factor (BDNF), another neurotrophin using the trk-B receptor to enter and influence preganglionic spinal nerves. However, we found that the expression of BDNF mRNA in celiac ganglia, located in the peritoneum, of our control animals was not consistently detectable, despite extending the RT-PCR amplification. The frequency of detection increased in celiac ganglia harvested from animals 5 or more days after 6-OHDA, suggesting increased not decreased BDNF expression after nerve terminal destruction. To validate our ability to measure BDNF expression, we sought to confirm earlier reports of BDNF mRNA in another sympathetic ganglia, the superior cervical ganglia (Wetmore and Olson, [1995]). In this cervical sympathetic ganglion, we consistently detected BDNF mRNA, suggesting that ganglionic BDNF expression is regionally specific. We also found that BDNF expression in the superior cervical ganglia tended to increase after 6-OHDA, supporting our finding in the celiac ganglia. Because NT-4 and BDNF are taken up by trk-B receptors on preganglionic nerves and because neither NT-4 nor BDNF decreases in the celiac ganglia after 6-OHDA, it is unlikely that nerve terminal damage produces trk-B-mediated defects in the preganglionic cell bodies, axons, or synaptic contacts sufficient to impair neurotransmission.
In contrast, trk-A-mediated defects in principal ganglionic neurons are likely because nerve growth factor (NGF) is taken up into ganglionic neurons by this receptor on its terminals and because the NGF protein content in the celiac ganglia was decreased following 6-OHDA. However, the nadir of celiac ganglia NGF content was only 37% of the control, which is much less than the 3% of the control found in the superior cervical ganglia following axotomy (Zhou et al., [1994]). Thus, although it has been demonstrated that the near-total NGF deficit after axotomy was sufficient to impair ganglionic function, studies were needed to determine if the smaller NGF deficiency in the celiac ganglia following sympathetic nerve terminal damage was sufficient to do the same.
Ganglionic NGF deficiency has the ability to produce two specific postsynaptic defects that could contribute to impaired ganglionic neurotransmission. The first is dendritic retraction (Snider, [1988]), a mechanism not investigated in the present study. The second is down-regulation of nicotinic acetylcholine receptors, which we did investigate. The near-total decrease of NGF content in the superior cervical ganglia following axotomy and explantation is associated with a decrease in both ganglionic mRNA (Zhou et al., [1998]) and ganglionic protein content (Yeh et al., [2001]; Zhou et al., [2001]) of several subunits of the nicotinic acetylcholine receptor. Furthermore, the in vitro addition of NGF to the media of explanted ganglia partially prevents the decrease in the number of alpha-3 subunits of the nicotinic acetylcholine receptor (A3-NR; Yeh et al., [2001]), a subunit required for neurotransmission in sympathetic ganglia (Xu et al., [1999]; Yeh et al., [2001]). Our finding that expression of A3-NR was decreased in the celiac ganglia following treatment with 6-OHDA suggests that the less severe NGF deficit produced in the celiac ganglia following nerve terminal damage was sufficient to decrease A3-NR production. However, our data show only an associative relationship between decreased NGF and decreased A3-NR production, and an interventional study demonstrating that ganglionic NGF treatment can prevent the decrease of A3-NR is needed to prove causality. To determine if this degree of A3-NR deficiency actually impairs celiac ganglia neurotransmission, we compared the acute responses of principal ganglia neurons to nicotine-stimulated neurotransmission in saline- and 6-OHDA pretreated rats. We chose fos mRNA expression as the index of neuronal activation in these studies, as opposed to the more traditional Fos protein, because RT-PCR analysis of whole ganglia homogenates can be more precisely quantified, is representative of the average response in a ganglia, and avoids the subjective factors involved in quantifying immunohistochemical staining.
However, before we could use fos mRNA as an index of neuronal activation, we needed to perform additional control studies to determine (1) the time course of acute fos mRNA responses to nicotine and (2) the magnitude of chronic fos mRNA responses to 6-OHDA pretreatment per se, independent of acute stimulation. It is well established that the nuclear Fos protein response in both the central and peripheral (Koistinaho, [1991]; Mei et al., [2001]) nervous systems occurs approximately 2 hr after neuronal activation. This interval includes time for fos transcription and translation, as well as for translocation of Fos protein from the endoplasmic reticulum to the nucleus. Because the appearance of fos mRNA requires only transcription, the maximum fos mRNA response to nicotine in our first control study was reached earlier than that of Fos protein: fos mRNA in the celiac ganglia increased rapidly to and quickly recovered from its maximum 30 min following nicotine injection. Therefore, in the remaining studies measuring acute fos mRNA responses, we harvested ganglia 30 min after stimulation. Our second control study showed that 6-OHDA alone has a chronic effect of elevating fos mRNA in the celiac ganglia, independent of any acute stimulation. We then used immunohistochemistry to identify the cell populations responsible for this increased fos expression. 6-OHDA induced chronic Fos expression only in small, nonneuronal cells. In theory, these could be satellite cells, Schwann cells, and/or macrophages. However, we observed no Fos staining around axonal trunks exiting the ganglia, suggesting little if any Schwann cell activation. Although macrophages migrate into sympathetic ganglia following treatment with 6-OHDA (Schreiber et al., [1995]), it is not known if they express Fos at this stage. Last, we regularly observed nonneuronal staining clearly “ringing” principal neurons, suggesting that at least some of the activated cells after 6-OHDA are satellite cells. The finding that 6-OHDA chronically elevates Fos in what we believe are satellite cells is consistent with the known effects of distal axon injury to chronically elevate Fos protein production in Schwann cells adjacent to the insult (Plantinga et al., [1994]); we extend this concept to include satellite cells, the specialized Schwann cells surrounding principal ganglionic neurons. The chronically elevated Fos in these presumed satellite cells following 6-OHDA is also consistent with the finding that satellite cell protrusions grow into the synaptic cleft following axotomy, finalizing the physical separation of sympathetic ganglionic neurons from their presynaptic innervation (Matthews and Nelson, [1975]). However, the most important point for our studies is that Fos protein production following 6-OHDA alone was restricted to nonneuronal cells; it was not elevated in principal ganglia neurons. Therefore, in later studies where rats received acute nicotine stimulation after pretreatment with 6-OHDA, we subtracted the chronic nonneuronal fos mRNA response because of 6-OHDA alone. In doing so, we generated an index for acute fos mRNA responses that is specific to principal ganglia cells.
Having performed these necessary control studies, we tested our major hypothesis that nerve terminal damage and the resultant A3-NR deficiency are sufficient to impair ganglionic neurotransmission in vivo. To do so, we took advantage of the delay between treatment with 6-OHDA and the decrease in A3-NR mRNA observed in the celiac ganglia. Specifically, we looked for impaired ganglionic neurotransmission 5 days after 6-OHDA, when A3-NR mRNA was reduced, and a lack of impairment 2 days after 6-OHDA, when A3-NR mRNA was not yet reduced. We chose to directly stimulate ganglia neurons with nicotine administration not only to examine the function of their nicotinic acetylcholine receptors but also to avoid any impairment because of dendritic retraction (i.e., increased synaptic distance) that may have been produced by ganglionic NGF deficiency. As expected, fos mRNA responses to nicotine in the celiac ganglia were impaired only when A3-NR expression was reduced. Thus, we conclude that nerve terminal damage impairs ganglionic neurotransmission, and it is likely that nicotinic receptor deficiency is a mediator.
Although the index of fos mRNA of whole ganglia homogenates has the benefits of precise quantification and avoids intraganglionic variations, the alternative index of Fos immunohistochemistry is superior in defining the cell type that has been activated. Therefore, in our last two studies, we used Fos immunohistochemistry in principal ganglia cells to verify impaired ganglionic neurotransmission following nerve terminal damage. Indeed, the degree of impaired Fos protein response to nicotine following 6-OHDA was at least as large as that defined using fos mRNA. In our last study, we chose the stimulus of 2-deoxyglucose (2-DG)-induced glucopenia because it is known to reflexively activate the celiac ganglia (Mei et al., [2001]), especially those neurons projecting to the pancreas (Havel et al., [1988]) and liver (Mundinger et al., [1997]), and because it is a physiological activator of principal ganglia cells. The Fos protein response in the celiac ganglia to 2-DG-induced glucopenia was markedly impaired by pretreatment with 6-OHDA 5 days earlier, a time when A3-NR is decreased. Because the epinephrine response to peripheral 2-DG was equivalent in rats with or without nerve terminal damage, we conclude that 6-OHDA did not impair central sympathetic outflow. This conclusion is consistent with previous reports demonstrating that 6-OHDA does not cross the blood-brain barrier. In addition, 6-OHDA does not damage either the adrenal medulla or cholinergic nerves (Kostrzewa and Jacobowitz, [1974]), such as those innervating principal neurons in sympathetic ganglia. In a separate experiment, we administered 2-DG into the third cerebral ventricle and reproduced both adrenal medullary activation and the celiac ganglia Fos response seen during peripheral 2-DG. Therefore, the Fos responses in the celiac ganglia to glucopenia were a result of a central action of 2-DG that leads to activation of the preganglionic nerves innervating the celiac ganglia. The impaired Fos response following nerve terminal damage was a result of defective neurotransmission at principal nerve cell bodies. Our nicotine studies suggest that nicotine receptor deficiencies contribute to impaired neurotransmission during glucopenia, but further studies are needed to determine if decreased synaptic contact secondary to NGF deficiency and dendritic retraction also contributes.
We conclude that damage to sympathetic nerve terminals is sufficient to impair ganglionic neurotransmission in vivo and that this impairment is mediated, at least in part, by a decrease in functional nicotinic acetylcholine receptors in the ganglia. Neither NT-4 nor BDNF deficiency mediates this ganglionic impairment following nerve terminal damage. These studies imply that the ial loss of sympathetic nerve terminals seen in several diseases (Mathes et al., [1971]; Kozlovskis et al., [1986]; Konttinen et al., [1992]; Langer et al., [1995]; Schmid et al., [1999]; Camargos et al., [2000]; Jacob et al., [2000]) including autoimmune type 1 diabetes (Mei et al., [2002], [2006a]; Mundinger et al., [2003]) may induce an additional defect in ganglionic neurotransmission that contributes to total sympathetic neuropathy. Consequently, treatments designed to alleviate sympathetic dysfunction in these diseases may benefit from treating the nicotinic receptor deficiency in their respective ganglia in addition to treating the nerve terminal loss.
Acknowledgements
We thank Daryl Hackney for ganglionic Fos quantification; Jira Wade for catecholamine measurements; Dottie Winch, Kate Cueno, and Liz Terrill for ELISA measurements; Breanne Barrow and Emily Haines for RTPCR measurements; and Dr. Al Sipols for 3cv cannulae implantation and central infusion of 2-DG.
Funded by:
■ Medical Research Service of the Department of Veterans Affairs
■ National Institute of Health; Grant Number: R01 DK 50,154, P30 DK 17,047
REFERENCES
- Camargos ER, Franco DJ, Garcia CM, Dutra AP, Teixeira AL, Jr, Chiari E, Concei, Machado CR. Infection with different Trypanosoma cruzi populations in rats: myocarditis, cardiac sympathetic denervation, and involvement of digestive organs. Am J Trop Med Hyg. 2000;62:604–612. doi: 10.4269/ajtmh.2000.62.604. [DOI] [PubMed] [Google Scholar]
- Causing C, Gloster A, Aloyz R, Bamji S, Chang E, Fawcett J, Kuchel G, Miller F. Synaptic innervation density is regulated by neuron-derived BDNF. Neuron. 1997;18:257–267. doi: 10.1016/s0896-6273(00)80266-4. [DOI] [PubMed] [Google Scholar]
- Cryer PE. Hypoglycemia begets hypoglycemia in IDDM. Diabetes. 1993;42:1691–1693. doi: 10.2337/diab.42.12.1691. [DOI] [PubMed] [Google Scholar]
- De Castro F, Sanchez Vives MV, Munoz Martinez EJ, Gallego R. Effects of postganglionic nerve section on synaptic transmission in the superior cervical ganglion of the guinea-pig. Neuroscience. 1995;67:689–695. doi: 10.1016/0306-4522(95)00079-x. [DOI] [PubMed] [Google Scholar]
- Gatzinsky KP, Haugland RP, Thrasivoulou C, Orike N, Budi-Santoso AW, Cowen T. p75 and TrkA receptors are both required for uptake of NGF in adult sympathetic neurons: use of a novel fluorescent NGF conjugate. Brain Res. 2001;920:226–238. doi: 10.1016/s0006-8993(01)03099-2. [DOI] [PubMed] [Google Scholar]
- Habecker BA, Gritman KR, Willison BD, Van Winkle DM. Myocardial infarction stimulates galanin expression in cardiac sympathetic neurons. Neuropeptides. 2005;39:89–95. doi: 10.1016/j.npep.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Havel PJ, Veith RC, Dunning BE, Taborsky GJ., Jr. Pancreatic noradrenergic nerves are activated by neuroglucopenia but not hypotension or hypoxia in the dog: Evidence for stress-specific and regionally-selective activation of the sympathetic nervous system. J Clin Invest. 1988;82:1538–1545. doi: 10.1172/JCI113763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibbert AP, Morris SJ, Seidah NG, Murphy RA. Neurotrophin-4, alone or heterodimerized with brain-derived neurotrophic factor, is sorted to the constitutive secretory pathway. J Biol Chem. 2003;278:48129–48136. doi: 10.1074/jbc.M300961200. [DOI] [PubMed] [Google Scholar]
- Hyatt-Sachs H, Bachoo M, Schreiber R, Vaccariello SA, Zigmond RE. Chemical sympathectomy and postganglionic nerve transection produce similar increases in galanin and VIP mRNA but differ in their effects on peptide content. J Neurobiol. 1996;30:543–555. doi: 10.1002/(SICI)1097-4695(199608)30:4<543::AID-NEU9>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Ip NY, Li Y, Yancopoulos GD, Lindsay RM. Cultured hippocampal neurons show responses to BDNF, NT-3, and NT-4, but not NGF. J Neurosci. 1993;13:3394–3405. doi: 10.1523/JNEUROSCI.13-08-03394.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob G, Costa F, Shannon JR, Robertson RM, Wathen M, Stein M, Biaggioni I, Ertl A, Black B, Robertson D. The neuropathic postural tachycardia syndrome. N Engl J Med. 2000;343:1008–1014. doi: 10.1056/NEJM200010053431404. [DOI] [PubMed] [Google Scholar]
- Koistinaho J. Nicotine-induced Fos-like immunoreactivity in rat sympathetic ganglia and adrenal medulla. Neurosci Lettr. 1991;128:47–51. doi: 10.1016/0304-3940(91)90757-k. [DOI] [PubMed] [Google Scholar]
- Konttinen YT, Hukkanen M, Kemppinen P, Segerberg M, Sorsa T, Malmstrom M, Rose S, Itescu S, Polak JM. Peptide-containing nerves in labial salivary glands in Sjogren’s syndrome. Arthritis Rheum. 1992;35:815–820. doi: 10.1002/art.1780350717. [DOI] [PubMed] [Google Scholar]
- Korsching S, Thoenen H. Nerve growth factor in sympathetic ganglia and corresponding target organs of the rat: correlation with density of sympathetic innervation. Proc Natl Acad Sci U S A. 1983a;80:3513–3516. doi: 10.1073/pnas.80.11.3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsching S, Thoenen H. Quantitative demonstration of the retrograde axonal transport of endogenous nerve growth factor. Neurosci Lett. 1983b;39:1–4. doi: 10.1016/0304-3940(83)90155-6. [DOI] [PubMed] [Google Scholar]
- Korsching S, Thoenen H. Treatment with 6-hydroxydopamine and colchicine decreases nerve growth factor levels in sympathetic ganglia and increases them in the corresponding target tissues. J Neurosci. 1985;5:1058–1061. doi: 10.1523/JNEUROSCI.05-04-01058.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostrzewa RM, Jacobowitz DM. Pharmacological actions of 6-hydroxydopamine. Pharmacological Reviews. 1974;26:199–288. [PubMed] [Google Scholar]
- Kozlovskis PL, Fieber LA, Bassett AL, Cameron JS, Kimura S, Myerburg RJ. Regional reduction in ventricular norepinephrine after healing of experimental myocardial infarction in cats. J Mol Cell Cardiol. 1986;18:413–422. doi: 10.1016/s0022-2828(86)80904-x. [DOI] [PubMed] [Google Scholar]
- Landry M, Holmberg K, Zhang X, Hokfelt T. Effect of axotomy on expression of NPY, galanin, and NPY Y1 and Y2 receptors in dorsal root ganglia and the superior cervical ganglion studied with double-labeling in situ hybridization and immunohistochemistry. Exp Neurol. 2000;162:361–384. doi: 10.1006/exnr.1999.7329. [DOI] [PubMed] [Google Scholar]
- Langer A, Freeman MR, Josse RG, Armstrong PW. Metaiodobenzylguanidine imaging in diabetes mellitus: assessment of cardiac sympathetic denervation and its relation to autonomic dysfunction and silent myocardial ischemia. J Am Coll Cardiol. 1995;25:610–618. doi: 10.1016/0735-1097(94)00459-4. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Mathes P, Cowan C, Gudbjarnason S. Storage and metabolism of norepinephrine after experimental myocardial infarction. Am J Physiol. 1971;220:27–32. doi: 10.1152/ajplegacy.1971.220.1.27. [DOI] [PubMed] [Google Scholar]
- Matthews MR, Nelson VH. Detachment of structurally intact nerve endings from chromatolytic neurones of rat superior cervical ganglion during the depression of synaptic transmission induced by post-ganglionic axotomy. J Physiol. 1975;245:91–135. doi: 10.1113/jphysiol.1975.sp010837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei Q, Foulis AK, Fligner C, Hull R, H N, Gilliam L, Taborsky GJ., Jr. Selective Loss of Sympathetic Nerves from the Islet in Human Type 1 Diabetes: A Potential Mechanism for Impaired Glucagon Responses to Hypoglycemia. Diabetes. 2006a;55:A15. [Google Scholar]
- Mei Q, Mundinger TO, Kung D, Baskin DG, Taborsky GJ., Jr. Fos expression in rat celiac ganglion: an index of the activation of postganglionic sympathetic nerves. Am J Physiol Endocrinol Metab. 2001;281:E655–E664. doi: 10.1152/ajpendo.2001.281.4.E655. [DOI] [PubMed] [Google Scholar]
- Mei Q, Mundinger TO, Lernmark A, Taborsky GJ., Jr. Early, Selective, and Marked Loss of Sympathetic Nerves From the Islets of BioBreeder Diabetic Rats. Diabetes. 2002;51:2997–3002. doi: 10.2337/diabetes.51.10.2997. [DOI] [PubMed] [Google Scholar]
- Mei Q, Mundinger TO, Lernmark A, Taborsky GJ., Jr. Increased galanin expression in the celiac ganglion of BB diabetic rats. Neuropeptides. 2006b;40:1–10. doi: 10.1016/j.npep.2005.08.005. [DOI] [PubMed] [Google Scholar]
- Mundinger TO, Boyle MR, Taborsky GJ., Jr. Activation of hepatic sympathetic nerves during hypoxic, hypotensive and glucopenic stress. J Auton Nerv Syst. 1997;63:153–160. doi: 10.1016/s0165-1838(97)00004-0. [DOI] [PubMed] [Google Scholar]
- Mundinger TO, Mei Q, Figlewicz DP, Lernmark A, Taborsky GJ., Jr. Impaired glucagon response to sympathetic nerve stimulation in the BB diabetic rat: effect of early sympathetic islet neuropathy. Am J Physiol Endocrinol Metab. 2003;285:E1047–E1054. doi: 10.1152/ajpendo.00136.2003. [DOI] [PubMed] [Google Scholar]
- Nja A, Purves D. The effects of nerve growth factor and its antiserum on synapses in the superior cervical ganglion of the guinea-pig. JPhysiol. 1978;277:53–75. [PMC free article] [PubMed] [Google Scholar]
- Plantinga LC, Verhaagen J, Wong SL, Edwards PM, Bar PR, Gispen WH. The neurotrophic peptide Org 2766 does not influence the expression of the immediate early gene c-fos following sciatic nerve crush in the rat. Int J Dev Neurosci. 1994;12:117–125. doi: 10.1016/0736-5748(94)90004-3. [DOI] [PubMed] [Google Scholar]
- Purves D. Functional and structural changes in mammalian sympathetic neurones following interruption of their axons. J Physiol. 1975;252:429–463. doi: 10.1113/jphysiol.1975.sp011151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purves D, Snider WD, Voyvodic JT. Trophic regulation of nerve cell morphology and innervation in the autonomic nervous system. Nature. 1988;336:123–128. doi: 10.1038/336123a0. [DOI] [PubMed] [Google Scholar]
- Quinson N, Robbins HL, Clark MJ, Furness JB. Locations and innervation of cell bodies of sympathetic neurons projecting to the gastrointestinal tract in the rat. Arch Histol Cytol. 2001;64:281–294. doi: 10.1679/aohc.64.281. [DOI] [PubMed] [Google Scholar]
- Roosen A, Schober A, Strelau J, Bottner M, Faulhaber J, Bendner G, McIlwrath SL, Seller H, Ehmke H, Lewin GR, Unsicker K. Lack of neurotrophin-4 causes selective structural and chemical deficits in sympathetic ganglia and their preganglionic innervation. J Neurosci. 2001;21:3073–3084. doi: 10.1523/JNEUROSCI.21-09-03073.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruit KG, Osborne PA, Schmidt RE, Johnson EM, Jr, Snider WD. Nerve growth factor regulates sympathetic ganglion cell morphology and survival in the adult mouse. J Neurosci. 1990;10:2412–2419. doi: 10.1523/JNEUROSCI.10-07-02412.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid H, Forman LA, Cao X, Sherman PS, Stevens MJ. Heterogeneous cardiac sympathetic denervation and decreased myocardial nerve growth factor in streptozotocin-induced diabetic rats: implications for cardiac sympathetic dysinnervation complicating diabetes. Diabetes. 1999;48:603–608. doi: 10.2337/diabetes.48.3.603. [DOI] [PubMed] [Google Scholar]
- Schmidt RE, Dorsey DA, Roth KA, Parvin CA, Hounsom L, Tomlinson DR. Effect of streptozotocin-induced diabetes on NGF, P75 (NTR) and TrkA content of prevertebral and paravertebral rat sympathetic ganglia. Brain Research. 2000;867:149–156. doi: 10.1016/s0006-8993(00)02281-2. [DOI] [PubMed] [Google Scholar]
- Schreiber RC, Shadiack AM, Bennett TA, Sedwick CE, Zigmond RE. Changes in the macrophage population of the rat superior cervical ganglion after postganglionic nerve injury. J Neurobiol. 1995;27:141–153. doi: 10.1002/neu.480270203. [DOI] [PubMed] [Google Scholar]
- Sipols AJ, Baskin DG, Schwartz MW. Effect of intracerebroventricular insulin infusion on diabetic hyperphagia and hypothalamic neuropeptide gene expression. Diabetes. 1995;44:147–151. doi: 10.2337/diab.44.2.147. [DOI] [PubMed] [Google Scholar]
- Snider WD. Nerve growth factor enhances dendritic arborization of sympathetic ganglion cells in developing mammals. J Neurosci. 1988;8:2628–2634. doi: 10.1523/JNEUROSCI.08-07-02628.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veneman T, Mitrakou A, Mokan M, Cryer P, Gerich G. Effect of hyperketonemia and hyperlacticacidemia on symptons, cognitive dysfunction and counterregulatory hormone responses during hypoglycemia in normal humans. Diabetes. 1994;43:1311–1317. doi: 10.2337/diab.43.11.1311. [DOI] [PubMed] [Google Scholar]
- Wetmore C, Olson L. Neuronal and nonneuronal expression of neurotrophins and their receptors in sensory and sympathetic ganglia suggest new intercellular trophic interactions. J Comp Neurol. 1995;353:143–159. doi: 10.1002/cne.903530113. [DOI] [PubMed] [Google Scholar]
- Wyatt S, Pinon LG, Ernfors P, Davies AM. Sympathetic neuron survival and TrkA expression in NT3-deficient mouse embryos. EMBO J. 1997;16:3115–3123. doi: 10.1093/emboj/16.11.3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Gelber S, Orr-Urtreger A, Armstrong D, Lewis RA, Ou CN, Patrick J, Role L, De Biasi M, Beaudet AL. Megacystis, mydriasis, and ion channel defect in mice lacking the alpha3 neuronal nicotinic acetylcholine receptor. Proc Natl Acad Sci U S A. 1999;96:5746–5751. doi: 10.1073/pnas.96.10.5746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yawo H. Changes in the dendritic geometry of mouse superior cervical ganglion cells following postganglionic axotomy. J Neurosci. 1987;7:3703–3711. doi: 10.1523/JNEUROSCI.07-11-03703.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh J, Ferreira M, Ebert S, Yasuda RP, Kellar KJ, Wolfe BB. Axotomy and nerve growth factor regulate levels of neuronal nicotinic acetylcholine receptor alpha3 subunit protein in the rat superior cervical ganglion. J Neurochem. 2001;79:258–265. doi: 10.1046/j.1471-4159.2001.00545.x. [DOI] [PubMed] [Google Scholar]
- Zhang SH, Rush RA. Neurotrophin 3 is increased in the spontaneously hypertensive rat. J Hypertens. 2001;19:2251–2256. doi: 10.1097/00004872-200112000-00019. [DOI] [PubMed] [Google Scholar]
- Zhou XF, Zettler C, Rush RA. An improved procedure for the immunohistochemical localization of nerve growth factor-like immunoreactivity. J Neurosci Methods. 1994;54:95–102. doi: 10.1016/0165-0270(94)90163-5. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Deneris E, Zigmond RE. Differential regulation of levels of nicotinic receptor subunit transcripts in adult sympathetic neurons after axotomy. J Neurobiol. 1998;34:164–178. doi: 10.1002/(sici)1097-4695(19980205)34:2<164::aid-neu6>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Deneris E, Zigmond RE. Nicotinic acetylcholine receptor subunit proteins alpha7 and beta4 decrease in the superior cervical ganglion after axotomy. J Neurobiol. 2001;46:178–192. doi: 10.1002/1097-4695(20010215)46:3<178::aid-neu1001>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Zigmond RE. LIF, NGF and the cell body response to axotomy. Neuroscientist. 1997;3:176–185. [Google Scholar]